

Abstract
We evaluated the pharmacokinetics (PK), safety, and tolerability of a novel oral CRTh2 antagonist, fevipiprant (QAW039), in healthy subjects. Peak concentrations of fevipiprant in plasma were observed 1‒3 hours postdosing. Concentrations declined in a multiexponential manner, followed by an apparent terminal phase (t1/2, ∼20 hours). Steady state was achieved in 4 days with <2‐fold accumulation. Elimination was partly by renal excretion (≤30% of the dose) and glucuronidation. Food had minimal impact on the PK of fevipiprant, and it was well tolerated at single and multiple oral doses up to 500 mg/day. No dose‐dependent adverse events were observed, and all the events were mild or moderate in severity. Systemic concentrations were sufficiently high to achieve relevant target occupancy, considering in vitro pharmacology data. In summary, the data support further development as a once‐daily oral therapy for allergic diseases.
Keywords: fevipiprant, QAW039, pharmacokinetics, safety, healthy subjects
Prostaglandin D2 (PGD2) is an arachidonic acid metabolite that is released largely by activated mast cells in high concentrations and plays a key role in inflammatory response in allergic conditions.1, 2, 3 PGD2 exerts its actions through interaction with G‐protein‐coupled receptors, which include the classical prostanoid receptor DP1 and the more recently discovered DP2 or CRTh2 (chemoattractant receptor homologous molecule expressed on Th2 cells) receptor.4, 5 The DP1 receptor mediates the vascular effects of PGD2 such as stimulation of vasodilation and inhibition of platelet aggregation,6 whereas the CRTh2 receptor primarily mediates the inflammatory effects of PGD2.4, 7
CRTh2 receptors, expressed on eosinophils, basophils, T‐helper 2 cells, macrophages, and neutrophils are known to be involved in chemotaxis and activation of these cells, which form the key events initiating the inflammatory response in allergic diseases.7 There is growing interest in developing CRTh2 antagonists to counteract the pathophysiological effects of PGD2 and alleviate inflammatory responses in allergic diseases.8 Placebo‐controlled clinical studies showed that the CRTh2 antagonist OC000459 reduces eosinophil counts in asthmatic patients and nasal and ocular allergic symptoms in patients with allergic rhinitis exposed to grass pollen in an allergen challenge chamber model.9, 10 These studies confirmed a role for CRTh2 antagonists in allergic diseases, and, accordingly, there has been a significant increase in the number of CRTh2 antagonists in development.11, 12 Fevipiprant (QAW039; [(2‐[2‐methyl‐1‐(4‐[methylsulfonyl]‐2‐[trifluoromethyl]benzyl)‐1H‐pyrrolo(2,3‐b)pyridin‐3‐yl] acetic acid)]) is a selective, potent, reversible competitive CRTh2 antagonist with an in vitro dissociation constant KD value of 1.1 nM at the CRTh2 receptor and an IC50 value of 0.44 nM for inhibition of PGD2‐induced eosinophil shape change in human whole blood.13 It is currently under clinical development as an oral therapy for allergic conditions such as asthma.14 We report data from 2 phase 1 studies that evaluated the pharmacokinetics (PK), safety, and tolerability of fevipiprant on administration of single and multiple ascending doses in healthy subjects.
Methods
Both studies were randomized, double‐blind, and placebo‐controlled and were conducted at SGS‐Life Sciences Services (SGS Belgium NV‐SGS House, Antwerp, Belgium). The single‐dose, first‐in‐human, single‐center study employed an ascending‐alternate‐dose design. Sixteen healthy adult subjects were randomized to receive fevipiprant (n = 6) or placebo (n = 2). Subjects received 2 sequential doses of fevipiprant or placebo with a 10‐day washout period between the 2 doses (either 10 and 100 mg or 30 and 300 mg; Supplementary Figure 1). Subjects were admitted to the study center 1 day prior to dosing for baseline assessment, remained at the center until the morning of day 4 and returned on days 5 and 6 for further sample collection and safety assessment. The starting dose was estimated in accordance with the European and the US health authority guidance. The human dose equivalent to the no‐observed‐adverse‐effect level (NOAEL) dose in the most sensitive species (rat) of 200 mg/kg (4 weeks repeated‐dose toxicity study) is 32.2 mg/kg, based on body surface area scaling. The actual starting dose of 10 mg was 225‐fold lower for a 70‐kg subject. This low starting dose was chosen because the predicted plasma exposure and the high potency of fevipiprant biological effects were considered possible at low doses.
The multiple‐dose study randomized 32 subjects to receive fevipiprant (100 or 300 mg once daily or 500‐mg single dose in fed and fasting conditions or 250 mg twice daily; n = 6 for each dose level) or placebo (n = 2 for each dose level); see Supplementary Figure 2. Dose escalation was performed after assessing safety and PK data of the previous dose levels. Subjects receiving multiple doses were confined to the study center from day ‐1 to day 12 and dosed in the mornings for a period of 7 days, with the end‐of‐study evaluation being conducted on day 12. To explore the effect of food at single dose, the subjects received 500 mg fevipiprant or matching placebo in 2 sequential periods (fasting and fed). During the fed condition, subjects consumed the “FDA high‐fat breakfast,” with the end‐of‐study evaluation being conducted on day 6 of period 2 (fed) and an intertreatment washout period of 10 days.
Study Population (Single‐ and Multiple‐Dose Studies)
Healthy male (using acceptable methods of contraception) and female subjects aged 18 to 55 years with a body mass index of 18 to 29 kg/m2 were eligible for participation. Eligibility was assessed at screening and baseline by medical history, current medical condition, physical examination, and serum chemistry, hematology, and quantitative cardiac troponin I/T as a measure of cardiac muscle damage.
The key exclusion criteria included a history of alcohol or drug abuse; acute or chronic bronchospastic disease; use of any prescription drugs within 4 weeks or over‐the‐counter medications within 2 weeks before dosing; clinically significant electrocardiogram (ECG) abnormalities; and photosensitive reactions to medications, topical creams, or other chemical products. For both studies, the sample size was customary for phase 1 studies evaluating safety, tolerability, and PK and not powered to support inferential hypothesis testing. Subjects that fulfilled all inclusion/exclusion criteria were randomized. All the patients were included in the PK and safety analysis sets.
The protocols, consent forms, and other study materials were reviewed and approved by the responsible institutional review board (ZNA — Campus Middelheim, Antwerpen, Belgium). All subjects provided written informed consent prior to participation in the study.
Statistics
Subjects who received at least 1 dose of study drug and with quantifiable plasma concentrations were included in the PK analysis set. The safety analysis set included all subjects who received at least 1 dose of the study drug. The PK parameters for each subject were determined by noncompartmental analysis using WinNonlin Pro, version 5.2 (Pharsight Corporation, Mountain View, California).
PK Assessment and Analysis
The PK parameters were determined for both fevipiprant and its major acylglucuronide metabolite (AG‐metabolite). The following PK parameters were estimated: maximum plasma concentration (Cmax); apparent terminal half‐life (t1/2); area under the plasma concentration–time curve (AUC) from time zero to the last measurable concentration (AUC0–last), or extrapolated to infinity (AUC0–∞). PK parameters determined in the urine included renal clearance (CLr), amount of drug excreted (Ae) in urine from time zero to t (Ae0–t), and maximum excretion rate in urine (ERmax). Time to Cmax (tmax) was estimated in both plasma and urine.
Blood samples were collected into tubes containing anticoagulant at predefined times (Supplementary Table 1) for PK analysis. After centrifugation at 3000g for 15 minutes, 1 mL of plasma was withdrawn and acidified to stabilize the metabolite(s) and avoid back‐conversion to the parent compound. Urine samples were collected for predefined collection periods (Supplementary Table 1); 5 mL of urine sample per collection period was transferred to a tube and acidified to stabilize the metabolite(s) and avoid back‐conversion to the parent compound.
Plasma and urine sample processing consists of a solid‐phase extraction using a 96‐well plate (OASIS HLB) and analysis by liquid chromatography (LC)–tandem mass spectrometry.
Chromatographic separation was performed at a flow rate of 0.5 mL/min on Symmetry C18 column (30 × 2.1 mm, 3.5 μm). A binary gradient of 0.2% formic acid in water (mobile phase A) and methanol (mobile phase B) was used for LC separation. A triple quadrupole mass spectrometer (API 4000 from AB Sciex) was used for detection. The system was operating in electrospray positive ion mode using MRM transitions 427.1/145.0 for fevipiprant, 603.0/427.0 for AG‐metabolite, and 433.2/145.0 for [13CD5]fevipiprant (internal standard).
The lower limit of quantification (LLOQ) for fevipiprant was 1.00 ng/mL in plasma and urine. LLOQ for AG‐metabolite was 2.00 ng/mL in plasma and 5.00 ng/mL in urine. Intrarun and interrun precision and accuracy within the acceptance criteria were achieved during 3 validation runs. The absence of matrix effect was demonstrated, and recovery, stabilities (3 freeze‐thaw cycles, long‐term, bench‐top, incurred samples, etc.), and incurred samples reanalysis were also validated. Concentrations below the limit of quantification were treated as zero in summary statistics and were not considered for calculation of PK parameters. Summary statistics of PK parameters are provided.
In the single‐dose study, the dose‐proportionality relationship was assessed by log‐transformed PK parameters (AUC0–last and Cmax) using a power model: ln (PK) = μ + β × ln(dose) + subject, where subject was a random effect.
In the multiple‐dose study, for subjects receiving fevipiprant 100 and 300 mg once daily and 250 mg twice daily, individual ratios for AUCtau on day 7/day 1 were calculated and summarized to assess the accumulation ratio. Steady‐state attainment was determined graphically and by using a repeated‐measures linear‐mixed model on log‐transformed trough concentrations (day 2 onward). The model included day as a factor, log‐dose as a continuous covariate, and subject as a random effect.
Food Effect Model
The effect of food on the PK for the 500‐mg single dose was evaluated. Log‐transformed AUC and Cmax were analyzed using a linear mixed‐effects model with period (fed or fasted) as a fixed effect and subject as a random effect. The least‐squares mean estimate of the period contrast and corresponding 90% confidence interval (CI) was back‐transformed onto the original scale to estimate the geometric mean ratio (fed vs fasted) and its associated 90%CI.
Safety Assessments
In both the studies, safety assessments included recording of all AEs and serious AEs (SAEs) with their severity and relationship to the study drug. Clinical safety assessments included regular monitoring of hematology, blood chemistry, and urine chemistry. Regular assessments of vital signs, physical condition, ECG, and body weight were also performed. Continuous heart rate monitoring by telemetry and Holter monitoring was performed for 12 hours postdose. Safety data were summarized descriptively by treatment.
Selected ECG and vital sign parameters (body temperature, blood pressure, pulse rate) were analyzed using a repeated‐measures analysis of covariance model including time as a factor, dose and predose value as covariates, and dose by time as an interaction term. The mean difference of fevipiprant versus placebo was estimated at the highest dose along with 90% confidence interval (CI). All statistical analyses were performed using SAS.
Ethical Considerations
All subjects provided written informed consent before screening. The protocols were approved by the local ethics committee, and the studies were conducted in accordance with the declaration of Helsinki and Good Clinical Practices guidance.
Results
Patient Disposition and Demographic Characteristics
All subjects were white, and they were well balanced with regard to demographic characteristics (Table 1). All subjects completed the study and were included in the safety, tolerability, and PK analyses.
Table 1.
Baseline and Demographic Characteristics
| Fevipiprant Single‐Dose Study | Fevipiprant Multiple‐Dose Study | ||||||
|---|---|---|---|---|---|---|---|
| Parameters | 10/100 mg n = 6/Placebo n = 2 | 30/300 mg n = 6/placebo n = 2 | 100 mg Once Daily | 300 mg Once Daily | 500 mga | 250 mg Twice Daily | Placebo |
| n | 8 | 8 | 6 | 6 | 6 | 6 | 8 |
| Age, years | 46.6 (5.8) | 45.4 (9.9) | 41.3 (10.7) | 40.2 (8.1) | 38.0 (11.5) | 42.0 (13.0) | 44.9 (6.8) |
| Sex, male, n (%) | 7 (87.5) | 2 (25.0) | 6 (100.0) | 5 (83.3) | 6 (100.0) | 4 (66.7) | 3 (37.5) |
| BMI, kg/m2 | 25.5 (3.1) | 26.2 (2.2) | 23.7 (2.1) | 24.9 (0.9) | 24.9 (1.9) | 24.0 (2.6) | 24.5 (2.8) |
In the single‐dose study, each of the 8 subjects was randomized (6:2) to receive the treatments (active:placebo).
Data are expressed as mean (standard deviation) unless otherwise indicated.
BMI, body mass index.
aSingle dose of fevipiprant 500 mg in fed and fasting conditions sequentially.
Pharmacokinetics
Plasma concentration–time profiles following single ascending oral doses of fevipiprant are presented in Figure 1, and the corresponding PK parameters are presented in Table 2. Fevipiprant peak plasma concentrations were observed 1 to 3 hours (median) following single and multiple doses. The concentration–time profiles were similar across the different doses, and concentrations declined in a multiexponential manner. The mean apparent terminal half‐life (t1/2) was 18.6 and 20.2 hours following 100‐ and 300‐mg doses, respectively. In the single ‐dose study, systemic exposure increased over the 30‐fold dose range (from 10 to 300 mg), but dose proportionality could only be observed for a 2‐fold increase in dose, possibly because of underlying variability in data (Supplementary Table 2).
Figure 1.
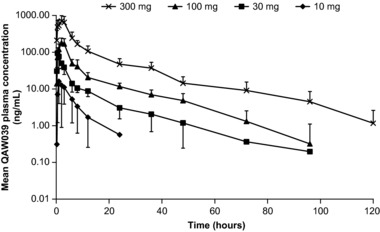
Mean ± SD plasma concentration–time curves of QAW039 following single ascending doses.
Table 2.
Summary of Pharmacokinetic (PK) Parameters of Fevipiprant (PK Analysis Set) for the Single‐ and Multiple‐Dose Studies
| Dose | tmax a (h) | Cmax (ng/mL) | AUC0–24 (ng·h/mL) | AUC0–last (ng·h/mL) | t1/2 (h) |
|---|---|---|---|---|---|
| Fevipiprant Single‐Dose Study | |||||
| 10 mg | 1 (1–3) | 21 (8) | 133c (60) | 104 (58) | NC |
| 30 mg | 1 (0.5–3) | 105 (87) | 382 (120) | 445 (166) | NC |
| 100 mg | 2 (1–3) | 201 (68) | 1114 (342) | 1369 (408) | 19 (11) |
| 300 mg | 2 (1–6) | 803 (253) | 4857 (1219) | 6170 (1186) | 20 (6) |
| Dose | tmax a (h) | Cmax Day 7 (ng/mL) | AUCtau Day 7 (ng·h/mL) | Racb (AUCtau) | AG‐Metabolite/Fevipiprant AUCtau molar ratiod |
| Fevipiprant Multiple‐Dose Study | |||||
| 100 mg once daily | 2 (1–6) | 524 (217) | 2873 (783) | 1.3 (0.4) | 1.4 (0.1) |
| 300 mg once daily | 3 (1–6) | 1183 (404) | 6993 (2155) | 1.4 (0.5) | 1.3 (0.2) |
| 250 mg twice daily | 3 (2–6) | 978 (484) | 4569 (1797) | 1.8 (0.4) | 1.2 (0.1) |
Data are expressed as mean (SD) unless otherwise specified.
AUC, area under the plasma concentration–time curve; AUC0–24 h, AUC from 0 to 24 hours postdose; AUC0–last, AUC from 0 to last measurable plasma concentration; AUCtau, AUC in the dosing interval; Cmax, maximum plasma concentration; CLr, renal clearance; NC, not calculated (because of lack of sufficient data in the terminal phase); tmax, time to reach Cmax; t1/2, elimination half‐life.
atmax, expressed as median (min–max). bRac, accumulation ratio, AUCtau (day 7)/AUCtau (day 1). cThree subjects had concentrations below the limit of quantification (1 ng/mL) at 24 hours postdose. dAUCtau AG‐metabolite day 1 (μM·h)/AUCtau fevipiprant day 1 (μM·h).
The mean total amount of fevipiprant recovered within 72 hours postdose was between 12.7% and 16.6% of the dose in the single‐dose study. In the multiple‐dose study 28.3%–33.8% of the dose was recovered in urine (means of different doses; Table 3). Renal clearance of fevipiprant determined in both studies ranged from 8.2 to 10.9 L/h.
Table 3.
Urine Pharmacokinetic (PK) Parameters for Fevipiprant (PK Analysis Set)
| Dose | Ae0–72 (μg) | Ae0–72 (% of Dose) | CLr (L/h) | ERmax (μg/h) |
|---|---|---|---|---|
| Fevipiprant Single‐Dose Study | ||||
| 10 mg | 1274 (255) | 12.7 (2.6) | 8.2 (3.6) | 104 (20) |
| 30 mg | 3997 (779) | 13.3 (2.6) | 8.6 (2.8) | 323 (87) |
| 100 mg | 13 420 (2248) | 13.4 (2.2) | 10.1 (2.9) | 1113 (286) |
| 300 mg | 49 640 (12 612) | 16.6 (4.2) | 8.7 (2.1) | 4513 (1715) |
| Dose | Aetau | Aetau (% Dose) | CLr | Molar Ratioa |
| Fevipiprant Multiple‐Dose Study | ||||
| 100 mg once daily | 33 820 (5689.5) | 33.8 (5.7) | 10.9 (4.9) | 0.8 (0.1) |
| 300 mg once daily | 96 780 (13 736.0) | 32.3 (4.6) | 10.9 (1.7) | 0.7 (0.1) |
| 250 mg twice daily | 70 770 (12 505.0) | 28.3 (5.0) | 9.9 (3.1) | 0.9 (0.1) |
Data are expressed as mean (SD) unless otherwise indicated.
Ae0–72, amount of drug excreted into the urine from 0 to 72 hours postdose; Cmax, maximum plasma concentration; CLr, renal clearance; ERmax, maximum (peak) observed excretion rate of drug into urine; Rac, accumulation ratio; tmax, time to reach Cmax; t1/2, elimination half‐life.
aAetau AG‐metabolite (day 7)/Aetau fevipiprant (day 7).
Comparison of the trough concentrations of fevipiprant in the multidose study, from days 2 to 6 indicated that steady state was achieved by day 4. The tmax of the AG‐metabolite was similar to fevipiprant, indicating rapid glucuronidation. The AUC molar ratios (AG‐metabolite/fevipiprant) of 1.4–1.5 indicate higher systemic exposure to the metabolite (Table 2). Fevipiprant showed a small degree of accumulation on repeat dosing, with mean accumulation ratios (day 7/day 1) for AUCtau of 1.3–1.4 and 1.8 for once daily and twice daily, respectively.
Food Effect
The rate of absorption of fevipiprant and AG‐metabolite following 500 mg oral administration was minimally affected by a high‐fat breakfast as recommend by the FDA for food effect investigations (Figure 2). The median tmax in fed subjects was 2.5 hours, with an increased variability range of 2–8 hours compared with the fasted state, for which it was 2 hours with a range of 1–3 hours. The estimated geometric mean ratios (fed/fasted) along with the 90%CI for the various PK parameters were: AUCinf, 1.0 (0.93–1.07); AUClast and AUCtau, 0.95 (0.90–1.01); and Cmax, 0.90 (0.63–1.26).
Figure 2.
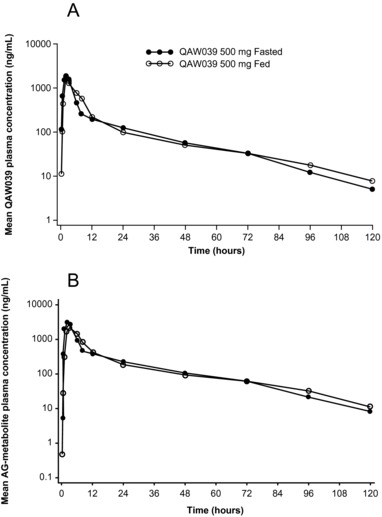
Mean plasma concentration–time profiles of (A) QAW039 and (B) the acyl glucuronide metabolite (AG‐metabolite) on log‐linear scale in fed and fasted states.
The amount of fevipiprant recovered in urine was 129 and 146 mg in fed and fasted states, respectively. The mean CLr of fevipiprant was 9.37 and 10.3 L/h in the fed and fasted state, respectively. The Ae0–120 molar ratio of metabolite to fevipiprant was 0.8–1.2 in fed and fasted states, confirming the importance of urinary excretion as a route of elimination for both fevipiprant and its glucuronide metabolite and that CLr is unaffected by food. These results indicate that food had a minimal impact on the PK of fevipiprant.
Safety and Tolerability
A total of 5 subjects (31.3%) and 17 subjects (53.1%) in the fevipiprant treatment arm, and 2 subjects (25%) and 7 subjects (87.5%) in the placebo arm experienced at least 1 AE during the single‐ and multiple‐dose studies, respectively (Table 4). There were no SAEs or deaths reported. All AEs were either mild or moderate, and the incidence of AEs was similar across the different fevipiprant groups and the placebo group, except in the treatment arm exploring the fevipiprant 500‐mg dose in the fed condition, during which no AEs were observed.
Table 4.
Incidence of AEs (>1 Subject) by Preferred Term
| Fevipiprant Single‐Dose Study | Fevipiprant Multiple‐Dose Study | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Preferred Term | 10 mg (n = 6) | 30 mg (n = 6) | 100 mg (n = 6) | 300 mg (n = 6) | Placebo (n = 8) | 100 mg (n = 6) | 300 mg (n = 6) | 500 mg Fasteda (n = 6) | 500 mg Feda (n = 6) | 250 mg Twice Daily (n = 6) | Placebo (n = 8) |
| Subjects with AEs, n (%) | 3 (50.0) | 1 (16.7) | — | 1 (16.7) | 2 (25.0) | 3 (50.0) | 4 (66.7) | 5 (83.3) | — | 5 (83.3) | 7 (87.5) |
| Headache | 1 (16.7) | — | — | — | 1 (12.5) | 1 (16.7) | 3 (50.0) | 2 (33.3) | — | 2 (33.3) | 5 (62.5) |
| Nasopharyngitis | ‐ | — | — | — | — | 2 (33.3) | — | 2 (33.3) | — | 2 (33.3) | 3 (37.5) |
| Constipation | ‐ | — | — | — | — | 1 (16.7) | — | — | — | — | 3 (37.5) |
| Dry mouth | ‐ | — | — | — | — | — | — | 2 (33.3) | — | — | 1 (12.5) |
| Back pain | ‐ | — | — | — | — | — | — | 1 (16.7) | — | — | 2 (25.0) |
Data are expressed as n (%).
AE, adverse event.
a500 mg was given as a single oral dose. AE starting in 1 period and continuing into the second period was counted only once for the 500‐mg single dose.
The most frequent AEs were headache and nasal congestion; all other AEs occurred as single events in individual subjects (Table 4). There were no clinically relevant changes with respect to clinical chemistry, hematology, ECG parameters, vital signs, and physical examinations.
Discussion
The research interest in CRTh2 receptor antagonists is based on their ability to prevent the PGD2‐mediated inflammatory response in allergic diseases, as demonstrated in various preclinical models15 and clinical reports.10, 16 Allergic conditions are characterized by PGD2‐mediated recruitment of Th2 cells and eosinophils that express CRTh2 receptors. Binding of PGD2 to CRTh2 receptors stimulates Th2 cells to secrete cytokines (IL4, IL5, and IL13) and eosinophils to release tissue destructive mediators, leading to tissue remodeling and poor symptom control. Fevipiprant, a novel CRTh2 antagonist, inhibits in vitro activation, migration, and release of mediators by effector cells and thereby should counteract the pathophysiological effects of PGD2 and alleviate the inflammatory response. Fevipiprant has earlier demonstrated good in vitro potency (IC50 for the whole‐blood eosinophil shape change assay of 0.44 nM) during the preclinical studies.13 In comparison with other published structurally related CRTh2 antagonists, it exhibits improved affinity combined with a significantly slower receptor dissociation time.17 We report results from 2 phase 1 studies for fevipiprant that assessed the PK, safety, and tolerability of single and multiple ascending doses in healthy subjects.
On administration of single and multiple oral doses, fevipiprant peak concentrations in plasma were observed 1–3 hours postdose. The initial decline of the plasma concentration occurred in a multiexponential manner; the apparent terminal phase had a t1/2 of approximately 20 hours. Steady state was achieved within 4 days with <2‐fold accumulation. Dose proportionality was only confirmed for a 2‐fold increase in dose, possibly because of the variability in the data.
Fevipiprant was metabolized to an inactive AG‐metabolite; the tmax of 2.5–3.0 hours for the metabolite indicates rapid conversion. Renal clearance was an important route of elimination, with up to one‐third of the dose excreted as fevipiprant in the urine; in addition, AG‐metabolite was found at similar molar amounts in urine. Fevipiprant was secreted actively into urine, as the renal clearance (∼170 mL/min) was >10‐fold higher than the product of the unbound fraction in plasma for fevipiprant (0.118) and the glomerular filtration rate (GFR, 125 mL/min*0.118 = 14.75 mL/min). The PK of fevipiprant at a 500‐mg single dose was little affected by food. The tmax and AUC0–120 for the fed and fasted dose groups were 2.5 versus 2.0 hours and 14.0 versus 14.8 μg·h/mL, respectively.
Based on the observed human PK, the dissociation constant (Kd) for fevipiprant at the human CRTh2 receptor of 1.1 nM as well as its unbound fraction in plasma of 11.8%, doses of 150 mg once daily or higher are expected to provide ≥90% CRTh2 receptor occupancy for the whole dosing interval at steady state. The human whole‐blood eosinophil shape change assay predicts a similar or even higher potency of fevipiprant.13 Therefore, clinical efficacy may plateau at doses of 150 mg once daily or lower, but the relevance of the in vitro pharmacology data to predict clinical efficacy is unclear.
Fevipiprant was well tolerated across the studied dose range (10‒500 mg) on oral administration as single and multiple doses. There were no discontinuations because of AEs. There were no SAEs and deaths reported; the overall incidence of AEs was not dose dependent, and the AE profile for the fevipiprant arms was similar to that of the placebo arms. These data are in line with previous safety data for CRTh2 antagonists, including OC000459,16 AZD1981,18 and setipiprant.19 The observed plasma exposure to QAW039 (AUC and Cmax) has safety margins of >25 compared with exposure in the nonclinical models at the NOAEL doses. The major human metabolite (AG‐metabolite) was also formed in the species used for nonclinical safety assessments with similar or higher exposure observed at NOAEL compared with human exposure at 500 mg once daily. This metabolite was shown to be inactive as a CRTh2 antagonist (data on file at Novartis).
Fevipiprant has shown good in vitro potency and a suitable PK profile (high systemic exposure following oral dosing and relevant concentrations up to and beyond 24 hours postdose) with an acceptable degree of variability in systemic exposure. These characteristics of fevipiprant are expected to deliver a therapeutic dose that is safe and allows for once‐a‐day dosing, all of which are highly desirable features for clinical development. These findings support further exploration of the potential of fevipiprant as a treatment option for patients with allergic diseases.
Multiple CRTh2 receptor antagonists are under clinical investigation,20 and there has also been a significant degree of turnover of CRTh2 antagonists in clinical development (AZD1981, setipiprant, AZD5985, RG7185), the main reasons disclosed being low clinical efficacy (setipiprant) or undesirable PK profiles.20 For example, MK7246 was discontinued because of high intersubject variability,21 and studies on AM211 also reported high intersubject variability and enterohepatic recirculation.22 In contrast, fevipiprant has demonstrated efficacy in phase 2 studies in patients with asthma and is now proceeding toward development phase 3. Clinical efficacy was observed as well as strong mechanistic effects, including a reduction in lung eosinophils that was demonstrated in a recently published mechanistic study.22, 23, 24
Conclusions
The peak levels and steady state for fevipiprant were achieved rapidly with limited accumulation. Food had a marginal impact on exposure, and elimination was dependent on metabolism and renal clearance, limiting its sensitivity to pharmacokinetic drug interactions. In addition, fevipiprant was safe and well tolerated across the dose range studied (10–500 mg) for both single and multiple doses. Fevipiprant has the potential to be developed as a convenient once‐daily oral dose therapy for patients with allergic diseases.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Material
Acknowledgments
We thank all the clinical investigators and study coordinators at the participating centers and all the subjects who participated in the study. This work was funded by Novartis Institutes for BioMedical Research. We acknowledge Amit Garg and Praveen Kaul, Novartis Healthcare Pvt. Ltd., Hyderabad, India, for medical writing support.
Declaration of Conflicting Interests
V.J.E., M.W., M.L., and W.E. are employees of Novartis Pharma AG, Basel, Switzerland. E.V. and L.G. are employees of SGS life sciences, Antwerp. Belgium. W.O., G.D., and P.G. are former employees of Novartis Institutes for BioMedical Research, Horsham, United Kingdom. D.S. is an employee of Novartis Institutes for BioMedical Research Inc, Cambridge, Massachusetts. S.N. is an employee of Novartis Healthcare Pvt. Ltd., Hyderabad, India.
Author Contributions
As study investigators or study personnel V.J.E., M.W., M.L., E.V., L.G., W.O., G.D., and P.G. were involved in the concept and design of the studies, interpretation of data, and/or preparation of the manuscript. S.N. provided support for statistical analysis, W.E. conducted all bioanalytical work. All the authors had full access to raw data, contributed to each draft of the manuscript, take full responsibility for the content and approved the final manuscript for publication.
Funding
This study was funded by Novartis Pharma AG.
References
- 1. Lewis RA, Soter NA, Diamond PT, Austen KF, JA Oates, Roberts LJ 2nd. Prostaglandin D2 generation after activation of rat and human mast cells with anti‐IgE. J Immunol. 1982;129:1627‒1631. [PubMed] [Google Scholar]
- 2. Murray JJ, Tonnel AB, Brash AR, et al. Release of prostaglandin D2 into human airways during acute antigen challenge. N Engl J Med. 1986;315:800‒804. [DOI] [PubMed] [Google Scholar]
- 3. Doyle WJ, Boehm S, Skoner DP. Physiologic responses to intranasal dose‐response challenges with histamine, methacholine, bradykinin, and prostaglandin in adult volunteers with and without nasal allergy. J Allergy Clin Immunol. 1990;86:924‒935. [DOI] [PubMed] [Google Scholar]
- 4. Pettipher R. The roles of prostaglandin D2 receptors DP1 and CRTh2 in promoting allergic responses. Br J Pharmacol. 2008;153:S191‒S199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kostenis E, Ulven T. Emerging roles of DP and CRTH2 in allergic inflammation. Trends Mol Med. 2006;12:148‒158. [DOI] [PubMed] [Google Scholar]
- 6. Cheng K, Wu TJ, Wu KK, et al. Antagonism of the prostaglandin D2 receptor 1 suppresses nicotinic acid‐induced vasodilation in mice and humans. Proc Natl Acad Sci U S A. 2006;103:6682‒6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophiles, and basophiles via seven‐transmembrane receptor CRTH2. J Exp Med. 2001;193:225‒261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pettipher R, Hansel TT, Armer R. Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat Rev Drug Discov. 2007;6:313‒325. [DOI] [PubMed] [Google Scholar]
- 9. Barnes N, Pavord I, Chuchalin A, et al. A randomised, double‐blind, placebo‐controlled study of the CRTh2 receptor antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy. 2012;42:38‒44. [DOI] [PubMed] [Google Scholar]
- 10. Horak F, Zieglmayer P, Zieglmayer R, et al. The CRTh2 antagonist OC000459 reduces nasal and ocular symptoms in allergic subjects exposed to grass pollen, a randomised, placebo‐controlled, double‐blind trial. Allergy. 2012;67:1572‒1579. [DOI] [PubMed] [Google Scholar]
- 11. Ly TW, Bacon KB. Small‐molecule CRTH2 antagonists for the treatment of allergic inflammation: an overview. Expert Opin Investig Drugs. 2005;14:769‒773. [DOI] [PubMed] [Google Scholar]
- 12. Ulven T, Kostenis E. Novel CRTH2 antagonists: a review of patents from 2006 to 2009. Expert Opin Ther Pat. 2010;20:1505‒1530. [DOI] [PubMed] [Google Scholar]
- 13. Willard L, Brown Z, Owen C, Dubois G. Characterization of QAW039 and QAV680, two novel, potent and selective CRTh2 antagonists. Eur Resp J. 2014;44(suppl. 58):P4072. [Google Scholar]
- 14. Gonem S, Berair R, Singapuri A, et al. Phase 2a randomised placebo‐controlled trial of the oral prostaglandin D2 receptor (DP2/CRTh2) antagonist QAW039 in eosinophilic asthma. Eur Resp J. 2014;44(Suppl 58):2908. [Google Scholar]
- 15. Tasaki M, Kobayashi M, Tenda Y, et al. Inhibition of antigen‐induced airway inflammation and hyperresponsiveness in guinea pigs by a selective antagonist of "chemoattractant receptor homologous molecule expressed on Th2 cells" (CRTH2). Eur J Pharm Sci. 2013;49:434‒440. [DOI] [PubMed] [Google Scholar]
- 16. Barnes N, Pavord I, Chuchalin A, et al. A randomized, double‐blind, placebo‐controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy. 2012; 42:38‒48. [DOI] [PubMed] [Google Scholar]
- 17. Sykes D, Bradley M, Riddy D, et al. QAW039 a slowly dissociating CRTh2 antagonist with the potential for improved clinical efficacy. Eur Resp J. 2014;44(suppl 58):2908 [DOI] [PubMed] [Google Scholar]
- 18. Snell N, Foster M, Vestbo J. Efficacy and safety of AZD1981, a CRTH2 receptor antagonist, in patients with moderate to severe COPD. Respir Med. 2013;107(11):1722‒1730. [DOI] [PubMed] [Google Scholar]
- 19. Sidharta PN, Diamant Z, Dingemanse J. Single‐ and multiple‐dose tolerability and pharmacokinetics of the CRTH2 antagonist setipiprant in healthy male subjects. Fundam Clin Pharmacol. 2014;28(6):690‒699. [DOI] [PubMed] [Google Scholar]
- 20. Norman P. Update on the status of DP2 receptor antagonists; from proof of concept through clinical failures to promising new drugs. Expert Opin Investig Drugs. 2014;23:55‒66. [DOI] [PubMed] [Google Scholar]
- 21. Wang YH, Trucksis M, McElwee JJ, et al. UGT2B17 genetic polymorphisms dramatically affect the pharmacokinetics of MK‐7246 in healthy subjects in a first‐in‐human study. Clin Pharmacol Ther. 2012;92:96‒102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bain G, King CD, Brittain J, et al. Pharmacodynamics, pharmacokinetics, and safety of AM211: a novel and potent antagonist of the prostaglandin D2 receptor type 2. J Clin Pharmacol. 2012;52:1482‒1493. [DOI] [PubMed] [Google Scholar]
- 23. Gonem S, Berair R, Singapuri A, et al. Phase 2a randomized placebo‐controlled trial of the oral prostaglandin D2 receptor (DP2/ CRTh2) antagonist QAW039 in eosinophilic asthma. Eur Resp J. 2014;44:(suppl 58):2908. [Google Scholar]
- 24. Erpenbeck V, Popov T, Miller D, et al. QAW039 (fevipiprant) improves lung function and control of asthma symptoms in patients with more severe air flow limitation: a proof‐of‐concept study. Abstract # 851370. Poster presented at the European Respiratory Society Annual Congress, September 36–30, 2015, Amsterdam, The Netherlands. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Material