

Abstract
Aggressiveness is a behavioral trait that has the potential to be harmful to individuals and society. With an estimated heritability of about 40%, genetics is important in its development. We performed an exploratory genome‐wide association (GWA) analysis of childhood aggressiveness in attention deficit hyperactivity disorder (ADHD) to gain insight into the underlying biological processes associated with this trait. Our primary sample consisted of 1,060 adult ADHD patients (aADHD). To further explore the genetic architecture of childhood aggressiveness, we performed enrichment analyses of suggestive genome‐wide associations observed in aADHD among GWA signals of dimensions of oppositionality (defiant/vindictive and irritable dimensions) in childhood ADHD (cADHD). No single polymorphism reached genome‐wide significance (P < 5.00E‐08). The strongest signal in aADHD was observed at rs10826548, within a long noncoding RNA gene (beta = −1.66, standard error (SE) = 0.34, P = 1.07E‐06), closely followed by rs35974940 in the neurotrimin gene (beta = 3.23, SE = 0.67, P = 1.26E‐06). The top GWA SNPs observed in aADHD showed significant enrichment of signals from both the defiant/vindictive dimension (Fisher's P‐value = 2.28E‐06) and the irritable dimension in cADHD (Fisher's P‐value = 0.0061). In sum, our results identify a number of biologically interesting markers possibly underlying childhood aggressiveness and provide targets for further genetic exploration of aggressiveness across psychiatric disorders. © 2016 The Authors. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics Published by Wiley Periodicals, Inc.
Keywords: ADHD, aggression, GWAS
INTRODUCTION
Aggressiveness can be defined as any behavior directed toward an individual with the immediate intent to cause harm [Anderson and Bushman, 2002]. Violence, which is strongly related to aggressiveness, is the sixth leading cause of burden of disease for people aged 15–44 years worldwide [WHO, 2008]. To date, most interventions designed to reduce violence risk typically have small effects, reflecting our limited understanding of its causes and stressing the need for further studies [Moffitt, 2005; McGuire, 2008].
As a complex phenomenon, aggressiveness spans across numerous facets of human behavior, ranging from emotional lability and temperamental traits (e.g., hot‐tempered, short fuse, irritable) to physical violence [Lesch et al., 2012]. These traits are frequently found among youth with attention deficit hyperactivity disorder (ADHD), a common child and adolescent psychiatric disorder with a prevalence of about 5% and a rate of persistence into adulthood of about 50% [Faraone et al., 2015]. ADHD is defined by symptoms of inattention and hyperactivity/impulsivity, and youth with ADHD often have co‐existing disorders, some of which are closely related to aggressiveness and violence, such as conduct disorder (CD) and/or oppositional defiant disorder (ODD) and disorders characterized by symptoms defined within the broader term of antisocial behavior [Dalsgaard et al., 2002]. These disorders put youth with ADHD at high risk of problems associated with aggressiveness in adulthood [Klassen et al., 2010], especially when the aggressive behavior has an early onset [Hofvander et al., 2009]. This can be illustrated by the fact that around 30% of youth and 25% of adult prison inmates are found to qualify for an ADHD diagnosis [Young et al., 2014]. Studies of childhood aggressiveness in adults can, therefore, be of great importance to improve our understanding of adult ADHD.
The etiology of ADHD as well as traits of aggressiveness is complex, with genetics playing an important role. The heritability of ADHD has been estimated to be up to 88% across the lifespan [Larsson et al., 2013], whereas the estimates of genetic influence on aggression vary across studies, collectively reaching about 40–50% [Brendgen et al., 2006; Tuvblad and Baker, 2011]. Such diversity in the estimation of aggression heritability may result from inconsistency in measures across studies. Several different aggression measures have been utilized to assess the genetic and environmental influences on its development [Veroude et al., 2015], reflecting that there is no consensus regarding its definition [Ramirez and Andreu, 2006]. Furthermore, the estimates of aggressiveness are influenced by the age of the study participants. The literature reports stability of aggressiveness between childhood and adulthood, with adolescence as a transient period with little stability in this trait [Moffitt, 2005]. Genes seem to explain little variation in adolescent aggression, but are likely to account for individual differences in childhood and adult aggression [Lyons et al., 1995]. Also, given higher levels of aggression in males and higher genetic load in males with antisocial behavior compared to females, it is an open question whether genetic propensity is of greater importance in one sex over the other [Miles and Carey, 1997; Tuvblad and Baker, 2011]. Interestingly, similar considerations of age and sex effects are also present in studies of ADHD as well as when ADHD is co‐morbid with aggressive behavior [Faraone et al., 2015, 1991].
Given that ADHD and aggression often co‐occur and that both traits are heritable, twin studies have noted the possibility of shared genetic etiology between ADHD and aggression. A common genetic factor has been reported among ADHD and symptoms of aggression in 9–10‐year‐old children [Tuvblad et al., 2009]. Likewise, it has been suggested that impulsivity and aggression are genetically mediated to a similar extent [Seroczynski et al., 1999].
Influenced by major theories on neuronal circuits, genetic association studies of ADHD and/or aggression have been dominated by candidate gene studies, focusing on the regulation of monoaminergic transmission [Faraone et al., 2015; Veroude et al., 2015]. In line with twin studies, these candidate gene analyses have provided further support toward a shared genetic component between ADHD and aggression. Many genes associated with ADHD point toward the same biological mechanisms as those associated with aggressive behavior, including genes that are involved in the synthesis, binding, transport and degradation of neurotransmitters, especially dopamine and serotonin [Faraone et al., 2015; Veroude et al., 2015]. It has been reported, for example, that the genes MAOA, DRD2, DRD4, COMT, SLC6A4, TPH1, and TPH2 may contribute to the development of ADHD as well as aggressive behaviors [Gizer et al., 2009; Vassos et al., 2014]. However, these candidate gene studies suffer from the lack of replication in independent samples (where available) and small effect sizes suggest that some of these genes play a more limited role in the susceptibility to ADHD and/or aggressive behavior, or that their involvement may be limited to rare familial cases [McKinney et al., 2008; Halmoy et al., 2010; Tiihonen et al., 2014]. Thus, the overall genetic architecture of ADHD and/or aggression remains largely unknown and warrants studies using a hypothesis‐free approach [Vassos et al., 2014].
Genome‐wide association (GWA) studies allow interrogation of the entire genome to generate new hypotheses. To date, few GWA studies have been performed for ADHD and/or aggressiveness, with no finding passing the stringent Bonferroni‐corrected genome‐wide significance level (P < 5.00E‐08) for either phenotype [Dick et al., 2011; Tielbeek et al., 2012; Mick et al., 2014; Salvatore et al., 2015]. Nonetheless, as these studies were generally underpowered, some understanding of biological processes behind ADHD and/or aggressiveness may emerge from the convergence of identified nominally significant loci. Previous GWA studies on aggressive behaviors in ADHD have noted a number of suggestive association signals, generating biological hypotheses regarding the etiology of ADHD and/or aggression [Anney et al., 2008; Aebi et al., 2015]. In addition, a recent GWA study revealed a positive linear correlation between ADHD polygenic scores and comorbid aggression scores, indicating that the presence of aggressive symptoms in ADHD is likely to index a greater genetic load [Hamshere et al., 2013]. Similarly hypothesis‐free, genome‐wide linkage analyses have also reported evidence of significant co‐segregation between ADHD and disruptive behavior [Jain et al., 2007].
The lack of robust genetic association signals may be explained by the modest sample sizes and the complex nature of both ADHD and aggressiveness, where genetic factors are intertwined with environmental influences [Brendgen et al., 2006]. In addition, heterogeneity in genetic susceptibility, phenotypic manifestation, and operationalization of aggressiveness may depress association signals [Cross‐Disorder Group of the Psychiatric Genomics et al., 2013]. The phenotypic heterogeneity in ADHD may potentially be exacerbated by its high rates of comorbidity with not only aggressive behaviors, but also mood and anxiety disorders [Biederman et al., 1992]. Another possible reason behind the lack of replicable genetic findings is the limited annotation of the human genome. The annotation has mostly been focused on protein‐coding genes that represent only ∼1% of our genome, making it difficult to evaluate possible biological pathways involved in ADHD and/or aggressiveness, as the majority of GWA findings tend to reside outside the traditional protein‐coding regions [Dick et al., 2011; Schizophrenia Working Group of the Psychiatric Genomics, 2014].
In the present study, we aimed to perform exploratory genome‐wide association tests to shed light on the genetic susceptibility loci and biological processes possibly involved in the etiology of childhood aggressiveness in ADHD. We utilized the GWA method to analyze childhood aggressiveness in adults with ADHD gathered in studies across Europe. To minimize phenotypic heterogeneity between samples, we derived our measure of childhood aggressiveness in adult ADHD (aADHD) from the Wender Utah Rating Scale (WURS). This questionnaire was used as part of the assessment procedure at all sites. As the WURS reflects childhood recollections, we also explored a possible genetic overlap of association signals observed in aADHD with those of irritable and defiant/vindictive dimensions of ODD in youth with ADHD (cADHD) [Aebi et al., 2015]. Finally, we performed an examination of non‐protein coding genes in order to obtain a better understanding of the biological processes underlying childhood aggressiveness in aADHD.
MATERIALS AND METHODS
Subjects
aADHD samples
Recruitment of adult ADHD patients was conducted at three sites within an international multi‐center persistent ADHD collaboration (IMpACT, http://www.impactadhdgenomics.com): Germany, Norway, and Spain. All individuals were of Caucasian ancestry. Only participants who gave written informed consent were enrolled in the studies, which complied with the Declaration of Helsinki.
German sample
Patients with a diagnosis of aADHD were recruited by experienced psychiatrists at the University of Würzburg (Würzburg, Germany). Unrelated in‐ and outpatients of self‐reported central‐European descent completed a semi‐structured clinical interview according to DSM‐IV. Inclusion criteria were onset before the age of 7 years, lifelong persistence, current diagnosis and age of recruitment between 18 and 65 years. Exclusion criteria were the appearance of symptoms restricted to the duration of any other Axis I disorder; current diagnosis of active alcohol or other drug abuse or dependence; lifetime diagnosis of bipolar I disorder, schizophrenia, or any other psychotic disorder; and an IQ score below 80. For a more detailed sample description, please confer previous publications [Reif et al., 2009; Franke et al., 2010]. The study was approved by the Ethic Committee of the University of Würzburg (Würzburg, Germany).
Norwegian sample
Participants were recruited at the University of Bergen (UiB, Bergen, Norway) as described elsewhere [Halmoy et al., 2009]. In short, adult patients with ADHD were recruited through a Norwegian national medical registry as well as by psychologists and psychiatrists working at outpatient clinics. All patients had been previously diagnosed with ADHD using either DSM‐IV or ICD‐10. The ICD‐10 criteria were adapted to the DSM‐IV criteria by allowing the inattentive subtype as sufficient for the ADHD diagnosis. Individuals with other neuropsychiatric disorders were not excluded as long as the ADHD criteria were fulfilled. Individuals with IQ below 70 were excluded from the study. All participants provided either blood or saliva samples for DNA extraction. The study was approved by the regional committee for medical and health research ethics, western Norway.
Spanish sample
Participants were recruited at the Department of Psychiatry from the Hospital Universitari Vall d'Hebron (HUVH, Barcelona, Spain) as described elsewhere [Sanchez‐Mora et al., 2015]. Patients were adults of Caucasian origin and met Diagnostic and Statistical Manual for Mental Disorders‐IV (DSM‐IV) criteria for ADHD. The diagnosis of ADHD was evaluated with the Structured Clinical Interview for DSM‐IV Axis I and II Disorders (SCID‐I and SCID‐II) and the Conner's Adult ADHD Diagnostic Interview for DSM‐IV (CAADID Parts I and II). Consensus eligibility criteria for the current study were a diagnosis of ADHD according to the diagnostic criteria of DSM‐IV, onset before the age of 7 years via retrospective diagnosis (which was confirmed by a family member, wherever possible), lifelong persistence and current diagnosis. DNA was extracted from either peripheral blood or saliva samples. The study was approved by the ethics committee of the institution.
cADHD sample
Youth with ADHD were participants in the International Multicentre ADHD Genetics (IMAGE) study, recruited in 12 children and adolescent psychiatry clinics representing eight countries across Europe. Approval was obtained by the Institutional Review Board of SUNY Upstate Medical University and from ethical review boards within each country. A detailed description of the study design and assessment procedures has been provided in previous publications [Muller et al., 2011a,2011b]. In short, entry criteria for probands were a clinical diagnosis of ADHD according to DSM‐IV‐based structured interviews and access to one or both biological parents and one or more full siblings for DNA collection and clinical assessment. Exclusion criteria included autism, epilepsy, IQ < 70, brain disorders, and any genetic or medical disorder associated with externalizing behaviors that might mimic ADHD.
Measures of Aggressiveness
aADHD samples
The adult measure of childhood aggressiveness in the aADHD samples was derived from the Wender Utah Rating Scale (WURS) [Ward et al., 1993]. The WURS is a questionnaire used for retrospective assessment of childhood symptoms of ADHD in adults. An exploratory factor analysis (EFA) was run to determine the latent structure of the WURS. The EFA consisted of a principal component analysis with Varimax rotation and yielded three factors with Eigen values above one. From the main factor explaining the greatest amount of variance in responses to the WURS (30.7%), the top six items with the highest loadings (0.74–0.82) all represented prototypical elements of aggressiveness: “temper outburst/tantrums,” “angry,” “hot‐ or short‐tempered/low boiling point,” “disobedient with parents/rebellious/sassy,” “losing control of myself,” and “irritable,” For each item, the participant was asked to evaluate if she/he as a child was (or had) a specific symptom and to rate it according to the following four response categories: “not at all/very slightly” (0), “mildly” (1), “moderately” (2), quite a bit” (3), or “very much” (4). The arithmetic sum of the responses of the aforementioned items was adopted as a continuous measure of aggressiveness, ranging from 0 to 24. Supplementary Figure S1 shows the distribution of this measure across genders in the three aADHD datasets.
cADHD sample
The dimensions of oppositionality were assessed using the long form of the revised Conners Parent Rating Scale (CPRS‐R:L) [Conners et al., 1998]. The defiant/vindictive and irritable dimensions of ODD were defined on theoretical grounds as described elsewhere [Aebi et al., 2015], and reflect two previously described dimensions of ODD [Stringaris et al., 2012; Aebi et al., 2013].
Genotype Data
Genotyping of each sample was performed by each of the four participating groups, individually. To maximize available genetic information among examined datasets, genetic imputation was carried out independently at each site.
aADHD Samples
German sample
Genotyping of participants was performed on Illumina's PsychChip array (Illumina, San Diego, CA) at the Broad Institute (Cambridge, MA) using the PsychChip 15048346 B manifest. Genotypes were assigned in Illumina's GenomeStudio v2010.3, using the calling algorithm/genotyping module version 1.8.4. Quality control procedures were performed as described previously, with lightly modified exclusion criteria (SNPs exhibiting missingness above 98%; minor allele frequency below 5%; failing Hardy–Weinberg equilibrium test [P < 10−4]) [Zayats et al., 2015]. Genotype imputation was performed with SHAPEIT/IMPUTE2 pipeline as described elsewhere, using 1000 Genomes Phase 3 data as a reference [Marchini et al., 2007; Howie et al., 2009; Cross‐Disorder Group of the Psychiatric Genomics, 2013].
Norwegian sample
Participants were genotyped on Human OmniExpress‐12v1‐1_B (Illumina, San Diego, CA) platform at the deCODE Genetics facility (Reykjavik, Iceland). Genotyping and quality control procedures are described elsewhere [Zayats et al., 2015]. Imputation was performed utilizing IMPUTE software as previously detailed [Marchini et al., 2007; Howie et al., 2009; Cross‐Disorder Group of the Psychiatric Genomics, 2013].
Spanish sample
Genome‐wide genotyping was performed with the Illumina HumanOmni1‐Quad BeadChip platform. Quality control was implemented at the individual and SNP level using PLINK and included filtering subjects with low call rate (<98%) or gender discrepancy followed by filtering SNPs with minor allele frequency (MAF) < 0.01, Hardy–Weinberg equilibrium test P‐values < 1e‐06 or call rate < 0.99 in either cases or controls. Imputation was performed using BEAGLE software [Browning and Browning, 2007].
cADHD sample
Sample collection and DNA isolation has been described previously [Brookes et al., 2006]. Genome‐wide genotyping and quality control was performed as part of the GAIN study using the Perlegen 600 K genotyping platform, as previously described [Neale et al., 2008]. The imputation was performed using MACH and the Hapmap 2 (Release 22 Build 36) reference data set [Li et al., 2010]. Quality control was performed on the imputed data, and SNPs with imputation quality scores lower than 0.30, a minor allele frequency lower than 0.01, and those failing the Hardy–Weinberg equilibrium test at a threshold of P ≤ 10−5 were excluded. SNPs and subjects with missingness rates higher than 0.05 were removed from the data.
Statistical Analyses
The age and gender distributions between the aADHD and cADHD samples were assessed using χ2 for gender and ANOVA for age.
Genome‐wide association (GWA) of aggressiveness
In the aADHD sample, single nucleotide polymorphisms (SNPs) were tested for association with the WURS‐derived measure of aggressiveness in the form of linear regression carried out in PLINK using post‐imputation dosage data [Purcell et al., 2007]. Regression models were adjusted for age and sex. Genotype data of each site were first processed individually. The results were then combined with the use of fixed‐effects inverse variance meta‐analysis in METAL [Willer et al., 2010]. Only SNPs with minor allele frequency (MAF) equal to or above 1% and imputation INFO measure equal to or above 0.6 were included in the analyses. Genomic control, QQ plotting, and regional plotting of top loci were applied to check the integrity of test statistics [Devlin and Roeder, 1999; Cuellar‐Partida et al., 2015]. The genomic inflation factor was calculated using METAL [Willer et al., 2010]. A genome‐wide significance threshold of 5.00E‐08 was adopted to correct for multiple testing.
GWA analyses of irritable and defiant/vindictive dimensions of ODD in cADHD sample was performed in PLINK software in the form of linear regression adjusted for sex and age [Purcell et al., 2007]. Details of the analyses are described elsewhere [Aebi et al., 2015].
Gene‐based and Gene‐set association of aggressiveness in the aADHD meta‐analyzed sample
Gene‐based and gene‐set pathway analysis were performed in the aADHD sample carried out in MAGMA software [de Leeuw et al., 2015]. First, a degree of association was calculated for each gene based on METAL‐derived individual SNPs’ P‐values, using 1000 Genomes CEU dataset as a reference panel to correct for linkage disequilibrium (LD) [Genomes Project et al., 2012]. To evaluate each gene's contribution to examined gene‐sets (gene‐set pathway analysis), the P‐value of each gene was converted to a Z‐value and used as an outcome variable in a regression model with gene‐set membership as a predictor. Gene size and gene‐sets’ gene density were added as covariates to adjust for possible confounding effects and prevent spurious association.
For gene‐based tests, we assessed the association with both protein and non‐protein‐coding genes. The protein‐coding gene list was curated from the catalog of known genes downloaded from the Genome Browser of the University of California Santa Cruz (UCSC, CA). The non‐protein‐coding genes were examined in the form of long non‐coding RNA (lncRNA) genes detailed in the aforementioned catalog. For gene‐set pathway analysis, we examined structural categories of gene ontology (GO, http://geneontology.org), with respect to cellular function, biological process and cellular compartments. To achieve meaningful statistics and interpretation of the results, we restricted our pathway analysis to those GO terms that contained SNPs in at least 10 genes per term in our aADHD data.
Genome‐wide enrichment analyses between GWA results in aADHD and cADHD samples
Prior to performing enrichment analyses, the genetic data in both aADHD and cADHD samples were pruned to remove correlated loci in linkage disequilibrium (LD) with each other. A pairwise correlation coefficient (r2) threshold of 0.2 and the 1000 Genomes CEU reference dataset were used to identify independent SNPs, as previously described [Lindgren et al., 2009; Genomes Project et al., 2012].
Enrichment was examined by means of Fisher's test performed in the R software, assessing the difference in proportion of SNPs revealing association P‐values below 0.05 in the cADHD sample according to suggestive association in the aADHD sample (P‐value below or equal to 1.00E‐03 versus P‐value above 1.00E‐03) [Rahmioglu et al., 2015]. Consistency in directionality of SNP effects with indication of enrichment between aADHD and cADHD samples was tested as linear regression on the effect (beta) of each SNP for aADHD as an outcome and for cADHD (either irritable or defiant/vindictive dimensions of ODD, respectively) as predictor variables [Do et al., 2013].
Examination of previously reported aggressiveness‐related candidate GWA loci
We assembled a list of previously reported candidate GWA loci associated with aggressive behavior by systematic literature search the catalog of published genome‐wide association studies provided by National Human Genome Research Institute (NHGRI) (https://www.genome.gov/26525384), using key words of “aggression,” “anger,” “violence,” as well as “conduct disorder” and “antisocial personality disorder.” Each identified candidate GWA locus was then looked up in meta‐analyzed aADHD sample.
RESULTS
Subjects, Measure of Aggressiveness, and GWA Analyses
In total, 1,060 adult patients as well as 750 children and adolescents with ADHD were available for the analyses. The age ranges in the aADHD samples were 17–75 in the German sample, 18–57 in the Norwegian sample, and 17–60 in the Spanish sample. In the cADHD sample, the age range was 5–17. Details of the final samples are summarized in Table I. Supplementary Figure S1 presents the distribution of the selected measure of aggressiveness in each aADHD dataset.
Table I.
Details of the ADHD Patient Samples
| aADHD samples | ||||
|---|---|---|---|---|
| IMpACT site | Number of participants | Females (%) | Age (mean ± SD) | Aggressiveness score (mean ± SD) |
| Germany | 368 | 53.0 | 35.18 ± 10.53 | 11.33 ± 5.17 |
| Norway | 293 | 52.6 | 32.61 ± 11.00 | 12.10 ± 6.39 |
| Spain | 399 | 32.3 | 31.31 ± 12.39 | 10.19 ± 6.15 |
| Total | 1,060 | 45.1 | 33.01 ± 11.51 | 11.11 ± 5.94 |
| cADHD sample | |||||
|---|---|---|---|---|---|
| ODD scores (mean ± SD) | |||||
| Number of participants | Females a (%) | Age b (mean ± SD) | Irritable | Defiant/vindictive | |
| IMAGE | 750 | 12.3 | 10.67 ± 2.77 | 7.75 ± 3.06 | 8.95 ± 4.18 |
SD, standard deviation.
Aggressiveness score was derived from WURS in the aADHD sample. In the cADHD sample, dimensions of oppositionality (irritable and defiant/vindictive dimensions) were examined [Aebi et al., 2015].
Difference in the proportion of females between the aADHD and cADHD samples: P < 2.2E‐16 (χ2 test).
Difference in age between the aADHD and cADHD samples: P < 2.2E‐16 (ANOVA).
After quality control of imputed SNPs in the adult samples, 9.301.568 SNPs were available for the analyses in the German sample, 8.910.491 SNPs in the Norwegian sample, and 6.683.176 SNPs in the Spanish sample. Among these three datasets, 7.576.458 autosomal SNPs were present in at least two and, thus, were meta‐analyzed to assess genetic architecture of childhood aggressiveness in aADHD. In cADHD sample, 1.871.025 autosomal SNPs were available for the analyses.
Individual GWA analyses revealed no genome‐wide significant hits (P ≤ 5.00E‐08) in either aADHD sample (not shown) nor in the cADHD sample (Supplementary Table SI and Fig. S2). None of the variants in the meta‐analysis reached the Bonferroni‐corrected genome‐wide significance level (P ≤ 5.00E‐08) either. The strongest signal was observed at rs10826548 on chromosome 10 located within the transcript of a long noncoding RNA (lncRNA) (beta = −1.66, standard error (SE) = 0.34, P‐value = 1.07E‐06) (Fig. 1), closely followed by rs35974940 in the neurotrimin (NTM) gene (beta = 3.23, SE = 0.67, P‐value = 1.26E‐06) (Fig. 2). Top associated markers (P ≤ 1.00E‐05) are summarized in Supplementary Table SII. The genomic inflation factor was close to one for all individual and meta‐GWA analyses in aADHD. QQ plots of GWA analyses in aADHD are presented in Supplementary Figure S3.
Figure 1.
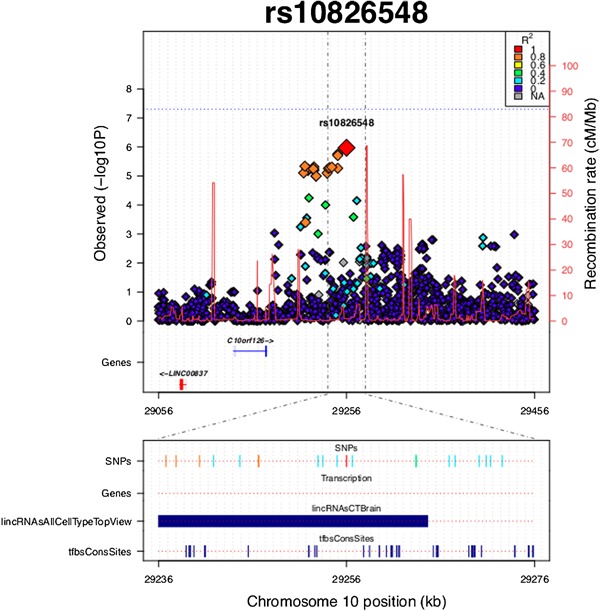
Plot of the locus surrounding rs10826548. SNPs are plotted by position on chromosome 10 against GWA P‐values for aggressive behavior measure in aADHD. Estimated recombination rates from HapMap are plotted in bright red to reflect local LD structure. The SNPs surrounding rs10826548 are color‐coded to reflect their LD with it (according to pair‐wise r2 values from the HapMap CEU database). SNPs with LD r2 ≥ 0.2 are plotted at the bottom of the graph with LD color‐coding specified in the top right corner. “Genes” refers to protein‐coding genes in the presented region. “lincRNAsAllCellTypeTopView” reflects the data from lncRNA USCS track in brain tissue. “tfbsConsSites” reflects the TFBS UCSC track.
Figure 2.
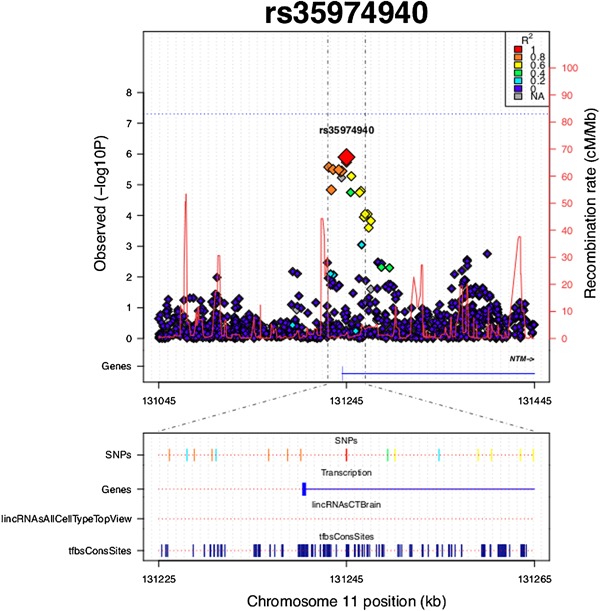
Plot of the locus surrounding rs35974940. SNPs are plotted by position on chromosome 11 against GWA P‐values for aggressive behavior measure in aADHD. Estimated recombination rates from HapMap are plotted in bright red to reflect local LD structure. The SNPs surrounding rs35974940 are color‐coded to reflect their LD with it (according to pair‐wise r2 values from the HapMap CEU database). SNPs with LD r2 ≥ 0.2 are plotted at the bottom of the graph with LD color‐coding specified in the top right corner. “Genes” refers to protein‐coding genes in the presented region. “lincRNAsAllCellTypeTopView” reflects the data from lncRNA USCS track in brain tissue. “tfbsConsSites” reflects the TFBS UCSC track.
Gene‐Based and Gene‐Set Association of Aggressiveness in the aADHD Meta‐Analyzed Sample
Among annotated protein‐coding genes, 17.595 had more than one SNP present in the aADHD data. The strongest signal was noted for the WD repeat domain 62 (WDR62) gene (P‐value = 4.84E‐05). Supplementary Table SIII summarizes the top protein‐coding genes (P ≤ 1.00E‐03) observed in aADHD sample. None of the protein‐coding gene‐based tests survived the correction for multiple testing.
Among lncRNA genes, 22.696 had more than one SNP present in our aADHD data. The strongest association was observed for ENST00000427806 (P‐value = 3.04E‐05). The top lncRNA genes (P ≤ 1.00E‐03) detected in this study are reported in Supplementary Table SIV. None of the non‐protein‐coding gene‐based tests survived the correction for multiple testing.
Among GO pathways, 1.945 terms contained SNPs in at least 10 genes per term in the aADHD data. The most prominent association was observed for negative regulation of I‐kappaB kinase/NF‐kappaB signaling pathway (GO:0043124 term, P‐value = 7.26E‐04). Supplementary Table SV reports top GO terms (P ≤ 0.01) recognized in this study. None of the GO pathways survived the correction for multiple testing.
Genome‐Wide Enrichment Analyses Between GWA Results in aADHD and cADHD Samples
To assess potential genome‐wide overlap of association signals between measures of childhood aggressiveness in aADHD and cADHD, we investigated the independent (r2 < 0.2) GWA signals of suggestive significance (P ≤ 1.00E‐03) in aADHD for enrichment in GWA signals of either defiant/vindictive or irritable dimensions in cADHD. Given our modest sample size, only those SNPs were considered in cADHD results that revealed a P‐value below or equal to 0.05 to avoid the examination of effects with a wide confidence interval. The top GWA SNPs of WURS‐derived childhood aggressiveness in aADHD showed significant enrichment of signals from both the defiant/vindictive dimension (Fisher's P‐value = 2.28E‐06) and the irritable dimension in cADHD GWA analysis (Fisher's P‐value = 0.0061; Fig. 3A).
Figure 3.
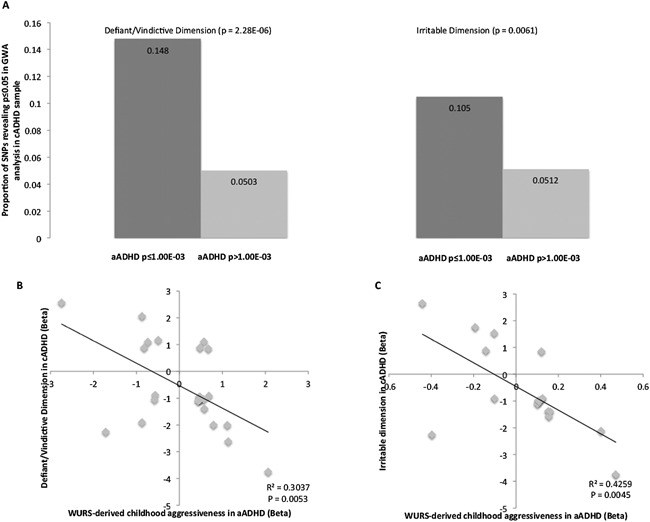
Enrichment and direction of effect among GWA signals of oppositional dimensions in cADHD and WURS‐derived childhood aggressiveness in aADHD. Part A reflects the proportion of SNPs nominally associated (P < 0.05) with each examined oppositional dimension in cADHD (defiant/vindictive and irritable) among suggestive signals (P ≤ 1.00E‐03) of association with childhood aggressiveness in aADHD. Reported P‐values are those of Fisher's exact test. Parts B and C reflect directions of effect of 24 independent nominally significant loci in GWA analyses of defiant/vindictive dimension in cADHD and childhood aggressiveness in aADHD (part B) as well as 17 independent nominally significant loci in GWA analyses of irritable dimension in cADHD and childhood aggressiveness in aADHD (part C). Linear regression r2 measures and P‐values are shown.
Next, we examined the directionality of effects of variants with association signals in both aADHD and cADHD samples (P ≤ 1.00E‐03 in aADHD and P < 0.05 in cADHD). Significant correlation between betas was observed in assessment of both oppositional dimensions in cADHD and childhood aggressiveness in aADHD (P = 0.0053 and 0.0045 for defiant/vindictive and irritable dimensions respectively), but the direction of the relationship was negative (Fig. 3B and C). Supplementary Table SVII summarizes the top hits (P ≤ 1‐00E‐05) observed in GWA meta‐analysis of childhood aggressiveness in aADHD and their corresponding statistics observed in cADHD.
Examination of Previously Reported Aggressiveness‐Related Candidate Genes and GWA Loci
Among previously reported aggressiveness‐related GWA loci, several SNPs noted to be associated with anger, conduct disorder and adult anti‐social personality disorder revealed P‐values below 0.05 in our study (Supplementary Table SVIII). The strongest signal in the GWA analysis of childhood aggressiveness in aADHD among the aforementioned loci was observed for rs4889240 in the PKD1L2 (polycystic kidney disease 1‐like 2) gene (beta = −0.73, SE = 0.25, P‐value = 0.0039), previously reported to be associated with CD symptom count in ADHD patients. The same SNP also revealed nominally significant association in the same direction with the defiant/vindictive dimension (beta = −0.54, SE = 0.21, P‐value = 0.0094), but not with the irritable dimension in cADHD. In this result, one should keep in mind that the cADHD described here is a subsample of the sample in which the original finding for rs4889240 was described [Aebi et al., 2015]. Full results of our literature search are presented in Supplementary Table SVIII.
DISCUSSION
In this study, we performed a genome‐wide exploration of childhood aggressiveness as reported retrospectively by adult patients with ADHD (aADHD), examining both conventional protein‐coding and lncRNA genes. We also explored the overlap with parent‐reported oppositional behavior in youth with ADHD (cADHD) and evaluated previously reported aggression‐related GWA loci. Given our modest sample size (1060 aADHD patients) and the anticipated small effect of common polymorphisms in complex traits, it is not surprising that we did not observe any genome‐wide significant SNPs (P < 5.00E‐08). Nonetheless, we were able to identify several nominally significant variants (P ≤ 1.00E‐05) in biologically interesting genes for follow‐up studies of aggressiveness in ADHD, a feature of the disorder that has received little attention so far.
The strongest signal in the performed single‐point GWA tests of childhood aggressiveness in aADHD was noted for rs10826548 (beta = −1.16, SE = 0.34, P = 1.07E‐06, Supplementary Table SI). This variant resides in the transcript of a lncRNA with uncertain coding potential (TCONS_00018147) (Fig. 1). Non‐protein coding RNAs play a critical role in the regulation of gene expression and have been previously associated with neuropsychiatric disorders, including ADHD [Perkins et al., 2005; Gonzalez‐Giraldo et al., 2015; Zayats et al., 2015]. In addition, it has recently been observed that SNPs previously associated with neurological and psychiatric conditions may be highly concentrated in the regions of long non‐protein coding RNA genes [Ning et al., 2014].
The second most significant locus identified in this study is located within the neurotrimin (NTM) gene (intronic rs35974940, P = 1.26E‐06, Supplementary Table SI and Fig. S2). NTM is a protein‐coding gene, encoding a member of glycosylphosphatidylinositol (GPI)‐anchored cell adhesion molecules, containing immunoglobulin (Ig) domain. These proteins are predominantly expressed in the central nervous system (CNS) [Struyk et al., 1995].
Among the association signals observed in NTM gene, several have the potential to alter its expression. As determined in the TRANSFAC database implemented in the SNPinfo server of the National Institute of Environmental Health Sciences (http://snpinfo.niehs.nih.gov), rs34588147 and rs35665773 (GWA P‐values of 3.59E‐06 and 3.25E‐06, respectively, Supplementary Table SI) are transcription factor binding sites (TFBS) (Fig. 2). Moreover, two other SNPs in high linkage disequilibrium with the aforementioned ones (rs12804059 and rs7119590, r2 = 1 in CEU population) also represent TFBS. Notably, differential expression of NTM between two major brain regions linked to aggression subtypes—prefrontal cortex and amygdala—was observed in early prenatal stage of human brain development (P = 0.015, http://www.brainspan.org).
Gene expression regulation during neuronal development as one of the possible mechanisms behind aggressiveness in aADHD was further affirmed by our top associated lncRNA gene—ENST00000427806 (P = 3.04E‐05, Supplementary Table SIV). The target gene of this lncRNA has been predicted to be the protein‐coding ST6 (alpha‐N‐acetyl‐neuraminyl‐2,3‐beta‐galactosyl‐1,3)‐N‐acetylgalactosaminide alpha‐2,6‐sialyltransferase 5 (ST6GALNAC5) gene [Vucicevic et al., 2015]. The protein encoded by ST6GALNAC5 is a member of sialyltransferases, with reported function in cell adhesion through cell–cell and cell–extracellular matrix interactions [Tsuchida et al., 2003]. Intriguingly, ST6GALNAC5, similarly to NTM, also revealed differential expression in the aggression‐related structures of prefrontal cortex and amygdala in early prenatal stages of human brain development (P = 0.013; http://www.brainspan.org).
As the adult measure of aggressiveness was derived from self‐reported experiences in childhood, we examined the possibility of overlap of its GWA signals with those from GWA analyses of two oppositional dimensions in a cADHD sample. We observed a slight enrichment of association signals between the nominally associated loci in aADHD and those observed in the GWA of both the defiant/vindictive and the irritable ODD dimensions examined in cADHD (Fig. 3). However, it is noteworthy that the aADHD and cADHD samples were imputed using different reference panels with disparate genomic coverage.
Surprisingly, the correlation between the direction of effects of the aforementioned SNPs was negative (Fig. 3B and C). Such an inverse relationship in effect directionality between parent‐reported ODD dimensions and adult retrospective report of childhood aggressiveness is most likely a chance finding due to our study being under‐powered. It might also be related to phenotypic and genetic heterogeneity of the examined samples. There were considerable differences in the percentage of females between the aADHD and the cADHD samples (Table I), which could indicate such mechanisms. It has been shown that both age and sex are important factors in genetic influences in ADHD and aggression [Lyons et al., 1995; Miles and Carey, 1997; Tuvblad and Baker, 2011; Faraone et al., 2015]. In addition, the aggressiveness in the cADHD sample was determined by parent‐report, whereas in the aADHD sample, it was based on retrospective self‐report. The correlation between parent‐report and self‐report has been shown to be generally poor [Achenbach et al., 1987], as also discussed in a recent study that found little overlap between samples of cADHD and aADHD [Moffitt et al., 2015]. Hence, the measures of aggressiveness in the cADHD and the aADHD samples are different. Furthermore, the youth and adult ADHD samples may also be heterogeneous because childhood ADHD does not always persist into adulthood [Faraone et al., 2006; Moffitt et al., 2015]. Thus, to gain better understanding of the genetic overlap between childhood aggression in aADHD and oppositional dimensions in cADHD, this relationship should be examined in larger sample using more rigorous statistical methods, such as those developed to test specifically for genetic correlation among various traits [Yang et al., 2011; Bulik‐Sullivan et al., 2015a,2015b]. This was not possible to implement in the current study due to our modest sample size.
Examination of previously reported aggressiveness‐related GWA loci revealed modest commonality in genetic architecture between the childhood measures of aggressiveness in both cADHD and aADHD, as well as in CD and anti‐social personality disorder (Supplementary Table SVIII). This observation may be in line with formerly reported phenotypic overlap between these conditions, although to which extent this overlap can be transmitted to various subtypes of aggressiveness remains to be determined [Storebo and Simonsen, 2013].
This study should be viewed in light of its limitations. One explanation for not observing any genome‐wide significant loci (P < 5.00E‐08) could be our relatively modest sample size and examination of common variants only (MAF > 1%). This study had 63% power to detect common variants with small effect size of explaining 0.5% of variability under an additive model and an alpha level of 0.05 (http://genome.sph.umich.edu/wiki/Power_Calculations:Quantitative_Traits). This may also be observed in the distribution of the QQ plots (Supplementary Fig. S3).
Another explanation for the lack of significant findings may lay in phenotypic variability. Clinical heterogeneity may weaken true association signals due to the use of different assessment protocols or real genetic heterogeneity among subtypes of ADHD [McClellan and King, 2010]. There are several methodological caveats to assessing aggressiveness [Moffitt et al., 2015]. As our samples consist of outpatients, we investigate a broader and perhaps “softer” aspect of aggressiveness than say, for example, if we were to study prison inmates and/or juvenile offenders. However, this approach provides us with access to the vast majority of aggressive behaviors, which may not come to be written in official records [Moffitt, 2005]. Furthermore, we lack assessment of different subtypes of aggressive behavior that may be related to different genotypes.
Considering the different direction of effects and different measures of aggression in the cADHD and the aADHD samples, analyzing the adult samples and the youth sample together could potentially have obscured the genetic association signal. This is why we refrained from performing meta‐analysis across all samples. Nonetheless, the WURS includes a host of symptoms related to various elements of aggressiveness, which, based on our factor analysis as well as previous research [Ward et al., 1993] seem to be of key importance to the phenotype of aADHD, and the ODD measures have also been validated in previous studies of cADHD [Stringaris et al., 2012; Aebi et al., 2013]. Our approach may add to the discussion of the Negative Valence System in the Research Domain Criteria (RDoC) of the National Institute of Mental Health (NIMH) of how to conceptualize and operationalize aggressiveness as a dimension across different samples and disorders [Verona and Bresin, 2015; Veroude et al., 2015].
We lacked information on current substance abuse in our aADHD sample. Substance abuse is known to be frequently comorbid with ADHD and may confound the relationship between ADHD and current aggressiveness. However, we utilized a retrospective measure of childhood aggressiveness that is likely to reflect behavior over a longer period of time and should, thus, be less affected by volatile environmental influences [Gulberg‐Kjär and Johansson, 2009].
Finally, since the genome‐wide genotyping arrays consist of SNPs only, we were not able to assess the contribution of previously reported variable tandem repeats (e.g., those in MAOA) that were noted to be associated with aggressive behaviors and/or ADHD.
Taken together with evidence from previous studies, our results implicate mechanisms of cell adhesion as well as regulation of gene expression in the etiology of childhood aggressiveness in ADHD. As there is a substantial degree of overlap in aggressiveness among neuropsychiatric disorders, it could be beneficial to analyze conditions where aggression is present together in order to pinpoint biological processes in dysfunctional forms of aggressiveness. Further studies including samples of both children, adolescents and adults, adopting multimodal measures and longitudinal designs are warranted. Such studies may help our understanding as to which extent various subtypes of aggression are mediated by different mechanisms.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Table SI
Supplementary Table SII
Supplementary Table SIII
Supplementary Table SIV
Supplementary Table SV
Supplementary Table SVI
Supplementary Table SVII
Supplementary Table SVIII
supplementary figure S1
supplementary figure S2
supplementary figure S3
ACKNOWLEDGMENTS
This work was supported by the KG Jebsen Foundation for Medical Research, University of Bergen, the Western Norwegian Health Authorities (Helse Vest), European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement no 602805 and H2020 Research and Innovation Program under the Marie Sklodowska‐Curie grant agreement number 643051 (MiND).
The sample used in this study is part of the international multicentre persistent ADHD collaboration (IMpACT). IMpACT unites major research centres working on the genetics of ADHD persistence across the lifespan and has participants in the Netherlands, Germany, Spain, Norway, the United Kingdom, the United States of America, Brazil, and Sweden. Principal investigators of IMpACT are Barbara Franke (chair), Andreas Reif, Stephen V. Faraone, Jan Haavik, Bru Cormand, Antoni Ramos Quiroga, Philip Asherson, Klaus‐Peter Lesch, Jonna Kuntsi, Claiton Bau, Jan Buitelaar, Stefan Johansson, Henrik Larsson, Alysa Doyle, and Eugenio Grevet.
This study was also funded by Deutsche Forschungsgemeinschaft (DFG: KFO 125 and TRR 58/A5 to KPL; DFG RE1632/5‐1 to AR), Fritz Thyssen Foundation (Az.10.13.1185 to KPL and OR), and Jacobs Foundation (to OR).
Dr. Cormand received financial support from the Spanish “Ministerio de Economía y Competitividad” (SAF2012‐33484) and AGAUR (2014SGR932).
Marta Ribasés is a recipient of a Miguel de Servet contract from the “Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación,” Spain. Financial support was received from “Instituto de Salud Carlos III‐FIS,” Spain (PI11/00571, PI11/01629, PI12/01139, PI14/01700), “Agència de Gestió d'Ajuts Universitaris i de Recerca‐AGAUR Generalitat de Catalunya” (2014SGR1357), and “Departament de Salut,” Government of Catalonia, Spain.
The International Multisite ADHD Genetics (IMAGE) project is a multi‐site, international effort. Funding support for the IMAGE project was provided by NIH grants R01MH62873 and R01MH081803 to SV. Faraone and the genotyping of samples was provided through the Genetic Association Information Network (GAIN). The dataset used for the analyses described in this manuscript were obtained from the database of Genotypes and Phenotypes (dbGaP) found at http://www.ncbi.nlm.nih.gov/gap through dbGaP accession number #20726‐2. Samples and associated phenotype data for the IMAGE Project were provided by the principal investigators Stephen V. Faraone, Philip Asherson, Tobias Banaschewski, Jan Buitelaar, Richard P. Ebstein, Michael Gill, Ana Miranda, Fernando Mulas, Robert D. Oades, Herbert Roeyers, Aribert Rothenberger, Joseph Sergeant, Edmund Sonuga‐Barke, and Hans‐Christoph Steinhausen, the chief investigators at each site, Rafaela Marco, Nanda Rommelse, Wai Chen, Henrik Uebel, Hanna Christiansen, Ueli C. Mueller, Cathelijne Buschgens, Barbara Franke, and Margaret Thompson, as well as the statistical analysis team, M. Daly, C. Lange, N. Laird, J. Su, and B. Neale.
We wish to thank the support received from the European College of Neuropsychopharmacology (ECNP network: “ADHD across the lifespan”).
Brevik EJ, van Donkelaar MMJ, Weber H, Sánchez‐Mora C, Jacob C, Rivero O, Kittel‐Schneider S, Garcia‐Martínez I, Aebi M, van Hulzen K, Cormand B, Ramos‐Quiroga JA, IMAGE Consortium . Lesch K‐P, Reif A, Ribasés M, Franke B, Posserud M‐B, Johansson S, Lundervold AJ, Haavik J, Zayats T. 2016. Genome‐Wide Analyses of Aggressiveness in Attention‐Deficit Hyperactivity Disorder. Am J Med Genet Part B 171B:733–747.
REFERENCES
- Achenbach TM, McConaughy SH, Howell CT. 1987. Child/adolescent behavioral and emotional problems: Implications of cross‐informant correlations for situational specificity. Psychol Bull 101(2):213–232. [PubMed] [Google Scholar]
- Aebi M, Plattner B, Metzke CW, Bessler C, Steinhausen HC. 2013. Parent‐ and self‐reported dimensions of oppositionality in youth: Construct validity, concurrent validity, and the prediction of criminal outcomes in adulthood. J Child Psychol Psychiatry 54(9):941–949. [DOI] [PubMed] [Google Scholar]
- Aebi M, vanDonkelaar MM, Poelmans G, Buitelaar JK, Sonuga‐Barke EJ, Stringaris A, I Consortium, Faraone SV, Franke B, Steinhausen HC, vanHulzen KJ. 2015. Gene‐set and multivariate genome‐wide association analysis of oppositional defiant behavior subtypes in attention‐deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 9999:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CA, Bushman BJ. 2002. Human aggression. Annu Rev Psychol 53(1):27–51. [DOI] [PubMed] [Google Scholar]
- Anney RJ, Lasky‐Su J, O'Dushlaine C, Kenny E, Neale BM, Mulligan A, Franke B, Zhou K, Chen W, Christiansen H, Arias‐Vasquez A, Banaschewski T, Buitelaar J, Ebstein R, Miranda A, Mulas F, Oades RD, Roeyers H, Rothenberger A, Sergeant J, Sonuga‐Barke E, Steinhausen H, Asherson P, Faraone SV, Gill M. 2008. Conduct disorder and ADHD: Evaluation of conduct problems as a categorical and quantitative trait in the international multicentre ADHD genetics study. Am J Med Genet B Neuropsychiatr Genet 147B(8):1369–1378. [DOI] [PubMed] [Google Scholar]
- Biederman J, Faraone SV, Keenan K, Benjamin J, Krifcher B, Moore C, Sprich‐Buckminster S, Ugaglia K, Jellinek MS, Steingard R, et al. 1992. Further evidence for family‐genetic risk factors in attention deficit hyperactivity disorder. Patterns of comorbidity in probands and relatives psychiatrically and pediatrically referred samples. Arch Gen Psychiatry 49(9):728–738. [DOI] [PubMed] [Google Scholar]
- Brendgen M, Vitaro F, Boivin M, Dionne G, Perusse D. 2006. Examining genetic and environmental effects on reactive versus proactive aggression. Dev Psychol 42(6):1299–1312. [DOI] [PubMed] [Google Scholar]
- Brookes K, Xu X, Chen W, Zhou K, Neale B, Lowe N, Anney R, Franke B, Gill M, Ebstein R, Buitelaar J, Sham P, Campbell D, Knight J, Andreou P, Altink M, Arnold R, Boer F, Buschgens C, Butler L, Christiansen H, Feldman L, Fleischman K, Fliers E, Howe‐Forbes R, Goldfarb A, Heise A, Gabriels I, Korn‐Lubetzki I, Johansson L, Marco R, Medad S, Minderaa R, Mulas F, Muller U, Mulligan A, Rabin K, Rommelse N, Sethna V, Sorohan J, Uebel H, Psychogiou L, Weeks A, Barrett R, Craig I, Banaschewski T, Sonuga‐Barke E, Eisenberg J, Kuntsi J, Manor I, McGuffin P, Miranda A, Oades RD, Plomin R, Roeyers H, Rothenberger A, Sergeant J, Steinhausen HC, Taylor E, Thompson M, Faraone SV, Asherson P. 2006. The analysis of 51 genes in DSM‐IV combined type attention deficit hyperactivity disorder: Association signals in DRD4, DAT1 and 16 other genes. Mol Psychiatry 11(10):934–953. [DOI] [PubMed] [Google Scholar]
- Browning SR, Browning BL. 2007. Rapid and accurate haplotype phasing and missing‐data inference for whole‐genome association studies by use of localized haplotype clustering. Am J Hum Genet 81(5):1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik‐Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, ReproGen C, C Psychiatric Genomics, C Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control, Duncan L, Perry JR, Patterson N, Robinson EB, Daly MJ, Price AL, Neale BM. 2015a. An atlas of genetic correlations across human diseases and traits. Nat Genet 47(11):1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik‐Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, C Schizophrenia Working Group of the Psychiatric Genomics, Patterson N, Daly MJ, Price AL, Neale BM. 2015b. LD Score regression distinguishes confounding from polygenicity in genome‐wide association studies. Nat Genet 47(3):291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conners CK, Sitarenios G, Parker JD, Epstein JN. 1998. The revised Conners’ parent rating scale (CPRS‐R): Factor structure, reliability, and criterion validity. J Abnorm Child Psychol 26(4):257–268. [DOI] [PubMed] [Google Scholar]
- Cross‐Disorder Group of the Psychiatric Genomics C. 2013. Identification of risk loci with shared effects on five major psychiatric disorders: A genome‐wide analysis. Lancet 381(9875):1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross‐Disorder Group of the Psychiatric Genomics C, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, Witte JS, Absher D, Agartz I, Akil H, Amin F, Andreassen OA, Anjorin A, Anney R, Anttila V, Arking DE, Asherson P, Azevedo MH, Backlund L, Badner JA, Bailey AJ, Banaschewski T, Barchas JD, Barnes MR, Barrett TB, Bass N, Battaglia A, Bauer M, Bayes M, Bellivier F, Bergen SE, Berrettini W, Betancur C, Bettecken T, Biederman J, Binder EB, Black DW, Blackwood DH, Bloss CS, Boehnke M, Boomsma DI, Breen G, Breuer R, Bruggeman R, Cormican P, Buccola NG, Buitelaar JK, Bunney WE, Buxbaum JD, Byerley WF, Byrne EM, Caesar S, Cahn W, Cantor RM, Casas M, Chakravarti A, Chambert K, Choudhury K, Cichon S, Cloninger CR, Collier DA, Cook EH, Coon H, Cormand B, Corvin A, Coryell WH, Craig DW, Craig IW, Crosbie J, Cuccaro ML, Curtis D, Czamara D, Datta S, Dawson G, Day R, De Geus EJ, Degenhardt F, Djurovic S, Donohoe GJ, Doyle AE, Duan J, Dudbridge F, Duketis E, Ebstein RP, Edenberg HJ, Elia J, Ennis S, Etain B, Fanous A, Farmer AE, Ferrier IN, Flickinger M, Fombonne E, Foroud T, Frank J, Franke B, Fraser C, Freedman R, Freimer NB, Freitag CM, Friedl M, Frisen L, Gallagher L, Gejman PV, Georgieva L, Gershon ES, Geschwind DH, Giegling I, Gill M, Gordon SD, Gordon‐Smith K, Green EK, Greenwood TA, Grice DE, Gross M, Grozeva D, Guan W, Gurling H, De Haan L, Haines JL, Hakonarson H, Hallmayer J, Hamilton SP, Hamshere ML, Hansen TF, Hartmann AM, Hautzinger M, Heath AC, Henders AK, Herms S, Hickie IB, Hipolito M, Hoefels S, Holmans PA, Holsboer F, Hoogendijk WJ, Hottenga JJ, Hultman CM, Hus V, Ingason A, Ising M, Jamain S, Jones EG, Jones I, Jones L, Tzeng JY, Kahler AK, Kahn RS, Kandaswamy R, Keller MC, Kennedy JL, Kenny E, Kent L, Kim Y, Kirov GK, Klauck SM, Klei L, Knowles JA, Kohli MA, Koller DL, Konte B, Korszun A, Krabbendam L, Krasucki R, Kuntsi J, Kwan P, Landen M, Langstrom N, Lathrop M, Lawrence J, Lawson WB, Leboyer M, Ledbetter DH, Lee PH, Lencz T, Lesch KP, Levinson DF, Lewis CM, Li J, Lichtenstein P, Lieberman JA, Lin DY, Linszen DH, Liu C, Lohoff FW, Loo SK, Lord C, Lowe JK, Lucae S, MacIntyre DJ, Madden PA, Maestrini E, Magnusson PK, Mahon PB, Maier W, Malhotra AK, Mane SM, Martin CL, Martin NG, Mattheisen M, Matthews K, Mattingsdal M, McCarroll SA, McGhee KA, McGough JJ, McGrath PJ, McGuffin P, McInnis MG, McIntosh A, McKinney R, McLean AW, McMahon FJ, McMahon WM, McQuillin A, Medeiros H, Medland SE, Meier S, Melle I, Meng F, Meyer J, Middeldorp CM, Middleton L, Milanova V, Miranda A, Monaco AP, Montgomery GW, Moran JL, Moreno‐De‐Luca D, Morken G, Morris DW, Morrow EM, Moskvina V, Muglia P, Muhleisen TW, Muir WJ, Muller‐Myhsok B, Murtha M, Myers RM, Myin‐Germeys I, Neale MC, Nelson SF, Nievergelt CM, Nikolov I, Nimgaonkar V, Nolen WA, Nothen MM, Nurnberger JI, Nwulia EA, Nyholt DR, O'Dushlaine C, Oades RD, Olincy A, Oliveira G, Olsen L, Ophoff RA, Osby U, Owen MJ, Palotie A, Parr JR, Paterson AD, Pato CN, Pato MT, Penninx BW, Pergadia ML, Pericak‐Vance MA, Pickard BS, Pimm J, Piven J, Posthuma D, Potash JB, Poustka F, Propping P, Puri V, Quested DJ, Quinn EM, Ramos‐Quiroga JA, Rasmussen HB, Raychaudhuri S, Rehnstrom K, Reif A, Ribases M, Rice JP, Rietschel M, Roeder K, Roeyers H, Rossin L, Rothenberger A, Rouleau G, Ruderfer D, Rujescu D, Sanders AR, Sanders SJ, Santangelo SL, Sergeant JA, Schachar R, Schalling M, Schatzberg AF, Scheftner WA, Schellenberg GD, Scherer SW, Schork NJ, Schulze TG, Schumacher J, Schwarz M, Scolnick E, Scott LJ, Shi J, Shilling PD, Shyn SI, Silverman JM, Slager SL, Smalley SL, Smit JH, Smith EN, Sonuga‐Barke EJ, StClair D, State M, Steffens M, Steinhausen HC, Strauss JS, Strohmaier J, Stroup TS, Sutcliffe JS, Szatmari P, Szelinger S, Thirumalai S, Thompson RC, Todorov AA, Tozzi F, Treutlein J, Uhr M, vandenOord EJ, VanGrootheest G, VanOs J, Vicente AM, Vieland VJ, Vincent JB, Visscher PM, Walsh CA, Wassink TH, Watson SJ, Weissman MM, Werge T, Wienker TF, Wijsman EM, Willemsen G, Williams N, Willsey AJ, Witt SH, Xu W, Young AH, Yu TW, Zammit S, Zandi PP, Zhang P, Zitman FG, Zollner S, C International Inflammatory Bowel Disease Genetics, Devlin B, Kelsoe JR, Sklar P, Daly MJ, O'Donovan MC, Craddock N, Sullivan PF, Smoller JW, Kendler KS, Wray NR. 2013. Genetic relationship between five psychiatric disorders estimated from genome‐wide SNPs. Nat Genet 45(9):984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar‐Partida G, Renteria ME, MacGregor S. 2015. LocusTrack: Integrated visualization of GWAS results and genomic annotation. Source Code Biol Med 10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard S, Mortensen PB, Frydenberg M, Thomsen PH. 2002. Conduct problems, gender and adult psychiatric outcome of children with attention‐deficit hyperactivity disorder. Br J Psychiatry 181:416–421. [DOI] [PubMed] [Google Scholar]
- de Leeuw CA, Mooij JM, Heskes T, Posthuma D. 2015. MAGMA: Generalized gene‐set analysis of GWAS data. PLoS Comput Biol 11(4):e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin B, Roeder K. 1999. Genomic control for association studies. Biometrics 55(4):997–1004. [DOI] [PubMed] [Google Scholar]
- Dick DM, Aliev F, Krueger RF, Edwards A, Agrawal A, Lynskey M, Lin P, Schuckit M, Hesselbrock V, Nurnberger J Jr., Almasy L, Porjesz B, Edenberg HJ, Bucholz K, Kramer J, Kuperman S, Bierut L. 2011. Genome‐wide association study of conduct disorder symptomatology. Mol Psychiatry 16(8):800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg‐Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkila K, Hypponen E, Isaacs A, Jackson AU, Johansson A, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikainen LP, Magnusson PK, Mangino M, Mihailov E, Montasser ME, Muller‐Nurasyid M, Nolte IM, O'Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney AS, Doring A, Elliott P, Epstein SE, Eyjolfsson GI, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJ, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimaki T, Lin SY, Lindstrom J, Loos RJ, Mach F, McArdle WL, Meisinger C, Mitchell BD, Muller G, Nagaraja R, Narisu N, Nieminen TV, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stancakova A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YD, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrieres J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Jarvelin MR, Jula A, Kahonen M, Kaprio J, Kesaniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, Marz W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njolstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PE, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BH, Altshuler D, Ordovas JM, Boerwinkle E, Palmer CN, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Mohlke KL, Ingelsson E, Abecasis GR, Daly MJ, Neale BM, Kathiresan S. 2013. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet 45(11):1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraone SV, Asherson P, Banaschewski T, Biederman J, Buitelaar J, Ramos‐Quiroga JA, Rohde LA, Sonuga‐Barke E, Tannock R, Franke B. 2015. Attention‐deficit/hyperactivity disorder. Nat Rev Dis Primers 1:1–23. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Keenan K, Tsuang MT. 1991. A family‐genetic study of girls with DSM‐III attention deficit disorder. Am J Psychiatry 148(1):112–117. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Mick E. 2006. The age‐dependent decline of attention deficit hyperactivity disorder: A meta‐analysis of follow‐up studies. Psychol Med 36(2):159–165. [DOI] [PubMed] [Google Scholar]
- Franke B, Vasquez AA, Johansson S, Hoogman M, Romanos J, Boreatti‐Hummer A, Heine M, Jacob CP, Lesch KP, Casas M, Ribases M, Bosch R, Sanchez‐Mora C, Gomez‐Barros N, Fernandez‐Castillo N, Bayes M, Halmoy A, Halleland H, Landaas ET, Fasmer OB, Knappskog PM, Heister AJ, Kiemeney LA, Kooij JJ, Boonstra AM, Kan CC, Asherson P, Faraone SV, Buitelaar JK, Haavik J, Cormand B, Ramos‐Quiroga JA, Reif A. 2010. Multicenter analysis of the SLC6A3/DAT1 VNTR haplotype in persistent ADHD suggests differential involvement of the gene in childhood and persistent ADHD. Neuropsychopharmacology 35(3):656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491(7422):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizer IR, Ficks C, Waldman ID. 2009. Candidate gene studies of ADHD: A meta‐analytic review. Hum Genet 126(1):51–90. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Giraldo Y, Camargo A, Lopez‐Leon S, Adan A, Forero DA. 2015. A functional SNP in MIR124‐1, a brain expressed miRNA gene, is associated with aggressiveness in a Colombian sample. Eur Psychiatry 30(4):499–503. [DOI] [PubMed] [Google Scholar]
- Gulberg‐Kjär T, Johansson B. 2009. Old people reporting childhood AD/HD symptoms: Retrospectively self‐rated AD/HD symptoms in a population‐based Swedish sample aged 65–80. Nordic J Psychiatry 63(5):375–382. [DOI] [PubMed] [Google Scholar]
- Halmoy A, Fasmer OB, Gillberg C, Haavik J. 2009. Occupational outcome in adult ADHD: Impact of symptom profile, comorbid psychiatric problems, and treatment: A cross‐sectional study of 414 clinically diagnosed adult ADHD patients. J Atten Disord 13(2):175–187. [DOI] [PubMed] [Google Scholar]
- Halmoy A, Johansson S, Winge I, McKinney JA, Knappskog PM, Haavik J. 2010. Attention‐deficit/hyperactivity disorder symptoms in offspring of mothers with impaired serotonin production. Arch Gen Psychiatry 67(10):1033–1043. [DOI] [PubMed] [Google Scholar]
- Hamshere ML, Langley K, Martin J, Agha SS, Stergiakouli E, Anney RJ, Buitelaar J, Faraone SV, Lesch KP, Neale BM, Franke B, Sonuga‐Barke E, Asherson P, Merwood A, Kuntsi J, Medland SE, Ripke S, Steinhausen HC, Freitag C, Reif A, Renner TJ, Romanos M, Romanos J, Warnke A, Meyer J, Palmason H, Vasquez AA, Lambregts‐Rommelse N, Roeyers H, Biederman J, Doyle AE, Hakonarson H, Rothenberger A, Banaschewski T, Oades RD, McGough JJ, Kent L, Williams N, Owen MJ, Holmans P, O'Donovan MC, Thapar A. 2013. High loading of polygenic risk for ADHD in children with comorbid aggression. Am J Psychiatry 170(8):909–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofvander B, Ossowski D, Lundstrom S, Anckarsater H. 2009. Continuity of aggressive antisocial behavior from childhood to adulthood: The question of phenotype definition. Int J Law Psychiatry 32(4):224–234. [DOI] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. 2009. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 5(6):e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Palacio LG, Castellanos FX, Palacio JD, Pineda D, Restrepo MI, Munoz JF, Lopera F, Wallis D, Berg K, Bailey‐Wilson JE, Arcos‐Burgos M, Muenke M. 2007. Attention‐deficit/hyperactivity disorder and comorbid disruptive behavior disorders: Evidence of pleiotropy and new susceptibility loci. Biol Psychiatry 61(12):1329–1339. [DOI] [PubMed] [Google Scholar]
- Klassen LJ, Katzman MA, Chokka P. 2010. Adult ADHD and its comorbidities, with a focus on bipolar disorder. J Affect Disord 124(1–2):1–8. [DOI] [PubMed] [Google Scholar]
- Larsson H, Chang Z, D'Onofrio BM, Lichtenstein P. 2013. The heritability of clinically diagnosed attention deficit hyperactivity disorder across the lifespan. Psychol Med 44(10):2223–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch KP, Araragi N, Waider J, van den Hove D, Gutknecht L. 2012. Targeting brain serotonin synthesis: Insights into neurodevelopmental disorders with long‐term outcomes related to negative emotionality, aggression and antisocial behaviour. Philos Trans R Soc Lond B Biol Sci 367(1601):2426–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer J, Ding J, Scheet P, Abecasis GR. 2010. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34(8):816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren CM, Heid IM, Randall JC, Lamina C, Steinthorsdottir V, Qi L, Speliotes EK, Thorleifsson G, Willer CJ, Herrera BM, Jackson AU, Lim N, Scheet P, Soranzo N, Amin N, Aulchenko YS, Chambers JC, Drong A, Luan J, Lyon HN, Rivadeneira F, Sanna S, Timpson NJ, Zillikens MC, Zhao JH, Almgren P, Bandinelli S, Bennett AJ, Bergman RN, Bonnycastle LL, Bumpstead SJ, Chanock SJ, Cherkas L, Chines P, Coin L, Cooper C, Crawford G, Doering A, Dominiczak A, Doney AS, Ebrahim S, Elliott P, Erdos MR, Estrada K, Ferrucci L, Fischer G, Forouhi NG, Gieger C, Grallert H, Groves CJ, Grundy S, Guiducci C, Hadley D, Hamsten A, Havulinna AS, Hofman A, Holle R, Holloway JW, Illig T, Isomaa B, Jacobs LC, Jameson K, Jousilahti P, Karpe F, Kuusisto J, Laitinen J, Lathrop GM, Lawlor DA, Mangino M, McArdle WL, Meitinger T, Morken MA, Morris AP, Munroe P, Narisu N, Nordstrom A, Nordstrom P, Oostra BA, Palmer CN, Payne F, Peden JF, Prokopenko I, Renstrom F, Ruokonen A, Salomaa V, Sandhu MS, Scott LJ, Scuteri A, Silander K, Song K, Yuan X, Stringham HM, Swift AJ, Tuomi T, Uda M, Vollenweider P, Waeber G, Wallace C, Walters GB, Weedon MN, C Wellcome Trust Case Control, Witteman JC, Zhang C, Zhang W, Caulfield MJ, Collins FS, Davey Smith G, Day IN, Franks PW, Hattersley AT, Hu FB, Jarvelin MR, Kong A, Kooner JS, Laakso M, Lakatta E, Mooser V, Morris AD, Peltonen L, Samani NJ, Spector TD, Strachan DP, Tanaka T, Tuomilehto J, Uitterlinden AG, van Duijn CM, Wareham NJ, Hugh W, Procardis C, Waterworth DM, Boehnke M, Deloukas P, Groop L, Hunter DJ, Thorsteinsdottir U, Schlessinger D, Wichmann HE, Frayling TM, Abecasis GR, Hirschhorn JN, Loos RJ, Stefansson K, Mohlke KL, Barroso I, McCarthy MI, Giant C. 2009. Genome‐wide association scan meta‐analysis identifies three loci influencing adiposity and fat distribution. PLoS Genet 5(6):e1000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MJ, True WR, Eisen SA, Goldberg J, Meyer JM, Faraone SV, Eaves LJ, Tsuang MT. 1995. Differential heritability of adult and juvenile antisocial traits. Arch Gen Psychiatry 52(11):906–915. [DOI] [PubMed] [Google Scholar]
- Marchini J, Howie B, Myers S, McVean G, Donnelly P. 2007. A new multipoint method for genome‐wide association studies by imputation of genotypes. Nat Genet 39(7):906–913. [DOI] [PubMed] [Google Scholar]
- McClellan J, King MC. 2010. Genetic heterogeneity in human disease. Cell 141(2):210–217. [DOI] [PubMed] [Google Scholar]
- McGuire J. 2008. A review of effective interventions for reducing aggression and violence. Philos Trans R Soc Lond B Biol Sci 363(1503):2577–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney J, Johansson S, Halmoy A, Dramsdahl M, Winge I, Knappskog PM, Haavik J. 2008. A loss‐of‐function mutation in tryptophan hydroxylase 2 segregating with attention‐deficit/hyperactivity disorder. Mol Psychiatry 13(4):365–367. [DOI] [PubMed] [Google Scholar]
- Mick E, McGough J, Deutsch CK, Frazier JA, Kennedy D, Goldberg RJ. 2014. Genome‐wide association study of proneness to anger. PLoS ONE 9(1):e87257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles DR, Carey G. 1997. Genetic and environmental architecture of human aggression. J Pers Soc Psychol 72(1):207–217. [DOI] [PubMed] [Google Scholar]
- Moffitt TE. 2005. The new look of behavioral genetics in developmental psychopathology: Gene‐environment interplay in antisocial behaviors. Psychol Bull 131(4):533–554. [DOI] [PubMed] [Google Scholar]
- Moffitt TE, Houts R, Asherson P, Belsky DW, Corcoran DL, Hammerle M, Harrington H, Hogan S, Meier MH, Polanczyk GV, Poulton R, Ramrakha S, Sugden K, Williams B, Rohde LA, Caspi A. 2015. Is adult ADHD a childhood‐onset neurodevelopmental disorder? Evidence from a four‐decade longitudinal cohort study. Am J Psychiatry 172(10):967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller UC, Asherson P, Banaschewski T, Buitelaar JK, Ebstein RP, Eisenberg J, Gill M, Manor I, Miranda A, Oades RD, Roeyers H, Rothenberger A, Sergeant JA, Sonuga‐Barke EJ, Thompson M, Faraone SV, Steinhausen HC. 2011a. The impact of study design and diagnostic approach in a large multi‐centre ADHD study: Part 2: Dimensional measures of psychopathology and intelligence. BMC Psychiatry 11:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller UC, Asherson P, Banaschewski T, Buitelaar JK, Ebstein RP, Eisenberg J, Gill M, Manor I, Miranda A, Oades RD, Roeyers H, Rothenberger A, Sergeant JA, Sonuga‐Barke EJ, Thompson M, Faraone SV, Steinhausen HC. 2011b. The impact of study design and diagnostic approach in a large multi‐centre ADHD study. Part 1: ADHD symptom patterns. BMC Psychiatry 11:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Lasky‐Su J, Anney R, Franke B, Zhou K, Maller JB, Vasquez AA, Asherson P, Chen W, Banaschewski T, Buitelaar J, Ebstein R, Gill M, Miranda A, Oades RD, Roeyers H, Rothenberger A, Sergeant J, Steinhausen HC, Sonuga‐Barke E, Mulas F, Taylor E, Laird N, Lange C, Daly M, Faraone SV. 2008. Genome‐wide association scan of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 147B(8):1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning S, Zhao Z, Ye J, Wang P, Zhi H, Li R, Wang T, Li X. 2014. LincSNP: A database of linking disease‐associated SNPs to human large intergenic non‐coding RNAs. BMC Bioinformatics 15:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DO, Jeffries C, Sullivan P. 2005. Expanding the ‘central dogma’: The regulatory role of nonprotein coding genes and implications for the genetic liability to schizophrenia. Mol Psychiatry 10(1):69–78. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. 2007. PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmioglu N, Macgregor S, Drong AW, Hedman AK, Harris HR, Randall JC, Prokopenko I, TGC International Endogene Consortium, Nyholt DR, Morris AP, Montgomery GW, Missmer SA, Lindgren CM, Zondervan KT. 2015. Genome‐wide enrichment analysis between endometriosis and obesity‐related traits reveals novel susceptibility loci. Hum Mol Genet 24(4):1185–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez JM, Andreu JM. 2006. Aggression, and some related psychological constructs (anger, hostility, and impulsivity); some comments from a research project. Neurosci Biobehav Rev 30(3):276–291. [DOI] [PubMed] [Google Scholar]
- Reif A, Jacob CP, Rujescu D, Herterich S, Lang S, Gutknecht L, Baehne CG, Strobel A, Freitag CM, Giegling I, Romanos M, Hartmann A, Rosler M, Renner TJ, Fallgatter AJ, Retz W, Ehlis AC, Lesch KP. 2009. Influence of functional variant of neuronal nitric oxide synthase on impulsive behaviors in humans. Arch Gen Psychiatry 66(1):41–50. [DOI] [PubMed] [Google Scholar]
- Salvatore JE, Edwards AC, McClintick JN, Bigdeli TB, Adkins A, Aliev F, Edenberg HJ, Foroud T, Hesselbrock V, Kramer J, Nurnberger JI, Schuckit M, Tischfield JA, Xuei X, Dick DM. 2015. Genome‐wide association data suggest ABCB1 and immune‐related gene sets may be involved in adult antisocial behavior. Transl Psychiatry 5:e558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Mora C, Ramos‐Quiroga JA, Bosch R, Corrales M, Garcia‐Martinez I, Nogueira M, Pagerols M, Palomar G, Richarte V, Vidal R, Arias‐Vasquez A, Bustamante M, Forns J, Gross‐Lesch S, Guxens M, Hinney A, Hoogman M, Jacob C, Jacobsen KK, Kan CC, Kiemeney L, Kittel‐Schneider S, Klein M, Onnink M, Rivero O, Zayats T, Buitelaar J, Faraone SV, Franke B, Haavik J, Johansson S, Lesch KP, Reif A, Sunyer J, Bayes M, Casas M, Cormand B, Ribases M. 2015. Case‐control genome‐wide association study of persistent attention‐deficit hyperactivity disorder identifies FBXO33 as a novel susceptibility gene for the disorder. Neuropsychopharmacology 40(4):915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics C. 2014. Biological insights from 108 schizophrenia‐associated genetic loci. Nature 511(7510):421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seroczynski AD, Bergeman CS, Coccaro EF. 1999. Etiology of the impulsivity/aggression relationship: Genes or environment? Psychiatry Res 86(1):41–57. [DOI] [PubMed] [Google Scholar]
- Storebo OJ, Simonsen E. 2013. The association between ADHD and antisocial personality disorder (ASPD): A review. J Atten Disord XX(X):1–10. [DOI] [PubMed] [Google Scholar]
- Stringaris A, Zavos H, Leibenluft E, Maughan B, Eley TC. 2012. Adolescent irritability: Phenotypic associations and genetic links with depressed mood. Am J Psychiatry 169(1):47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struyk AF, Canoll PD, Wolfgang MJ, Rosen CL, D'Eustachio P, Salzer JL. 1995. Cloning of neurotrimin defines a new subfamily of differentially expressed neural cell adhesion molecules. J Neurosci 15(3 Pt 2):2141–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tielbeek JJ, Medland SE, Benyamin B, Byrne EM, Heath AC, Madden PA, Martin NG, Wray NR, Verweij KJ. 2012. Unraveling the genetic etiology of adult antisocial behavior: A genome‐wide association study. PLoS ONE 7(10):e45086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiihonen J, Rautiainen MR, Ollila HM, Repo‐Tiihonen E, Virkkunen M, Palotie A, Pietilainen O, Kristiansson K, Joukamaa M, Lauerma H, Saarela J, Tyni S, Vartiainen H, Paananen J, Goldman D, Paunio T. 2014. Genetic background of extreme violent behavior. Mol Psychiatry 20:786–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida A, Okajima T, Furukawa K, Ando T, Ishida H, Yoshida A, Nakamura Y, Kannagi R, Kiso M, Furukawa K. 2003. Synthesis of disialyl Lewis a (Le(a)) structure in colon cancer cell lines by a sialyltransferase, ST6GalNAc VI, responsible for the synthesis of alpha‐series gangliosides. J Biol Chem 278(25):22787–22794. [DOI] [PubMed] [Google Scholar]
- Tuvblad C, Baker LA. 2011. Human aggression across the lifespan: Genetic propensities and environmental moderators. Adv Genet 75:171–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuvblad C, Zheng M, Raine A, Baker LA. 2009. A common genetic factor explains the covariation among ADHD ODD and CD symptoms in 9‐10 year old boys and girls. J Abnorm Child Psychol 37(2):153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassos E, Collier DA, Fazel S. 2014. Systematic meta‐analyses and field synopsis of genetic association studies of violence and aggression. Mol Psychiatry 19(4):471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verona E, Bresin K. 2015. Aggression proneness: Transdiagnostic processes involving negative valence and cognitive systems. Int J Psychophysiol 98(2 Pt 2):321–329. [DOI] [PubMed] [Google Scholar]
- Veroude K, Zhang‐James Y, Fernandez‐Castillo N, Bakker MJ, Cormand B, Faraone SV. 2015. Genetics of aggressive behavior: An overview. Am J Med Genet B Neuropsychiatr Genet 171B:3–43. [DOI] [PubMed] [Google Scholar]
- Vucicevic D, Corradin O, Ntini E, Scacheri PC, Orom UA. 2015. Long ncRNA expression associates with tissue‐specific enhancers. Cell Cycle 14(2):253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MF, Wender PH, Reimherr FW. 1993. The Wender Utah Rating Scale: An aid in the retrospective diagnosis of childhood attention deficit hyperactivity disorder. Am J Psychiatry 150(6):885–890. [DOI] [PubMed] [Google Scholar]
- WHO. 2008. The global burden of disease: 2004 update. Geneva: World Health Organisation. [Google Scholar]
- Willer CJ, Li Y, Abecasis GR. 2010. METAL: Fast and efficient meta‐analysis of genomewide association scans. Bioinformatics 26(17):2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. 2011. GCTA: A tool for genome‐wide complex trait analysis. Am J Hum Genet 88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S, Moss D, Sedgwick O, Fridman M, Hodgkins P. 2014. A meta‐analysis of the prevalence of attention deficit hyperactivity disorder in incarcerated populations. Psychol Med 45(2):247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayats T, Athanasiu L, Sonderby I, Djurovic S, Westlye LT, Tamnes CK, Fladby T, Aase H, Zeiner P, Reichborn‐Kjennerud T, Knappskog PM, Knudsen GP, Andreassen OA, Johansson S, Haavik J. 2015. Genome‐wide analysis of attention deficit hyperactivity disorder in norway. PLoS ONE 10(4):e0122501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Table SI
Supplementary Table SII
Supplementary Table SIII
Supplementary Table SIV
Supplementary Table SV
Supplementary Table SVI
Supplementary Table SVII
Supplementary Table SVIII
supplementary figure S1
supplementary figure S2
supplementary figure S3