

ABSTRACT
Genome‐wide association studies (GWAS) have led to the discovery of over 200 single nucleotide polymorphisms (SNPs) associated with type 2 diabetes mellitus (T2DM). Additionally, East Asians develop T2DM at a higher rate, younger age, and lower body mass index than their European ancestry counterparts. The reason behind this occurrence remains elusive. With comprehensive searches through the National Human Genome Research Institute (NHGRI) GWAS catalog literature, we compiled a database of 2,800 ancestry‐specific SNPs associated with T2DM and 70 other related traits. Manual data extraction was necessary because the GWAS catalog reports statistics such as odds ratio and P‐value, but does not consistently include ancestry information. Currently, many statistics are derived by combining initial and replication samples from study populations of mixed ancestry. Analysis of all‐inclusive data can be misleading, as not all SNPs are transferable across diverse populations. We used ancestry data to construct ancestry‐specific human phenotype networks (HPN) centered on T2DM. Quantitative and visual analysis of network models reveal the genetic disparities between ancestry groups. Of the 27 phenotypes in the East Asian HPN, six phenotypes were unique to the network, revealing the underlying ancestry‐specific nature of some SNPs associated with T2DM. We studied the relationship between T2DM and five phenotypes unique to the East Asian HPN to generate new interaction hypotheses in a clinical context. The genetic differences found in our ancestry‐specific HPNs suggest different pathways are involved in the pathogenesis of T2DM among different populations. Our study underlines the importance of ancestry in the development of T2DM and its implications in pharmocogenetics and personalized medicine.
Keywords: complex disease, East Asian populations, GWAS, human phenotype network, type 2 diabetes
Introduction
Type 2 diabetes mellitus (T2DM) is a complex disease characterized by hyperglycemia, impaired insulin secretion from pancreatic beta cells, and insulin resistance of peripheral target tissues [Ashcroft & Rorsman, 2012; Hara et al., 2014; Samuel & Shulman, 2012]. It leads to a reduced quality of life and a long list of comorbidities, including heart disease, stroke, renal disease, blindness, and amputation [Freeman & Cox, 2006]. In 2013, the International Diabetes Federation reported that 382 million people worldwide have T2DM, representing 8.3% of the total adult population [Sun et al., 2014]. The alarming prevalence of T2DM is already a major global challenge for both population health and the economic stability of healthcare systems, but it is projected to get even worse. T2DM incidence is expected to increase by 69% in developing countries and 20% in developed countries by 2030 [Shaw et al., 2010; Zhao et al., 2012].
The rapid rise of T2DM in East Asian countries in particular has been unprecedented. In China, for example, the prevalence of T2DM has surged from 1% of adults in 1980 to 11.6% in 2013 [Xu & Wang, 2013]. This dramatic rise in T2DM incidence has been mirrored in India, Japan, and many other East Asian countries over the last 20 years [Ma & Chan, 2013]. Mysteriously, the enormous increase in T2DM incidence has not been accompanied by a similar increase in body mass index (BMI) for East Asians. Although T2DM in Caucasian populations is strongly associated with obesity, East Asian populations show a weaker association between T2DM and BMI [Chan et al., 2009]. Previous studies have shown that people of East Asian ancestry have increased risk of T2DM and other cardiometabolic risk factors starting at a BMI of 23 kg/m2, well within the normal healthy range specified by the World Health Organization [Chan et al., 2009]. East Asian diabetic patients are also characterized by early beta cell dysfunction, increased insulin resistance, lower waist circumference, and increased adiposity [Ma & Chan, 2013]. These unexplained observations suggest there may be different genetic architectures behind the pathogenesis of T2DM across ancestries. This hypothesis has been the motivation for an increased effort over the last 5 years to study interethnic differences in genetic variants associated with T2DM.
It is already known that the risk of developing T2DM is determined by a strong genetic component, as well as environmental factors. A study on Finnish twins in 1992 revealed that the concordance rate of T2DM in monozygotic twins is around 70%, while the concordance rate in dizygotic twins is only 20–30% [Kaprio et al., 1992]. Additionally, significant disparities in T2DM prevalence and risk allele frequencies [Chen et al., 2012] across different ancestries suggest genetic involvement in determining disease risk [Ahlqvist et al., 2011].
Over the last decade, linkage analysis, candidate gene approach, large‐scale association studies, and genome‐wide association studies (GWAS) have been performed to identify loci that contribute to T2DM susceptibility. To date, over 65 susceptibility loci have been identified for T2DM, almost 40% of which were first identified in East Asian population studies [Hara et al., 2014; Sun et al., 2014]. Many of the genetic variants were found to be transferrable across ancestries, but many others were unable to replicate in other ancestry groups [Sim et al., 2011]. The significant number of ancestry‐specific susceptibility loci and the extreme directional differentiation of risk allele frequencies across human populations suggested that the manifestation of T2DM may have different intermediate mechanisms in different ancestry populations [Chen et al., 2012].
Despite the progress in identifying susceptibility loci, the underlying pathophysiology and causal variants of T2DM remain largely unknown [Sun et al., 2014] and a molecular explanation for the disparities in T2DM incidence and phenotypic differences in patients of different ancestries has yet to be discovered. This is in large part because many of the characteristics of complex disease—epistasis, heterogeneity, polygenicity, and pleiotropy—have obscured the true relationship between genotype and phenotype [Zhou et al., 2014]. Complex diseases such as T2DM are not the consequence of a single gene mutation, but reflect the nonlinear additive and epistatic effects of many interdependent genetic variants of modest effect [Moore, 2003].
To study the complex interaction between phenotype and genotype, we propose the use of networks, an intuitive and powerful approach built on mathematical and statistical foundations. Network models are robust tools to study the epistatic and pleiotropic effects of a number of common complex diseases [Darabos et al., 2014a; Hu et al., 2014]. These networks allow us to systematically explore and visualize the shared biology of diseases and their interactions at the gene and biological pathway level. This approach has proven successful at establishing de novo relationships between phenotypes previously thought to be unrelated [Darabos et al., 2013, 2014b,c; Goh et al., 2007]. Identifying common genetic backgrounds in seemingly unrelated diseases helps with hypothesis generation about clinically relevant biological pathways and particularly useful drug targets.
In this study, we curated data from over 1,800 GWAS, extracting ancestry information along with genetic variants strongly associated with T2DM. This allows us to construct ancestry‐specific human phenotype networks (HPN) centered on T2DM. We analyze these networks to better characterize the genetic variants, genes, and pathways involved in T2DM for both European and East Asian populations, looking for elements that transfer across ancestries as well as elements specific to ancestry groups. By doing this, we aim to gain a better understanding of genetic contributions to T2DM development and identify possible clinical implications of the networks.
Methods
In this section, we present the methods developed to obtain T2DM‐centered HPN specific to each ancestry group considered.
Data Collection and Curation
GWAS identify single nucleotide polymorphisms (SNPs) associated with phenotypical traits (physical or behavioral). We accessed the catalog of published GWAS literature from the National Human Genome Research Institute (NHGRI) and considered hundreds of T2DM‐associated studies (http://www.genome.gov/gwastudies/, March 2014). The GWAS catalog reports over 1,800 studies and 900+ phenotypes associated with 7,000+ genes and 12,000+ SNPs. For each study, it lists the key information retrieved from PubMed, including associations between SNPs, gene(s), and traits (including genetic disorders and physical and behavioral traits). We extracted ancestry‐specific data from each relevant study by surveying the full text, figures, tables, and supplementary material, recording all SNPs with a P‐value < 10−4. (Note: Because of this this study, the NHGRI GWAS catalog has been moved to the European Molecular Biology Laboratory‐European Bioinformatics Institute [EMBL‐EBI] at http://www.ebi.ac.uk/gwas, which features a new search interface and updated content.)
For each associated SNP, we recorded the risk allele frequency (RAF), odds ratio (OR), P‐value, initial study size, and ancestry of the study subjects. We manually curated a comprehensive database of ancestry‐specific data for 3,815 SNPs associated with T2DM and more than 70 other phenotypic traits. Ethnicities, such as Han Chinese, Korean, Iceland, Scandinavian, etc., were collapsed into three broad ancestry groups: European, East Asian, and African. Specific ethnic groups that did not fit into any of these three categories—such as South Asians, Pima Native Americans, and Micronesians—were excluded from the analysis. Studies that were conducted in mixed ancestry populations or unspecified populations were also excluded from the study. Because of the strong bias of GWAS performed in European and East Asian populations, there is significantly more data for these populations compared to African populations.
We recorded the P‐value and OR from the replication stages whenever possible. When there was no available replication stage data, we recorded the P‐value and OR from the initial GWAS discovery stage. When multiple GWAS replicated the same SNP in the same population, the data from the largest GWAS study was recorded and used to construct the network. Data from GWAS that did not specify the ancestry of their study subjects or used mixed ethnicities for their study were excluded from network construction.
Human Phenotype Network
HPNs [Darabos et al., 2013, 2014c] are general mathematical graph models in which the nodes represent human genetic disorders, physical traits, or behavioral traits. The edges represent shared attributes, such as shared genetic variants, genes, pathways, or protein‐protein interactions, to name a few. HPNs rely on GWAS data for genetic information on diseases, behavioral traits, and physical attributes. The underlying connections of the HPN contribute to the understanding of the basis of disorders, which in turn leads to a better understanding of human disease.
In their seminal work, Goh et al. [2007] explored the human disease network, limiting its analysis to the genes shared by different diseases. In 2009, Suthram et al. [2010] analyzed diseases by their related messenger RNA in combination with the human protein interaction network. They found significant genetic similarities between certain diseases, suggesting shared drug treatments and targets. In 2014, Zhou et al. [2014] presented yet another way of finding overlap in disease commonalities by linking disorders that share symptoms.
In the present work, we build on the phenotype‐to‐SNP HPN presented in our previous study [Darabos et al., 2014a] and construct ancestry‐specific phenotype‐to‐pathway HPNs. The most recent version of the GWAS catalog (last accessed June 5, 2014) was used as the primary phenotype‐genotype association dataset. To maximize coverage, we included unique genotype‐phenotype associations found in the database of genotypes and phenotypes (dbGaP). To map SNPs to genes, we used data from the GWAS catalog, which includes an automated pipeline from the National Center for Biotechnology Information (NCBI) that provides each SNP's mapped genes. To map genes to pathways, we used the Kyoto Encyclopedia of Genes and Genomes (KEGG), “a collection of manually curated databases dealing with genomes, biological pathways, diseases, drugs, and chemical substances” [Kanehisa & Goto, 2000] and Reactome, a “free, open‐source, curated and peer reviewed pathway database” (http://www.reactome.org/). Even though the gene‐to‐pathway mappings are not ancestry‐specific, the pathways found using ancestry‐specific genes are relevant to that particular ancestry group. Because GWAS and dbGaP phenotype labels are not standardized, we used the International Classification of Diseases ninth revision (ICD‐9) codes to manually identify redundancies in the phenotype data and merge them into single nodes. For example, reported traits such as “diabetic retinopathy in type 2 diabetes,” and “neuropathic pain in type 2 diabetes” were all grouped under the broader phenotype category of “type 2 diabetes mellitus.”
The pathway‐based HPNs are constructed with the following extraction and annotation method, illustrated in supplementary Figure S1:
extract all phenotypes from the NHGRI GWAS catalog and link them to their mapped SNPs and genes using the GWAS catalog and dbGaP. Phenotypes with no mapped SNPs or genes are omitted,
manually merge redundant phenotypes with guidance of ICD‐9 codes,
extract all genes in the database and link them to their associated pathways using KEGG and Reactome, and
connect phenotypes with overlapping pathways with an undirected edge.
From the NHGRI GWAS catalog and dbGaP, we extracted a total of 1,252 phenotypic traits, annotated with 37,681 SNPs in 16,411 loci. By merging redundant phenotype labels, we reduce the number of phenotypic traits to 986 (supplementary Table S1). With the method presented above, we constructed the “raw” unfiltered bipartite network from these traits and 1,424 associated pathways. Bipartite means that the network is built from two distinct sets of vertices: phenotypes and pathways (Figs. 1 and 2). The bipartite network is projected in the space of phenotype vertices to obtain the HPN. Supplementary Figure S2 depicts an example of the mechanism used to identify pathway overlap between phenotypes. In the resulting HPN, the nodes represent phenotypes and the connecting edges represent shared biological pathways based on the SNPs associated with each phenotype. We associate a strength, or weight, to each edge by computing the Jaccard similarity coefficient. The Jaccard index is defined as “the size of the intersection divided by the size of the union of the sample sets” [Anderberg, 2014].
where A and B are the sets of biological pathways associated with the traits at the endpoints of the considered edge. Isolated phenotypes are removed, as our focus remains on the connection between phenotypes. By building these associations, we are able to link phenotypes by shared biological pathways based on genetics. The result of the projection is depicted in Figure 3.
Figure 1.
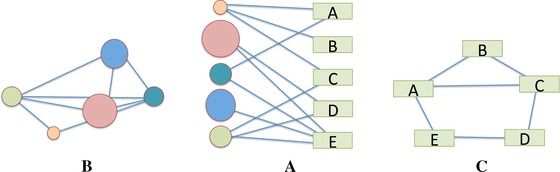
A schematic representation of a bipartite network (middle) and its projections in each vertex space. In our example, the circles are phenotypes and the rectangles are pathways, or vice versa.
Figure 2.
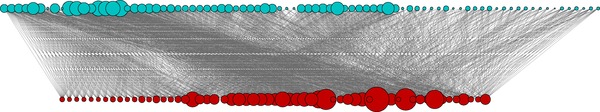
An example of a subset of the phenotype‐SNP bipartite network, filtered to increase readability (edge weight cutoff = 0.01). Phenotypes are denoted with blue and pathways are denoted with red. Vertex sizes are proportional to the number of associated biological pathways.
Figure 3.
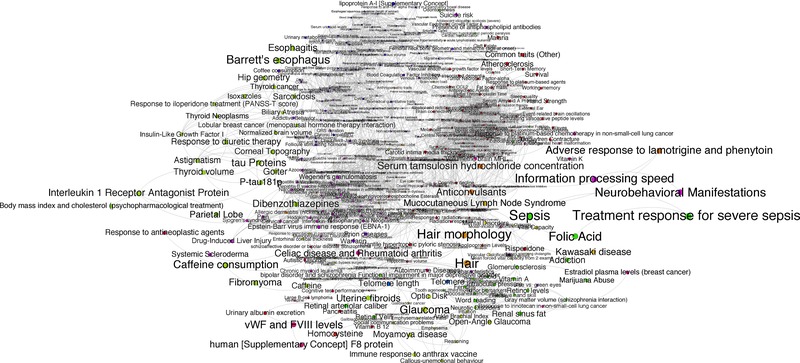
The projected HPN filtered (edge weight cutoff = 0.01). Vertices are colored by “‘modules”’ [Darabos et al., 2014c] to increase readability and are proportional to the number of associated biological pathways.
The HPN encompasses all phenotypes listed in the GWAS catalog and dbGaP, provided that they are connected to at least one other trait. The unfiltered HPN contains 985 phenotypic traits and over 26,000 edges, with an average connectivity of 500+. It is clearly a very dense network, even after filtering, and suffers from the “hairball” effect. Thus, we use a simple global edge‐weight minimum threshold approach to filter out the weakest links within the HPN. This method is self‐explanatory: all edges with a Jaccard similarity coefficient below a predefined threshold are removed. Beyond the scope of this work, it is worth mentioning smart, topology‐based filtering methods such as Longbaugh's [2012] combing method or Serrano et al.’s [2009] multiscale backbone. Biological networks are generally expected to have heterogeneous connectivity with a “heavy‐tailed” degree distribution, placing them in the scale‐free family. This means that the degree distribution follows a power law, or exponential decay. Within the network, this translates into the presence of “hubs”—a minority of highly connected nodes. When the degree distribution of a scale‐ free network is plotted on a logarithmic scale, the resulting curve is approximately linear across the top [Newman, 2010]. According to the degree distribution presented in Figures 4 and 5, on a linear and logarithmic scale, respectively, the filtered HPN presented in this work is no exception.
Figure 4.
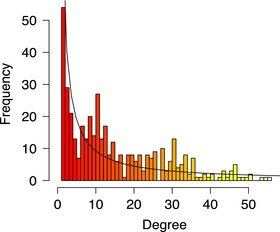
The degree distribution postfiltering on a linear scale.
Figure 5.
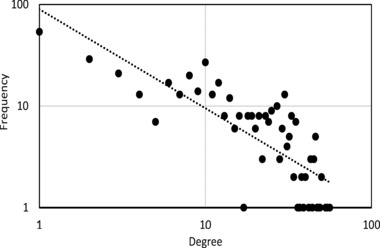
The degree distribution postfiltering on a logarithmic scale.
T2DM‐Centric HPN
Although previous studies on HPNs, diseasomes, and interactomes were thorough, none included the ancestry‐specific aspects of genetic diseases. It is clear, however, that genetic disorder susceptibility is often closely related to ancestry [Marigorta & Navarro, 2013], for instance in the case of Parkinson's disease [Heckman et al., 2013] or Crohn's disease [Nakagome et al., 2010]. In the present work, we focus on T2DM, comparing the subnetworks of different populations.
The data collection and curation stage described earlier enables us to determine SNPs associated with T2DM for each major ancestry group. We use this information to build individual T2DM‐centered HPN subnetworks, using the same method described in the previous section. For each ancestry population, we retain only the phenotypes that share at least one SNP associated with T2DM for that population. When constructing the subnetwork, we also preserve the immediate neighbors of each trait in order to build simple communities of phenotypes that are related through shared biological pathways and that have been associated with the selected SNPs.
We first built a global T2DM‐centered HPN that was not ancestry‐specific and encompassed all available GWAS data. The global T2DM‐HPN generated from combined ancestry data may result in misleading conclusions about the pathogenesis of T2DM, especially if the intermediary steps vary for different populations. Thus, we used our ancestry‐specific data to generate T2DM‐centered HPNs specific to European, East Asian, and African populations. The four networks are depicted in Figure 6A–D.
Figure 6.
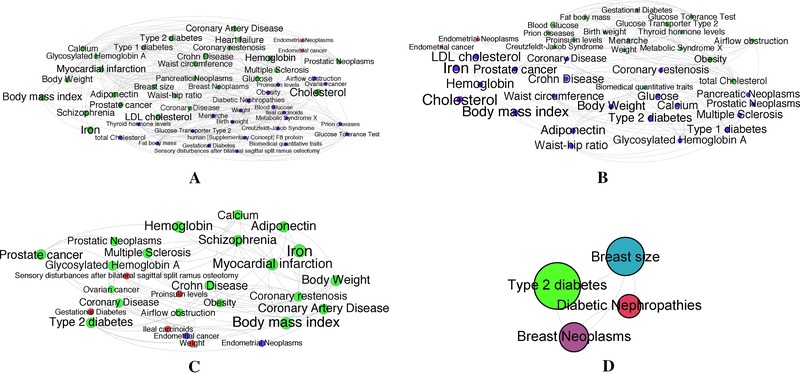
T2DM‐centric ancestry‐specific HPN subnetworks. (A) The global T2DM‐HPN encompassing all ancestry backgrounds. (B) The European ancestry T2DM‐HPN. (C) The East Asian ancestry T2DM‐HPN. (D) The African ancestry T2DM‐HPN. All networks have been filtered using a minimal edge‐weight threshold (cutoff = 5 × 10−4) to increase the readability.
Results
In this section, we first present the results of the literature survey to identify genetic variations associated with T2DM in different populations. We then analyze the resulting population‐specific T2DM‐specific HPNs. Finally, we discuss the clinical and biomedical implications of our findings.
Variations Associated With T2DM
Using the results of our methodical survey of GWAS literature detailed in the Methods section, we categorized the SNPs associated with T2DM into three major ancestry groups: Europeans, East Asians, and Africans. The number of T2D‐associated SNPs and loci we identified for each ancestry are reported in Table 1. We report the number of unique SNPs for each ancestry in parenthesis. The complete list of the unique SNPs for each population can be found in supplementary Table S3.
Table 1.
The number of SNPs and loci associated with T2D in each specific ancestry
| EU | EA | AF | |
|---|---|---|---|
| SNPs | 142 (106) | 147 (99) | 14 (10) |
| Loci | 95 | 115 | 14 |
In parenthesis, we report the number of SNPs unique to that ancestry (i.e., SNPs that cannot be found in other ancestry populations). The detailed breakdown of the unique SNPs for each ancestry is listed in Table S3.
The number of SNPs and loci associated with T2DM in populations of European and East Asian descent are comparable at ∼140 SNPs and ∼100 loci. About half of the SNPs reported to be associated with T2DM in the GWAS catalog were not used to generate our networks because their data came from studies that did not take into account the ancestry background of their subjects. Table 1 also reveals that the number of genetic variations and loci for African populations is approximately 10% that of either Europeans or East Asians. This major discrepancy in available data is due to the fewer number of GWAS conducted in African populations.
Ancestry‐Specific HPNs of T2DM
The networks resulting from the ancestry‐specific SNP filtering of the vertices of the general HPN are extremely densely connected (95+%). The networks presented in Figure 6 have been edge‐filtered according to the global edge‐weight threshold method introduced in the Methods section. From a visual inspection, we note that the expected phenotypes stand out in both Asian and the European networks (Fig. 6B and C): BMI, body weight, cholesterol, etc. Because of the lack of available African population data and the resulting sparseness of the African HPN, we have focused our analysis and discussion on the East Asian and European networks. A summary of the global statistical properties pre‐ and postfiltering of all four networks is presented in Table 2.
Table 2.
Global statistical properties of the T2DM‐specific HPNs
| EU | EA | AF | All populations | |
|---|---|---|---|---|
| Nodes/traits (N) | 40 | 27 | 4 | 51 |
| Edges (E) | 767/189 | 340/74 | 6 | 1250/232 |
| Average degree (K) | 38.35/9.45 | 25.185/5.481 | 3 | 49.02/12.67 |
| Average weighted degree (W) | 0.034/0.036 | 0.033/0.028 | 0.002 | 0.034/0.025 |
| Density (D) | 0.983/0.242 | 0.969/0.211 | 1 | 0.098/0.98 |
| Average clustering coefficient (CC) | 0.985/0.658 | 0.974/0.518 | 1 | 0.983/0.644 |
| Average path length (APL) | 1.017/2.597 | 1.031/2.741 | 1 | 1.02/2.416 |
Nodes and traits N is the count of vertices in the network. Edges E is the number of edges. The average K degree is the average number of edges connected to each node averaged over the entire network, K = 2E/N. The average weighted W degree is the sum of the weights associated to all the edges impinging each node averaged over the entire network. The density of the network D is the fraction of existing edges over all possible edges in the network (complete network). The average clustering coefficient CC is the probability that two neighboring nodes of any given vertex are also neighbors of each other. Vertices are called neighbors when an edge connects them. The average path length APL is the average minimal number of edges separating all pairs of vertices. Newman's [2010] textbook on networks contains a more complete mathematical definition of these properties. The values pre‐ and postfiltering are separated by a “/.”
The elevated (>0.5) average clustering coefficients and the short path lengths of all networks after filtering place these networks in the “nonrandom” category. Considering the network pair we are most interested in, we compare the East Asian subnetwork and the European subnetwork. Table 3 recapitulates the total number of nodes and edges, the number of overlapping nodes and edges, and the number of nodes and edges that are unique to each network. The actual unique and overlapping phenotypes between East Asian and European ancestry populations are presented in supplementary Table S2.
Table 3.
The unique and overlapping phenotype nodes and edges from the comparison of the East Asian ancestry and European ancestry HPNs
| East Asians | Europeans | |||
|---|---|---|---|---|
| Nodes | Edges | Nodes | Edges | |
| Unique to network | 6 (22%) | 135 (40%) | 19 (48%) | 562 (74%) |
| Overlapping | 21 (78%) | 205 (60%) | 21 (52%) | 205 (26%) |
| Total | 27 | 340 | 40 | 767 |
“Overlapping” refers to nodes or edges that are found in both networks. Our study focuses on the six phenotype nodes unique to the East Asian HPN. The corresponding phenotypes are displayed in Table S2. The African HPN was not compared to other networks because it is too small to depict any meaningful relationships.
From Table 3, we conclude that there is a significant overlap between the East Asian and European T2DM HPNs. However, each has an important number of unique vertices and edges that are worth exploring in more detail. The next section focuses on the clinical implications of unique edges within the East Asian network and how it can help us formulate novel hypotheses about phenotypic interactions based on biological pathways that are specific to each ancestry group.
Clinical and Biomedical Implications
We used 3,815 manually curated SNPs associated with over 70 phenotypes to construct ancestry‐specific subnetworks around T2DM. In comparing networks, we identified phenotypes that were linked to T2DM in the East Asian network, but not in the European network. GWAS performed in populations of European ancestry are significantly more abundant, as well as larger and more statistically powerful than GWAS done in East Asian populations. Because of this, many more SNPs, genes, and pathways are associated with T2DM in the European HPN compared to the East Asian HPN. Therefore, a link that appears on the European HPN that is not found on the East Asian HPN could be a result of publication bias rather than an actual difference in the network architecture. In contrast, a link found on the East Asian network likely denotes SNPs or loci that were unable to be replicated in European populations, despite larger study sizes. Out of the 27 phenotypes in the East Asian pathway network, 21 transferred across ancestries to appear in the European HPN (Table 3; supplementary Table S2). The other six phenotypes were myocardial infarction, ovarian cancer, ileal carcinoids, schizophrenia, metabolites, and glycemic traits. These phenotypes are unique to the East Asian HPN.
Myocardial Infarction
One edge found on the East Asian HPN that was not seen in the European network is a connection between T2DM and myocardial infarction (MI). Coronary artery disease (CAD) and T2DM have long been recognized as comorbidities. More than half of T2DM patients have signs of cardiovascular disease complications at diagnosis (http://www.diabetes.co.uk/diabetes‐complications/heart‐disease.html, July 2014) and patients with T2DM are at least two times more susceptible to myocardial infarction [Scherrer et al., 2011]. Yet, despite a clear association between these two complex diseases, the genetic causes of the link have been uncertain. Several GWAS have identified genetic variants in the chromosome locus 9p21 that contribute to the risk of both CAD and T2DM [Cheng et al., 2011; Helgadottir et al., 2008]. The majority of these studies, however, were conducted in European populations, and none were able to identify a common SNP that associated with both diseases. In other words, SNPs associated with T2DM did not associate with any arterial disease and vice versa [Helgadottir et al., 2008].
No risk variant was established to be associated with both CAD and T2DM until a 2011 study found SNPs rs10811661 and rs10757283 in the Chinese Han population [Cheng et al., 2011]. It is the only known study to report that the same SNPs confer to both T2DM and CAD, providing evidence that the genetics of T2DM and CAD may be varied between East Asian and European populations.
This postulate is further supported by a study that has associated the ALMS1 gene to early‐onset myocardial infarction in East Asian populations. ALMS1 is best known for its connection to Alstrom syndrome, a disease characterized by obesity, insulin resistance, cardiomyopathy, and T2DM [Ichihara et al., 2013]. The link between T2DM and MI in only the East Asian HPN and the observed incidence discrepancies between ancestries suggest that more population‐specific GWAS studying MI in the future would be valuable.
Ovarian Cancer
East Asian populations tend to have a significantly lower incidence rate than Europeans in most cancers, including ovarian cancer. The ovarian cancer incidence rate in females of European ancestry ranges from 17.4 to 18.1 per 100,000, while the incidence rate for East Asian females is much lower at 9.2 to 15.5 per 100,000 [Forman, 2009]. This disparity could be a cause of different environmental conditions, but it also suggests a heterogeneous genetic structure behind ovarian cancer across populations.
Indeed, HNF1B (hepatocyte nuclear factor 1 homeobox B), a gene encoded by TCF2 (transcription factor 2), is an East Asian specific link between ovarian cancer and T2DM. HNF1B is highly associated with maturity onset diabetes of the young (MODY), a rare form of diabetes that is characterized by beta‐cell dysfunction and insulin resistance [Bellanne‐Chantelot et al., 2005; Gardner & Tai, 2012]. This link provides a possible explanation for the significant disparities in incidence rate between ethnicities in ovarian cancer.
Clinical studies over the last few years have produced mounting epidemiological evidence that metformin, the most widely prescribed antidiabetic drug in the world, lowers the incidence rate and improves the survival rate of ovarian cancer [Kumar et al., 2013]. Libby et al. [2009] observed in a large observational cohort study of 8,000 T2DM patients that ovarian cancer was diagnosed among 7.3% of metformin users compared to 11.6% of nonmetformin users. A case‐study done by Kumar et al. [2013] showed that the 5‐year survival rate for women with ovarian cancer who had received metformin had a 67% survival rate compared to a 47% survival rate in women with ovarian cancer who had not received metformin treatment.
Metformin was originally used to treat T2DM because it increases insulin sensitivity and inhibits gluconeogenesis in the liver. Its function in lowering blood glucose levels leads to a decrease in insulin level, an effect that may indirectly inhibit cell growth in ovarian cancer because insulin is a growth‐promoting hormone [Ben Sahra et al., 2010]. Metformin also controls cell proliferation by activating AMPK, a kinase that negatively regulates the mTOR pathway that controls cell growth and proliferation [Ben Sahra et al., 2010; Rattan et al., 2011].
The success of metformin provides evidence that ovarian cancer development is significantly correlated with insulin level. Similar to metformin, HNF1B's function in impairing beta‐cell function lowers the amount of insulin in the body. The link in the East Asian HPN indicates that HNF1B may play a role in the decreased incidence rate of ovarian cancer in East Asians compared to Europeans. The ability of the HPN to detect this link shows its potential clinical value in identifying novel therapies and drug targets in genes and pathways. It provides us with more information about the pathophysiology behind both diseases, as well as clues about the mechanism of the drug.
Generating New Hypothesis
Ileal Carcinoids
So far, there has been no publication documenting a link between T2DM and ileal carcinoids. Because of the link between T2DM and ileal carcinoids in the East Asian network, however, a plausible hypothesis is that ileal carcinoids, like ovarian cancer, are particularly sensitive to insulin levels and AMPK activation. In other words, one could hypothesize that metformin treatments benefit patients with ileal carcinoids based on their shared genetic origins.
Schizophrenia
Another undocumented interaction found specifically in the East Asian network is the three‐way link between schizophrenia, ovarian cancer, and T2DM. Higher rates of schizophrenia are observed in East Asian populations, but the reasons are currently unknown. It has also been observed that patients with schizophrenia have an increased incidence of T2DM. Recent studies found that the co‐occurrence of T2DM and schizophrenia in Han Chinese patients might be partly explained by shared genetic variants, notably in the IGF2BP2 gene [Ripke et al., 2013; Suthram et al., 2010; Yuan et al., 2013]. This gene is responsible for an insulin‐like growth factor that stimulates beta‐cell proliferation and is widely acknowledged to be a susceptibility locus for T2DM. The appearance of this link on our East Asian HPN suggests that the genetic basis for the co‐occurrence of T2DM and schizophrenia in East Asian populations is worth studying more extensively.
Metabolite Levels
Metabolites are the intermediates and products of metabolic processes and include small molecules such as glucose, amino acids, insulin, and uric acid. They play a key role in many biological pathways but their genetic basis and physiological impact is mostly unknown [Kettunen et al., 2012]. The relationship between T2DM and metabolite levels has only recently been elucidated in a 2011 study, in which it was suggested that elevated levels of a panel of amino acids was both a marker and effector for insulin resistance as well as impaired insulin secretion [Wang et al., 2011]. A genetic link between metabolite levels and T2DM was unique to our East Asian HPN, suggesting that metabolite levels play a larger predictor role of T2DM in East Asian populations than in European populations.
Indeed, a recent discovery of a novel locus associated with T2DM supports this population‐specific link. The locus SLC16A11, a gene that codes for proteins that transport metabolites, was first associated with T2DM in Mexicans and Native Americans [Consortium TST 2 D, 2014]. The higher risk version of the gene has since been found to increase the likelihood of developing T2DM by 25%. Its presence in about 50% of Native Americans may be a large accounting factor for the extremely high prevalence of T2DM in Native populations [Consortium, 2014]. The gene is also found in about 20% of East Asians, but is very rare in European and African populations.
The ability of the East Asian HPN to detect this link suggests that there is indeed a genetic basis for the relationship between raised metabolite levels and the development of T2DM. Further research in this relationship may reveal more clues about the pathogenesis of T2DM.
Discussion and Conclusions
In this study, we constructed ancestry‐specific human phenotype subnetworks based on NHGRI GWAS data to compare and contrast the underlying genetic architecture of T2DM for European and East Asian populations. Most genetic variants initially discovered in Europeans have been confirmed by replication studies conducted in East Asian populations, although many of these genetic variants show significant differences in RAF and P values. This is most evident for rs7903146 in locus TCF7L2, a transcription factor involved in insulin secretion from pancreatic β‐cells and the strongest risk allele identified so far in Europeans. Although the P‐value for European ancestry populations is 2.0 × 10−51, the P‐value for East Asian ancestry populations is barely significant at 2.5 × 10−2 [Voight et al., 2010].
Conversely, rs2237892 in locus KCNQ1 was first identified in an East Asian GWAS and has a much more significant P‐value (2.5 × 10−40) in East Asian populations than it does in European ancestry populations (P‐value = 7.2 × 10−04) [Unoki et al., 2008]. The KCNQ1 gene encodes the pore‐forming subunit of a voltage‐gated potassium channel, which is a critical function for insulin‐secreting INS‐1 cells. The discovery of population‐specific genes such as UBE2E2 in East Asians brought about suggestions that different pathways may be involved in the pathogenesis of T2DM [Cho et al., 2012; Yamauchi et al., 2010].
The ancestry‐specific HPN allowed us to confirm the sharing of T2DM genetic variants across populations [Sim et al., 2011]. Perhaps more importantly, however, the networks were also able to visualize individual genes that are specific to different ancestries, adding to hypotheses that T2DM disease risk and pathophysiology may vary [Cho et al., 2012]. It is clear from the networks that populations of European ancestry make up only a subset of genetic variation and are thus insufficient in fully characterizing T2DM. Network visualization allowed us to identify comorbidities that may be genetically linked and generate hypotheses for underlying genes involved in both phenotypes. The networks also provided a genetic explanation for the observed comorbidity between T2DM and myocardial infarction, as well as the genetic basis for the usage of metformin for the treatment of ovarian cancer. It highlighted the role of insulin‐sensitivity in East Asian populations in T2DM pathogenesis as compared to Europeans, suggesting that targeting insulin resistance should be more heavily emphasized in East Asians.
Our networks are currently limited by lack of data. Despite breakthrough advances in GWAS technology in the last few years, the susceptibility genes that have been identified so far can only account for 10–15% of T2DM heritability [Bonnefond et al., 2010]. To date, most of the published GWAS have been performed in populations of European or East Asian ancestry. Chen et al. [2012] found that T2DM SNPs showed extreme differentiation of risk allele frequencies across human populations and our manual curation confirmed that many T2DM risk alleles showed significantly contrasting RAFs. The variance in RAF values demonstrates the importance of conducting genetic studies across different ancestry populations in the search for novel T2DM‐associated SNPs. This concept was successfully applied recently when variants in SLC16A11 were reported to increase risk by up to 50% in Mexican and other Latin American populations [Consortium, 2014]. As the field of translational genomics shifts toward next‐generation sequencing technologies, the possibilities of filling in the missing heritability gap and constructing a more complete network look promising [Qin et al., 2012]. In particular, the growing search for rare variants will be valuable to the ancestry‐specific HPN because rare variants are most likely to be unique to specific populations [Sim et al., 2011].
Interestingly, the genetic variants with large differences in allele frequencies had similar effect sizes between East Asian and European populations, suggesting that the biological consequences of these variants are similar across ancestry groups. A link that appears specifically in the East Asian network may not be a result of different biology behind the disease, but rather a result of a low RAF in European populations that left a genetic variant undetectable. Thus, these links may actually be transferrable across populations.
Another factor that may have influenced our network model is ancestry‐specific linkage disequilibrium (LD), which differs based on ancestry. Studies have shown, however, that significant LD differences across ancestries are not very common [Teo et al., 2009]. This is true especially between European and East Asian ancestry populations, which are known to have high haplotype sharing rates [Conrad et al., 2006]. Because of the small proportion of SNPs with significant LD differences between European and East Asian ancestries, ancestry‐specific linkage disequilibrium should have minimal effects on our network models, though we can make no guarantees. Additionally, because we map SNPs to biological pathways, the effect of one high LD block on our network model is greatly reduced. In subsequent studies, attempts should be made to quantify the impact of ancestry‐specific linkage disequilibrium.
Although ancestry and race categorizations have long been fiercely debated within scientific communities, the existing literature suggests that human genetic variation tends to be geographically structured. Because of humankind's extensive history of migration and gene flow, classification based on race includes individuals that are not genetically pure, resulting in boundaries that are somewhat inaccurate and arbitrary. Analysis of genetic variation has shown, however, that classification based on ancestry reliably observes three distinct clusters: African, Europeans, and East Asian populations [Jorde & Wooding, 2004]. We generated three population‐specific HPNs with African, European, and East Asian ancestry data for this reason. Our ancestry‐specific networks show that, similar to age and sex information, ancestry information may prove useful in biomedical contexts, with possible implications for pharmocogenetics and personalized medicine.
The clinical implications of heterogeneous underlying pathways are vast. Possible differences in metabolism and transport would greatly influence pharmaceutical targets, drug exposure times, and dosage [Man et al., 2010]. The network view of T2DM recognizes the complexities of the disease. Studying individual risk variants or genes is not enough to develop a full understanding of any complex disease because of the vast array of interactions at the molecular level. An integrated approach will help us unravel the intricate relationships involved in T2DM and ultimately find drug therapy cocktails that involve multiple targets.
One of the current limitations of our ancestry‐specific HPNs is there are multiple subpopulations such as Latin Americans/Mexicans, Pima Native Americans, or South Asians that do not fall into any of the three broad categories we used for our networks. At this time, we are not able to construct robust HPNs for these populations because there is simply not enough data available. Both Latin American and South Asian subpopulations have unusually high incidence rates of T2DM and will likely be the subjects of many future GWA studies. When enough data are gathered, ancestry‐specific HPNs could expand beyond the three populations chosen in this study. In future studies, we plan on implementing more sophisticated methods of filtering the network in order to improve the signal‐to‐noise ratio. Additionally, we are working on integrating environmental exposure data into the HPN and its subnetworks. Although high‐quality environmental exposure data are challenging to find, the potential of such integrated models is immense and would greatly contribute to the study of complex disease.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1
Figure S2
Table S1: The phenotypes we extracted from the NHGRI GWAS catalog and dbGaP are presented below. After merging redundant phenotypic labels, we have a total of 986 phenotypes considered for network construction. These phenotypes and their associated pathways are used to build the bipartite non‐ancestry‐specific (global) HPN.
Acknowledgments
This work was supported by NIH grants R01 EY022300, LM009012, LM010098, AI59694, GM103506, and GM103534 and by a PA CURE grant from the State of Pennsylvania. The authors greatly appreciate the help of Maria Cricco in producing the network figures and computing their statistical properties.
References
- Ahlqvist E, Ahluwalia TS, Groop L. 2011. Genetics of type 2 diabetes. Clin Chem 57(2):241–254. http://doi.org/10.1373/clinchem.2010.157016 [DOI] [PubMed] [Google Scholar]
- Anderberg MR. 2014. Cluster analysis for applications: probability and mathematical statistics: a series of monographs and textbooks. Vol. 19. Academic Press. [Google Scholar]
- Ashcroft FM, Rorsman P. 2012. Diabetes mellitus and the β cell: the last ten years. Cell 148(6):1160–1171. http://doi.org/http://dx.doi.org/10.1016/j.cell.2012.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanne‐Chantelot C, Clauin S, Chauveau D, Collin P, Daumont M, Douillard C, Dubois‐Laforgue D, Dusselier L, Gautier J, Jadoul M, Laloi‐Michelin M, and others. 2005. Large genomic rearrangements in the hepatocyte nuclear factor‐1beta (TCF2) gene are the most frequent cause of maturity‐onset diabetes of the young type 5. Diabetes 54(11):3126–3132. [DOI] [PubMed] [Google Scholar]
- Ben Sahra I, Le Marchand‐Brustel Y, Tanti J‐F, Bost F. 2010. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther 9(5):1092–1099. [DOI] [PubMed] [Google Scholar]
- Bonnefond A, Froguel P, Vaxillaire M. 2010. The emerging genetics of type 2 diabetes. Trends Mol Med 16(9):407–416. http://linkinghub.elsevier.com/retrieve/pii/S1471491410000973 [DOI] [PubMed] [Google Scholar]
- Chan JCN, Malik V, Jia W, Kadowaki T, Yajnik CS, Yoon K‐H, Hu FB. 2009. Diabetes in Asia: epidemiology, risk factors, and pathophysiology. J Am Med Assoc 301(20):2129–2140. http://doi.org/10.1001/jama.2009.726 [DOI] [PubMed] [Google Scholar]
- Chen R, Corona E, Sikora M, Dudley JT, Morgan AA, Moreno‐Estrada A, … Butte AJ. 2012. Type 2 diabetes risk alleles demonstrate extreme directional differentiation among human populations, compared to other diseases. PLoS Genet 8(4):e1002621 http://doi.org/10.1371/journal.pgen.1002621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Shi L, Nie S, Wang F, Li X, Xu C, Wang P, Yang B, Li Q, Pan Z, and others. 2011. The same chromosome 9p21.3 locus is associated with type 2 diabetes and coronary artery disease in a Chinese Han population. Diabetes 60(2):680–684. http://doi.org/10.2337/db10‐0185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Chen C‐H, Hu C, Long J, Hee Ong RT, Sim X, Seielstad M. 2012. Meta‐analysis of genome‐wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 44(1):67–72. http://dx.doi.org/10.1038/ng.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Jakobsson M, Coop G, Wen X, Wall JD, Rosenberg N a, Pritchard JK. 2006. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet 38(11):1251–1260. http://doi.org/10.1038/ng1911 [DOI] [PubMed] [Google Scholar]
- Consortium TST 2 D . 2014. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506(7486):97–101. http://dx.doi.org/10.1038/nature12828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabos C, Desai K, Cowper‐Sallari R, Giacobini M, Graham BE, Lupien M, Moore JH. 2013. Inferring human phenotype networks from genome‐wide genetic associations. Lect Notes Comput Sci 7833:23–34. [Google Scholar]
- Darabos C, Harmon SH, Moore JH. 2014a. Using the bipartite human phenotype network to reveal pleiotropy and epistasis beyond the gene. Pac Symp Biocomput:188–199. http://www.ncbi.nlm.nih.gov/pubmed/24297546 [PMC free article] [PubMed] [Google Scholar]
- Darabos C, Leung D, Moore JH. 2014b. Inferring human phenotype networks from pathway‐based International Genetic Epidemiology Society Vienna, Austria. http://www.geneticepi.org/iges‐2014/
- Darabos C, White MJ, Graham BE, Leung DN, Williams SM, Moore JH. 2014c. The multiscale backbone of the human phenotype network based on biological pathways. BioData Mining 7(1):1 http://doi.org/10.1186/1756‐0381‐7‐1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman D. 2009. Cancer Incidence and Survival By Major Ethnic Group, England, 2002–2006 National Cancer Intelligence Network. [Google Scholar]
- Freeman H, Cox RD. 2006. Type‐2 diabetes: a cocktail of genetic discovery. Hum Mol Genet 15(Suppl 2):R202–R209. http://doi.org/10.1093/hmg/ddl191 [DOI] [PubMed] [Google Scholar]
- Gardner DS, Tai ES. 2012. Clinical features and treatment of maturity onset diabetes of the young (MODY). Diabetes Metab Syndr Obes 5:101–108. http://doi.org/10.2147/DMSO.S23353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh K‐I, Cusick ME, Valle D, Childs B, Vidal M, Barabasi A‐L. 2007. The human disease network. Proc Natl Acad Sci 104(21):8685–8690. http://doi.org/10.1073/pnas.0701361104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Fujita H, Johnson TA, Yamauchi T, Yasuda K, Horikoshi M, Kadowaki T. 2014. Genome‐wide association study identifies three novel loci for type 2 diabetes. Hum Mol Genet 23(1):239–246. http://doi.org/10.1093/hmg/ddt399 [DOI] [PubMed] [Google Scholar]
- Heckman MG, Soto‐Ortolaza AI, Aasly JO, Abahuni N, Annesi G, Bacon JA, Ross OA. 2013. Population‐specific frequencies for LRRK2 susceptibility variants in the genetic epidemiology of Parkinson's disease (GEO‐PD) consortium. Mov Disord 28(12):1740–1744. http://doi.org/10.1002/mds.25600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, Stefansson K. 2008. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet 40(2):217–224. http://dx.doi.org/10.1038/ng.72 [DOI] [PubMed] [Google Scholar]
- Hu T, Pan Q, Andrew AS, Langer JM, Cole MD, Tomlinson CR, Karagas MR, Moore JH. 2014. Functional genomics annotation of a statistical epistasis network associated with bladder cancer susceptibility. BioData Min 7(1):5 http://doi.org/10.1186/1756‐0381‐7‐5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihara S, Yamamoto K, Asano H, Nakatochi M, Sukegawa M, Ichihara G, Yokota M. 2013. Identification of a glutamic acid repeat polymorphism of ALMS1 as a novel genetic risk marker for early‐onset myocardial infarction by genome‐wide linkage analysis. Circ Cardiovasc Genet 6(6):569–578. http://doi.org/10.1161/CIRCGENETICS.111.000027 [DOI] [PubMed] [Google Scholar]
- Jorde LB, Wooding SP. 2004. Genetic variation, classification and “race.” Nat Genet 36:S28–S33. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucl Acids Res 28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaprio J, Tuomilehto J, Koskenvuo M, Romanov K, Reunanen A, Eriksson J, Kesäniemi YA. 1992. Concordance for type 1 (insulin‐dependent) and type 2 (non‐insulin‐dependent) diabetes mellitus in a population‐based cohort of twins in Finland. Diabetologia 35(11):1060–1067. http://doi.org/10.1007/BF02221682 [DOI] [PubMed] [Google Scholar]
- Kettunen J, Tukiainen T, Sarin A‐P, Ortega‐Alonso A, Tikkanen E, Lyytikainen L‐P, Ripatti S. 2012. Genome‐wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet 44(3):269–276. http://dx.doi.org/10.1038/ng.1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Meuter A, Thapa P, Langstraat C, Giri S, Chien J, Rattan R, Cliby W, Shridhar V. 2013. Metformin intake is associated with better survival in ovarian cancer. Cancer 119(3):555–562. http://doi.org/10.1002/cncr.27706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JMM. 2009. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 32(9):1620–1625. http://doi.org/10.2337/dc08‐2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longabaugh WJR. 2012. Combing the hairball with BioFabric: a new approach for visualization of large networks. BMC Bioinform 13:275 http://doi.org/10.1186/1471‐2105‐13‐275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma RCW, Chan JCN. 2013. Type 2 diabetes in East Asians: similarities and differences with populations in Europe and the United States. Ann N Y Acad Sci 1281(1):64–91. http://doi.org/10.1111/nyas.12098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man M, Farmen M, Dumaual C, Teng CH, Moser B, Irie S, Hockett R. 2010. Genetic variation in metabolizing enzyme and transporter genes: comprehensive assessment in 3 major East Asian subpopulations with comparison to Caucasians and Africans. J Clin Pharmacol 50(8):929–940. http://doi.org/10.1177/0091270009355161 [DOI] [PubMed] [Google Scholar]
- Marigorta UM, Navarro A. 2013. High trans‐ethnic replicability of GWAS results implies common causal variants. PLoS Genet 9(6):e1003566 http://doi.org/10.1371/journal.pgen.1003566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JH. 2003. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered 56(1‐3):73–82. http://doi.org/73735 [DOI] [PubMed] [Google Scholar]
- Nakagome S, Takeyama Y, Mano S, Sakisaka S, Matsui T, Kawamura S, Oota H. 2010. Population‐specific susceptibility to Crohn's disease and ulcerative colitis; dominant and recessive relative risks in the Japanese population. Ann Hum Genet 74(2):126–136. http://doi.org/10.1111/j.1469‐1809.2010.00567.x [DOI] [PubMed] [Google Scholar]
- Newman M. 2010. Networks: An Introduction. New York, NY: Oxford University Press, Inc. [Google Scholar]
- Qin H‐D, Scott A, Wang HZ, Shugart YY. 2012. From GWAS to next‐generation sequencing on human complex diseases: the implications for translational medicine and therapeutics In: Shugart Y. Y., editor. Applied Computational Genomics. Netherlands: Springer, pp. 157–179. [Google Scholar]
- Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V. 2011. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 13(5):483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, Sullivan PF. 2013. Genome‐wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 45(10):1150–1159. http://dx.doi.org/10.1038/ng.2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VT Samuel, Shulman GI. 2012. Mechanisms for insulin resistance: common threads and missing links. Cell 148(5):852–871. http://doi.org/10.1016/j.cell.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer JF, Garfield LD, Chrusciel T, Hauptman PJ, Carney RM, Freedland KE, Lustman PJ. 2011. Increased risk of myocardial infarction in depressed patients with type 2 diabetes. Diabetes Care 34(8):1729–1734. http://doi.org/10.2337/dc11‐0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano MÁ, Boguñá M, Vespignani A. 2009. Extracting the multiscale backbone of complex weighted networks. Proc Natl Acad Sci 106(16):6483–6488. http://doi.org/10.1073/pnas.0808904106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw JE, Sicree RA, Zimmet PZ. 2010. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 87(1):4–14. http://linkinghub.elsevier.com/retrieve/pii/S016882270900432X?showall=true [DOI] [PubMed] [Google Scholar]
- Sim X, Ong RT‐H, Suo C, Tay W‐T, Liu J, Ng DP‐K, … Tai E‐S. 2011. Transferability of type 2 diabetes implicated loci in multi‐ethnic cohorts from southeast asia. PLoS Genet 7(4):e1001363 http://doi.org/10.1371/journal.pgen.1001363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Yu W, Hu C. 2014. Genetics of type 2 diabetes: insights into the pathogenesis and its clinical application. BioMed Res Int 14:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthram S, Dudley JT, Chiang AP, Chen R, Hastie TJ, Butte AJ. 2010. Network‐based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput Biol 6(2):e1000662 http://doi.org/10.1371/journal.pcbi.1000662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo YY, Small KS, Fry AE, Wu Y, Kwiatkowski DP, Clark TG. 2009. Power consequences of linkage disequilibrium variation between populations. Genet Epidemiol 33(2):128–135. http://doi.org/10.1002/gepi.20366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, Maeda S. 2008. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 40(9):1098–1102. http://doi.org/10.1038/ng.208 [DOI] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, McCarthy MI. 2010. Twelve type 2 diabetes susceptibility loci identified through large‐scale association analysis. Nat Genet 42(7):579–589. http://dx.doi.org/10.1038/ng.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Gerszten RE. 2011. Metabolite profiles and the risk of developing diabetes. Nat Med 17(4):448–453. http://dx.doi.org/10.1038/nm.2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wang L, He J, Bi Y, Li M, Wang T, Wang L, Jiang Y, Dai M, Lu J, and others. 2013. Prevalence and control of diabetes in chinese adults. J Am Med Assoc 310(9):948–959. http://doi.org/10.1001/jama.2013.168118 [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Hara K, Maeda S, Yasuda K, Takahashi A, Horikoshi M, Kadowaki T. 2010. A genome‐wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A‐C2CD4B. Nat Genet 42(10):864–868. http://dx.doi.org/10.1038/ng.660 [DOI] [PubMed] [Google Scholar]
- Yuan J, Jin C, Qin H‐D, Wang J, Sha W, Wang M, Shugart YY. 2013. Replication study confirms link between TSPAN18 mutation and schizophrenia in Han Chinese. PLoS ONE 8(3):e58785 http://doi.org/10.1371/journal.pone.0058785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Zhao F, Li Y, Zheng Z. 2012. Projected and observed diabetes epidemics in China and beyond. Curr Cardiol Rep 14(1):106–111. http://doi.org/10.1007/s11886‐011‐0227‐9 [DOI] [PubMed] [Google Scholar]
- Zhou X, Menche JJ, A‐L Barabasi, Sharma A. 2014. Human symptoms‐disease network. Nat Commun 5:4212 http://doi.org/10.1038/ncomms5212 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1
Figure S2
Table S1: The phenotypes we extracted from the NHGRI GWAS catalog and dbGaP are presented below. After merging redundant phenotypic labels, we have a total of 986 phenotypes considered for network construction. These phenotypes and their associated pathways are used to build the bipartite non‐ancestry‐specific (global) HPN.