

Abstract
Amyotrophic lateral sclerosis (ALS) is an adult‐onset motor neuron disease that has been associated with a diverse array of genetic changes. Prominent among these are mutations in RNA‐binding proteins (RBPs) or repeat expansions that give rise to toxic RNA species. RBPs are additionally central components of pathologic aggregates that constitute a disease hallmark, suggesting that dysregulation of RNA metabolism underlies disease progression. In the context of neuronal physiology, transport of RNAs and localized RNA translation in axons are fundamental to neuronal survival and function. Several lines of evidence suggest that axonal RNA translation is a central process perturbed by various pathogenic events associated with ALS. Dysregulated translation of specific RNA groups could underlie feedback effects that connect and reinforce disease manifestations. Among such candidates are RNAs encoding proteins involved in the regulation of microtubule dynamics. Further understanding of axonally dysregulated RNA targets and of the feedback mechanisms they induce could provide useful therapeutic insights. WIREs RNA 2016, 7:589–603. doi: 10.1002/wrna.1352
For further resources related to this article, please visit the WIREs website.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is the most common adult‐onset motor neuron disease and is characterized by the degeneration of cortical and spinal motor neurons. This degeneration induces progressive muscle atrophy that ultimately leads to respiratory failure within a few years from disease onset. On a cellular level, studies for many years have linked the disease with prominent manifestations including oxidative damage, excitotoxicity, protein aggregation, defective axonal transport, and mitochondrial dysfunction.1 Up until 2008, the only known genetic cause of the disease was mutations in the SOD1 gene. SOD1 encodes a Cu/Zn superoxide dismutase, a major antioxidant protein, which when mutated aggregates in association with mitochondria, thus providing some obvious links to the observed disease‐associated cellular malfunctions. More recently, however, advances in sequencing and genotyping technologies have significantly expanded the list of known genetic causes and have led to the identification of numerous additional disease‐causing mutations.2 The most prominent of them correspond to dominant mutations in genes encoding proteins involved in RNA biogenesis events (FUS, TARDBP) or repeat expansions that might give rise to toxic RNA species (C9ORF72). This has shifted the focus to defects in RNA metabolism as the underlying cause of disease and has raised the question of how very similar disease manifestations can arise from a seemingly quite diverse set of underlying genetic changes.3
In patients and in mouse models of the disease, the expression of ALS‐causing mutants specifically affects neuronal cells and spares most other somatic cell types. This suggests that some aspect of neuronal cell physiology underlies this selective vulnerability. Neurons exhibit an intricate morphology with a cell soma that gives rise to an elaborate network of dendrites and axons responsible for signal receipt and transmission. Axons of motor neurons can extend for distances up to 1 m, in humans, and contain a cytoplasmic volume up to a thousand times greater than the neuronal cell body itself.4 Maintaining this morphological and functional polarization is necessary for proper neuronal function and survival and requires a constant supply of proteins, lipids, and whole organelles from the soma to the periphery through active transport on cytoskeletal elements.
However, apart from simple maintenance of their morphology, both axons and dendrites exhibit the need to process and respond to stimulation in a local and segregated manner and can function with a remarkable degree of autonomy from the cell soma. A major way of achieving autonomous localized responses is through transport of a diverse set of RNA molecules, encoding enzymes, structural and signaling proteins, whose translation is tightly controlled.5 Indeed, localized translation is important for fundamental neuronal processes,6 including axon guidance during development and regeneration after injury, synaptic plasticity, and transmission of survival signals to the cell soma.7 Not surprisingly, defects in localized translation have been linked to various neuronal diseases.8
In the case of ALS, mounting evidence suggests that ‘dying‐back’ axonopathy and axonal degeneration are primary causes of pathology.4, 9 Pathological changes in nerve terminals appear to occur before detectable changes in the neuronal cell somas or onset of symptoms, indicating that an inability to sustain normal peripheral functions initiates the disease process.9, 10 Interestingly, the prominent cellular dysfunctions associated with ALS, namely oxidative damage, excitotoxicity, protein aggregation, defective axonal transport, and mitochondrial dysfunction, can all lead directly or indirectly to dysregulation of localized RNAs. We discuss here the idea that localized RNA translation is a central process affected by a series of interconnected events associated with ALS pathology (Figure 1). The existence of several interconnections and the potential for feedback effects would provide the basis for directing different initial pathogenic stimuli toward the same end response. Diverse stimuli, derived for example, from disparate genetic mutations or environmental triggers, could provide different points of entry to initiate a common cascade of pathogenic events, leading to dysregulation of axonal RNA translation and eventual axonal degeneration.
Figure 1.
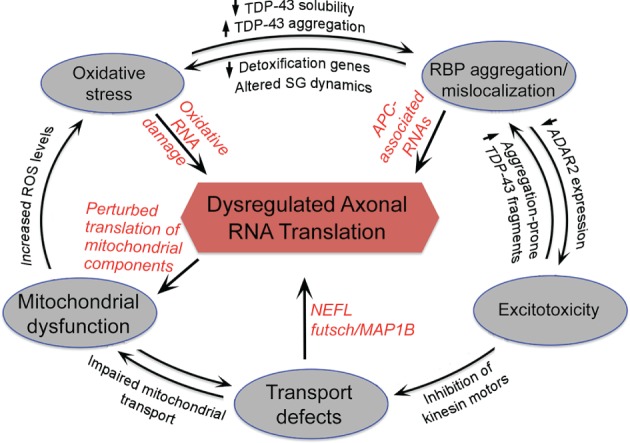
Axonal RNA translation as a central process dysregulated in amyotrophic lateral sclerosis (ALS). Potential interconnections between ALS‐associated pathogenic events and feedback effects discussed in the text. Disparate genetic mutations or environmental triggers could provide different points of entry to initiate cascades of events leading to dysregulation of RNA metabolism in axons through affecting RNA transport and/or translation. Red fonts indicate affected RNAs or events that could impact on axonal RNA translation.
GENETIC CAUSES OF ALS
ALS occurs mainly in adults (45–60 years of age) and most cases are sporadic. Only 5–10% of ALS cases are inherited in an autosomal dominant pattern. Genetic causes for the disease have been uncovered mostly for familial cases; however, given the quite similar phenotypic manifestations of the disease it is thought that understanding the underlying pathogenic mechanisms of familial cases would provide information also relevant to the spectrum of sporadic disease. Recent technological advances have resulted in the identification of an ever‐increasing number of genetic alterations associated with ALS. Some of the major ones are mapped to the genes for SOD1, TARDBP, FUS, C9ORF72, OPTN, VCP, UBQLN2, PFN1, HNRNPA2, and TUBA4A.2, 11 We provide, below, a brief overview of mutations in SOD1, TARDBP, FUS, and C9ORF72 genes, which are the focus of the majority of studies. We are referring to additional genetic alterations where relevant.
SOD1 (copper/zinc superoxide dismutase 1) was the first identified genetic cause of familial ALS. The SOD1 gene encodes an enzyme that catalyzes the removal of superoxide species. More than 180 mutations within SOD1 have been linked to ALS, with the majority resulting in amino acid substitutions throughout the length of the protein.12 Several of these mutations do not have an effect on SOD1 dismutase activity but are linked to misfolding and aggregation of the protein.12 The mitochondrial accumulation of SOD1 aggregates is thought to result in the mitochondrial dysfunction associated with ALS.13 Mutant SOD1 mouse models recapitulate many features of ALS including mitochondrial dysfunction, axonal degeneration, and defective axonal transport.13, 14, 15, 16 Interestingly, overexpression of wild‐type human SOD1 in mice also causes progressive motor neuron degeneration, supporting a pathogenic role for misfolded SOD1 protein.12
TDP‐43 (Tar DNA‐binding protein 43 kDa, encoded by the TARDBP gene) is a ubiquitously expressed and highly conserved RNA/DNA‐binding protein that is predominantly found in the nucleus, but also shuttles between the nucleus and cytoplasm.17 TDP‐43 has two RNA‐binding motifs and is involved in gene transcription, pre‐mRNA splicing, mRNA stability, and mRNA transport.3, 17 Identification of TDP‐43 RNA targets in a number of studies has indicated that TDP‐43 binds to thousands of RNA targets with a preference for UG‐rich RNA sequences.18, 19, 20, 21, 22 ALS mutations in TDP‐43 are mainly found within highly conserved amino acids of its Gly‐rich low‐complexity domain (LCD).3, 17 However, even in the absence of pathogenic mutations, TDP‐43 constitutes a major component of protein aggregates found in the vast majority of familial and sporadic ALS patients,3, 23, 24, 25 thus pointing to a central role for TDP‐43 protein aggregation, which can be triggered by diverse pathogenic stimuli.
FUS (fused in sarcoma; also known as TLS, translocated in liposarcoma) is another DNA/RNA‐binding protein that shares very similar structure and domain features to TDP‐43. FUS is also predominantly observed in the nucleus and has been ascribed a number of nuclear‐related functions such as in DNA damage and repair, transcription regulation, RNA splicing, and microRNA processing.3, 17, 26 FUS also shuttles to the cytoplasm and functions in RNA transport. FUS is a component of neuronal transport RNPs,27 it translocates to dendrites upon mGluR5 activation facilitating transport of mRNAs in these structures, and regulates spine morphology.28, 29 Identification of its RNA targets has revealed that, similar to TDP‐43, FUS binds to a large number of RNAs through a loosely defined consensus.30, 31, 32, 33 Mutations in FUS account for ~4% of familial ALS cases and are mostly found within the N‐terminal LCD of the protein or at a C‐terminal sequence comprising a nuclear localization signal.17 Both types of mutations are thought to lead to the formation of cytoplasmic inclusions either directly by increasing the aggregation propensity of the LCD or indirectly by increasing the concentration of the protein in the cytoplasm.34, 35 Supporting the importance of tightly regulating the concentration of FUS, overexpression of wild‐type FUS has been associated with disease in humans and in animal models.36, 37, 38 While FUS and TDP‐43 inclusions are associated with similar phenotypic consequences, they nevertheless appear to be distinct on a molecular level, with TDP‐43 being mostly absent from FUS‐positive inclusions.39 Potentially relevant to this observation, genetic studies in zebrafish indicate that FUS and TDP‐43 act on the same pathogenic pathway, with TDP‐43 acting upstream of FUS.40
The recent identification of a large hexanucleotide (G4C2) repeat expansion (HRE) in the C9ORF72 gene (chromosome 9 open reading frame 72) has provided a genetic explanation for ~40% of familial ALS cases.41, 42 The expansion can contain up to thousands of repeats and how it leads to ALS pathology is a subject of intense investigation. Sense and antisense transcription of the HRE locus leads to production of HRE RNA species that adopt G‐quadruplex structure.43 HRE transcription additionally causes a reduction in the amount of the normal protein product produced from the C9ORF72 gene. However, loss of function of C9ORF72 protein does not lead to motor neuron phenotypes,44 indicating that pathogenic effects arise from expression of the toxic HRE RNA species. Potential mechanisms for this include sequestration of RNA‐binding proteins (RBPs)45, 46, 47 and/or noncanonical, repeat‐associated non‐AUG (RAN) translation of HREs that give rise to toxic dipeptide‐repeat proteins.48 Independent concurrent reports have identified nucleocytoplasmic transport as a crucial cellular process affected by C9ORF72 mutations. Both nuclear import and RNA export were found to be affected in Drosophila and yeast models of HRE toxicity.49, 50, 51 These effects could be mediated through sequestration of the RanGAP regulator by HRE RNAs.49 Additionally, specific C9ORF72 protein isoforms associate with importin β1 and the Ran‐GTPase.52 Interestingly, TDP‐43 is one of the cellular proteins whose nucleocytoplasmic distribution is affected, both in Drosophila cells and in differentiated, patient‐derived neurons, providing a potential connection between C9ORF72 mutations and TDP‐43 pathology.49
EFFECTS OF ALS PATHOLOGIC EVENTS ON LOCALIZED RNA TRANSLATION
Our focus here is to discuss effects of genetic ALS mutations and pathogenic events on RNA translation in axons and to point out interconnections and potential feedback effects. We refer to other reviews for additional discussions on ALS pathogenesis.1, 53
RNA‐Binding Protein Aggregation
Pathogenic Potential and Mechanisms of RBP Aggregate Formation
A major hallmark of affected motor neurons in ALS is the presence of protein aggregates containing the RBPs FUS and TDP‐43. These proteins are found in aggregates not only in patients carrying mutations in the corresponding genes but also in patients carrying distinct or no known mutations, supporting a widespread role of RBP aggregation in neuronal degeneration.54 Consistent with that, both FUS and TDP‐43 need to aggregate in the cytoplasm in order to confer toxicity in yeast.55, 56 Additionally, in a Caenorhabditis elegans model of the disease, the abundance of insoluble FUS directly correlates with neurotoxicity and, importantly, increasing or decreasing these insoluble FUS assemblies exacerbates or ameliorates neurotoxicity, respectively.57 Supporting the pathogenic role of protein aggregation, other strategies of dissolving such aggregates have shown that this can be beneficial. The yeast protein Hsp104, a hexameric AAA+ ATPase that deconstructs various amyloids and fibrillar oligomers, has been engineered to target FUS and TDP‐43 aggregates. Dissolution of FUS and TDP‐43 aggregates, in yeast models, restored proper protein localization and suppressed toxicity.58, 59
Transgenic rodent models of FUS and TDP‐43 proteinopathy have demonstrated varying levels of inclusion formation, lethality, and motor defects. This variability could be due to differences in promoter usage, level and timing of transgene expression, or type of mutant protein expressed.60, 61 Another potential consideration is that pathogenic protein insolubility could occur in the absence of overt, microscopically visible inclusion formation.62 Indeed, in response to heat shock, submicroscopic, biochemically detectable aggregates are formed, despite the absence of obvious in vivo foci.63 Nevertheless, in motor neurons differentiated from patient‐derived induced pluripotent stem cells an increase in detergent‐insoluble TDP‐43 is observed,64, 65 while cytoplasmic levels of mutant FUS correlate with mutation severity66 and lead to spontaneous aggregation upon aging of the culture,67 supporting a role for protein aggregation also in these patient‐derived cell systems.
In recent years, there has been substantial progress in our understanding of the underlying mechanisms leading to pathologic protein aggregation. An important consideration is that both FUS‐ and TDP‐43‐containing aggregates appear to be related to stress granules,68 which are RNA–protein assemblies formed under conditions of stress and reduced translation initiation, and are thought to represent a pool of translationally stalled RNPs.69 The connection of pathologic inclusions to stress granules is based on the fact that pathologic inclusions contain several stress granule markers. Additionally, several pathogenic RBPs (such as FUS, TDP‐43, and hnRNPA1) associate normally with stress granules in response to stress and can induce their formation in cell culture systems.70
Formation of stress granules was originally shown to be driven by multimerization of the RBP TIA‐1 that contains a prion‐like domain.71 The last few years have led to the realization that such prion‐like LCDs are particularly abundant in RNA‐ and DNA‐binding proteins, several of which have been implicated in ALS and other neurodegenerative diseases.72 Interactions of LCDs as well as multivalent interactions mediated through RNA underlie a liquid–liquid phase separation that leads to the formation of various membrane‐less cellular compartments, including stress granules.73 However, under normal conditions, such liquid‐demixed phases are very dynamic, their components are recycled in short time scales, and are thus quite distinct from the solid aggregates associated with disease. A series of recent reports have addressed this question, showing that pathogenic mutations in the LCD‐containing proteins FUS and hnRNPA1 can accelerate the conversion of dynamic liquid phases into fibrillar aggregates that can further seed the assembly of additional amyloid‐like deposits.34, 74, 75
Apart from aggregation‐inducing mutations in RBPs, persistence of RNP granules could be caused by mutations in additional factors that regulate their dynamic properties or by blocking quality control pathways that are normally involved in their removal. Indeed, ALS mutations in the VCP/p97 protein, a AAA‐ATPase, have been associated with impaired autophagic clearance of stress granules,76 while mutations in Profilin1 similarly impair stress granule dynamics.77 The current hypothesis, therefore, suggests that pathologic inclusions arise from the perturbation of physiologic RNP remodeling events, which convert dynamic RNP assemblies into self‐propagating stable fibrous assemblies or prevent their efficient clearance.70
Effects of RBP Aggregates on RNA Localization and Translation
How are RBP aggregates mediating neurotoxic effects? Under conditions of stress or in response to toxic insults, dynamic formation of stress granules is important to promote cell survival. Stress granule formation accompanies polysome disassembly and translational arrest of most cellular mRNAs to ensure specific production of proteins involved in response to stress and eventual recovery. Inability to disassemble these structures could lead to a persistent reduction in protein production, which could be particularly detrimental to neuronal cells given their high energy and metabolic demands. Translational arrest, in response to several stressful stimuli, is mediated through phosphorylation of the initiation factor eIF2a.69 Phospho‐eIF2a cannot support formation of a functional eIF2‐GTP‐tRNAMet ternary complex, thus blocking translation at the initiation stage. Consistent with a pathogenic role for prolonged translational repression, eIF2a phosphorylation is upregulated by TDP‐43 toxicity and inhibition of this phosphorylation mitigates TDP‐43 toxicity in fly models and mammalian neurons.78 Underscoring the importance of correct translational regulation in neuronal physiology, sustained translational repression has been suggested to also have a causative role in prion‐mediated neurodegeneration.79 In the case of ALS, important RNA targets affected by translational repression include axonal RNAs. TDP‐43 proteinopathy prevents translation of futsch RNA in the Drosophila neuromuscular junction (NMJ) and restoring futsch expression rescues TDP‐43 toxicity and NMJ defects.80, 81
Despite their resemblance to stress granules, pathogenic aggregates could additionally have distinct properties. Imaging assays to detect newly synthesized proteins and translation sites have shown that cytoplasmic granules formed by mutant FUS can support translation of at least some RNA species.82 Some of the RNAs found in FUS granules are normally localized in cell protrusions and neuronal axons,82, 83 suggesting that in certain cases the pathogenic contribution might not be derived through loss of translation but rather through ectopic translation leading to an imbalance in the local protein production between the neuronal periphery and the cell soma.
Formation of persistent RNP aggregates could additionally trigger toxicity through sequestration of protein factors and reduction of their functional cytoplasmic pool (Figure 2). As detailed below, the integral components of pathogenic aggregates, FUS and TDP‐43, have roles in axonal RNA transport that are perturbed by disease mutations. Additionally, the adenomatous polyposis coli (APC) protein is recruited in cytoplasmic FUS granules in cell culture systems and in patient tissue.82 Interestingly, this association is observed in cytoplasmic granules induced by pathogenic mutations, but not in arsenite‐induced stress granules (K. Yasuda, unpublished observations). APC is an RBP that functions in targeting a large group of RNAs in cellular protrusions and neuronal axons.83, 84 Sequestration of APC in cytoplasmic aggregates could thus impact a number of axonal functions relying on localized RNA translation, including microtubule dynamics and organization84 (see below). Indeed, APC has important functions in the nervous system, which have, however, been investigated mostly in the context of development. Conditional loss of APC leads to disrupted cortical layer formation, aberrant axon tract development, and microtubule disorganization.85 In cell culture systems, APC is important for neurite extension, axon specification, and outgrowth and localized APC inactivation leads to growth cone collapse.86, 87, 88, 89 Additionally, however, APC is abundantly expressed in neuronal cells of the adult central nervous system where it distributes within dendritic and axonal processes.90 Apart from its function related to RNA metabolism, APC also has a well‐established role as a regulator of the β‐catenin transcription factor in the context of the Wnt signaling pathway. It will be interesting to determine how these two APC functions are coordinated and how APC association with pathogenic granules affects Wnt signaling, a pathway that, intriguingly, has been implicated in other neurodegenerative diseases.91, 92
Figure 2.
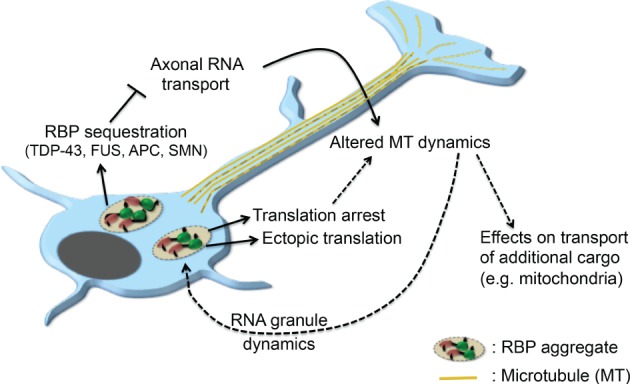
RNA‐binding protein (RBP) aggregation and connections to microtubule dynamics. RBP aggregates associated with amyotrophic lateral sclerosis (ALS) can lead to translation arrest, ectopic translation, or sequestration of RBPs and subsequent perturbation in axonal RNA transport. Some of the affected RNAs encode microtubule regulators thus leading to alterations in microtubule dynamics. Such RNAs include the futsch/MAP1B RNA affected by TDP‐43 mutations or APC‐associated RNAs that can be affected by APC sequestration in FUS granules. Changes of the microtubule cytoskeleton could subsequently affect transport of additional cargo, such as mitochondria, or could have feedback effects on the dynamics of RNA granules.
Another FUS‐binding partner, the RNP‐assembly protein SMN, has revealed potential connections between ALS and the motor neuron disease spinal muscular atrophy caused by reduced levels of the SMN protein. Formation of mutant FUS aggregates leads to redistribution of SMN within cytoplasmic granules, which trap SMN and impede its dynamic release.57, 93 Among other functions in splicing and snRNP biogenesis, SMN additionally localizes to axons and regulates axonal mRNA transport and local translation.94 Interestingly, overexpression of an SMN fragment, known to mediate its axonal functions, rescues axonal defects induced by mutant FUS.93
Axonal RNA Transport Defects and the Dynamics of the Microtubule Cytoskeleton
Motor neurons are highly polarized cells with long axons, and transport along the axonal length is required for delivery of essential components, such as RNAs, proteins, and organelles, to the axonal compartment. Axonal transport is primarily carried out on microtubules through the action of molecular motors, kinesins, and cytoplasmic dynein that mediate transport in the anterograde and retrograde direction, respectively.95 Apart from maintenance of the axonal compartment, retrograde transport is additionally important for relay of survival signals from the periphery to the neuronal soma. RNAs encoding nuclear import and transcription factors are locally translated within axons in response to injury or trophic factor stimulation, and this local protein production is required for efficient retrograde signaling, survival, and recovery.96, 97, 98, 99
Disturbances in axonal transport are observed in ALS patients as well as in ALS mouse models. Mutant SOD1 mice exhibit defects in axonal transport, which occur early in the disease process.95 The exact mechanisms are unknown, but likely involve several pathways involving modification of microtubule‐dependent motors or of cargoes themselves. For example, TNF elevation disrupts kinesin function through p38 activation, while glutamate reduces axonal transport of neurofilaments through phosphorylation.95 There is also evidence, in SOD1 mice, for the existence of microtubules with altered, hyperdynamic properties.100 Given that kinesin motors exhibit preferences for specific sets of stable, modified microtubules,101 such a change in microtubule dynamics could affect their recognition by motor proteins and thus alter the efficiency of transport of specific cargoes.
Which cargoes are relevant to pathogenicity is not clear. Studies of mutant SOD1 mice have focused on transport of mitochondria and organelles.102, 103, 104 However, impairment of mitochondrial transport does not appear to be a direct cause of motor neuron degeneration,105, 106 suggesting that transport of other cargoes might better correlate with axonal degeneration. In this regard, it is interesting that both the ALS‐associated RBPs FUS and TDP‐43 have been implicated in RNA transport events in neuronal cells.
TDP‐43 is trafficked in neurons107, 108 and is found in cytoplasmic RNP granules along the length of the axon and at the presynaptic axon terminals in the NMJ both in mice and Drosophila motor neurons.109, 110 TDP‐43 axonal granules are transported bidirectionally in a microtubule‐dependent manner. By contrast TDP‐43 mutants (M337V and A315T) accumulate in the soma and proximal axons and exhibit impaired anterograde movement in Drosophila motor neurons, in primary mouse cortical neurons, and in patient iPS‐derived motor neurons.110 One of the RNAs associated with TDP‐43 granules is the neurofilament L (Nefl) mRNA. Association with TDP‐43 promotes its anterograde transport, which is impaired in the presence of pathologic TDP‐43 mutants.110 At least during early days in culture the transport defect is specific for TDP‐43 RNP granules and axonal transport of mitochondria is not affected. However, TDP‐43 mutations affect mitochondrial transport in transgenic mice or after longer periods of in vitro culture, suggesting that RNP transport defects precede, and might cause (see below), more generalized transport abnormalities.110, 111
Coyne et al.81 have used a Drosophila ALS model based on TDP‐43 to uncover potential RNAs whose defective transport underlies TDP‐43 toxicity. They focused on the futsch RNA, which encodes a protein homologous to the mammalian MAP1B and which regulates microtubule organization at synapses. In the context of TDP‐43 proteinopathy, futsch localization to the NMJ and its translation were found to be impaired. Interestingly, MAP1B distribution is also altered in spinal cord motor neurons from ALS patients and restoring Futsch expression in Drosophila motor neurons was neuroprotective by restoring microtubule acetylation and stability at the NMJ and, additionally, reducing TDP‐43 aggregation. Another TDP‐43 target linked to tubulin modification is HDAC6,112 a tubulin deacetylase, suggesting that local microtubule acetylation and stabilization could be an important factor mediating TDP‐43 toxicity. Stable microtubules could potentially affect transport of additional cargo requiring specific sets of microtubule filaments.101 This could offer a potential explanation for the late, secondary effects of TDP‐43 mutations on transport of mitochondrial cargo, mentioned above. Additionally, microtubules or microtubule motors have been implicated in determining the dynamics of RNP aggregates such as stress granules113, 114 and could thus play a role in modifying the aggregation state of pathogenic inclusions. These data suggest the existence of an interplay, whereby functional TDP‐43 RNP transport granules direct correct expression of microtubule regulators, ensuring the formation of a microtubule network that can support transport of additional cargo. At the same time, microtubules ensure the functionality of TDP‐43 granules by modulating their aggregation propensity (Figure 2).
Interestingly, FUS targets could similarly include microtubule regulators. FUS is a component of localized RNPs associated with the APC tumor‐suppressor protein.82 HITS‐CLIP of APC, from mouse brain tissue, identified its neuronal RNA interactome and revealed that APC targets were enriched, among others, for RNAs encoding tubulin isotypes and microtubule regulators. Preventing the interaction of APC with one of these targets, the β2B‐tubulin mRNA, prevented its axonal localization and expression and disrupted dynamic microtubules within axonal growth cones.84 ALS mutations and aggregation of FUS lead to reduced overall translation in axon terminals57 and can prevent correct localization of APC‐associated RNAs. Interestingly, the mechanism underlying this mislocalization appears to involve additional ways through which FUS can impact on the stable microtubule network (Yasuda et al., in preparation). It seems, therefore, that mutations in both TDP‐43 and FUS can commonly lead to disruption of local RNA translation and can affect microtubule structure and dynamics, potentially consequently affecting transport of additional cargo (Figure 2).
Supporting the involvement of the microtubule cytoskeleton in ALS pathogenesis, an exome‐wide rare variant burden analysis of familial ALS cases has revealed mutations in the gene encoding the tubulin alpha‐4A isoform (TUBA4A). The identified TUB4A4 mutants affect microtubule dynamics and destabilize the microtubule network.11 Hyperdynamic microtubules have additionally been detected in an SOD1 mouse model and pharmacologic treatments to reduce the dynamicity and stabilize neuronal microtubules prevented neuronal death, restored axonal transport, and delayed disease onset.100 Such effects on microtubule dynamics would link ALS to several other neurodegenerative diseases characterized by mutations in tubulin isoforms or microtubule‐interacting proteins.11, 115
Effects of Oxidative Stress on RNA Translation and RBP Distribution
Oxidative stress, resulting from an accumulation of reactive oxygen species (ROS), has been documented in a large number of ALS cases.116 Particular interest has been shown in the role of oxidative stress in ALS, because mutations in SOD1, which encodes a major antioxidant protein, account for a significant proportion of familial ALS cases.2 Elevated levels of oxidative damage to proteins, lipids, and DNA have been found in ALS patient tissue.1 Oxidative damage to RNA species has also been shown in ALS patient tissue as well as in mSOD1 mouse models where it has been shown to occur early during disease pathology and to promote degeneration.117 mRNA oxidation is primarily found in motor neurons and oligodendrocytes. Interestingly, some mRNA species are more susceptible to oxidative damage and include RNAs encoding proteins involved in mitochondrial electron transport, protein biosynthesis, myelination, protein folding, and degradation.117 Oxidatively damaged RNA negatively affects the fidelity of translation and leads to an overall reduction in the amount of functional polypeptides produced.118, 119
Apart from direct effects on translation through RNA oxidation, oxidative stress can indirectly affect RNA metabolism through affecting RBP aggregation. Specifically, oxidative stress can affect TDP‐43 in multiple ways. It induces TDP‐43 crosslinking through cysteine oxidation and disulfide bond formation leading to reduced solubility.120 It further induces TDP‐43 acetylation, impairing its binding to RNA and enhancing its aggregation propensity.121
On the other hand, expression of mutant TDP‐43 in motor neuron cell lines can induce oxidative stress,122 suggesting the existence of feedback effects. Potential mechanisms could be through preventing expression of detoxification genes such as HO‐1 122 or through affecting formation and dynamics of stress granules,123 which exhibit an antioxidant activity by reducing ROS production124 (Figure 1).
Effects of Excitotoxicity on RBP Distribution and Axonal Transport
Excitotoxicity, the phenomenon of neuronal degeneration induced by overstimulation of glutamate receptors, has been widely associated with ALS. Excitotoxicity can be caused from increased synaptic levels of glutamate, the main neurotransmitter mediating excitatory neurotransmission in motor neurons, or by increased sensitivity of the postsynaptic neuron to glutamate as a result of changes in glutamate receptor expression.125 The main receptors mediating glutamate neurotransmission are the α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptors, consisting of four types of subunits (GluR1–R4/GluA1–A4). The tetrameric receptor forms a channel, whose permeability and response rates are controlled posttranscriptionally. A to I RNA editing of the GluA2 subunit converts a glutamine (Q) residue to arginine (R) and prevents Ca++ entering through the channel pore. Under normal conditions almost all GluA2 subunits are edited and the prevention of calcium entry is proposed to guard against excitotoxicity. The rate of desensitization or resensitization of the receptor can additionally be modified through alternative splicing of its subunits. Editing and splicing of AMPA receptor subunits is also coordinated with trafficking of their mRNAs at dendritic sites, where they are locally translated in response to synaptic stimulation.126, 127
Expression of ADAR2, an RNA editing enzyme catalyzing A to I conversion, as well as RNA editing of GluA2 mRNA are significantly downregulated in the motor neurons of the majority of sporadic ALS patients exhibiting TDP‐43 but not SOD1 pathology. Consistently, loss of ADAR2 in mice is linked to TDP‐43 pathology.128 The consequent assembly of unedited GluA2 subunits into Ca‐permeable AMPA receptors has been linked to other manifestations of ALS pathogenicity. First, increased Ca++ permeability leads to accumulation of aggregation‐prone N‐terminal TDP‐43 fragments generated through proteolytic cleavage by the Ca‐dependent protease calpain.128 Additionally, excitotoxicity can lead to transport defects.95 Glutamate can activate JNK and p38 kinases. JNK phosphorylates the kinesin motor domain and inhibits kinesin from binding to microtubules. P38, which is also induced by mutant SOD1, can phosphorylate the kinesin light chain and inhibit cargo binding.95 Therefore, deregulated transmission through AMPA receptors can provide another entry point for initiating the cascade of pathogenic events including RBP aggregation and defective axonal transport associated with ALS.
Whether other ALS mutations can lead to defects in AMPA receptor editing is not known. FUS can regulate stability of GluA1 RNA likely through controlling its polyA tail length, and FUS depletion affects synaptic transmission both in vitro and in vivo.129 GGGGCC RNA foci formed by a repeat expansion of the C9ORF72 gene have been shown to sequester another member of the ADAR family (ADAR3/ADARB2), which was also required for their formation or maintenance.45 However, ADAR3 does not exhibit RNA editing activity suggesting that, in this case, its role could be mostly mediated through its ability to bind RNAs.
Mitochondrial Dysfunction—Roles of RBPs and Axonal Translation
Mitochondrial impairment has been extensively associated with ALS and shown to contribute significantly to disease symptoms.130 Mutant SOD1 alters mitochondrial morphology and distribution within motor neuron axons.103, 104 Expression of mutant FUS leads to mitochondrial fragmentation and damage131; while mutant TDP‐43 induces mitochondrial morphology and transport abnormalities in transgenic mice and cultured neurons.111, 132, 133 Although mitochondrial defects are a common occurrence, in vivo tracking of mitochondria showed that SOD1 and TDP43 mutant mice differ in the temporal and spatial characteristics of these abnormalities, suggesting that different genetic causes lead to the same outcome through different underlying mechanisms.111
Potential mechanisms could include direct association of the mutant proteins with mitochondria as described for aggregated, misfolded SOD1.13 Fus also associates with mitochondria in an interaction mediated by hsp60. Downregulating hsp60 partially rescues Fus‐induced mitochondrial defects and degeneration in flies.131 A similar association with axonal mitochondria has been reported for TDP‐43.132 Another mechanism leading to mitochondrial dysfunction could rely on effects on microtubule‐dependent motors and microtubule dynamics. As mentioned above, ALS mutations can lead to dysregulated transport and translation of RNAs encoding microtubule regulators. Alteration of microtubule dynamics could potentially affect transport of mitochondria to the periphery as mitochondrial transport requires the kinesin‐1 protein, Kif5B,134 which belongs to a family of motors exhibiting preference for movement along detyrosinated microtubule tracks,135 a subset of posttranslationally modified stable microtubules. Indeed, redistribution of mitochondria to the periphery in response to nutrient starvation requires microtubule detyrosination.136
Despite its prevalence, the contribution of defective mitochondrial transport to disease progression has been questioned, as increasing mitochondrial mobility, in G93A‐SOD1 mice, did not rescue ALS‐like symptoms106 and furthermore, defects in mitochondrial transport were uncoupled from axonal degeneration.105 It is likely that other aspects of mitochondrial damage, affecting for example membrane conductance or calcium homeostasis, could make more significant contributions to motor neuron loss.106, 137
In this regard, it is interesting that localized axonal RNA translation is important for maintaining mitochondrial functionality. The RNA encoding the intermediate filament protein lamin B2 (LB2) is found in axons and its translation is induced upon axonal stimulation. The locally synthesized axonal LB2 associates with mitochondria and loss of axonal LB2 leads to defects in mitochondrial morphology and function.138 LB2 is not the only nuclear‐encoded mitochondrial protein that is locally synthesized in nerve terminals. Several others are locally translated, including cytochrome c oxidase IV (CoxIV) or ATP synthase 9 (ATP5G1), which encode key subunits of the oxidative phosphorylation complexes.139, 140, 141 Interfering with the localized production of mitochondrial components results in compromised mitochondrial membrane potential, reduced local ATP levels, and enhanced production of local ROS in both cultured neurons and mice.141, 142 Interestingly, increased ROS levels could lead to oxidative RNA damage, to which, as mentioned above, RNAs encoding mitochondrial components exhibit increased susceptibility.117 This could therefore indicate the existence of feedback effects between translation of mitochondrial components, mitochondrial dysfunction, and ROS production that enhances and perpetuates disease‐related phenotypes (Figure 1).
Whether local translation of mitochondrial RNAs is affected by ALS mutations is not known. It is, however, potentially interesting that RNAs encoding mitochondrial proteins or mitochondrial RNA regulators have been identified in at least some screens looking for FUS and TDP‐43 RNA targets.19, 22, 33
CONCLUSION
Transport of RNAs along axons and axonal RNA translation are important for survival and regeneration of neuronal cells. RNA transport and translation are disrupted by diverse genetic mutations and pathologic events associated with ALS, suggesting that local RNA translation is a central event dysregulated during disease progression. Multiple interconnections and feedback effects can propagate an initial pathogenic stimulus and reinforce deleterious effects, providing a framework for understanding how diverse genetic mutations or environmental factors can lead to the common phenotypic manifestations associated with ALS. While the particular localized RNAs, which participate in these pathogenic mechanisms, are not fully identified, some emerging groups are providing useful insights. Aggregation of the RBPs TDP‐43 and FUS can impact on transport and translation of RNAs encoding components or regulators of the microtubule cytoskeleton. Such changes in microtubule dynamics could subsequently disrupt transport of additional relevant cargo, such as mitochondria, and could further promote RBP aggregation through altering the dynamic properties of RNA granules. Interestingly, reduced stability and changes in the dynamic properties of microtubules have been observed in other neurodegenerative diseases and approaches to restore microtubule stability can have therapeutic potential.143, 144, 145, 146 Another potentially important group of localized RNAs encodes mitochondrial components and its dysregulation could contribute to the observed mitochondrial dysfunction phenotypes. Approaches to globally identify mislocalized RNAs and changes in axonal translation would be important to further understand affected pathways and processes that could be therapeutically modulated.
Conflict of interest: The authors have declared no conflicts of interest for this article.
The copyright line for this article was changed on 12 August 2016 after original online publication.
REFERENCES
- 1. Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 2011, 7:616–630. [DOI] [PubMed] [Google Scholar]
- 2. Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014, 17:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 2013, 79:416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fischer‐Hayes LR, Brotherton T, Glass JD. Axonal degeneration in the peripheral nervous system: implications for the pathogenesis of amyotrophic lateral sclerosis. Exp Neurol 2013, 246:6–13. [DOI] [PubMed] [Google Scholar]
- 5. Deglincerti A, Jaffrey SR. Insights into the roles of local translation from the axonal transcriptome. Open Biol 2012, 2:120079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holt CE, Schuman EM. The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 2013, 80:648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jung H, Yoon BC, Holt CE. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 2012, 13:308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu‐Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST, Wolozin B. Local RNA translation at the synapse and in disease. J Neurosci 2011, 31:16086–16093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dadon‐Nachum M, Melamed E, Offen D. The "dying‐back" phenomenon of motor neurons in ALS. J Mol Neurosci 2011, 43:470–477. [DOI] [PubMed] [Google Scholar]
- 10. Vaughan SK, Kemp Z, Hatzipetros T, Vieira F, Valdez G. Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis‐causing mutations. J Comp Neurol 2015, 523:2477–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, Scotter EL, Kost J, Keagle P, Miller JW, et al. Exome‐wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 2014, 84:324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rotunno MS, Bosco DA. An emerging role for misfolded wild‐type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci 2013, 7:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tafuri F, Ronchi D, Magri F, Comi GP, Corti S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front Cell Neurosci 2015, 9:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res 1999, 819:120–131. [DOI] [PubMed] [Google Scholar]
- 15. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264:1772–1775. [DOI] [PubMed] [Google Scholar]
- 16. Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS‐linked SOD1 mutants to motor neurons. Nat Neurosci 1999, 2:50–56. [DOI] [PubMed] [Google Scholar]
- 17. Lagier‐Tourenne C, Polymenidou M, Cleveland DW. TDP‐43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 2010, 19:R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Polymenidou M, Lagier‐Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. Long pre‐mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP‐43. Nat Neurosci 2011, 14:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, Ratti A. TDP‐43 and FUS RNA‐binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post‐transcriptional fate in motoneuron‐like cells. J Biol Chem 2012, 287:15635–15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiao S, Sanelli T, Dib S, Sheps D, Findlater J, Bilbao J, Keith J, Zinman L, Rogaeva E, Robertson J. RNA targets of TDP‐43 identified by UV‐CLIP are deregulated in ALS. Mol Cell Neurosci 2011, 47:167–180. [DOI] [PubMed] [Google Scholar]
- 21. Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, et al. Characterizing the RNA targets and position‐dependent splicing regulation by TDP‐43. Nat Neurosci 2011, 14:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, et al. Identification of neuronal RNA targets of TDP‐43‐containing ribonucleoprotein complexes. J Biol Chem 2011, 286:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, et al. Pathological TDP‐43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 2007, 61:427–434. [DOI] [PubMed] [Google Scholar]
- 24. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314:130–133. [DOI] [PubMed] [Google Scholar]
- 25. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006, 351:602–611. [DOI] [PubMed] [Google Scholar]
- 26. Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. J Mol Cell Biol 2009, 1:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA‐transporting granule. Neuron 2004, 43:513–525. [DOI] [PubMed] [Google Scholar]
- 28. Fujii R, Takumi T. TLS facilitates transport of mRNA encoding an actin‐stabilizing protein to dendritic spines. J Cell Sci 2005, 118:5755–5765. [DOI] [PubMed] [Google Scholar]
- 29. Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 2005, 15:587–593. [DOI] [PubMed] [Google Scholar]
- 30. Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep 2012, 2:603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakaya T, Alexiou P, Maragkakis M, Chang A, Mourelatos Z. FUS regulates genes coding for RNA‐binding proteins in neurons by binding to their highly conserved introns. RNA 2013, 19:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A, Urano F, Sobue G, Ohno K. Position‐dependent FUS‐RNA interactions regulate alternative splicing events and transcriptions. Sci Rep 2012, 2:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T. RNA targets of wild‐type and mutant FET family proteins. Nat Struct Mol Biol 2011, 18:1428–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162:1066–1077. [DOI] [PubMed] [Google Scholar]
- 35. Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, et al. ALS‐associated fused in sarcoma (FUS) mutations disrupt Transportin‐mediated nuclear import. EMBO J 2010, 29:2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D, Wei X, Xia XG. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet 2011, 7:e1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, Tudor EL, Smith BN, Klasen C, Miller CC, et al. Overexpression of human wild‐type FUS causes progressive motor neuron degeneration in an age‐ and dose‐dependent fashion. Acta Neuropathol 2013, 125:273–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sabatelli M, Moncada A, Conte A, Lattante S, Marangi G, Luigetti M, Lucchini M, Mirabella M, Romano A, Del Grande A, et al. Mutations in the 3' untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum Mol Genet 2013, 22:4748–4755. [DOI] [PubMed] [Google Scholar]
- 39. Mackenzie IR, Rademakers R, Neumann M. TDP‐43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 2010, 9:995–1007. [DOI] [PubMed] [Google Scholar]
- 40. Kabashi E, Bercier V, Lissouba A, Liao M, Brustein E, Rouleau GA, Drapeau P. FUS and TARDBP but not SOD1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet 2011, 7:e1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Renton AE, Majounie E, Waite A, Simon‐Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 2011, 72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 2011, 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reddy K, Zamiri B, Stanley SY, Macgregor RB Jr, Pearson CE. The disease‐associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length‐dependent uni‐ and multimolecular RNA G‐quadruplex structures. J Biol Chem 2013, 288:9860–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lagier‐Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A 2013, 110:E4530–E4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee YB, Chen HJ, Peres JN, Gomez‐Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, et al. Hexanucleotide repeats in ALS/FTD form length‐dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 2013, 5:1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cleary JD, Ranum LP. Repeat‐associated non‐ATG (RAN) translation in neurological disease. Hum Mol Genet 2013, 22:R45–R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW III, Sun S, Herdy JR, Bieri G, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 2015, 18:1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Freibaum BD, Lu Y, Lopez‐Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525:129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J. Isoform‐specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol 2015, 78:568–583. [DOI] [PubMed] [Google Scholar]
- 53. Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013, 14:248–264. [DOI] [PubMed] [Google Scholar]
- 54. Blokhuis AM, Groen EJ, Koppers M, van den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol 2013, 125:777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol 2011, 9:e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP‐43 proteinopathy model: exploring the molecular determinants of TDP‐43 aggregation and cellular toxicity. Proc Natl Acad Sci U S A 2008, 105:6439–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, et al. ALS/FTD mutation‐induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 2015, 88:678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jackrel ME, Shorter J. Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease‐linked proteins. Dis Model Mech 2014, 7:1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jackrel ME, DeSantis ME, Martinez BA, Castellano LM, Stewart RM, Caldwell KA, Caldwell GA, Shorter J. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2014, 156:170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Philips T, Rothstein JD. Rodent models of amyotrophic lateral sclerosis. Curr Protoc Pharmacol 2015, 69:5.67.1–5.67.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McGoldrick P, Joyce PI, Fisher EM, Greensmith L. Rodent models of amyotrophic lateral sclerosis. Biochim Biophys Acta 1832, 2013:1421–1436. [DOI] [PubMed] [Google Scholar]
- 62. Shelkovnikova TA. Modelling FUSopathies: focus on protein aggregation. Biochem Soc Trans 2013, 41:1613–1617. [DOI] [PubMed] [Google Scholar]
- 63. Wallace EW, Kear‐Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM, et al. Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 2015, 162:1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, et al. Drug screening for ALS using patient‐specific induced pluripotent stem cells. Sci Transl Med 2012, 4:145ra104. [DOI] [PubMed] [Google Scholar]
- 65. Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP‐43 proteinopathies and reveal cell‐specific vulnerability. Proc Natl Acad Sci U S A 2012, 109:5803–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lenzi J, De Santis R, de Turris V, Morlando M, Laneve P, Calvo A, Caliendo V, Chio A, Rosa A, Bozzoni I. ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell‐derived motoneurons. Dis Model Mech 2015, 8:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Japtok J, Lojewksi X, Naumann M, Klingenstein M, Reinhardt P, Sterneckert J, Putz S, Demestre M, Boeckers TM, Ludolph AC, et al. Stepwise acquirement of hallmark neuropathology in FUS‐ALS iPSC models depends on mutation type and neuronal aging. Neurobiol Dis 2015, 82:420–429. [DOI] [PubMed] [Google Scholar]
- 68. Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol 2013, 201:361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci 2008, 33:141–150. [DOI] [PubMed] [Google Scholar]
- 70. Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA‐protein granules in degenerative disorders. Cell 2013, 154:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion‐like aggregation of TIA‐1. Mol Biol Cell 2004, 15:5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA‐binding proteins with prion‐like domains in neurodegenerative disease. Brain Res 2012, 1462:61–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Weber SC, Brangwynne CP. Getting RNA and protein in phase. Cell 2012, 149:1188–1191. [DOI] [PubMed] [Google Scholar]
- 74. Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lin Y, Protter DS, Rosen MK, Parker R. Formation and maturation of phase‐separated liquid droplets by RNA‐binding proteins. Mol Cell 2015, 60:208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153:1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Figley MD, Bieri G, Kolaitis RM, Taylor JP, Gitler AD. Profilin 1 associates with stress granules and ALS‐linked mutations alter stress granule dynamics. J Neurosci 2014, 34:8083–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2α phosphorylation rescues TDP‐43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 2014, 46:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, et al. Sustained translational repression by eIF2α‐P mediates prion neurodegeneration. Nature 2012, 485:507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Coyne AN, Yamada SB, Siddegowda BB, Estes PS, Zaepfel BL, Johannesmeyer JS, Lockwood DB, Pham LT, Hart MP, Cassel JA, et al. Fragile X protein mitigates TDP‐43 toxicity by remodeling RNA granules and restoring translation. Hum Mol Genet 2015, 24:6886–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Coyne AN, Siddegowda BB, Estes PS, Johannesmeyer J, Kovalik T, Daniel SG, Pearson A, Bowser R, Zarnescu DC. Futsch/MAP1B mRNA is a translational target of TDP‐43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci 2014, 34:15962–15974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yasuda K, Zhang H, Loiselle D, Haystead T, Macara IG, Mili S. The RNA‐binding protein Fus directs translation of localized mRNAs in APC‐RNP granules. J Cell Biol 2013, 203:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mili S, Moissoglu K, Macara IG. Genome‐wide screen reveals APC‐associated RNAs enriched in cell protrusions. Nature 2008, 453:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Preitner N, Quan J, Nowakowski DW, Hancock ML, Shi J, Tcherkezian J, Young‐Pearse TL, Flanagan JG. APC is an RNA‐binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell 2014, 158:368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yokota Y, Kim WY, Chen Y, Wang X, Stanco A, Komuro Y, Snider W, Anton ES. The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron 2009, 61:42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Barth AI, Caro‐Gonzalez HY, Nelson WJ. Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin Cell Dev Biol 2008, 19:245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Koester MP, Muller O, Pollerberg GE. Adenomatous polyposis coli is differentially distributed in growth cones and modulates their steering. J Neurosci 2007, 27:12590–12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF‐induced axon growth is mediated by localized inactivation of GSK‐3β and functions of the microtubule plus end binding protein APC. Neuron 2004, 42:897–912. [DOI] [PubMed] [Google Scholar]
- 89. Shi SH, Cheng T, Jan LY, Jan YN. APC and GSK‐3β are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr Biol 2004, 14:2025–2032. [DOI] [PubMed] [Google Scholar]
- 90. Brakeman JS, Gu SH, Wang XB, Dolin G, Baraban JM. Neuronal localization of the Adenomatous polyposis coli tumor suppressor protein. Neuroscience 1999, 91:661–672. [DOI] [PubMed] [Google Scholar]
- 91. Berwick DC, Harvey K. The importance of Wnt signalling for neurodegeneration in Parkinson's disease. Biochem Soc Trans 2012, 40:1123–1128. [DOI] [PubMed] [Google Scholar]
- 92. Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci 2010, 11:77–86. [DOI] [PubMed] [Google Scholar]
- 93. Groen EJ, Fumoto K, Blokhuis AM, Engelen‐Lee J, Zhou Y, van den Heuvel DM, Koppers M, van Diggelen F, van Heest J, Demmers JA, et al. ALS‐associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet 2013, 22:3690–3704. [DOI] [PubMed] [Google Scholar]
- 94. Fallini C, Bassell GJ, Rossoll W. Spinal muscular atrophy: the role of SMN in axonal mRNA regulation. Brain Res 2012, 1462:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 2013, 14:161–176. [DOI] [PubMed] [Google Scholar]
- 96. Rishal I, Fainzilber M. Axon‐soma communication in neuronal injury. Nat Rev Neurosci 2014, 15:32–42. [DOI] [PubMed] [Google Scholar]
- 97. Perry RB, Doron‐Mandel E, Iavnilovitch E, Rishal I, Dagan SY, Tsoory M, Coppola G, McDonald MK, Gomes C, Geschwind DH, et al. Subcellular knockout of importin β1 perturbs axonal retrograde signaling. Neuron 2012, 75:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ben‐Yaakov K, Dagan SY, Segal‐Ruder Y, Shalem O, Vuppalanchi D, Willis DE, Yudin D, Rishal I, Rother F, Bader M, et al. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J 2012, 31:1350–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, Jaffrey SR. Intra‐axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol 2008, 10:149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Fanara P, Banerjee J, Hueck RV, Harper MR, Awada M, Turner H, Husted KH, Brandt R, Hellerstein MK. Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J Biol Chem 2007, 282:23465–23472. [DOI] [PubMed] [Google Scholar]
- 101. Sirajuddin M, Rice LM, Vale RD. Regulation of microtubule motors by tubulin isotypes and post‐translational modifications. Nat Cell Biol 2014, 16:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bilsland LG, Sahai E, Kelly G, Golding M, Greensmith L, Schiavo G. Deficits in axonal transport precede ALS symptoms in vivo. Proc Natl Acad Sci U S A 2010, 107:20523–20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, et al. Familial amyotrophic lateral sclerosis‐linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet 2007, 16:2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Vande Velde C, McDonald KK, Boukhedimi Y, McAlonis‐Downes M, Lobsiger CS, Bel Hadj S, Zandona A, Julien JP, Shah SB, Cleveland DW. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One 2011, 6:e22031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Marinkovic P, Reuter MS, Brill MS, Godinho L, Kerschensteiner M, Misgeld T. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2012, 109:4296–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhu YB, Sheng ZH. Increased axonal mitochondrial mobility does not slow amyotrophic lateral sclerosis (ALS)‐like disease in mutant SOD1 mice. J Biol Chem 2011, 286:23432–23440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fallini C, Bassell GJ, Rossoll W. The ALS disease protein TDP‐43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum Mol Genet 2012, 21:3703–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wang IF, Wu LS, Chang HY, Shen CK. TDP‐43, the signature protein of FTLD‐U, is a neuronal activity‐responsive factor. J Neurochem 2008, 105:797–806. [DOI] [PubMed] [Google Scholar]
- 109. Narayanan RK, Mangelsdorf M, Panwar A, Butler TJ, Noakes PG, Wallace RH. Identification of RNA bound to the TDP‐43 ribonucleoprotein complex in the adult mouse brain. Amyotroph Lateral Scler Frontotemporal Degener 2013, 14:252–260. [DOI] [PubMed] [Google Scholar]
- 110. Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, et al. Axonal transport of TDP‐43 mRNA granules is impaired by ALS‐causing mutations. Neuron 2014, 81:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet 2014, 23:1413–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kim SH, Shanware NP, Bowler MJ, Tibbetts RS. Amyotrophic lateral sclerosis‐associated proteins TDP‐43 and FUS/TLS function in a common biochemical complex to co‐regulate HDAC6 mRNA. J Biol Chem 2010, 285:34097–34105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bartoli KM, Bishop DL, Saunders WS. The role of molecular microtubule motors and the microtubule cytoskeleton in stress granule dynamics. Int J Cell Biol 2011, 2011:939848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Loschi M, Leishman CC, Berardone N, Boccaccio GL. Dynein and kinesin regulate stress‐granule and P‐body dynamics. J Cell Sci 2009, 122:3973–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dubey J, Ratnakaran N, Koushika SP. Neurodegeneration and microtubule dynamics: death by a thousand cuts. Front Cell Neurosci 2015, 9:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med 2010, 48:629–641. [DOI] [PubMed] [Google Scholar]
- 117. Chang Y, Kong Q, Shan X, Tian G, Ilieva H, Cleveland DW, Rothstein JD, Borchelt DR, Wong PC, Lin CL. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS One 2008, 3:e2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Tanaka M, Chock PB, Stadtman ER. Oxidized messenger RNA induces translation errors. Proc Natl Acad Sci U S A 2007, 104:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Shan X, Chang Y, Lin CL. Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression. FASEB J 2007, 21:2753–2764. [DOI] [PubMed] [Google Scholar]
- 120. Cohen TJ, Hwang AW, Unger T, Trojanowski JQ, Lee VM. Redox signalling directly regulates TDP‐43 via cysteine oxidation and disulphide cross‐linking. EMBO J 2012, 31:1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Cohen TJ, Hwang AW, Restrepo CR, Yuan CX, Trojanowski JQ, Lee VM. An acetylation switch controls TDP‐43 function and aggregation propensity. Nat Commun 2015, 6:5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Duan W, Li X, Shi J, Guo Y, Li Z, Li C. Mutant TAR DNA‐binding protein‐43 induces oxidative injury in motor neuron‐like cell. Neuroscience 2010, 169:1621–1629. [DOI] [PubMed] [Google Scholar]
- 123. McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande VC. TAR DNA‐binding protein 43 (TDP‐43) regulates stress granule dynamics via differential regulation of G3BP and TIA‐1. Hum Mol Genet 2011, 20:1400–1410. [DOI] [PubMed] [Google Scholar]
- 124. Takahashi M, Higuchi M, Matsuki H, Yoshita M, Ohsawa T, Oie M, Fujii M. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol Cell Biol 2013, 33:815–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Van Damme P, Dewil M, Robberecht W, Van Den Bosch L. Excitotoxicity and amyotrophic lateral sclerosis. Neurodegener Dis 2005, 2:147–159. [DOI] [PubMed] [Google Scholar]
- 126. La Via L, Bonini D, Russo I, Orlandi C, Barlati S, Barbon A. Modulation of dendritic AMPA receptor mRNA trafficking by RNA splicing and editing. Nucleic Acids Res 2013, 41:617–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Smith WB, Starck SR, Roberts RW, Schuman EM. Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron 2005, 45:765–779. [DOI] [PubMed] [Google Scholar]
- 128. Yamashita T, Kwak S. The molecular link between inefficient GluA2 Q/R site‐RNA editing and TDP‐43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. Brain Res 2014, 1584:28–38. [DOI] [PubMed] [Google Scholar]
- 129. Udagawa T, Fujioka Y, Tanaka M, Honda D, Yokoi S, Riku Y, Ibi D, Nagai T, Yamada K, Watanabe H, et al. FUS regulates AMPA receptor function and FTLD/ALS‐associated behaviour via GluA1 mRNA stabilization. Nat Commun 2015, 6:7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cozzolino M, Ferri A, Valle C, Carri MT. Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci 2013, 55:44–49. [DOI] [PubMed] [Google Scholar]
- 131. Deng J, Yang M, Chen Y, Chen X, Liu J, Sun S, Cheng H, Li Y, Bigio EH, Mesulam M, et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet 2015, 11:e1005357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, Wang X. The ALS disease‐associated mutant TDP‐43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet 2013, 22:4706–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP‐43 transgenic mice. Proc Natl Acad Sci U S A 2010, 107:16325–16330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 1998, 93:1147–1158. [DOI] [PubMed] [Google Scholar]
- 135. Dunn S, Morrison EE, Liverpool TB, Molina‐Paris C, Cross RA, Alonso MC, Peckham M. Differential trafficking of Kif5c on tyrosinated and detyrosinated microtubules in live cells. J Cell Sci 2008, 121:1085–1095. [DOI] [PubMed] [Google Scholar]
- 136. Herms A, Bosch M, Reddy BJ, Schieber NL, Fajardo A, Ruperez C, Fernandez‐Vidal A, Ferguson C, Rentero C, Tebar F, et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun 2015, 6:7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cozzolino M, Rossi S, Mirra A, Carri MT. Mitochondrial dynamism and the pathogenesis of amyotrophic lateral sclerosis. Front Cell Neurosci 2015, 9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yoon BC, Jung H, Dwivedy A, O'Hare CM, Zivraj KH, Holt CE. Local translation of extranuclear lamin B promotes axon maintenance. Cell 2012, 148:752–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Aschrafi A, Natera‐Naranjo O, Gioio AE, Kaplan BB. Regulation of axonal trafficking of cytochrome c oxidase IV mRNA. Mol Cell Neurosci 2010, 43:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gioio AE, Eyman M, Zhang H, Lavina ZS, Giuditta A, Kaplan BB. Local synthesis of nuclear‐encoded mitochondrial proteins in the presynaptic nerve terminal. J Neurosci Res 2001, 64:447–453. [DOI] [PubMed] [Google Scholar]
- 141. Natera‐Naranjo O, Kar AN, Aschrafi A, Gervasi NM, Macgibeny MA, Gioio AE, Kaplan BB. Local translation of ATP synthase subunit 9 mRNA alters ATP levels and the production of ROS in the axon. Mol Cell Neurosci 2012, 49:263–270. [DOI] [PubMed] [Google Scholar]
- 142. Kar AN, Sun CY, Reichard K, Gervasi NM, Pickel J, Nakazawa K, Gioio AE, Kaplan BB. Dysregulation of the axonal trafficking of nuclear‐encoded mitochondrial mRNA alters neuronal mitochondrial activity and mouse behavior. Dev Neurobiol 2014, 74:333–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Brunden KR, Trojanowski JQ, Smith AB 3rd, Lee VM, Ballatore C. Microtubule‐stabilizing agents as potential therapeutics for neurodegenerative disease. Bioorg Med Chem 2014, 22:5040–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhang B, Maiti A, Shively S, Lakhani F, McDonald‐Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, et al. Microtubule‐binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A 2005, 102:227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Barten DM, Fanara P, Andorfer C, Hoque N, Wong PY, Husted KH, Cadelina GW, Decarr LB, Yang L, Liu V, et al. Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule‐stabilizing agent BMS‐241027. J Neurosci 2012, 32:7137–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Yoshiyama Y, Zhang B, Bruce J, Trojanowski JQ, Lee VM. Reduction of detyrosinated microtubules and Golgi fragmentation are linked to tau‐induced degeneration in astrocytes. J Neurosci 2003, 23:10662–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]