

Abstract
Following heart transplantation, alloimmune responses can cause graft rejection by damaging donor vascular and parenchymal cells. However, it remains unclear whether cardiomyocytes are also directly killed by immune cells. Here, we used two‐photon microscopy to investigate how graft‐specific effector CD8+ T cells interact with cardiomyocytes in a mouse heart transplantation model. Surprisingly, we observed that CD8+ T cells are completely impaired in killing cardiomyocytes. Even after virus‐mediated preactivation, antigen‐specific CD8+ T cells largely fail to lyse these cells although both cell types engage in dynamic interactions. Furthermore, we established a two‐photon microscopy‐based assay using intact myocardium to determine the susceptibility of cardiomyocytes to undergo apoptosis. This feature, also known as mitochondrial priming reveals an unexpected weak predisposition of cardiomyocytes to undergo apoptosis in situ. These observations together with the early exhaustion phenotype of graft‐infiltrating specific T cells provide an explanation why cardiomyocytes are largely protected from direct CD8+ T‐cell‐mediated killing.
Keywords: Cardiomyocytes, Cytotoxic T cells, Mitochondrial priming, T‐cell exhaustion, Two‐photon microscopy
Introduction
Allograft rejection is a result of innate and adaptive immune responses against the transplanted tissue. In general, T cells are believed to be central to this process, with CD8+ cytotoxic T cells (CTLs) substantially contributing to acute rejection as well as chronic allograft vasculopathy 1, 2. Following heart transplantation, loss of graft parenchymal cells like cardiomyocytes (CMs) is correlated with the outcome of transplantation, and severe damage of the myocardium results in graft failure 3. Strikingly, it remains unknown to what extent graft‐specific CTLs can contact, recognize, damage, or directly kill CMs Thus, some central immunological mechanisms of heart graft parenchymal cell damage are not well understood.
Recent advances in imaging techniques provide new tools to study the process of graft rejection and to further understand the complexity of the immune response during rejection 4. In an ear skin transplantation model, the CTLs activity toward graft cells were recently visualized by two‐photon microscopy (TPM), providing insights into the spatiotemporal regulation of immune cell dynamic migration and function post transplantation (p.t.) 5. However, CTL migration and killing in the heart graft during rejection has not been reported.
Target cell apoptosis is one possible outcome of CTL‐mediated attack 6. However, it remains unknown how easily apoptosis can be induced in different heart graft parenchymal cells. Importantly, different cell types might differ in their susceptibility to undergo apoptosis 7, 8. So far, the relative efficiency of pro‐apoptotic signaling in cardiomyocytes has not been determined in situ.
In the present study, we used a mouse ovalbumin (OVA)‐heart transplantation model to investigate the killing capacity of graft‐specific CTLs. Using two‐photon microscopy (TPM), we found that graft‐specific CTLs could infiltrate the graft myocardium and locally migrate and contact cardiomyocytes. However, CTLs possessed an early exhaustion phenotype with high expression of PD‐1 and were neither able to kill OVA‐expressing CMs in vivo nor peptide‐pulsed targets ex vivo. Even following virus‐mediated preactivation, graft‐infiltrating CTLs did not rapidly lyse CMs, despite antigen‐dependent contact formation. Furthermore, we established a novel TPM‐based assay using intact myocardium to determine the level of pro‐apoptotic signaling of CMs, revealing that the balance between pro‐ and anti‐apoptotic proteins in CMs is highly in favor of anti‐apoptotic factors rendering these cells highly resistant to CTL‐induced killing.
Results
Antigen‐specific CD8+ and CD4+ T cells cooperate to trigger early rejection of murine heart grafts
To investigate T‐cell‐mediated immune responses that lead to allograft rejection, we applied a mouse OVA‐heart transplantation (HTx) model together with adoptively transferred OVA‐specific T cells. Hearts from transgenic C57BL/6 mice that systemically express OVA 9 were transplanted into C57BL/6 (wild‐type, WT) recipients one day after T cells transfer (Fig. 1A). In agreement with earlier observations 9, 10, rejection of heart grafts requires both antigen‐specific CD8+ (OT‐1) and CD4+ (OT‐2) T cells (Fig. 1B). Therefore we always transferred both OT‐1 and OT‐2 cells to all recipients throughout this study.
Figure 1.
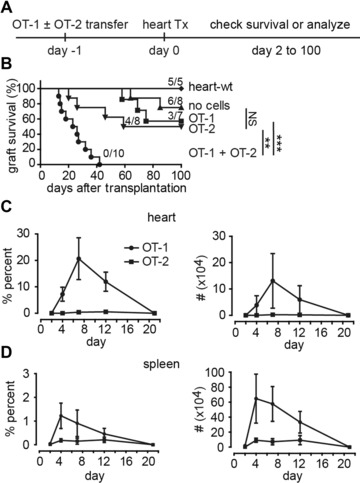
OT‐1 and OT‐2 T cells are required for rejection of murine heart grafts in an OVA‐heart transplantation model. (A) Experimental design: 1–2 × 106 naive OT‐1 and/or 1–2 × 106 OT‐2 T cells were transferred i.v. into C57BL/6 recipients. One day later, hearts from OVA‐expressing C57BL/6 donors were transplanted and monitored. (B) Following adoptive transfer of T cells indicated, survival was determined by palpation of graft contraction. The numbers of mice with surviving grafts are given for each group. NS, not significant; **p<0.01 and ***p<0.0001, log‐rank test. Data are pooled from seven independent experiments with 38 mice used in total. (C, D) Kinetics of OT‐1 and OT‐2 T cells in the heart graft (C) and spleen (D) from recipients transferred with OT‐1 and OT‐2 T cells. Percentage of all leukocytes (left panels) and absolute numbers (right panels). (C, D) Data are shown as mean ± SD and are pooled from two independent experiments, with two to three mice per experiment with a total of 17 mice used.
Notably, we found a rapid increase of OT‐1 T‐cell frequencies and numbers during the first 4–7 days p.t., followed by a gradual decrease (Fig. 1C and D), allowing us to study the CTL activities in vivo. In contrast, OT‐2 T cells neither proliferated nor infiltrated as efficiently as OT‐1 T cells (Fig. 1C and D).
To allow visualization of CTLs and CMs, we transferred GFP+ OT‐1 and nonfluorescent OT‐2 cells, and used donor hearts that in addition to OVA also expressed CFP under the chicken actin promotor (CFP‐OVA hearts). Control CFP+ grafts (not expressing OVA) appeared normal, lacked any OT‐1 T‐cell infiltration, or other overt pathological changes (Fig. 2A, top panel left). In contrast, as early as 4 days p.t., the OVA‐expressing transplants were infiltrated by immune cells. The significant loss of intact CMs, as indicated by lack of CFP+ cells, became apparent from day 7 p.t. on (Fig. 2A). Early during rejection, damaged CM were found at circumscribed lesions (Fig. 2A, bottom panel) that were often associated with the epicardium, resembling the endocardial “Quilty” lesions in human heart transplantation pathology 11 and around larger myocardial blood vessels 12.
Figure 2.
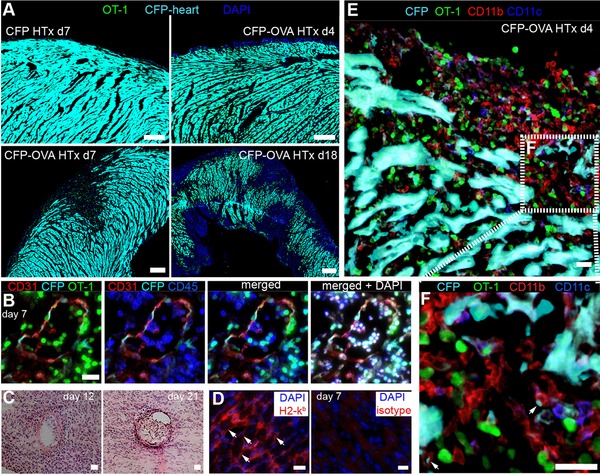
Inflammatory infiltrates of the heart graft prior to rejection. (A) Overview composite pictures of OVA‐CFP heart grafts (cyan), from recipients transferred with GFP+ OT‐1 T cells (green) plus nonfluorescent OT‐2 cells analyzed at the time points indicated. (B) Massive immune cell infiltrates and damaged endothelium in the graft. (C) Severe perivascular and intraluminal immune cells infiltrations in graft. (D) White arrows point to CMs expressing H2‐Kb. (E) Immunofluorescence of a CFP‐OVA graft reveals CD11b+ and CD11c+ cells in the infiltrated myocardium. (F) Zoom‐in view of area indicated by the white box in (E). White arrows point to CFP+ fragments taken up by CD11b+ cells. Scale bar: (A), 100 μm; (B–F), 20 μm. Images are representative of two independent experiments with eight mice per experiment.
Furthermore, histology revealed severe vascular inflammation and endothelial damage, characterized by perivascular and intraluminal infiltration of immune cells (Fig. 2B and C), which is known to be associated with thrombus formation and vessel occlusion that could lead to ischemic infarction and subsequently death of CMs 13, 14. Importantly, we found graft CMs express MHC I (Fig. 2D), which might facilitate the recognition of graft‐specific CTLs. Detailed analysis revealed a massive infiltration of OT‐1 cells as well as CD11b+ and CD11c+ cells next to damaged or disrupted CMs (Fig. 2E and F).
Graft‐specific effector CD8+ T cells in the heart cannot rapidly kill cardiomyocytes
We next applied TPM to characterize cellular migration dynamics and killing capacity of effector OT‐1 T cells present in the heart graft. From the explanted hearts, the apex or a defined region of the anterosuperior right ventricle were placed in a custom build imaging chamber and superfused with oxygenated medium at 37°C. This imaging setup has previously been described for two‐photon microscopy of lung sections 15. Control CFP‐heart transplants (not expressing OVA) were devoid of infiltrating OT‐1 T cells, showed a regular morphology of CMs, and showed only fine CFP− interstitial spaces (Fig. 3A, left panel). During prolonged TPM (1–2.5 h movies), these CMs stayed intact and showed little spontaneous contractility (Fig. 3A, left panel and Supporting Information Movie 1).
Figure 3.
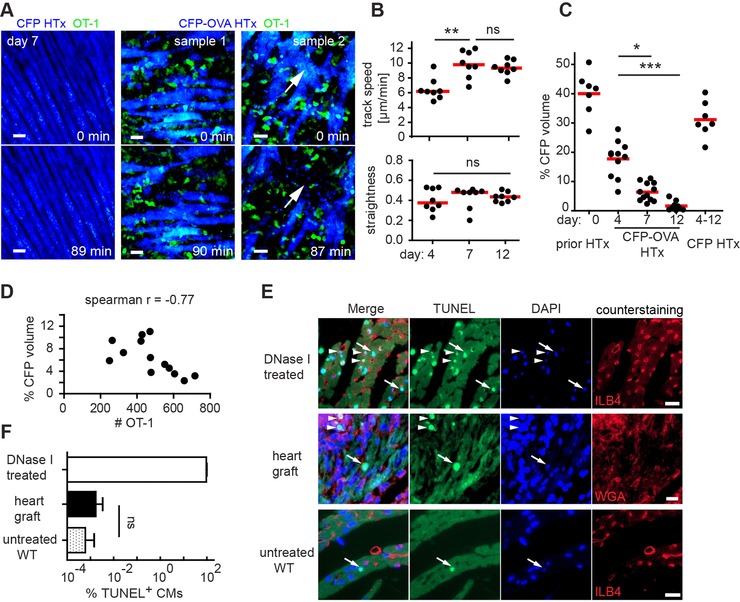
TPM imaging of effector OT‐1 T cells migration during heart graft rejection. (A) Representative frames from ex vivo time‐lapse TPM of a control CFP (left panel) or a CFP‐OVA heart graft (middle and right panel). Blue, CFP+ CMs; green, OT‐1 T cells. CMs frequently escape from killing during the observation period (sample 1). CMs are killed only very rarely (sample 2, white arrows, and Supporting Information Movie 2). Scale bar, 20 μm. (B) Track speed (upper panel) and straightness (lower panel) of effector OT‐1 T‐cell during migration. (C) Quantification of CMs loss during rejection. (B and C) Dots represent median value from (B) one short movie or (C) one snapshot; bars represent median; *p<0.05, **p<0.01, ns, ***p<0.001; not significant; Kruskal–Wallis and Dunn's test for CFP‐OVA groups. Dots represent snapshots (C). (D) Correlation between number of effector OT‐1 T cells and percentage of CFP+ voxels of movies taken at day 7 p.t. Spearman's correlation coefficient (r = −0.77). (E) TUNEL staining (green) shows few apoptotic nuclei in graft CMs; green, TUNEL‐positive apoptotic nuclei (white arrows point to apoptotic CM nuclei; arrowheads indicate apoptotic nuclei in non‐CMs); blue, DAPI; red, counterstaining; Scale bar, 10 μm. (F) Percentage of apoptotic CMs revealed by TUNEL staining is shown as mean + SD. Data in A–D are pooled from seven experiments with two to four mice used per time point. Data in F are pooled from two experiments with two mice per experiment.
In contrast, as early as 4 days (data not shown) and 7 days p.t. of CFP‐OVA hearts, massive local accumulations of OT‐1 T cells were observed (Fig. 3A, middle panel and Supporting Information Movie 2). OT‐1 T cells were present beneath the epicardial surface and between CFP+ CMs. We found that antigen‐specific effector OT‐1 T cells showed a random walk‐like migration with a relatively low average migration speed (6.48 μm/min) at day 4, increasing to approximately 10 μm/min at day 7 and day 12, both accompanied by consistent high track straightness (Fig. 3B). Importantly, killing of CMs, evidenced by loss of CFP signal (Fig. 3A, right panel and Supporting Information Movie 2), was observed only in three CMs in 25 movies analyzed (with a cumulative observation time of 42.5 h), demonstrating overall fast T‐cell migration but low cytotoxic activity of graft infiltrating effector OT‐1 CD8+ T cells.
To quantify the loss of CMs during rejection, we applied surface rendering of the CFP+ CMs within a standard imaging region sized 400 × 400 × 80 μm3 (Supporting Information Fig. 1). Prior to HTx, approximately 40% of these imaging regions contained CFP+ voxels (Fig. 3C, day 0). This was reduced to 17.7% at day 4 and to 1.62% at day 12 p.t. (Fig. 3C). As expected, the percentage of CFP+ voxels did not change significantly in the OVA‐negative CFP control grafts (Fig. 3C). Notably, we found that the percentage of CFP+ voxels correlated with the number of GFP+ effector OT‐1 T cells present in the same volume (Fig. 3D).
To address how frequently CMs undergo apoptosis during rejection, heart grafts were stained using the TdT‐mediated dUTP‐biotin nick end labeling (TUNEL) assay to visualize and quantify TUNEL+ nuclei with fragmented DNA, a hallmark of apoptosis. As expected, in DNase I‐treated positive control sections, all nuclei were TUNEL positive (Fig. 3E). In contrast, hardly any CMs were TUNEL+ in untreated WT hearts (Fig. 3E). In the heart grafts, we observed some TUNEL+ nuclei. However, these nuclei were not located inside CMs but rather appear to represent TUNEL+ graft‐infiltrating cells (Fig. 3E). Overall, we found that the density of TUNEL+ CMs in the transplanted hearts was very low and not significantly different from WT hearts (Fig. 3F). Despite the presence of high numbers of effector T cells CM cell death was rarely observed by both TUNEL assay as well as TPM. Thus, taken together our data suggest that effector T cells are not fully functional and/or that CMs are rather resistant to effector CD8+ T cell‐induced cell death.
Virus‐mediated preactivation of CD8+ T cells induces hyperacute antigen‐specific graft rejection
To address whether the low killing efficiency of effector OT‐1 T cells is associated with the improper priming of T cells post HTx, we next designed an experimental system in which fully primed effector CD8+ T cells were present in the recipient prior to HTx (Fig. 4A). To this end, OT‐1 and OT‐2 T cells were transferred into recipients that received 3 days prior to transplantation (day ‐3) the poxvirus‐modified vaccinia virus Ankara (MVA; 106 PFU i.p.) or a genetically engineered MVA strain that also encoded OVA (MVA‐OVA). Immunization with MVA‐OVA has been shown to elicit robust antigen‐specific cellular immune responses and to fully activate naive OT‐1 T cells 15. Accordingly, at the day of transplantation, most MVA‐OVA‐primed effector OT‐1 cells showed an activated CD44+CD62L− phenotype (Fig. 4B). In the MVA‐OVA as well as in an OVA plus poly(I:C) priming model, extremely rapid rejection of CFP‐OVA hearts was observed (Fig. 4C). To address a potential role of OVA‐specific antibodies in rapid graft rejection, we also immunized μMT mice that lack B cells with MVA‐OVA and observed that such recipients also rapidly rejected grafts (Fig. 4C).
Figure 4.
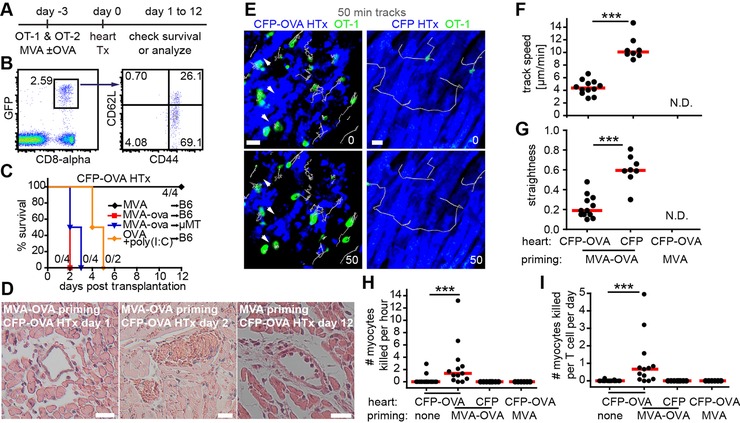
Virus‐mediated activation of OT‐1 T cells induces rapid antigen‐specific rejection of heart grafts. (A) Experimental design. (B) At day 0, most GFP+CD8+ effector OT‐1 in the blood show a mature CD44+CD62L+ phenotype. (C) Heart graft survival after MVA or MVA‐OVA or OVA plus poly(I:C) (i.p. at day 1 p.t.) immunization shows acute antigen‐specific rejection. (D) H&E staining revealed diffuse interstitial edema, hemorrhage, thrombosis, and perivascular infiltrates in grafts from MVA‐OVA immunized recipients (left and middle panels). Few perivascular infiltrates and signs of damage were present in grafts from MVA immunized recipients (right panel). (E), TPM image of virus‐primed effector OT‐1 cells (green) and killing of CMs at day 1 p.t. White arrows point to disappearing CMs (blue; left panels; Supporting Information Movie 3). TPM of CFP grafts following MVA‐OVA immunization identifies rapidly migrating effector OT‐1 T cells (right panels; Supporting Information Movie 4) indicating that OVA‐negative hearts do not trigger OT‐1 T‐cell arrest. Track speed (F) and straightness (G) of effector OT‐1 T cells (day 1 p.t.) in CFP‐OVA or CFP grafts after MVA or MVA‐OVA immunization. N.D., no cells detectable; ***p<0.001, Mann–Whitney test. (H‐I) Quantification of the number of CMs killed per hour (H) or number of CMs killed per T cell per day (I); ***p<0.001, Kruskal–Wallis and Dunn's test. Data are pooled from two experiments with two mice used for each experiments (B); from two experiments, with six mice used for each experiments (C); and poly (I:C) from one experiment with two mice (C); from two experiments and four mice for each experiment (D); from four experiments, with three to four mice for each experiment (E‐G); from seven experiments, with two to four mice used for each experiment (H‐I). Scale bar, 20 μm.
Histological analysis of CFP‐OVA grafts of MVA‐OVA immunized recipients revealed diffuse interstitial edema, hemorrhage, and perivascular infiltrates at day 1 (Fig. 4D, left), followed by severe thrombosis at day 2 (Fig. 4D, middle). As expected, limited damage with perivascular infiltrates was found in CFP‐OVA grafts from recipients immunized with MVA (Fig. 4D, right). These observations suggest that virus‐primed OT‐1 cell‐induced damage of endothelial cells leads to vascular occlusion causing hypoxic cell death of CMs.
Virus‐mediated activation of CD8+ T cells enhance antigen‐specific contact‐dependent lysis of CMs
To observe the behavior of virus‐activated CTLs in the graft by TPM, we explanted grafts from MVA‐OVA‐immunized recipients at day 1, when most CMs were still intact at the start of imaging. Following immunization with MVA‐OVA, graft‐infiltrating OT‐1 effector T cells showed a confined migratory behavior and formed long lasting contacts with CFP‐OVA‐expressing CMs (Fig. 4E–G). In contrast, OVA‐negative hearts were infiltrated by only few OT‐1 T cells showing straight and quick migration (Fig. 4E–G, and Supporting Information Movie 4). These findings show that virus‐activated, graft‐specific CTLs can rapidly home to transplanted hearts, make contacts, and attach to antigen‐expressing CMs. The higher contact formation probability of MVA‐OVA preactivated OT‐1 effector T cells translated into an increased number of observed killing‐events (Fig. 4H and I, and Supporting Information Movie 3). CTL‐mediated killing efficiency was quantified by automatically determining T‐cell numbers over time and manual counting the number of killed CMs per movie. Only in case of CFP‐OVA HTx with prior MVA‐OVA immunization a median killing rate > 0 could be determined (Fig. 4H).Taking the number of specific CTLs present during killing into account, we calculated a median killing rate of 0.67 CMs killed /T cell/24 h in mice preimmunized with MVA‐OVA receiving CFP‐OVA hearts (Fig. 4I). We tested multiple time points following transplantation and observed comparable CTL killing rates (data not shown). Thus, even following full CTL priming by prior vaccination, CTL‐mediated killing of antigen‐presenting cardiomyocytes appears to be less efficient than previously thought.
Graft‐specific effector CD8+ T cells primed by the grafted heart show an early exhaustion phenotype
For functional and phenotypic characterization, OT‐1 T cells were harvested from OVA HTx recipients, while effector OT‐1 T cells from mice immunized with MVA‐OVA were used as control. Strikingly, OT‐1 T cells isolated from HTx recipients, but not of those immunized with MVA‐OVA, expressed the exhaustion markers PD‐1, Tim‐3, and LAG‐3 (Supporting Information Fig. 2A and B, and Fig. 5A and B). KLRG1 was only expressed following MVA‐OVA priming while CD137 (4‐1BB) was not detected in both setups (Supporting Information Fig. 2C and D).
Figure 5.
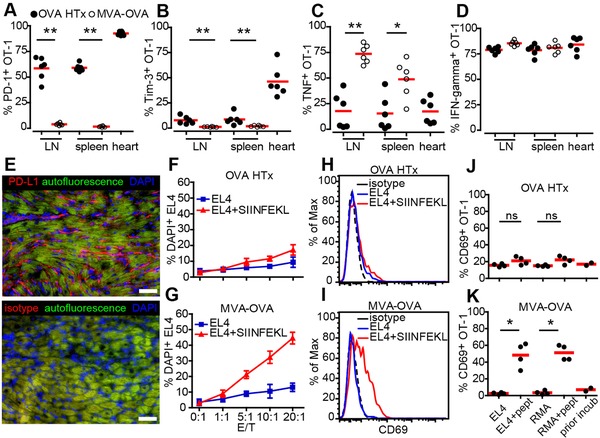
Low killing capacity of heart‐OVA primed effector OT‐1 T cells is associated with high expression of PD‐1, reduced cytokine responses, and impaired in vitro cytotoxicity. (A, B) Quantification of PD‐1+ (A) and Tim‐3+ (B) OT‐1 T cells harvested from the organs indicated of transplanted (OVA HTx) or immunized (MVA‐OVA) mice. (C, D) Following in vitro re‐stimulation, the percentage of TNF‐α (C) or IFN‐γ+ (D) OT‐1 T cells has been analyzed. Each dot represents an individual mouse. (E) Expression of PD‐L1 in the grafts; scale bar, 50 μm. (F, G) OT‐1 T cells isolated from lymph nodes and spleen of transplanted (F) or MVA‐OVA‐immunized (G) recipients were cultured with SIINFEKL peptide‐loaded or unloaded EL4 cells in vitro for 4 h and the percent of DAPI+ killed targets was determined. (H‐K) OT‐1 T cells isolated from recipients post OVA HTx (H, J) or from mice challenged with MVA‐OVA (I, K) show pronounced differences regarding CD69 upregulation following culture with peptide‐loaded targets. Means ± SD (F,G); bars, means (A–D, J‐K), *p<0.05, **p<0.01, Mann–Whitney test. Data are pooled from two experiments with six mice used per experiment (A–D); from two experiments with four mice per experiment (E); from four experiments with duplicate wells (F‐G); from two experiments with duplicate wells (H–K).
Significantly less OT‐1 T cells producing TNF‐α were found in OVA HTx recipients than in mice primed with MVA‐OVA (Fig. 5C) while IFN‐γ production was intact in both groups (Fig. 5D). Notably, PD‐L1 was expressed by both CMs and infiltrating cells in the graft (Fig. 5E). Thus, already 1 week after transplantation CTLs activated by antigen present in the transplanted heart show an exhaustion phenotype 16, 17 with high expression of negative regulatory surface proteins but still intact IFN‐γ secretion.
To functionally test CTL‐mediated killing, we isolated OT‐1 T cells from OVA HTx‐ or MVA‐OVA‐primed recipients and cultured them with SIINFEKL peptide‐loaded or unloaded EL4 tumor cells in vitro. OT‐1 T cells primed by OVA HTx showed little antigen‐specific killing activity (Fig. 5F) while MVA‐OVA primed T cells robustly killed SIINFEKL‐loaded EL4 cells (Fig. 5G). The same effect was observed for peptide‐loaded RMA target cells (data not shown). Notably, OT‐1 T cells activated by OVA HTx failed to significantly upregulate CD69, an early T‐cell activation marker, following exposure to peptide‐loaded targets (Fig. 5H and J) while MVA‐OVA primed T cells showed a massive upregulation of this activation marker (Fig. 5I and K). Taken together, these data suggest that cytotoxic T cells are already largely exhausted 1 week after transplantation inside the graft and show low direct killing activity.
Cardiomyocytes show low mitochondrial priming and resist CTL and ABT‐737‐induced apoptosis
Compared to the rapid killing of peptide‐loaded B cells reported earlier 18, CMs seem to be highly resistant to CTL‐mediated killing even when virus‐activated CTLs serve as effectors (Fig. 4H and I). To compare the killing efficiency of virus‐activated CTLs toward CMs and B cells, we transferred fluorescent‐labeled B cells from WT and OVA mice at 1:1 ratio into μMT recipients that had been immunized with MVA‐OVA 4–5 day earlier. After 60–120 min, recipients were sacrificed and the frequency of the transferred B cells and OT‐1 T cells were determined in the spleen. Based on these data, we calculated a median killing rate of 22.4 ± 10.8 B cells killed per T cell per day, which is more than 33‐fold higher than the killing rate of CMs (Fig. 6A). These findings indicate that CMs are highly resistant to attacks of effector CD8+ T cells.
Figure 6.
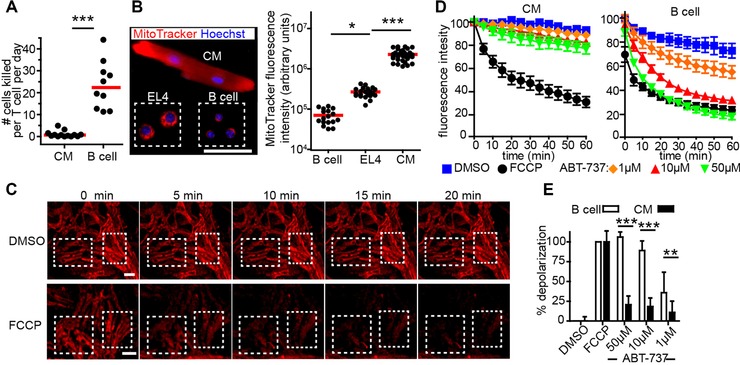
Cardiomyocytes are highly resistant to apoptosis and cytotoxic T‐cell killing. (A) B cells enriched from OVA (OVA+) and WT (OVA−) mice were injected i.v. at a 1:1 ratio into MVA‐OVA immunized μMT mice that had been transferred with GFP+ OT‐1 and OT‐2 cells 4–5 days earlier. The frequency of OVA+, OVA− B cells and OT‐1 cells was determined in spleen by FACS and the killing rate calculated. For comparison, CM data from Fig. 4I is shown. Dots represent mice; bars, median; p<0.0001, Mann–Whitney test. (B) Fluorescence microscopy of EL4 cells, B cells, and CMs stained with MitoTracker (red) and Hoechst (blue) and quantification of total cell MitoTracker fluorescence intensity. Dots represent cells; bars, mean; *p<0.05, ***p<0.001, Kruskal–Wallis and Dunn's test; scale bar 50 μm. (C) TPM of explanted CFP+ myocardium stained with JC‐1, incubated with DMSO or FCCP and imaged at time points indicated, two‐three regions of interest (white boxes) were analyzed per movie. Scale bar, 30 μm. (D) Time course of mitochondrial potential following treatment with the substances indicated. JC‐1 fluorescence intensity of CM and B cell has been determined by TPM or plate fluorescence reader, respectively. Means ± SEM are shown (E) Differences in the loss of mitochondria potential were calculated at 60 min. Mean ±SD, ***p<0.001, **p<0.01, two‐way ANOVA. Data for CMs are pooled from seven experiments with two mice used per experiment (A); data for B cells are pooled from three experiments with three mice used for each experiment; from two experiments with three mice per experiment (B); from three experiments with two to three mice per experiment (D‐E).
Since mitochondria are known to play an important role in cytotoxic T lymphocyte‐induced cell death 19, we further characterized these compartments in B cells and CMs asking whether the state of mitochondrial priming might be different in these cell types. Using the fluorescent dye MitoTracker, we found that CMs possess approximately 100‐fold more mitochondrial volume than B cells (Fig. 6B). To determine the state of mitochondrial priming of CMs, intact myocardium was stained with mitochondrial potential‐indicating dye JC‐1 and exposed to the pro‐apoptotic, cell‐permeable BH3‐mimetic, ABT‐737. This compound binds to and thereby neutralizes the anti‐apoptotic proteins Bcl‐2, Bcl‐XL, and Bcl‐w 20. As a positive control, the depolarizing compound FCCP was used (Fig. 6C). By TPM we observed that CMs in the intact myocardium hardly lost any mitochondrial potential over time, as revealed by constant red fluorescence of JC‐1, when incubated with DMSO (Fig. 6C; negative control) or with either 1, 10, or 50 μM ABT‐737 (Fig. 6D, and Supporting Information Movie 5). In contrast, B cell depolarized very fast after exposure to 10 and 50 μM ABT‐737 and also showed some depolarization at 1 μM (Fig. 6D and E). These findings indicate that the balance between pro‐ and anti‐apoptotic proteins in CMs is highly in favor of anti‐apoptotic factors and can help to explain why CMs are resistant to cytotoxic T lymphocyte‐induced cell death.
Discussion
In the present study, we used two‐photon microscopy to visualize the interactions of graft‐specific CTLs with cardiomyocytes following heart transplantation. We found that CTLs could migrate on cardiomyocytes, establish cell‐to‐cell contacts but failed to lyse cardiomyocytes. Furthermore, we observed an early exhaustion phenotype (PD‐1+Tim3+LAG‐3+/−Klrg1−) of graft‐specific CTLs, and found that cardiomyocytes in the intact heart muscle show a low tendency to undergo apoptosis.
In the first 5 days after transplantation, we observe early inflammatory lesions mainly close to the epicardium. These sites of graft‐specific T cells and myeloid cell infiltration resemble so‐called endocardial “Quilty” lesion found in humans heart grafts 11. Notably, human heart grafts are screened by endocardial biopsy, and thus early lesion at the epicardial surface of the graft cannot be detected easily.
Heart graft rejection can be triggered by a full mismatch of MHC between donor and recipient. In that setup, the foreign MHC molecules can trigger a robust and rapid cell‐mediated rejection within 7–9 days of transplantation 21. In the present study we used a minor MHC‐mismatch heart transplantation model to investigate how graft‐specific CTLs interact with and kill CMs. Interestingly, the overall rejection kinetics are rather similar in our minor‐mismatch model, when compared to the full‐MHC mismatch setup reported by others 22. For example, bioluminescence imaging of heart grafts expressing luciferase in cardiomyocytes revealed loss of graft‐derived signals between 4 and 7 days after full‐MHC mismatch transplantation 22. Together with our observation of limited inflammatory cell infiltration during the first 2–7 days after transplantation, these data indicate that limited numbers of activated immune cells within the parenchyma are probably not responsible for transplanted organ dysfunction. Interestingly, fully MHC‐mismatched heart graft rejection was observed in perforin‐deficient recipients 23 suggesting that classical perforin‐mediated killing by CTLs is seemingly also not important in the rejection of mismatched grafts. This finding fits well with our observation that CTLs are not able to kill cardiomyocytes in a cell‐contact dependent manner and argues that other processes such as cytokine‐mediated inflammatory responses or hypoxic events in the graft essentially contribute the rejection of the transplanted organ.
Prolonged antigen stimulation or antigen persistence is thought to be a key contributing factor for induction of exhaustion, and has been initially observed in chronic infections or during cancer progression. In these diseases, T‐cell exhaustion is caused by high antigenic load and antigen persistence but this is also the case following organ transplantation 24. Exhaustive differentiation of alloreactive CD8+ T cells exhibiting hypo‐responsiveness to donor alloantigens might also be beneficial for transplantation success 25 or for successful engraftment of stem cell‐derived cellular therapies. In the present study, we observed the highest level of the inhibitory receptor PD‐1 on antigen‐specific CD8+ T cells residing in the transplanted heart. Notably, the graft itself also expressed PD‐L1, the ligand for PD‐1. Expression of PD‐1, together with the observed reduced expression of TNF‐α but still intact IFN‐γ expression is considered to represent an early phase of the hierarchical stages leading to T‐cell exhaustion 16. Furthermore, following isolation these CTLs were also impaired to kill target cells, and failed to upregulate CD69 expression while interacting with their targets. In vivo, exhausted T cells were not only impaired in killing of CMs but were also highly motile and showed nonconfined migration toward their targets. Thus, graft‐specific effector CD8+ T cells, positioned in the graft, show a general impairment of the T‐cell receptor triggered activation program, consistent with efficient negative regulation mediated by PD‐1.
Although improved killing of CMs was observed following viral immunization, the median CTL‐mediated killing rate of CMs was extremely low. This is a rather unexpected finding since it is generally assumed that CTLs can swiftly kill target cells in vitro or in vivo 18, 26. However, the high killing rates reported in those studies (approx. 100 to > 1000 targets killed/T cell/24 h) 27 were calculated based on the killing of B cells loaded with specific peptide, and thus could be due to the high levels of peptide‐loaded MHC‐I molecules present on the surface of the target cells. Testing this hypothesis by using B cells from OVA mice, we found that the killing rate of B cells is still 33‐fold higher than that of CMs determined under the same experimental in vivo conditions. These findings suggest that CMs are highly resistant to T‐cell‐mediated apoptosis.
CTLs activate the intrinsic apoptotic pathway by granzymes that are deposited on and then internalized by the target cells during a perforin‐induced membrane repair response 28. The key initiation step of the intrinsic apoptotic pathway, the mitochondrial outer membrane permeabilization, MOMP, is under the control of Bcl‐2 family proteins 29. Anti‐apoptotic proteins including Bcl‐2, Bcl‐xL, and Mcl‐1, bind to and sequester activator proteins thereby preventing activation of the pore‐forming proteins Bax and Bak. Thus the balance of pro‐ and anti‐apoptotic proteins at the mitochondria determines the intrinsic potential of cells toward apoptosis, a situation that has been denoted as mitochondrial priming 7. The pro‐apoptotic BH3 mimetic drug ABT‐737 binds at nanomolar concentrations to anti‐apoptotic Bcl‐2 family members and thereby releases pro‐apoptotic activator proteins that can activate Bax and Bak to form pores leading to MOMP and changes of the mitochondrial membrane potential 30.
Previous approaches determining mitochondrial priming required cell isolation that could destroy cell integrity and induce stress 30, 31. In the present study, we established a novel TPM‐based assay to determine the state of mitochondrial priming of CMs in situ. Using intact myocardium instead of isolated CMs allowed us to measure mitochondrial priming in more physiological conditions with minimal stress and damage to CMs. Furthermore, this method allowed us to detect mitochondrial priming with cells that cannot be isolated or are sensitive to physical and chemical damage during isolation. In this assay described here, CMs hardly showed any changes in the mitochondrial potential even at highest concentrations of the pro‐apoptotic BH3 mimetic ABT‐737, while B cells rapidly lost mitochondrial membrane potential. Thus, the relative mitochondrial priming of CMs and B cells is paralleled by their different response to the cytotoxicity of effector T cells. CMs have been shown to possess low levels of caspases after birth 32, display low‐level expression of apoptotic protease activating factor 1 (Apaf1) but high levels of X‐linked inhibitor of apoptosis protein (XIAP) 33, 34. These findings clearly indicate that several mechanisms are in place to protect CMs from induced cell death. Altogether, it seems to be advantageous for an organ of vital importance and limited capacity for regeneration and repair to possess several mechanisms to counteract apoptotic signals.
Taken together, direct visualization of effector CD8+ T‐cell migration in heart grafts during rejection revealed that single CTLs cannot rapidly lyse cardiomyocytes. CTLs were able to sense antigen in the heart grafts, and showed a dramatic exhaustion phenotype within 7 days of transplantation. Furthermore, cardiomyocytes showed a low probability to undergo pro‐apoptotic signaling, suggesting that they are not easy targets for CTL‐mediated killing. Thus, heart graft rejection might only be mediated by direct CTL‐mediated killing of cardiomyocytes when extremely high densities of CTLs are present, when the antigenic stimulus is very strong, or when the tendency to undergo exhaustive differentiation is blocked in graft‐attacking T cells. Therefore, CTL‐mediated damage of graft endothelial cells and inflammation‐mediated secondary damaging effects might be chiefly responsible for heart graft malfunction during rejection.
Materials and methods
Mice
Mice were bred at the central animal facility of Hannover Medical School. The following mouse strains were used: C57BL/6N, OT‐1 CD45.1+ mice (derived from C57BL/6‐Tg(TcraTcrb)1100Mjb/J), OT‐2 mice: B6.Cg‐Tg(TcraTcrb)425Cbn/J, C57BL/6‐Tg(CAG‐OVA)916Jen/J expressing membrane‐bound chicken ovalbumin under control of the beta actin promoter (OVA mice) 9, 10, F1‐cross between OVA and C57BL/6‐Tg(CAG‐ECFP) (CFP; named CFP‐OVA mice), F1‐cross between OT‐1 CD45.1+, and C57BL/6‐Tg(CAG‐EGFP) (named GFP‐OT‐1 mice), B‐cell deficient mice: B6.129S2‐Ighmtm1Cgn/J (μMT). All animal experiments were conducted in accordance with the local animal welfare regulations.
T‐cell transfers
Cells for transfer were harvested from lymph nodes and spleens to enrich to a purity >90% using MACS CD8+ or CD4+ T‐cell negative selection kit. Recipient mice were intravenously (i.v.) transferred with 1–2 × 106 cells of one or each cell population.
Murine heart transplantation
Mice were anesthetized by intraperitoneal injection of 50 mg/kg ketamine and 10 mg/kg Xylazine (Bayer) in PBS buffer. Heterotopic HTx was performed as described earlier 35.
Modified Vaccinia virus Ankara (MVA) strains and immunization
To activate OT‐1 T cells, 106 PFU MVA‐OVA or MVA were given i.p. following T cells transfer. Alternatively, 5 mg OVA plus 50 μg poly(I:C) (both Sigma‐Aldrich) was injected i.p. at day 1 following OVA‐heart transplantation.
Organ preparation
For histology, tissue was either fixed in 2% paraformaldehyde, or fixed in ice‐cold acetone. Graft‐infiltrating cells were obtained by mechanical disruption of heart grafts followed by incubation with Collagenase D and DNase I (Roche) as described 36. To detect intracellular cytokines, cells were restimulated with 50 ng/mL PMA (Calbiochem, Darmstadt, Germany) and 2 μg/mL ionomycin (Invitrogen) for 1 h, followed by further 3‐h incubation in the presence of 1 μg/mL brefeldin A (Sigma‐Aldrich). For intracellular staining, cells were fixed and permeabilized using the BD Biosciences cytofix/cytoperm reagents. The following antibodies and reagents were used: CD11b‐PE (M1/70), CD11c‐APC (N418), CD8‐beta‐Cy5 (RMCD8‐2), CD8‐alpha‐PerCP‐Cy5.5 (53‐6.7), CD4‐PerCP (RM4‐5), CD31‐PE (MEC 13.3), CD45‐APC (30‐F11), CD62L‐APC (MEL‐14), CD44‐eFluor 450 (IM7), CD44‐APC (IM7), PD‐1‐PE‐Cy7 (J43), TNF‐APC‐Cy7 (MP6‐XT22), IFN‐gamma‐PE (XMG1.2), CD69‐PerCP‐Cy5.5 (H1.2F3), MHC Class I (H‐2Kb)‐APC (AF6‐88.5.5.3), PD‐L1‐PE (MIH5), Tim‐3‐PE (RMT3‐23), LAG‐3 (C9B7W), KLRG‐1 (2F1), CD137 (17B5), Hoechst 33258 (Sigma), DAPI (Sigma).
Analysis of apoptosis
Heart tissue was fixed in 4% formalin, and the TUNEL assay was performed as previously described 37. Apoptotic nuclei were detected by TUNEL and by nuclear morphology using DAPI staining (Sigma) 38. In addition, sections were counter‐stained with wheat germ agglutinin (Lectin from Triticum vulgaris, Sigma) or isolectin B4 (Life Technologies) to confirm apoptotic nuclei of CMs. 4–6 TUNEL‐stained sections from each heart were used to assess the frequency of TUNEL/DAPI positive nuclei.
In vitro killing assay
Lymph node and spleen CD8+ T cells were enriched and sorted (XDP, Beckman Coulter) from OVA‐heart transplanted or MVA‐OVA immunized recipients 4–5 days following transplantation or immunization. As target cells, EL4 C57BL/6N T‐cell lymphoma cells (ATCC TIB‐39) were used. Target cells were labeled with eFluor 670 (eBioscience) before culture, and half of the cells were incubated with 30 μM SIINFEKL (Sigma‐Aldrich) at 37°C for 30 min. GFP+ OT‐1 T cells were mixed with peptide‐loaded or unloaded target cells at the indicated effector‐target (E:T) ratios and incubated for 4 h. Each E:T ratio was assayed in duplicate and 3 × 103–5 × 103 target cells were used per well. Following incubation, cells were harvested and stained with antibodies and DAPI. The percent of DAPI+ killed targets was calculated.
B cell in vivo killing assay
B cells were enriched from OVA (OVA+) or WT (OVA−) mice by B‐cell negative selection (MCAS, Miltenyi Biotec) and labeled with either 5 μM efluor670 (ebioscience) or 10 μM TAMRA (Life Technologies). 20–25 × 106 cells were injected i.v. at a 1:1 ratio into MVA‐OVA immunized μMT mice that had been transferred with GFP+ OT‐1 and OT‐2 cells 4–5 days earlier. After 1–2 h, the frequencies of OVA+, OVA− B cells, and OT‐1 cells were determined in spleen by FACS. Killing of B cells was calculated as follows:
With OVA+/− out meaning the frequencies of B cells expressing OVA (or control B cells) in the spleen during analysis, and OVA+/− in meaning the frequency of B cells in the injected cell mixture. Teff stands for the frequency of OT‐1 T cells in the spleen and t denotes the duration in hours of the individual experiment.
Two‐photon microscopy
Explanted heart grafts were gently cut into small pieces by a fine blade. The apex of the heart and the anterosuperior right ventricular wall were placed in a custom‐built incubation chamber using tissue adhesive (Surgibond) and superfused with oxygenated (95% O2/5% CO2) RPMI 1640 medium (Invitrogen) containing 5 g/l glucose (Sigma‐Aldrich) as described earlier 15. The temperature was monitored and maintained manually at 37°C. TPM was performed using an Olympus BX51 upright microscope equipped with a 20 × 0.95 NA water immersion objective (Olympus) and a MaiTai Ti:Sapphire laser (Spectra‐Physics) tuned to 920 nm for excitation of GFP and CFP. To generate time‐lapse series, Z‐stacks of up to 15–22 images were acquired every 20–60 s. From explanted grafts usually one long movie (60–150 min) was recorded to assess the killing capacity of CTLs and one or more short movies (20–30 min) were recorded to determine CTL migration behavior. Furthermore 3D, snapshots were collected to determine the cardiomyocyte volume within a standard imaging volume (usually, 400 μm × 400 μm × 80 μm). Imaris 7.5‐7.6 (Bitplane) was used for image analysis and automated tracking of cells. The accuracy of the automated tracking was manually controlled.
Isolation of cardiomyocytes
Adult mouse ventricular CMs were isolated from 8 to 12 week old C57BL/6N mice as described 39. Briefly, the excised heart was perfused in a Langendorff system and digested with trypsin and liberase DH (Roche). The isolated CMs were subsequently subjected to a recalcification procedure and seeded on laminin‐coated coverslips.
Visualization of mitochondria
Cells were stained with 200 nM MitoTracker Deep Red FM (Life Technologies) in RPMI 1640 without phenol red at 37°C for 30 min. Images were acquired with identical exposure time with a Zeiss Axiovert fluorescence microscope and adjusted identically with AxioVision 4.6 software. Fluorescence intensity of staining obtained with MitoTracker was measured with Fiji software (http://fiji.sc/Fiji) using the integrated density parameter.
Mitochondrial priming
B cells and small intact heart tissue were stained at 37°C for 30 min with 2 μM JC‐1 (Life Technologies) in DTEB buffer (135 mM trehalose, 50 mM KCl, 20 μM EDTA, 20 μM EGTA, 0.1% BSA, 5 mM succinate, 10 mM HEPES‐KOH, pH 7.5) 30. 105 labeled B cells in 50 μL were added to 50 μL of 100 μM or 20 μM or 2 μM ABT‐737 (Biozol) in DTEB buffer preloaded on a 96‐well plate as previously described 8. Mitochondrial potential was measured by JC‐1 red fluorescence emission intensity (545 ±9 nm excitation and 590 ± 20 nm emission) during 1 h of incubation at 37°C with measurements obtained every 5 min (Tecan infinite M200 plate reader). As controls, DMSO (negative control) and FCCP (Sigma, 20 μM; positive control) were added instead of ABT‐737. JC‐1‐labeled heart tissue were placed in a small chamber maintained at 37°C containing ABT‐737 in DTEB buffer, and images were taken every 5 min over 1 h by TPM. JC‐1 fluorescence intensity of CMs was measured by Fiji software using the integrated density parameter and the values were normalized to 100 at 0 min. The following equation was used to calculate percent depolarization at 60 min: Depolarization = 1 – ((Sample ‐ FCCP)/(DMSO ‐ FCCP)) as described 30.
Statistics
GraphPad Prism 4.03 was used for statistical analysis. The individual mouse was considered to be the experimental unit. No power calculation was performed. Statistical significance was determined with the Kruskal–Wallis and Dunn's test, Mann–Whitney test, and two‐way ANOVA. Survival curve data were tested using the log‐rank test. In figures, p values are indicated as follows: not significant when p>0.05 (ns), *p<0.05, **p<0.01, and ***p<0.0001. Mean and standard deviation (SD) or median and interquantile range (IQR) are used to indicate data spread, as described in the figure legends.
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Abbreviations
- Bcl‐2
B‐cell lymphoma 2
- BH3
Bcl‐2 Homology 3
- CFP
cyan fluorescent protein
- CM
cardiomyocyte
- HTx
heart transplantation
- MOMP
mitochondrial outer membrane permeabilization
- MVA
modified vaccinia virus Ankara
- p.t.
post transplantation
- TPM
two‐photon microscopy
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting information
Movie 1
Movie 2
Movie 3
Movie 4
Movie 5
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft grants (SFB738‐B5 and EXC62 to R.F.), ERC advanced grant (322645 to R.F.), H.W.& J. Hector Stiftung (to M.S. und M.E), and by Hannover Biomedical Research School (HBRS; to X.Z.) and the Center for Infection Biology (ZIB; to X.Z.). We thank Mathias Herberg for excellent animal care, Andreas Krueger, Christian Koenecke, Immo Prinz, and Gregor Warnecke for helpful discussions and critical comments; and Jingqing Sun for help in drafting figures.
[The copyright line of this article has been changed since first published on 11 March 2016 from the standard copyright to CC‐BY‐NC‐ND]
References
- 1. Russell, P. S. , Chase, C. M. , Winn, H. J. , Colvin, R. B. , Coronary atherosclerosis in transplanted mouse hearts. I. Time course and immunogenetic and immunopathological considerations. Am. J. Pathol. 1994. 144: 260–274. [PMC free article] [PubMed] [Google Scholar]
- 2. Bolinger, B. , Engeler, D. , Krebs, P. , Miller, S. , Firner, S. , Hoffmann, M. , Palmer, D. C. et al, IFN‐gamma‐receptor signaling ameliorates transplant vasculopathy through attenuation of CD8+ T‐cell‐mediated injury of vascular endothelial cells. Eur. J. Immunol. 2010. 40: 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stewart, S. , Winters, G. L. , Fishbein, M. C. , Tazelaar, H. D. , Kobashigawa, J. , Abrams, J. , Andersen, C. B. et al, Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J. Heart Lung Transplant. 2005. 24: 1710–1720. [DOI] [PubMed] [Google Scholar]
- 4. Camirand, G. , New perspectives in transplantation through intravital microscopy imaging. Curr. Opin. Organ Transplant. 2013. 18: 6–12. [DOI] [PubMed] [Google Scholar]
- 5. Celli, S. , Albert, M. L. , Bousso, P. , Visualizing the innate and adaptive immune responses underlying allograft rejection by two‐photon microscopy. Nat. Med. 2011. 17: 744–749. [DOI] [PubMed] [Google Scholar]
- 6. Barry, M. , Bleackley, R. C. , Cytotoxic T lymphocytes: all roads lead to death. Nat. Rev. Immunol. 2002. 2: 401–409. [DOI] [PubMed] [Google Scholar]
- 7. Ni Chonghaile, T. , Sarosiek, K. A. , Vo, T.‐T. , Ryan, J. A. , Tammareddi, A. , Moore, V. D. G. , Deng, J. et al, Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011. 334: 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vo, T.‐T. , Ryan, J. , Carrasco, R. , Neuberg, D. , Rossi, D. J. , Stone, R. M. , Deangelo, D. J. et al, Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012. 151: 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ehst, B. D. , Ingulli, E. , Jenkins, M. K. , Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am. J. Transplant. 2003. 3: 1355–1362. [DOI] [PubMed] [Google Scholar]
- 10. Filatenkov, A. A. , Jacovetty, E. L. , Fischer, U. B. , Curtsinger, J. M. , Mescher, M. F. , Ingulli, E. , CD4 T cell‐dependent conditioning of dendritic cells to produce IL‐12 results in CD8‐mediated graft rejection and avoidance of tolerance. J. Immunol. 2005. 174: 6909–6917. [DOI] [PubMed] [Google Scholar]
- 11. Joshi, A. , Masek, M. A. , Brown, B. W. , Weiss, L. M. , Billingham, M. E. , “Quilty” revisited: a 10‐year perspective. Hum. Pathol. 1995. 26: 547–557. [DOI] [PubMed] [Google Scholar]
- 12. Huibers, M. M. H. , Gareau, A. J. , Vink, A. , Kruit, R. , Feringa, H. , Beerthuijzen, J. M. T. , Siera‐de Koning, E. et al, The composition of ectopic lymphoid structures suggests involvement of a local immune response in cardiac allograft vasculopathy. J. Heart Lung Transplant. 2015. 34: 734–745. [DOI] [PubMed] [Google Scholar]
- 13. Ensminger, S. M. , Spriewald, B. M. , Witzke, O. , Morrison, K. , Pajaro, O. E. , Morris, P. J. , Rose, M. L. et al, Kinetics of transplant arteriosclerosis in MHC‐Class I mismatched and fully allogeneic mouse aortic allografts. Transplantation 2002. 73: 1068–1074. [DOI] [PubMed] [Google Scholar]
- 14. Shi, C. , Lee, W. S. , He, Q. , Zhang, D. , Fletcher, D. L. , Newell, J. B. , Haber, E. , Immunologic basis of transplant‐associated arteriosclerosis. Proc. Natl. Acad. Sci. USA 1996. 93: 4051–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Halle, S. , Dujardin, H. C. , Bakocevic, N. , Fleige, H. , Danzer, H. , Willenzon, S. , Suezer, Y. et al, Induced bronchus‐associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J. Exp. Med. 2009. 206: 2593–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yi, J. S. , Cox, M. A. , Zajac, A. J. , T‐cell exhaustion: characteristics, causes and conversion. Immunology. 2010. 129: 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wherry, E. J. , T cell exhaustion. Nat. Immunol. 2011. 131: 492–499. [DOI] [PubMed] [Google Scholar]
- 18. Mempel, T. R. , Pittet, M. J. , Khazaie, K. , Weninger, W. , Weissleder, R. , von Boehmer, H. , von Andrian, U. H. , Regulatory T cells reversibly suppress cytotoxic T cell function independent of effector differentiation. Immunity 2006. 25: 129–141. [DOI] [PubMed] [Google Scholar]
- 19. Martinvalet, D. , Zhu, P. , Lieberman, J. , Granzyme A induces caspase‐independent mitochondrial damage, a required first step for apoptosis. Immunity 2005. 22: 355–370. [DOI] [PubMed] [Google Scholar]
- 20. Oltersdorf, T. , Elmore, S. W. , Shoemaker, A. R. , Armstrong, R. C. , Augeri, D. J. , Belli, B. A. , Bruncko, M. et al, An inhibitor of Bcl‐2 family proteins induces regression of solid tumours. Nature 2005. 435: 677–681. [DOI] [PubMed] [Google Scholar]
- 21. Peugh, W. N. , Superina, R. A. , Wood, K. J. , Morris, P. J. , The role of H‐2 and non‐H‐2 antigens and genes in the rejection of murine cardiac allografts. Immunogenetics 1986. 23: 30–37. [DOI] [PubMed] [Google Scholar]
- 22. Tanaka, M. , Swijnenburg, R.‐J. , Gunawan, F. , Cao, Y.‐A. , Yang, Y. , Caffarelli, A. D. , de Bruin, J. L. et al, In vivo visualization of cardiac allograft rejection and trafficking passenger leukocytes using bioluminescence imaging. Circulation 2005. 112: I105–I110. [DOI] [PubMed] [Google Scholar]
- 23. Schulz, M. , Schuurman, H. J. , Joergensen, J. , Steiner, C. , Meerloo, T. , Kägi, D. , Hengartner, H. et al, Acute rejection of vascular heart allografts by perforin‐deficient mice. Eur. J. Immunol. 1995. 25: 474–480. [DOI] [PubMed] [Google Scholar]
- 24. Valujskikh, A. , Li, X. C. , Memory T cells and their exhaustive differentiation in allograft tolerance and rejection. Curr. Opin. Organ Transplant. 2012. 17: 15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Steger, U. , Denecke, C. , Sawitzki, B. , Karim, M. , Jones, N. D. , Wood, K. J. , Exhaustive differentiation of alloreactive CD8+ T cells: critical for determination of graft acceptance or rejection. Transplantation 2008. 85: 1339–1347. [DOI] [PubMed] [Google Scholar]
- 26. Wiedemann, A. , Depoil, D. , Faroudi, M. , Valitutti, S. , Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc. Natl. Acad. Sci. USA 2006. 103: 10985–10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Regoes, R. R. , Yates, A. , Antia, R. , Mathematical models of cytotoxic T‐lymphocyte killing. Immunol. Cell Biol. 2007. 85: 274–279. [DOI] [PubMed] [Google Scholar]
- 28. Keefe, D. , Shi, L. , Feske, S. , Massol, R. , Navarro, F. , Kirchhausen, T. , Lieberman, J. , Perforin triggers a plasma membrane‐repair response that facilitates CTL induction of apoptosis. Immunity 2005. 23: 249–262. [DOI] [PubMed] [Google Scholar]
- 29. Chipuk, J. E. , Green, D. R. , How do BCL‐2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008. 18: 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ryan, J. , Letai, A. , BH3 profiling in whole cells by fluorimeter or FACS. Methods 2013. 61: 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ryan, J. A. , Brunelle, J. K. , Letai, A. , Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Natl. Acad. Sci. USA 2010. 107: 12895–12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bahi, N. , Zhang, J. , Llovera, M. , Ballester, M. , Comella, J. X. , Sanchis, D. , Switch from caspase‐dependent to caspase‐independent death during heart development: essential role of endonuclease G in ischemia‐induced DNA processing of differentiated cardiomyocytes. J. Biol. Chem. 2006. 281: 22943–22952. [DOI] [PubMed] [Google Scholar]
- 33. Potts, M. B. , Vaughn, A. E. , McDonough, H. , Patterson, C. , Deshmukh, M. , Reduced Apaf‐1 levels in cardiomyocytes engage strict regulation of apoptosis by endogenous XIAP. J. Cell Biol. 2005. 171: 925–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chiong, M. , Wang, Z. V. , Pedrozo, Z. , Cao, D. J. , Troncoso, R. , Ibacache, M. , Criollo, A. et al, Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2011. 2: e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Corry, R. J. , Winn, H. J. , Russell, P. S. , Primarily vascularized allografts of hearts in mice. The role of H‐2D, H‐2K, and non‐H‐2 antigens in rejection. Transplantation 1973. 16: 343–350. [DOI] [PubMed] [Google Scholar]
- 36. Liu, X. , Mishra, P. , Yu, S. , Beckmann, J. , Wendland, M. , Kocks, J. , Seth, S. et al, Tolerance induction towards cardiac allografts under costimulation blockade is impaired in CCR7‐deficient animals but can be restored by adoptive transfer of syngeneic plasmacytoid dendritic cells. Eur. J. Immunol. 2011. 41: 611–623. [DOI] [PubMed] [Google Scholar]
- 37. Wollert, K. C. , Heineke, J. , Westermann, J. , Lüdde, M. , Fiedler, B. , Zierhut, W. , Laurent, D. et al, The cardiac Fas (APO‐1/CD95) Receptor/Fas ligand system: relation to diastolic wall stress in volume‐overload hypertrophy in vivo and activation of the transcription factor AP‐1 in cardiac myocytes. Circulation. 2000. 101: 1172–1178. [DOI] [PubMed] [Google Scholar]
- 38. Hilfiker‐Kleiner, D. , Hilfiker, A. , Fuchs, M. , Kaminski, K. , Schaefer, A. , Schieffer, B. , Hillmer, A. et al, Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004. 95: 187–195. [DOI] [PubMed] [Google Scholar]
- 39. Louch, W. E. , Sheehan, K. A. , Wolska, B. M. , Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell. Cardiol. 2011. 51: 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting information
Movie 1
Movie 2
Movie 3
Movie 4
Movie 5