

Abstract
The transcription factor Interferon Regulatory Factor 4 (IRF4) is essential for TH2 and TH17 cell formation and controls peripheral CD8+ T cell differentiation. We used Listeria monocytogenes infection to characterize the function of IRF4 in TH1 responses. IRF4−/− mice generated only marginal numbers of listeria-specific TH1 cells. After transfer into infected mice, IRF4−/− CD4+ T cells failed to differentiate into TH1 cells as indicated by reduced T-bet and IFN-γ expression, and showed limited proliferation. Activated IRF4−/− CD4+ T cells exhibited diminished uptake of the glucose analog 2-NBDG, limited oxidative phosphorylation and strongly reduced aerobic glycolysis. Insufficient metabolic adaptation contributed to the limited proliferation and TH1 differentiation of IRF4−/− CD4+ T cells. Our study identifies IRF4 as central regulator of TH1 responses and cellular metabolism. We propose that this function of IRF4 is fundamental for the initiation and maintenance of all TH cell responses.
The transcription factor Interferon Regulatory Factor 4 (IRF4) is expressed in various hematopoietic cells, including B and T cells but also different macrophage and dendritic cell subsets1,2,3,4,5,6,7. In B cells, IRF4 controls the germinal center reaction and high IRF4 expression is a prerequisite for plasma cell formation. As a consequence, antibodies are almost completely absent in IRF4-deficient mice8,9. Naive peripheral T cells express only low levels of IRF4. Upon T cell receptor stimulation, IRF4 is rapidly expressed and subsequently controls differentiation processes of these cells1,8,10,11. Deficiency of IRF4 in CD4+ T cells results in a complete block in the formation of TH2, TH9, TH17 and follicular TH (TFH) cells12,13,14,15,16,17,18,19,20. Although IRF4-deficiency allows the generation of Foxp3+ Treg cells, these cells are impaired in their suppressive functions21,22. IRF4 also controls peripheral CD8+ T cells differentiation. We and others could demonstrate that following antigen recognition, IRF4-deficient CD8+ T cells start to proliferate and to express effector molecules such as IFN-γ and granzyme B. However, IRF4-deficent cells cannot sustain proliferation and fail to upregulate effector molecules to the level observed in wild type CD8+ effector T cells. In line with these results, IRF4-deficient CD8+ T cells express reduced levels of transcription factors associated with CD8+ effector T cell formation including T-bet, BLIMP1 and ID28,11,23,24,25,26,27.
In contrast to other IRF family members, IRF4 binds interferon stimulated response elements (ISRE) with low affinity. However, in cooperation with transcription factors of the Ets or AP-1 families, IRF4 is able to strongly bind to Ets-IRF composite elements (EICE) or AP-1-IRF composite elements (AICE), respectively9,28. Cooperative binding with the Ets proteins PU.1 and SpiB to EICE has been demonstrated for B cells and myeloid cells. However, both transcription factors are usually not expressed in T cells, indicating that interaction of IRF4 with EICE does not commonly occur in T cells29,30. In contrast, T cells express the AP-1 proteins BATF, JunB, JunD and c-Jun, and cooperative binding of IRF4 with heterodimers of BATF and Jun family members was demonstrated for TH17 cells and CD8+ T cells29,30,31.
Using mRNA expression studies and chromatin immune precipitation (ChIP), target genes for IRF4 have been determined for TH17 and CD8+ T cells. These targets include a large number of genes involved in T cell activation and differentiation25,30,31,32. Interestingly, IRF4 and BATF frequently bind to regulatory DNA regions outside the promotors. Therefore, it was proposed that IRF4 and BATF might act as pioneering factors that promote and sustain chromatin remodeling and enhance accessibility of genes for other transcription factors, including lineage-specific factors such as T-bet or RORγt25,29,31,32. In CD8+ T cells, IRF4 controls expression of transcription factors involved in effector cell differentiation including Tbx21 (encoding T-bet), Prdm1 (encoding BLIMP1), Runx3 and Tcf7 (encoding TCF-1), as well as effector proteins such as cytokines and cytolytic proteins11,25,26. IRF4 is also involved in the metabolic changes of CD8+ T cells following activation. Naive T cells show basal levels of glucose and amino acid uptake and mainly use oxidative phosphorylation and fatty acid oxidation for energy production. T cell activation causes enhanced nutrient uptake as well as increased aerobic glycolysis and glutaminolysis. These changes in the metabolic profile are necessary to provide energy and substrates for de novo synthesis of proteins, nucleic acids and lipids required for proliferation and effector protein production33,34,35,36. Metabolic changes are controlled by different transcription factors including HIF1α, FOXO1 and FOXO3. IRF4 modulates the expression of these factors but also directly enhances expression of several proteins involved in nutrient uptake and glycolysis25,33. Impaired adaptation to metabolic demands can explain the failure of IRF4-deficient CD8+ T cells to sustain proliferation and to develop into mature effector cells25,33. IRF4 expression levels correlate with the strength of the TCR signal, thereby IRF4 links TCR affinity with the extent of metabolic changes following CD8+ T cell activation. It has been proposed that during immune responses this mechanism promotes the preferential expansion of high affinity CD8+ T cell populations25,27,33.
In contrast to its function in CD8+ T cells and CD4+ TH cell subsets described above, the role IRF4 in TH1 cell development is less clear. TH1 differentiation of IRF4−/− CD4+ T cells has been analyzed in vitro and in vivo using the Leishmania major infection model, with inconsistent results12,13,14,17. Although all studies provided evidence for TH1 differentiation of IRF4−/− CD4+ T cells, the efficacy of this process ranged from marginal to close to that observed in WT cells.
Here, we use the Listeria monocytogenes infection model to analyze the role of IRF4 in TH1 cell differentiation and function. We demonstrate that IRF4 is crucial for the generation of a TH1 response. IRF4−/− CD4+ T cells showed impaired TH1 differentiation and only limited proliferation after in vitro and in vivo activation. Compared to control cells, activated IRF4−/− CD4+ T cells exhibited impaired aerobic glycolysis and oxidative phosphorylation and this restricted metabolism could be responsible for the poor response of these cells.
Results
IRF4-deficient mice fail to mount a TH1 response to Listeria monocytogenes
Wild type (WT) and IRF4−/− mice were infected with LmOVA, a L. monocytogenes strain recombinant for ovalbumin37, and after 8 days the CD4+ TH1 response was analyzed. In WT mice, about half of the CD4+ T cells presented an activated CD44+CD62L− phenotype (Figure S1). IRF4−/− mice also accumulated CD62L− CD4+ T cells, however, these cells failed to up-regulate CD44. CD4+ T cells from infected mice were stimulated with the immunodominant listeriolysin O peptide LLO189-201 and the expression of CD40L (CD154)38, IFN-γ and TNF-α was determined by intracellular staining (Fig. 1 and Figures S2 and S3). Following LLO189-201 stimulation, approx. 2% of CD4+ T cells from infected WT mice responded with up-regulation of CD40L and more than half of these cells co-expressed high levels of IFN-γ and TNF-α, a hallmark of TH1 cells. LLO189-201–specific CD4+ T cells were significantly reduced in IRF4−/− mice, as indicated by the reduced frequencies of CD40L+ cells. Only a small fraction of these CD40L+ cells co-expressed IFN-γ and TNF-α and particularly IFN-γ+TNF-α+ cells were substantially reduced in IRF4−/− mice. In addition, cytokine-positive IRF4−/− CD4+ T cells displayed diminished staining intensity for IFN-γ and TNF-α, indicating that cells produced only low amounts of the cytokines (Fig. 1a,b).
Figure 1. IRF4−/− mice fail to mount a TH1 response against L. monocytogenes.
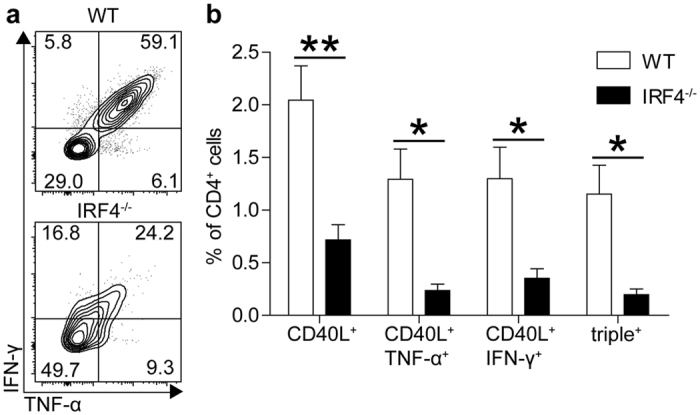
WT and IRF4−/− mice were i.v. infected with 5 × 104 LmOVA. Eight days post infection, spleen cells were stimulated for 4 h with 10−5 M LLO189-201 and intracellular expression of IFN-γ, TNF-α and CD40L was determined by flow cytometry. (a) Representative dot plots for CD4+CD40L+ spleen cells from infected mice stimulated with LLO189-201. (b) Frequencies of CD40L+, IFN-γ+ and TNF-α+ cells among CD4+ T cells from spleens of infected mice following stimulation. Bars represent mean ± SEM from 4 individually analyzed mice per group. The result is representative for 4 independent experiments.
IRF4−/− CD4+ T cells have an intrinsicTH1 differentiation defect
IRF4 is also expressed in subsets of dendritic cells and macrophages2,3,4,5,6,7 and absence of IRF4 in these cells could also impair CD4+ T cell responses. Therefore, CD4+ T cells were purified from WT and IRF4−/− mice on a CD90.1+CD90.2− and CD90.1−CD90.2+ background, respectively. Cells were mixed at a 1:1 ratio and co-transferred into RAG1−/− mice, which had been infected with Lm 4h earlier. After 8 days, WT CD4+ T cells had outnumbered the IRF4−/− CD4+ T cells by a factor of 4 (Fig. 2a,b). WT CD4+ T cells had uniformly acquired a CD44+CD62L−CD127− phenotype (Fig. 2c and Figure S4a) and significant subpopulations expressed KLRG1 and the chemokine receptor CXCR3, which have been associated with TH1 cells. In contrast, IRF4−/− CD4+ T cells showed reduced up-regulation of CD44 and CXCR3 and reduced down-regulation of CD62L and CD127. We failed to detect KLRG1+ IRF4−/− CD4+ T cells. Following polyclonal stimulation, approx. 70% of transferred WT CD4+ T cells responded with CD40L expression and about 30% of CD4+ T cells had differentiated into IFN-γ+TNF-α+ TH1 cells (Fig. 2d and Figure S4b). In contrast, the IRF4−/− CD4+ T cells showed reduced up-regulation of CD40L and only a small population was IFN-γ+TNF-α+. Finally, expression of the transcription factor T-bet was determined by intracellular staining (Fig. 2e,f). Whereas WT CD4+ T cells displayed elevated T-bet expression following infection, IRF4−/− CD4+ T cells presented with expression levels comparable to those of cells from uninfected mice.
Figure 2. Impaired TH1 differentiation of IRF4−/− CD4+ T cells.
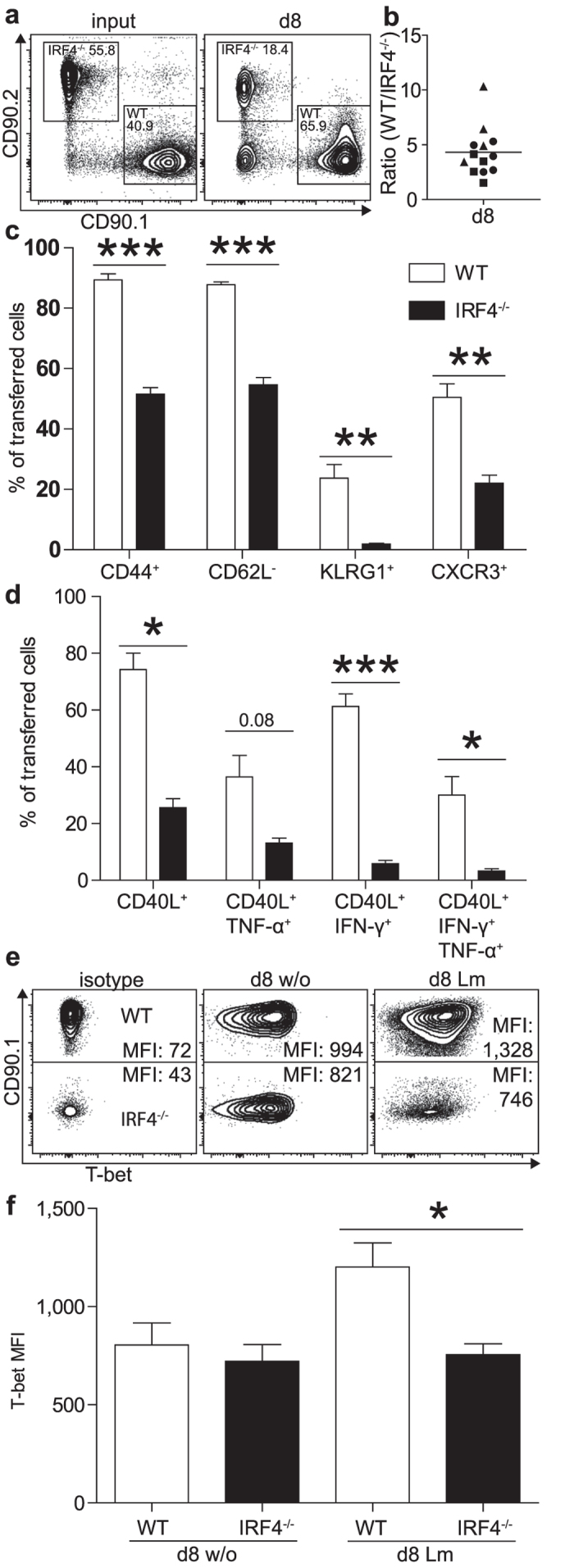
Purified CD4+ T cells from CD90.1+CD90.2− WT and CD90.1−CD90.2+ IRF4−/− mice were mixed in a 1:1 ratio and 2 × 106 cells were i.v. transferred into RAG1−/− mice, which had been infected with 105 Lm 4 h earlier. Eight days post infection, spleen cells of mice were analyzed. (a) Representative CD90.1/CD90.2 dot blots of CD4+ T cells at the time point of transfer (input) and at d8 post infection. (b) Normalized ratio of WT and IRF4−/− CD4+ T cells. The figure shows results for individual mice combined from 3 experiments and the mean. (c) Surface expression of CD44, CD62L, KLRG1 and CXCR3 on transferred CD4+ T cells. (d) CD40L, TNF-α, and IFN-γ expression of transferred CD4+ T cells from spleens of infected mice following stimulation with PMA/ionomycin. (e) Representative intracellular T-bet staining for transferred cells (MFI mean fluorescence intensity). (f) T-bet expression in WT and IRF4−/− CD4+ T cells following transfer and with or without infection. (c,d,f) Show mean ± SEM for 4 individually analyzed mice per group. Results are representative for 3 independent experiments (with the exception of results for CXCR3 which are derived from one experiment only).
To characterize the specific CD4+ T cell response in immune competent mice, ovalbumin-specific CD4+ T cells from WT and IRF4−/− OT-II mice (CD90.1+CD90.2+ and CD90.1−CD90.2+, respectively) were CFSE-labelled and competitively transferred into WT mice (CD90.1+CD90.2−). Recipients had either been infected with LmOVA or were treated with OVA323-339 peptide and LPS. Four days post transfer, WT OT-II cells had 2fold or 8fold outnumbered IRF4−/− OT-II cells in mice infected with LmOVA or treated with OVA323-339 peptide, respectively (Fig. 3a,b). Accordingly, transferred IRF4−/− OT-II cells remained to a larger extend CFSE-positive and this cell population failed to expand in infected or peptide-treated animals (Fig. 3c–e). In recipients infected with LmOVA, transferred WT OT-II cells expressed enhanced levels of IRF4 (Figure S5a). Compared to transferred WT OT-II cells, transferred IRF4−/− OT-II cells showed diminished CD44 and KLRG1 up-regulation and CD62L down-regulation (Figure S5b). Spleen cells from LmOVA infected recipients were stimulated either with OVA323-339 peptide or with PMA/ionomycin and analyzed for the expression of CD40L and cytokines (Fig. 4a,b). With both stimuli, we observed a population of CD40L+IFN-γ+TNF-α+ TH1 cells among transferred WT OT-II cells, which was hardly detectable in IRF4−/− OT-II cells. In conclusion, T cell transfer assays indicate that IRF4−/− CD4+ T cells failed to respond to different stimuli in vivo with the generation of TH1 cells.
Figure 3. Impaired T cell response of IRF4−/− CD4+ T cells following in vivo activation.
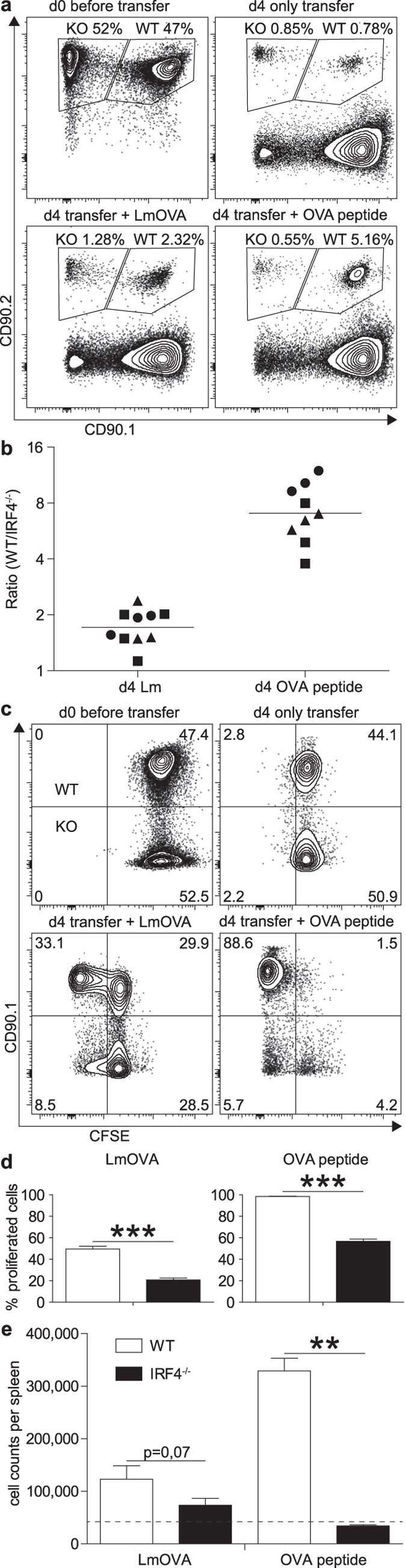
Purified CD4+ T cells from WT CD90.1+CD90.2+ OT-II mice and IRF4−/− CD90.1−CD90.2+ OT-II mice were mixed in a 1:1 ratio and labelled with CFSE. 3 × 106 cells were i.v. transferred into CD90.1+CD90.2− WT mice. Recipient mice had either been i.v. infected with 105 LmOVA one day before or were i.v. treated with 50 μg OVA323-339 peptide and 30 μg LPS 1 h post transfer. Mice were analyzed 4d post transfer. (a) Representative CD90.1/CD90.2 dot plots of CD4-gated cells. (b) Normalized ratio of WT and IRF4−/− CD4+ T cells. The figure shows results for individual mice combined from 3 experiments and the mean. (c) Representative CFSE/CD90.1 dot plots of CD4+CD90.2+ cells. (d) %-values of CFSElow cells among transferred OT-II cells. (e) Total numbers of transferred WT and IRF4−/− OT-II cells recovered from recipient mice. (dotted line: mean numbers of transferred cells recovered from mice without infection.) (d,e) Show mean ± SEM for 3 individually analyzed mice per group. Results are representative for 3 independent experiments.
Figure 4. Reduced TH1 differentiation of transferred IRF4−/− OT-II T cells.
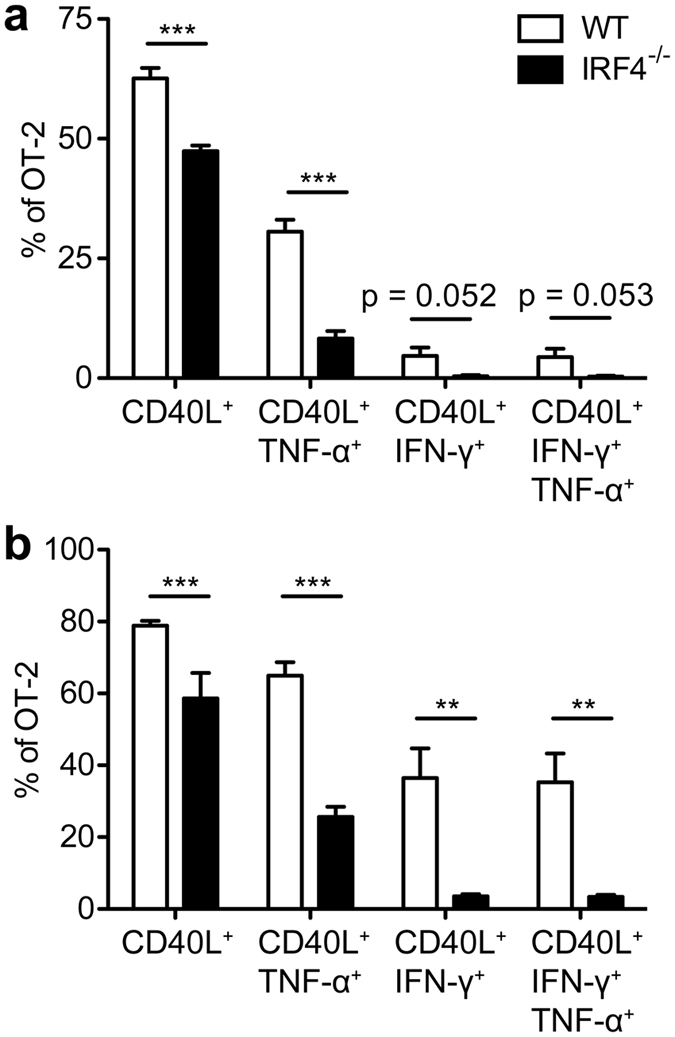
Purified CD4+ T cells from WT CD90.1+CD90.2+ or IRF4−/− CD90.1−CD90.2+ OT-II mice were mixed in a 1:1 ratio. 106 cells were i.v. transferred into CD90.1+CD90.2− WT mice which had been i.v. infected with 105 LmOVA one day before. On d4 post transfer, spleen cells from infected recipients were incubated for 4 h with 10−6M of OVA323-339 peptide or with PMA/ionomycin and stained intracellularly for CD40L, TNF-α, and IFN-γ. Figures show frequencies of CD40L+, IFN-γ+ and TNF-α+ cells among transferred OT-II T cells following stimulation with OVA323-339 (a) and PMA/ionomycin (b). Bars represent mean ± SEM from 6 individually analyzed mice per group. The result is representative for 4 independent experiments.
IRF4−/− CD4+ T cells show reduced TH1 differentiation in vitro
To test whether absence of IRF4 prevents TH1 differentiation, WT and IRF4−/− CD4+ T cells were mixed to obtain a ratio of 1:1, and polyclonally stimulated under TH1 cell inducing conditions. On day 4, cells were restimulated and tested for the expression of IFN-γ. Whereas 80% of WT CD4+ T cells expressed IFN-γ, only about 50% of IRF4−/− CD4+ T cells were IFN-γ+ (Fig. 5a), and in these cells the mean IFN-γ expression level was lower than in the respective WT cells. In accordance with the impaired IFN-γ production, we could also observe diminished up-regulation of T-bet in IRF4−/− CD4+ T cells (Fig. 5b). Thus, IRF4-deficiency impaired but did not generally prevent TH1 differentiation. Activated CD4+ T cells were also analyzed by quantitative PCR for the expression of Runx3 and Prdm1, coding for two transcription factors involved in T-cell differentiation and representing targets of IRF4 in CD8+ T cells25,26. In contrast to CD8+ T cells, we observed similar induction for both Runx3 and Prdm1 in WT and IRF4−/− CD4+ T cells (Figure S6a,b).
Figure 5. Impaired in vitro response of IRF4−/− CD4+ T cells.
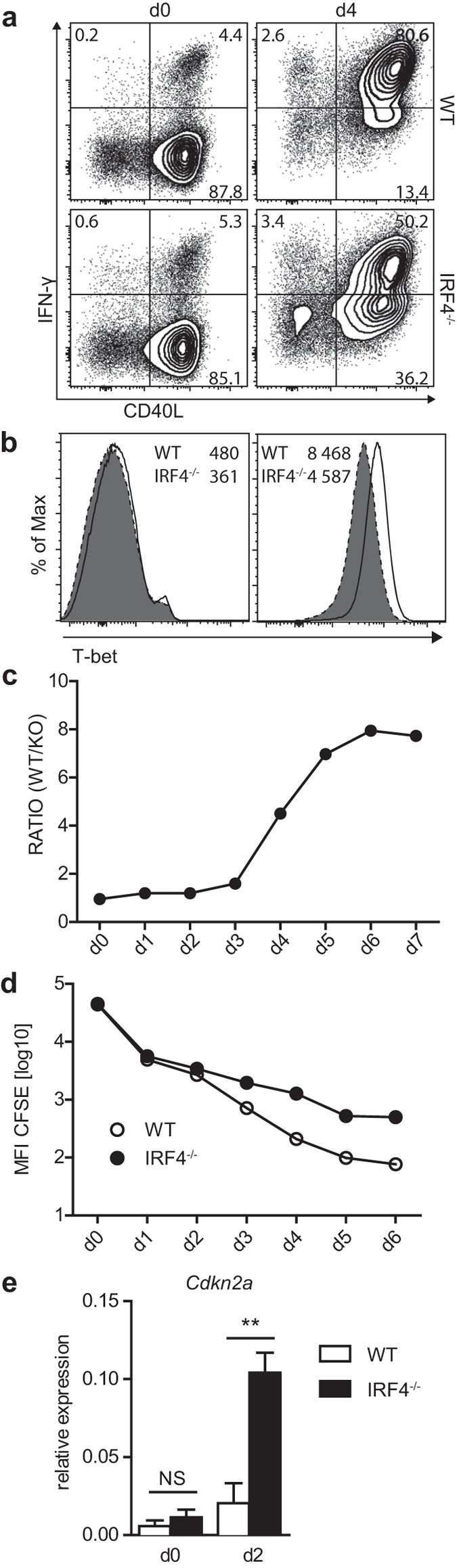
CD4+ T cells from WT (CD90.1+) and IRF4−/− mice (CD90.2+) were mixed with a 1:1 ratio. Cells were stimulated in vitro with anti-CD3 mAb, anti-CD28 mAb and IL-2 in the presence of IL-12 and anti-IL-4 mAb to induce TH1 differentiation. (a) After 4d, cells were stimulated for 4 h with PMA/ionomycin and IFN-γ and CD40L expression was determined by intracellular staining. Spleen cells from naive WT and IRF4−/− mice were stimulated and stained in parallel (d0). (b) T-bet expression in WT and IRF4−/− CD4+ T cells without stimulation and at d4 of stimulation (numbers give the MFIs). (c) Ratio of WT and IRF4−/− CD4+ T cells in co-culture at indicated days of stimulation. (d) Loss of CFSE staining intensity on viable WT and IRF4−/− CD4+ T cells. (e) Expression of Cdkn2a at d0 and d2 of purified WT and IRF4−/− CD4+ T cells stimulated in individual cultures. Blots and histograms in (a,b) show representative results for CD4-gated cells. Results are representative for 3 (a–c) or 2 (d) independent experiments. Bars in (e) represent the mean ± SEM of 4 individual samples from 2 independent experiments.
When co-cultures were analyzed for the ratio of WT to IRF4−/− CD4+ T cells, we realized that over time WT CD4+ T cells vastly outnumbered IRF4−/− CD4+ T cells (Fig. 5c). Compared to WT CD4+ T cells, IRF4−/− CD4+ T cells also presented with diminished CD25 expression and reduced blast formation as indicated by a lower forward scatter (Figure S7a,b). To determine proliferation, WT and IRF4−/− CD4+ T cells were mixed, labelled with CFSE, and the loss of CFSE staining was measured (Fig. 5d). Following activation, both WT and IRF4−/− CD4+ T cells showed initial reduction of CFSE staining intensity. After 2–3 days, decrease of CFSE staining was halted in IRF4−/− CD4+ T cells. In contrast, WT CD4+ T cells continued to lose staining intensity. A similar response was observed when WT and IRF4−/− CD4+ T cells were activated in individual cultures (Figure S7c). In accordance with these observations, activated IRF4−/− CD4+ T cells showed enhanced mRNA expression for the cyclin-dependent kinase inhibitor Cdkn2a, which has been identified as IRF4 target in CD8+ T cells26 (Fig. 5e).
In vitro activated CD4+ T cells were also analyzed for induction of apoptosis (Figure S8a). CD4+ T cells were stimulated and the proportion apoptotic was determined by measuring binding of the caspase substrate FLICA and uptake of propidium iodide (PI)11. Compared to WT controls, IRF4−/− CD4+ T cells showed slightly reduced frequencies of early apoptotic (FLICA+PI−) cells and slightly enhanced frequencies of late apoptotic (FLICA+PI+) cells. Determination of mRNA expression of Bcl2 and Bcl2l11 coding for the anti-apoptotic protein BCL2 and the pro-apoptotic protein BIM, respectively, revealed similar induction of both mRNA species in activated WT and IRF4−/− CD4+ T cells (Figure S8b,c). In summary, these results indicate that the impaired expansion of the IRF4−/− CD4+ T cell population was mainly due to reduced proliferation and only to a minor degree to enhanced apoptosis.
IRF4−/− CD4+ T cells are impaired in cellular metabolism
For CD8+ T cells, it has been shown that IRF4 controls expression of proteins involved in central metabolic pathways such as glucose uptake, glycolysis and oxidative phosphorylation and thereby promotes changes in cell metabolism necessary for T cell proliferation and effector functions25. Recently it was also demonstrated that TH1 differentiation of CD4+ T cells is associated with similar metabolic changes39. To test whether IRF4 controls the metabolism of TH1 cells, WT and IRF4−/− CD4+ T cells were activated and uptake of the fluorescent glucose analog 2-NBDG was measured by flow cytometry (Fig. 6a). Activation caused enhanced 2-NBDG uptake in WT and IRF4−/− CD4+ T cells, but uptake was reduced in IRF4−/− CD4+ T cells. The bioenergetics profile of IRF4−/− TH1 cells was then tested with extracellular flux assays (Fig. 6b,c). Following activation, basal oxygen consumption rates (OCR) were slightly lower in IRF4−/− than in WT CD4+ T cells (Fig. 6b). OCR was similar in both cell populations following treatment with the ATP synthase inhibitor oligomycin, however, IRF4−/− CD4+ T cells showed impaired OCR increase in response to disruption of the proton gradient with FCCP, indicating that the maximal respiratory capacity was reduced in comparison to WT CD4+ T cells. The glycolytic capacity was tested by measuring extracellular acidification rates (ECAR). Compared to activated IRF4−/− CD4+ T cells, activated WT CD4+ T cells showed an increase in ECAR after the addition of glucose. Blockade of ATP synthesis by oxidative phosphorylation with oligomycin further increased ECAR in WT CD4+ T cells, but had no effect on IRF4−/− CD4+ T cells. Thus, deficiency of IRF4 in CD4+ T cells resulted in diminished glucose uptake and reduced glycolysis and oxidative phosphorylation.Ex vivo analysis of OT-II CD4+ T cells from competitive transfer assays supported the in vitro results. Four days after transfer, IRF4−/− OT-II cells were smaller and expressed reduced levels of the transferrin receptor CD71 than WT OT-II cells (Fig. 6d). These IRF4−/− OT-II cells also showed diminished ability to take up 2-NBDG. In line with these results and similar to CD8+ T cells25, we observed reduced mRNA expression for the glucose transporter GLUT3 (Slc2a3) and the central glycolysis enzyme hexokinase 2 (Hk2) in in vitro activated IRF4−/− CD4+ T cells (Fig. 6e). Thus, IRF4 apparently controls metabolism in CD4+ T cells by regulating the expression of nutrient transporters and glycolysis enzymes. Interestingly and in contrast to IRF4−/− CD8+ T cells25, we did not observe altered mRNA expression for the transcription factor HIF1α, a central regulator of cellular metabolism.
Figure 6. Altered metabolism in IRF4−/− CD4+ T cells.
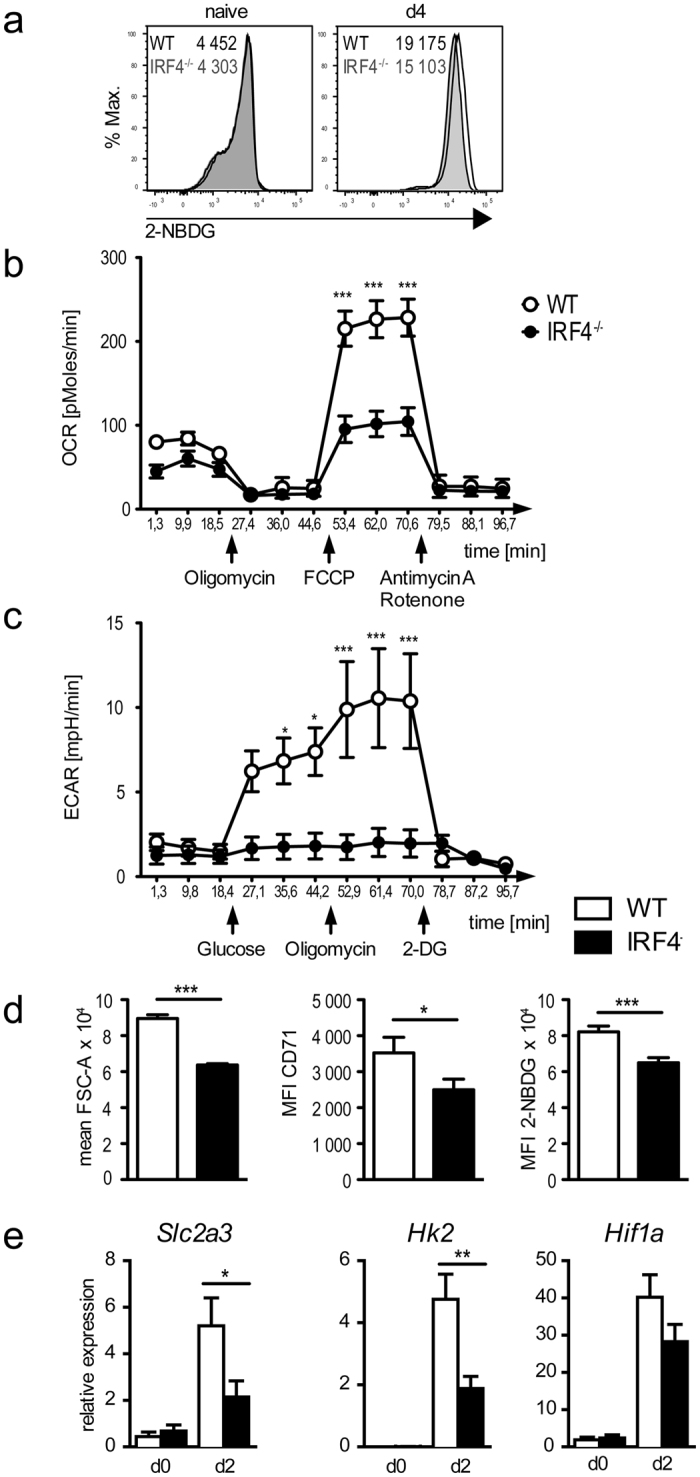
(a) CD4+ T cells from WT (black lines) and IRF4−/− mice (grey lines) were stimulated as described in Fig. 5. At day 4, cells were washed and in incubated with 30 μM 2-NBDG in glucose-free medium. After 2 h, 2-NBDG uptake was measured by flow cytometry. For comparison, spleen cells from naive mice (d0) were similarly analyzed (numbers give the MFIs). (b,c) CD4+ T cells were purified from spleens of WT and IRF4−/− mice and stimulated over night with anti-CD3 mAb and anti-CD28 mAb. Cells were washed and OCR (b) and ECAR (c) at baseline level and in response to glucose and to different inhibitors were determined as described in the material method section. (d) Competitive transfer of WT and IRF4−/− OT-II T cells was done as described in Fig. 4. Four days after transfer, OT-II cells from spleen were analyzed for size (FSC-A) and CD71 expression. Spleen cells were also in incubated with 30 μM 2-NBDG in glucose-free medium and after 2 h, 2-NBDG uptake was measured. (e) Expression of Slc2a3, Hk2 and Hif1a in activated WT and IRF4−/− CD4+ T cells. CD4+ T cells were purified and stimulated in individual cultures for 2d as described in Fig. 5. Histograms in (a) show CD4-gated cells and are representative for 3 independent experiments. Results in (b,c) are cumulative for 2 experiments with quintuplicate values for each cell subset. Bars in D represent mean ± SEM from 6 individually analyzed mice per group. Bars in (e) represent the mean ± SEM of 4 individual samples from 2 independent experiments.
In conclusion, our results indicate that IRF4 is crucial for the regulation of metabolism in TH1 cells which results in a failure to mount TH1 responses.
Discussion
Our study demonstrates that IRF4 is essential for effective formation of TH1 responses. Following L. monocytogenes-infection, listeria-specific IRF4−/− CD4+ T cells showed impaired TH1 differentiation and failed to expand to the level observed for WT CD4+ T cells. Published analyses of the role of IRF4 in TH1 differentiation revealed inconsistent results12,13,14. In vitro, IRF4-deficient CD4+ T cells showed either normal TH1 differentiation13,14 or were significantly impaired14. Following L. major infection, studies described normal13 or severely diminished IFN-γ production14 when cells from lesion draining lymph nodes were stimulated with leishmania-antigen. In our study, we observed limited accumulation of specific IRF4−/− CD4+ T cells in response to L. monocytogenes infection or peptide immunization. Remaining specific IRF4−/− CD4+ T cells were impaired in TH1 differentiation as indicated by reduced production of IFN-γ and TNF-α (both in terms of frequency of responding cells and of cytokine expression per cell), diminished expression of T-bet and a failure to upregulate the TH1 markers CXCR3 and KLRG1. In vitro, IFN-γ production could be induced in IRF4−/− CD4+ T cells, however, frequencies of IFN-γ+ cells were reduced and T-bet levels were diminished when compared to co-cultured WT cells. Thus, in contrast to the pronounced deficiency of IRF4−/− CD4+ T cells to differentiate into TH2, TH9, TH17 and TFH cells12,13,14,15,16,17,18,19,20, absence of IRF4 still allowed acquisition of some characteristics of TH1 cells. A more striking observation of our study was the pronounced failure of IRF4−/− CD4+ T cells to accumulate to large populations in vitro and in vivo. In response to L. monocytogenes infection, we detected reduced accumulation of CD4+ T cells responding to listeria-antigen with the up-regulation of CD40L, and in competitive transfer experiments, IRF4−/− CD4+ T cells were outnumbered by WT CD4+ T cells following infection or peptide immunization. Impaired accumulation of IRF4−/− CD4+ T cells was also observed in co-culture experiments in vitro. This failure of expansion of IRF4−/− CD4 T cells could also explain the inconsistent published results on IRF4 and TH1 differentiation12,13,14.
Impaired expansion of the IRF4+ CD4+ T cell population could be due to limited proliferation or a failure of survival and enhanced apoptosis. In contrast to a previous study40, we did not observe substantially enhanced apoptosis of IRF4−/− CD4+ T cells when compared to WT cells in in vitro assays. In this study, apoptosis was determined after re-stimulation of highly activated CD4+ T cells, which might explain the different outcome when compared to our study, where apoptosis was measured following primary stimulation of naive CD4+ T cells. The small difference in apoptosis in our study was also consistent with the similar induction of the anti-apoptotic BCL2 and pro-apoptotic BIM in WT and IRF4−/− CD4+ T cells. Our results rather indicate that IRF4−/− CD4+ T cells have a defect in differentiation and proliferation. We observed diminished blast formation, reduced expression of activation markers and limited proliferation, as evident by less pronounced loss of CFSE staining. The Cdkn2a gene was identified as direct IRF4 target in CD8+ T cells and it was postulated that IRF4 acts as repressor of Cdkn2a expression and thereby allows proliferation of CD8+ T cells26. A similar mechanism could also be active in CD4+ T cells and restrict the proliferation of IRF4−/− CD4+ T cells. In CD8+ T cells, IRF4 controls the adaption of the cellular metabolism to the requirements of proliferation and production of effector proteins following activation25,33. Activation of CD4+ TH cells also causes profound changes of metabolic pathways including up-regulation of aerobic glycolysis34,35,36,39. Our results reveal that IRF4 is important for these changes in CD4+ T cells. Activated IRF4−/− CD4+ T cells showed diminished surface expression of the transferrin receptor as well as reduced mRNA expression of the glucose transporter GLUT3 and impaired uptake of the glucose analog 2-NBDG. Cells had limited glycolytic capacity both at steady state and after blockade of mitochondrial ATP synthesis, which was associated with diminished hexokinase 2 expression. Finally, the maximal respiration capacity of activated IRF4−/− CD4+ T cells was reduced. Thus, metabolic performance of activated IRF4−/− CD4+ T cells was profoundly restricted and this limitation was likely responsible for the impaired response of IRF4−/− CD4+ T cells.
Our data for IRF4−/− TH1 cells resemble results for IRF4−/− CD8+ T cells. These cells become activated but cannot sustain proliferation and effector function which eventually results in the collapse of the response. A similar scenario might occur during TH1 responses. IRF4−/− CD4+ T cells become activated but fail to express genes required for proliferation and effector functions at adequate levels to allow effective TH1 differentiation and to sustain the response. We propose that this scenario is not restricted to TH1 differentiation but is also relevant for the deficiency of IRF4−/− CD4+ T cells to differentiate into other TH cell subsets. Thus, IRF4 is a fundamental transcription factor for all conventional T cells that controls adaptation of gene expression to the requirements of effector cells.
Material and Methods
Mice and infection
IRF4−/− mice8, RAG1−/− mice41, OT-II mice42 and CD90.1 congenic C57BL/6 mice (B6.PL-Thy1a/CyJ; Jackson, Bar Harbor, ME) were bred under specific pathogen-free conditions in the animal facility of the University Medical Center Hamburg-Eppendorf. Experiments were performed according to guidelines of the Deutsches Tierschutzgesetz and approved by the local committee for animal experiments of the City of Hamburg (permit number: 81/14). Mice were infected i.v. or i.p. with the indicated doses of either wild type Listeria monocytogenes (Lm) or of a L. monocytogenes strain recombinant for ovalbumin (LmOVA)11,37. Bacterial inocula were controlled by plating serial dilutions on tryptic soy broth (TSB) agar plates.
Cell isolation and cell culture
Cells from spleens were obtained by mashing the disintegrated organs through cell sieves into PBS followed by erythrocyte lysis with ACK lysing buffer (155 mM NH4Cl, 10 mM KHCO3, 100 μM EDTA, pH ~7.2). For induction of cytokine production, cells were incubated at 1–5 × 106 cells/ml in culture medium (RPMI 1640 medium supplemented with 5% fetal calf serum, glutamine, pyruvate, 2-mercaptoethanol and gentamicin) and stimulated for 4 h with 10−5 M ovalbumin peptide (OVA323-339; ISQAVHAAHAEINEAGR) or 10−5 M listeriolysin O peptide (LLO189-201; WNEKYAQAYPNVS) (both JPT Peptide Technologies, Berlin, Germany), or with 50 ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma Aldrich, St. Louis, MO) and 1 μM ionomycin (Sigma Aldrich). 10 μg/ml brefeldin A (Sigma Aldrich) was added for the last 3.5 h of culture to prevent cytokine secretion. Subsequently cells were analyzed by flow cytometry38,43.
For in vitro TH1 cell differentiation, CD4+ T cells were purified using Mouse CD4+ T Cell Isolation Kit (Easy Sep, Stemcell Technologies, Vancouver, Canada). Cells were incubated for up to 7 days in 24-well plates coated with anti-CD3 (clone 145 2C11, Biolegend, San Diego, CA). Cultures were supplemented with 2 μg/ml anti-CD28 (37.51), 20 U/ml rhIL-2 (Novartis, Basel, Switzerland), and 10 ng/ml IL-12 (Biolegend). The medium was replaced depending on the pH-indicator. In some experiments, 2 μg/ml anti-IL-4 (11B11) was added to prevent cells from TH2 differentiation. In competitive cultures, CD4+ T cells from congenic CD90.1+ WT mice and CD90.2+ IRF4−/− mice were mixed at a ratio of 1:1 and stimulated as described. For analysis of glucose uptake, CD4+ T cells from cell culture or directly isolated from infected mice were incubated for 2 h in glucose free RPMI 1640 medium (Life Technologies, Carlsbad, CA) with 30 μM 2-NBDG (2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose; Life Technologies). Cells were washed, stained for surface markers and analyzed by flow cytometry. Proliferation was measured by loss of CFSE staining intensity and apoptosis was determined by FLICA and propidium iodide staining as described11.
mRNA expression was determined by RT-PCR as described previously44. Primer sequences are available upon request. All samples were run in triplicates. Results were normalized to the expression of 18S rRNA and are presented as relative expression, which was calculated as 2−Δct × 1000.
Flow cytometric analysis
For surface staining, cells were first incubated with 10 μg/ml anti-FcγRII/III (2.4G2; BioXCell, West Lebanon, NH) and 1:100 rat serum (Jackson Laboratories, Bar Harbor, ME) in PBS to minimize unspecific antibody binding. Staining was performed on ice with fluorochrome-conjugated mAb according to standard methods. Dead cells were labelled with a fixable dead cell stain (Pacific Orange succinimidyl ester; Life Technologies). Cells were measured on a CantoII flow cytometer (BD Biosciences, San Jose, CA) and data was analyzed with the FlowJo software (Treestar, Ashland, OR).
For detection of intracellular cytokines, cells were surface stained and then incubated with a Pacific Orange succinimidyl ester to exclude dead cells from analysis. Cells were washed with PBS and fixed for 15 min with PBS, 2% paraformaldehyde at room temperature. Thereafter, cells were washed with PBS, 0.2% BSA, permeabilized with PBS, 0.1% BSA, 0.3% saponin (Sigma, Aldrich), and incubated in this buffer with 1% rat serum. After 5 min, fluorochrome-conjugated antibodies were added. After further 15 min, cells were washed with PBS and measured by flow cytometry. T-bet staining was performed using the buffers of the Foxp3 Transcription Factor Staining Set (eBioscience, San Diego, CA).
Fluorochrome-labelled antibodies anti-CD4 (clone RM4-5), anti-CD8α (53-6.7), anti-CD25 (7D4), anti-CD44 (IM7), anti-CD62L (MEL-14), anti-CD71 (R17217.1.4), anti-CD90.1 (His51), anti-CD90.2 (53-2.1), anti-CD127 (A7R34), anti-CD154/CD40L (MR1), anti-KLRG1 (2F1), anti-TCR Vα2 (B20.1), anti-CXCR3 (CXCR3-173), anti-IFN-γ (XMG1.2), anti-TNF-α (MP6-XT22), anti-T-bet (eBio4B10) and anti-IRF4 (3E4) were purchased from BD Biosciences, BioLegend or eBioscience.
Competitive T cell transfer assays
Spleen cells were isolated and CD4+ T cells were purified as described above. For competitive transfers in RAG1−/− mice, purified CD4+ T cells from CD90.1+ congenic WT mice and CD4+ T cells from IRF4−/− mice (CD90.2+) were mixed at a ratio of 1:1 and 2 × 106 cells were i.v. injected into the RAG1−/− mice, which had been infected i.p. 4 h before with 105 Lm. Mice were analyzed at day 8 after T cell transfer. For competitive OT-II cell transfer, CD4+ T cells were purified from CD90.1+CD90.2+ OT-II WT mice (OT-II × B6.PL-Thy1a/CyJ F1 mice) and from CD90.2+CD90.1− IRF4−/− OT-II mice. Donor T cells were mixed at a ratio of 1:1 and 3 × 106 cells were i.v. injected into CD90.1 congenic WT mice (CD90.1+CD90.2−). Recipient mice had either been i.v. infected with 105 LmOVA one day before or were i.v. treated with 50 μg OVA323-339 peptide and 30 μg LPS 1 h post T cell transfer. Spleen cells from recipients were analyzed on day 4 after transfer.
Extracellular flux assays
CD4+ T cells were purified from spleens of WT and IRF4−/− mice and incubated for 18 h with the T cell expansion and activation kit based on anti-CD3 and anti-CD28 stimulation according to the manufacturer’s protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). 4.5 × 105 cells/well were transferred to analysis plates (Seahorse Bioscience, North Billerica, MA) coated with CellTak (Corning, Corning, NY, USA) and plates were centrifuged to accumulate cells at the bottom of the plates. For determination of oxygen consumption rates (OCR), cells were washed and incubated in XF assay medium, 1 mM sodium pyruvate and 25 mM glucose (Seahorse Bioscience). Cells were analyzed using a XF-24 Extracellular Flux Analyzer (Seahorse Bioscience) according to the manufacturer’s protocols. During OCR analysis, cells were treated with 1 μM oligomycin, 0.9 μM fluorocarbonyl cyanide phenylhydrazone (FCCP), 1 μM rotenone and 1 μM antimycin A (Seahorse Bioscience). For the analysis of extracellular acidification rates (ECAR), cells were pretreated as described above and incubated in analysis plates in glucose- and pyruvate-free DMEM 5030 (Sigma Aldrich). Cells were treated with 10 mM glucose, 1 μM oligomycin and 100 mM 2-DG (2-deoxyglucose, Seahorse Bioscience) during the assay. OCR and ECAR values were calculated using the program provided by the manufacturer.
Statistical analysis
Statistical analyses were performed with Prism software (GraphPad Software Inc., La Jolla, CA, USA). Results were analyzed with the unpaired t test or in case of competitive cultures or transfers with a paired t test. A p-value of <0.05 was considered significant (*p < 0.05; **p < 0.01; ***p < 0.001; ns: not significant).
Additional Information
How to cite this article: Mahnke, J. et al. Interferon Regulatory Factor 4 controls TH1 cell effector function and metabolism. Sci. Rep. 6, 35521; doi: 10.1038/srep35521 (2016).
Supplementary Material
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, grant MI 476/3-1 and SFB841 to H.-W.M. and GRK841 to J.M.
Footnotes
Author Contributions J.M. designed, performed and analysed most experiments and drafted the manuscript; V.S., S.A., N.K. and L.M.F. performed and analysed experiments; J.R. and M.H. supervised experiments and wrote the manuscript; F.R. and H.-W.M. conceptualized the study, supervised the experiments, analysed the data, and wrote the manuscript.
References
- Huber M. & Lohoff M. IRF4 at the crossroads of effector T-cell fate decision. Eur. J. Immunol. 44, 1886–1895 (2014). [DOI] [PubMed] [Google Scholar]
- Suzuki S. et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8α− dendritic cell development. Proc. Natl. Acad. Sci. USA 101, 8981–8986 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T. et al. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J. Immunol. 174, 2573–2581 (2005). [DOI] [PubMed] [Google Scholar]
- Vander Lugt B. et al. Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat. Immunol. 15, 161–167 (2014). [DOI] [PubMed] [Google Scholar]
- Bajana S., Roach K., Turner S., Paul J. & Kovats S. IRF4 promotes cutaneous dendritic cell migration to lymph nodes during homeostasis and inflammation. J. Immunol. 189, 3368–3377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma K. et al. Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc. Natl. Acad. Sci. USA 102, 16001–16006 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi H. et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc. Natl. Acad. Sci. USA 102, 15989–15994 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittrücker H.-W. et al. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 275, 540–543 (1997). [DOI] [PubMed] [Google Scholar]
- Ochiai K. et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38, 918–929 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama T. et al. Molecular cloning of LSIRF, a lymphoid-specific member of the interferon regulatory factor family that binds the interferon-stimulated response element (ISRE). Nucleic Acid Res. 23, 2127–2136 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raczkowski F. et al. The transcription factor Interferon Regulatory Factor 4 is required for the generation of protective effector CD8+ T cells. Proc. Natl. Acad. Sci. USA 110, 15019–15124 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J. et al. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J. Exp. Med. 195, 1003–1012 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga N. et al. Development of Th1 and not Th2 immune responses in mice lacking IFN-regulatory factor-4. Int. Immunol. 15, 1–10 (2003). [DOI] [PubMed] [Google Scholar]
- Lohoff M. et al. Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc. Natl. Acad. Sci. USA 99, 11808–11812 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt V. et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 33, 192–202 (2010). [DOI] [PubMed] [Google Scholar]
- Tamiya T. et al. Smad2/3 and IRF4 play a cooperative role in IL-9-producing T cell induction. J. Immunol. 191, 2360–2371 (2013). [DOI] [PubMed] [Google Scholar]
- Brüstle A. et al. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 8, 958–966 (2007). [DOI] [PubMed] [Google Scholar]
- Huber M. et al. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc. Natl. Acad. Sci. USA 105, 20846–20851 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H. et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 31, 941–952 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollig N. et al. Transcription factor IRF4 determines germinal center formation through follicular T-helper cell differentiation. Proc. Natl. Acad. Sci. USA 109, 8664–8669 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretney E. et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nat. Immunol. 12, 304–311 (2011). [DOI] [PubMed] [Google Scholar]
- Grusdat M. et al. IRF4 and BATF are critical for CD8+ T-cell function following infection with LCMV. Cell Death Differ. 21, 1050–1060 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayar R. et al. TCR signaling via Tec kinase ITK and interferon regulatory factor 4 (IRF4) regulates CD8+ T-cell differentiation. Proc. Natl. Acad. Sci. USA 109, E2794–E2802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K. et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat. Immunol. 14, 1155–1165 (2013). [DOI] [PubMed] [Google Scholar]
- Yao S. et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity 39, 833–845 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayar R. et al. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J. Immunol. 192, 5881–5893 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy T. L., Tussiwand R. & Murphy K. M. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat. Rev. Immunol. 13, 499–509 (2013). [DOI] [PubMed] [Google Scholar]
- Li P. et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasmacher E. et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338, 975–980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M. et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 15, 373–383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M. et al. A validated regulatory network for Th17 cell specification. Cell 151, 289–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man K. & Kallies A. Synchronizing transcriptional control of T cell metabolism and function. Nat. Rev. Immunol. 15, 574–584 (2015). [DOI] [PubMed] [Google Scholar]
- MacIver N. J., Michalek R. D. & Rathmell J. C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi K. N. & Powell J. D. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 14, 435–446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce E. L., Poffenberger M. C., Chang C. H. & Jones R. G. Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds K. E. et al. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J. Immunol. 168, 1528–1532 (2002). [DOI] [PubMed] [Google Scholar]
- Mittrücker H.-W. et al. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc. Natl. Acad. Sci. USA 104, 12434–12439 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek R. D. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohoff M. et al. Enhanced TCR-induced apoptosis in Interferon-Regulatory-Factor 4-deficient CD4+ cells. J. Exp. Med. 200, 247–253 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P. et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 (1992). [DOI] [PubMed] [Google Scholar]
- Barnden M. J., Allison J., Heath W. R. & Carbone F. R. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell. Biol. 76, 34–40 (1988). [DOI] [PubMed] [Google Scholar]
- Kursar M. et al. Organ-specific CD4+ T cell response during Listeria monocytogenes infection. J. Immunol. 168, 6382–6387 (2002). [DOI] [PubMed] [Google Scholar]
- Paust H.-J. et al. The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J. Am. Soc. Nephrol. 20, 969–979 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.