

Abstract
The Adverse Outcome Pathway (AOP) framework provides a template that facilitates understanding of complex biological systems and the pathways of toxicity that result in adverse outcomes (AOs). The AOP starts with an molecular initiating event (MIE) in which a chemical interacts with a biological target(s), followed by a sequential series of KEs, which are cellular, anatomical, and/or functional changes in biological processes, that ultimately result in an AO manifest in individual organisms and populations. It has been developed as a tool for a knowledge-based safety assessment that relies on understanding mechanisms of toxicity, rather than simply observing its adverse outcome. A large number of cellular and molecular processes are known to be crucial to proper development and function of the central (CNS) and peripheral nervous systems (PNS). However, there are relatively few examples of well-documented pathways that include causally linked MIEs and KEs that result in adverse outcomes in the CNS or PNS. As a first step in applying the AOP framework to adverse health outcomes associated with exposure to exogenous neurotoxic substances, the EU Reference Laboratory for Alternatives to Animal Testing (EURL ECVAM) organized a workshop (March 2013, Ispra, Italy) to identify potential AOPs relevant to neurotoxic and developmental neurotoxic outcomes. Although the AOPs outlined during the workshop are not fully described, they could serve as a basis for further, more detailed AOP development and evaluation that could be useful to support human health risk assessment in a variety of ways.
Keywords: adverse outcome pathway, in vitro testing, key events, molecular initiating event, pathways of neurotoxicity, predictive toxicology
Introduction to AOP concept
Regulatory toxicology is undergoing a transformation from decision-making relying primarily on observing apical effects in animal models to biological pathway-based approaches that exploit understanding of cellular, tissue, and whole-organism dynamics. A key driver for this change is a collective desire of all stakeholders (academia, industry, and regulators, etc.) to better utilize the latest scientific thinking and tools within the safety assessment process (Vinken 2013). Success will improve protection of human health and the environment, considerably reduce animal testing, significantly increase the rate of data collection, and provide opportunities for industrial innovation and competitiveness. Central to this transformation is a shift toward knowledge-based weigh-of-evidence paradigm for hazard assessment. This transformative process must include development of cost efficient and less time consuming toxicity testing methods that predict the impact of chemicals on human and ecological health (Collins et al. 2008). The ultimate goal envisages routine toxicity testing conducted in human-cell based test systems (NRC 2007) combined with information derived from computational models to more accurately predict the potential adverse effects of chemicals rather than having to rely on in vivo animal models. Such predictive toxicology is still emerging and will require considerably more research and development before its principles and processes are mature enough to translate into mainstream regulatory practice (Thomas et al. 2013, Patlewicz and Lander 2013, Adeleye et al. 2014). One area that needs particular attention is how to actually harvest, curate, and manage relevant mechanistic knowledge so that it informs AOP development that serves regulatory needs.
Basic toxicological and biomedical research conducted over many decades has yielded a formidable body of scientific literature which contains an extraordinary amount of information on the mechanisms by which chemicals alter cellular signaling pathways and how such alterations can eventually lead to adverse health effects. Endeavoring to elucidate and describe such “toxic processes” (Aldridge 1996) from experimental investigation is thus nothing new in toxicology and has always required functional understanding of complex biological systems and how xenobiotic-induced perturbations can lead to their dysfunction and failure. However, generation of mechanistic information is only one step toward its use in decision making. For many years, the World Health Organization’s International Program on Chemical Safety has been demonstrating how to organize and apply information on a chemical’s mode of action to understand the human relevance of animal data (summarized in Meek and Klaunig 2010). More recently this mode of action/human relevance framework has been updated (Meek et al. 2014a) to reflect the experience gained by practitioners and to extend its application to other aspects of toxicological hazard assessment.
With a view to facilitating more widespread systematic use of mechanistic information for regulatory safety assessment for both human health and the environment, the Adverse Outcome Pathway (AOP) framework has been developed as a means to rationally combine data across multiple levels of biological organization to identify correlative and causal linkages between the sequence of events that lead to an adverse outcome due to excessive chemical exposure (Ankley et al. 2010). The AOP framework provides a means to apply mechanistic understanding into regulatory decision making, and serves as a toolbox for consolidating, managing, and exchanging knowledge among the research community. The concepts underlying the AOP structure encompass functional systems biology/toxicology thinking with the ultimate goal of predicting systems behavior. However, an AOP is not a description of a biological system per se, but is a higher-level depiction of the sequence of toxicological events that lead to dysfunction or failure of the system, given a certain set of circumstances or boundary conditions.
AOPs can vary in resolution and expanse and can include both qualitative and quantitative descriptions of key events (KE) and their interlinking causal relationships (Figure 1). Initial development and elucidation of an AOP begins with a proposed pathway (Ankley et al. 2010, Watanabe et al. 2011). Recently published guidance and template documents for development and assessment of AOPs (OECD 2013) suggest whenever possible anchoring the AOP at the molecular initiating event (MIE) and the adverse outcome (AO) at the individual or population level (i.e., the regulatory effect/endpoint of concern). However, AOP development may originate at any step in the pathway, whether nearer to the initial chemical-biological interaction, the MIE, or somewhere in-between initiation and AO. The AOP is then further developed by identifying and describing the intermediate KEs and the causal or correlative relationships between them. An additional aspect of this process is transitioning from a correlative-based AOP to one based on causative links between the MIE, KE, and AO within the pathway (Meek et al. 2014). This evidence can be drawn from various sources, but typically comprises relevant studies described in the literature, or results from experiments specifically designed for the purpose of AOP development to cover gaps of knowledge in the existing literature. Finally, an AOP can be enhanced with quantitative linkages that allow for more precision and surety in predicting outcomes from bio-markers of upstream key events or MIEs. This progress from correlative to causative and then quantitative descriptions of key event will decrease uncertainty in the model predictions and increase confidence for use by regulatory decision makers.
Figure 1.
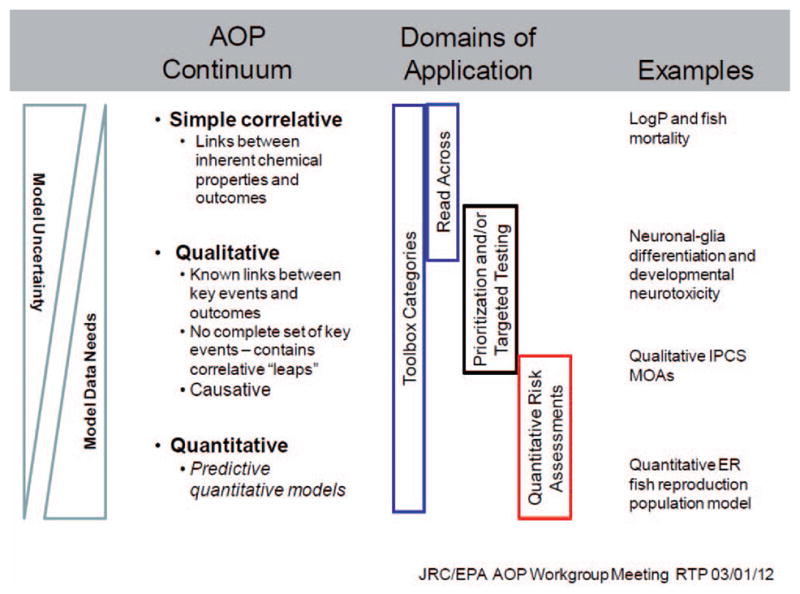
Relationships between the uncertainty in AOP models, the data and resources needed to develop the models and the regulatory domains of applicability.
The potential regulatory use of the proposed ten AOPs will be defined to a large extent by the scientific confidence with which they will facilitate extrapolation of data measured at low levels of biological organization (e.g., in cell culture) to predicted outcomes at higher levels of organization and the specificity with which they can link adverse outcome measurements to their specific causes. Application of evolved Bradford Hill considerations (Meek et al. 2014a) will provide the foundation for the weight of evidence evaluation of the AOP overall. Therefore, satisfying the most influential considerations such as biological concordance and essentiality of key events could be assessed as sufficient for screening applications (e.g., chemical category development). However, for application to hazard or risk assessment in higher tier assessments a higher certainty of the weight of evidence, including robust understanding of dose-response relationship will be required. Therefore, the use of AOP framework will be very much based on the principle fit-for-purpose, depending on the regulatory application.
In 2012 the Organisation for Economic Cooperation and Development (OECD) launched the AOP Development Program, which is coordinated by the OECD Extended Advisory Group on Molecular Screening and Toxicogenomics (EAG-MST). The OECD guidance document and template for developing and assessing AOPs aims to ensure consistency in approach and compliance with AOP standards related to content, structure, and presentation (OECD 2013). The AOP Knowledge Base (AOP-KB; https://aopkb.org) has also been recently developed which includes the AOP-Wiki, (https://aopkb.org/aopwiki). The AOP-Wiki is a web-accessible collaboration space for AOP development teams to work together in an efficient and convenient manner for the capture, curation and evaluation of AOPs. It also serves to crowd-source knowledge on a global scale to refine existing AOPs and trigger the development of new ones where gaps in the AOP landscape are identified. The AOP-KB is envisioned to be the primary hub for the regulatory science community to rapidly and efficiently access AOP knowledge to serve a variety of their needs.
As a first step in applying the AOP framework to adverse neurological outcomes caused by exposure to xenobiotics, the EU Reference Laboratory for Alternatives to Animal Testing (EURL ECVAM) joined forces with the Safety Evaluation Ultimately Replacing Animal Testing (SEURAT-1) consortium to organize a workshop (March 2013, Ispra). The goal of the workshop was to identify putative AOPs relevant to neurotoxicity and developmental neurotoxicity. The AOPs outlined during the workshop are by no means comprehensive for neurotoxicity, however, they do provide a starting point to stimulate discussion as to which information can and should be added in the future.
Challenges for neurotoxicity AOP development
Development and use of the AOP framework for neurological outcomes following developmental or adult exposure have been hampered by a number of serious challenges. A major concern for neurotoxicity is a general lack of understanding of the MIEs that are causally responsible for altered KEs triggering AOs. For example, the relationship between developmental lead (Pb2+) exposure and adverse cognitive outcomes in children is well described (Sanders et al. 2009); however, the initial molecular interactions between Pb2+ molecules and cellular targets that are causatively linked to adverse cognitive outcomes (e.g., IQ) are still not well understood. The lack of a known pathobiology of many neurodevelopmental disorders hampers AOPs development. For example, where is the locus of IQ loss from developmental lead exposure? The same can be said for a wide number of other well-known developmental and adult neurotoxicants (e.g., methylmercury, alcohol, polychlorinated biphenyls). Additionally, many human neurological disorders may have diverse pathophysiology that underlies similar clinical phenotypes, or conversely, diverse clinical outcomes that result from similar pathophysiology. For example, autism spectrum disorder (ASD) is a neurodevelopmental malady with an increasing incidence and is more prevalent in males (McDonald and Paul 2010). However, it is now clear that ASD is an umbrella term for multiple disorders with overlapping clinical symptoms, suggesting that there are shared and unique pathophysiological mechanisms which have yet to be identified.
There are, however, a limited number of neurotoxic outcomes that do have well-defined pathophysiological outcomes and MIEs. One example, developed as an AOP below, is acute neuronal sodium channel disruption and consequent behavioral effects, exemplified by p,p′-DDT and pyrethroids (Shafer et al. 2005) (see AOP on Acute neurotoxic effects of pyrethroids mediated by disruption of voltage-gated sodium channels). Another example are the well-known peripheral neuropathies induced by a number of chemicals, including organophosphates, carbon disulfide, pyridoxine (Vitamin B6), 2,5-hexandione, and acrylamide (LoPachin and DeCaprio 2005, Rao et al. 2014) (see AOP on Binding of certain organophosphates to NTE results in delayed neuropathy).
In these cases the AOs are well-described AOs in the peripheral systems of multiple species correlatively and/ or causatively linked to MIEs. For example, 2,5-hexandione forms irreversible covalent bonds (adducts) with proteins (LoPachin and DeCaprio 2005, Graham et al. 1995).
The lack of known pathophysiology for a specific AO does not make it difficult to propose an AOP or to hypothesize MIEs. However, it does pose challenges in developing the empirical data needed to move from a proposed AOP to a causal or quantitative AOP. This has important consequences for the development and acceptance of more efficient and predictive testing methods for detecting chemicals that may lead to AOs of concern. An AOP that contains good correlative and/or causative links between the MIE or early KEs provides risk managers an increasing level of confidence to make regulatory decisions (Ankley et al. 2010). For example, the known causative relationships between estrogen receptor binding, activation of downstream cellular ER-based signaling pathways, and adverse impacts on reproductive function facilitates the use of quantitative structure-activity relationships (QSAR) and chemical structure-based read-across models that can be used to make regulatory decisions (Schmieder et al. 2004, 2003). Indeed, this model is already in use as part of the OECD Tool-Box (Mombelli 2012).
Common key events for neurotoxic outcomes
Application of the AOP concept for hazard identification and characterization aims for faster, cheaper and more predictive neurotoxicity evaluation by including in vitro, preferably human-based, systems in the testing strategy (NRC 2007). Such an approach has been applied for assessing the potency of compounds to induce skin sensitization, a complex procedure involving a variety of KEs identified in specific types of skin and immune cells. However, in the case of skin sensitization, the MIE triggering the AOP is the same for many skin sensitizers, i.e., covalent interaction of electrophilic substances with cellular proteins (MacKay et al. 2013). The functional and structural heterogeneity of the nervous system, coupled with the dynamics of brain development, suggests that a broad array of MIEs may be involved in adverse neurological outcomes. This complexity coupled with the aforementioned dearth of well-accepted MIEs for developmental or adult neurotoxicity makes development of alternative test methods a serious challenge. One possible solution to this problem is the identification of converging KEs that are downstream from and common to multiple MIEs and pathways. On-going efforts to develop DNT screening methods based on common cellular phenotypes is an example of this approach (Coecke et al. 2007, Lein et al. 2007, Bal-Price et al. 2010, Crofton et al. 2011, Bal-Price et al. 2012). Alternatively, cellular signaling molecules common to multiple pathways could be utilized for assay development, which enables chemical testing in a medium- to high-throughput manner, enhancing the effectiveness and robustness of testing. This approach might seem too reductionist for reflecting the complex issue of neurotoxicity with its diverse MIEs and AOs, however, scientific data is needed to understand the degree to which this type of testing strategy based on common KEs linked to the one or more AOs is able to be predictive.
For example, common KEs were identified that might possibly serve as endpoints for in vitro neurotoxicity testing with a high predictivity for hazard potential. These include cytoskeleton alterations (AOP V, VI, VII, VIII), impaired mitochondrial function (AOP II, V), increased oxidative stress (AOP V, VI, VIII, X) and altered neuronal firing rates (AOP I, III, IV, V, VI, VII, IX, X). It is important to note that some of these KEs are common to many cell and tissue types, not just nervous system tissues, (e.g., markers of oxidative stress, mitochondrial function). Data is needed to demonstrate how these non-neuronal specific KEs are linked to nervous system specific adverse outcomes. A classic example is the generation of the toxic cation MPP+ from MPTP by glial cells and the subsequent destruction of neural dopaminergic neurons in a specific brain region, the substantia nigra, that results in Parkinson’s like symptoms. The exact reasons for the specificity of MPTP for these neurons remains controversial, with one explanation being that selective uptake of MPP+ by membrane transporters in dopaminergic cells is responsible for the targeting of dopamine neurons (Jenner and Marsden 1986, Tipton and Singer 1993).
Predictivity of assay endpoints based on common KEs, as well as variable combinations of them will need to be tested using a set of neurotoxic compounds (Crofton et al. 2011). This approach may lead to a defined test battery for neurotoxicity evaluation.
Within the proposed AOPs developed at the Workshop (see Appendices to be found online at http://informahealth-care.com/doi/abs/10.3109/10408444.2014.981331) two subgroups can be identified. The AOPs I, II, III, IV, V, and VI are strongly related to neurotransmitter receptor MIEs located in the cell membrane. These targets all have critical functions for neurotransmission. The AOPs VII, VIII, IX, and X are more related to events associated with general molecular and cellular support or defense mechanisms. Accordingly, there will be interlinks within as well as likely across these two groups. The process of neurotransmission is a fine-tuned, multi-event process involving various ion channels and receptors that finally depolarizes the membrane. Neurotoxicant-induced alterations in voltage-gated ion channels (see AOP IV) directly affects the functionality of neuronal N-methyl-D-aspartate (NMDA) receptors (see AOPs I, II). During depolarization the influx of Ca2+ ions is a crucial cellular process for multiple physiological functions, including synaptic plasticity (e.g., AOP II and IX). It is also a common intracellular process that may adversely affect the integrity of neural cells (see AOP V, VI, VII, VIII, IX, X). Therefore, within the description of the AOPs there are commonalities between pathways as a consequence of the complexity of the neurobiological processes related to normal functioning of the human brain.
Further development of the ten AOPs outlined here, as well as development of new AOPs for neurotoxicity, requires generation of new data as well as further mining of the existing data. In this regard it is of utmost importance that the test systems mimic human physiology as closely as possible (NRC 2007), that is, co-culture of neurons and glial cells, expression and sensitivity of receptors, presence/absence of signaling molecules and pathways especially in a spatio-temporal context. Development and use of these models must also account for inter-species differences in brain physiology as responses to the same compounds may differ between human and rodent in vitro models (Gassmann et al. 2010, Harrill et al. 2011).
Considering life stage-specific susceptibility in neurotoxicity AOPs
Ideally, AOPs for neurotoxicity should consider specific life stages such as development or aging as significant age-related susceptibilities in response to chemical exposure are well documented (Rice and Barone 2000, Landrigan et al. 2010). The development of AOPs that are based on life stage-specific KEs in nervous system development and aging, AOP-dependent hazard and risk assessment, should include not just the embryo, fetus and infants, but also juveniles and the elderly as major vulnerable subpopulations.
During brain development, several processes occur primarily or exclusively during this time. Key processes such as the commitment and differentiation of neural progenitor cells (NPCs) followed by glial and neuronal cell proliferation, migration, differentiation into specific neuronal and glial subtypes, axonal and dendritic outgrowth, formation and pruning of synapses, myelination, programmed cell death, ontogeny of neurotransmitters and receptors, and development of the blood brain barrier (BBB) are critical for functional brain development (Lein et al. 2005, Bal-Price et al. 2012, Stiles and Jernigan 2010). Disruption of any of these processes by neurotoxic compounds may modify neuronal/glial cell function leading to adverse alterations in neuroanatomy, neurophysiology, neurochemistry, and neurobehavior. Complex and dynamic glial/neuronal processes occur during discrete developmental windows that differ across brain regions, and this spatio-temporal variation influences the variable sensitivity of the developing brain to the same chemical exposure at different developmental stages (Barone et al. 2000, Rice and Barone 2000, Lein et al. 2005). Insults that occur early during CNS development have the potential to cause more widespread impacts throughout the brain, while those occurring later in development may only affect a specific structure or structures. For example, methylmercury has more widespread effects if exposure occurs early in CNS development, relative to exposures that occur later in development, which result in more focused insults in the cortex and cerebellum (Burbacher et al. 1990). Another critical instance with regard to life stage-specificities is given by the developmental switch of neuronal GABAergic responses from excitation to inhibition. This switch is dependent on GABA-induced GABAA receptor activation (Ganguly et al. 2001). Therefore, interference with GABA receptors during development and after brain maturation (see AOP III) is likely to cause distinctly different AOs (Westerholz et al. 2010).
Aging of the human brain is also characterized by processes that are unique or predominant at this life stage such as decreased neuronal cell volume, loss in cell numbers, reduced synaptic density/connectivity, and declines in cognitive function (Walhovd et al. 2014). In addition, neurons show evidence of DNA damage, elevated production of reactive oxygen species (ROS), Ca2+-signaling disturbances, mitochondrial dysfunction, and increased neuroinflammation with increasing age (Bishop et al. 2010). Moreover, declining hippocampal neurogenesis associated with aging of hippocampal neural stem/progenitor cells has been proposed to contribute to aging-related cognitive decline (van Wijngaarden and Franklin 2013). Identification of NPC dysfunction in the aging hippocampal regenerative niche suggests a parallel between aging and basic processes of neurodevelopment as NPC proliferation and neuronal differentiation, that are necessary for formation and maintenance of the brain, function throughout life (see AOP X). Importantly, these molecular biomarkers and biological pathways associated with aging are also implicated in neurodegenerative diseases, suggesting an overlap between biological pathways associated with ageing and neurodegenerative brain disorders.
A representative example of such life stage-dependent neurotoxicity is anesthetic exposure. There is growing concern that exposure to anesthetics causes learning impairment, memory deficits and behavioral abnormalities in young subjects, and accelerated cognitive decline in the elderly (Rohan et al. 2005, Johnson et al. 2002, Moller et al. 1998). Although the MIE(s) responsible for the AOs are not clear, it is suggested that the extent of neuroapoptosis, neuronal network assembly, and neuro- and synaptogenesis determine the qualitative and quantitative aspects of toxicity in the developing brain (reviewed in Jevtovic-Todorovic et al. 2013). In the elderly, clinical evidence for postoperative cognitive decline is present while underlying molecular mechanisms still need to be elucidated (Jevtovic-Todorovic et al. 2013).
An additional example of vulnerable life stages for neurotoxicity is adolescence. While developmental changes in the adolescent brain are more subtle than those in the first 4 years of life (Paus et al. 2001), magnetic resonance imaging (MRI) analyses of humans during juvenile and adolescence periods have clearly demonstrated structural and functional changes in synaptogenesis and connectivity in the human brain throughout adolescence and into adulthood (Giedd 2004, 2008). Ethanol acts on multiple processes occurring during adolescent development (reviewed in Guerri and Pascual 2010). It attenuates N-methyl-D-aspartate (NMDA)-mediated synaptic activity to a larger extent in the immature than in the mature hippocampus and thus, more potently inhibits the induction of long-term potentiation (LTP) in immature versus mature animals (Swartzwelder et al. 1995a, 1995b). MIEs, cellular processes and pathways critical for brain development during adolescence may overlap with those important for development and aging of the nervous system.
What is the relevance of life stage-susceptibility for neurotoxicity AOPs development? First, AOP developers need to think in a broader context for incorporating specific aspects of brain development and brain aging into the neurotoxicity mode of action (MoA) portfolio. This means that besides “classical” AOP for neurotoxicity like interference with neurotransmitter receptors or inhibition of acetylcholinesterase (AOP I–V), other pathways specific for brain development and aging should be considered when developing AOPs for neurotoxicity. As a result, life stage-specific key events will increase the number of AOPs for neurotoxicity. One example for such life stage-specific issues in AOP development is the notable increase in brain oxidative stress and neuroinflammation with age (Perluigi et al. 2014, Hsieh and Yang 2013). These suggest common key events in mitochondrial (AOP VII), inflammatory (AOP VIII), and epigenetic (AOP X) pathways within AOPs when addressing neurotoxicity in the elderly. Interestingly, two of the draft AOPs (AOP VII and AOP VIII) are interlinked and influence each other, rendering it difficult to separate them in a clear manner (Gemma et al. 2007). Importantly, in vitro models for KEs should mimic the relevant stage of neural/glial differentiation and maturation for developing AOPs specific for the developing, adolescent, adult, and aging nervous systems. As molecular targets, and thus AOPs, are numerous at all life stages and likely vary in importance across life stages, the AOPs presented here are just the initial step in a process that will continue to expand and be refined over the upcoming years as the science progresses.
Future directions for development of neurotoxicity AOPs
With few exceptions, development of AOPs for developmental and adult neurotoxicity is a relatively recent concept. There are many different directions that could be taken as work in this area proceeds. A goal of the workshop was to outline the directions that might prove most fruitful for the AOP concept to be employed effectively in environmental decision-making by risk assessors and others. The following directions should be considered as high priority:
Cataloguing the current state of knowledge regarding known or putative AOPs for neurotoxicty
Identifying AOPs specific for Developmental Neurotoxicology
Prioritizing AOP development
Identifying KEs that are amenable to High and Medium throughput screening
Demonstrating the utility of the AOP approach to risk assessors using case studies
Each of these priorities is discussed briefly below.
Cataloguing the current state of knowledge regarding known or putative AOPs
This workshop report presents examples of ten draft AOPs related to neurotoxicity and/or developmental neurotoxicity, but it was outside the scope of the workshop to attempt to identify and catalog all of the known or putative AOPs that are related to neurotoxicity. Indeed, even the AOPs presented here are to be considered first drafts that require additional data, description, and detail prior to use. These AOPs range from those that are mostly complete, such as those outlining pyrethroid effects on VGSCs or GABAA receptor mediated excitotoxicity and convulsions, to those that need substantial work to better establish linkages between proposed MIEs and KEs, such as the AOP for neuroinflammation leading to neurodegeneration. As mentioned previously, the AOP-Wiki will facilitate international collaboration in developing a knowledge base related to AOPs. As an important first step in utilizing AOPs for neurotoxicity and developmental neurotoxicity in risk decisions, it will be important for researchers to populate the AOP-Wiki with examples such as those developed at this workshop. This will help to identify the most complete AOPs, and allow identification and prioritization of data gaps that require additional data collection. In a time when resources are limited and it is not possible to address every data gap to have a “complete” AOP, prioritization of data gaps which are the most crucial for risk decision-making is necessary for effective and efficient prioritizing of research needs. Further, AOPs do not necessarily need to be complete to be useful and informative for decision-making, particularly if the data gaps and uncertainties are identified and understood.
Identifying AOPs for developmental neurotoxicology
As discussed above, it is challenging to develop AOPs for developmental neurotoxicity due to the complex symphony of events that occur during nervous system development. Still, development of AOPs following chemical exposure during development is crucial and should be a high priority given the increasing incidence of childhood neurological syndromes such as autism spectrum disorders, attention deficit hyperactivity disorder (ADHD), and others that have significant consequences for society (Bloom et al. 2010, McDonald and Paul 2010, Landrigan et al. 2012). However, as the etiology of these disorders, and in particular the role of environmental chemicals is not well understood, a more critical and useful initial goal would be to develop AOPs that are linked to adverse outcomes that traditionally have been used to make risk decisions for environmental chemicals. Doing so would allow researchers to take advantage of the existing databases in the public literature as well as publically available databases (e.g., ToxRefDB, http://www.epa.gov/ncct/toxrefdb/) to hypothesize and test putative AOPs for developmental neurotoxicity. Furthermore, it will facilitate adoption of the AOP concept by risk assessors, as they will be familiar with the described AOs.
Prioritizing AOP development
There are thousands of macromolecules (e.g., receptors, transporters, nucleic acids, lipids) in the brain that form a vast number of potential targets for xenobiotic chemicals. If even a small percentage of these function as MIEs, the task of developing AOPs for all of these poses an almost insurmountable hurdle. One way to address this challenge is to move beyond the linear approach to modeling AOPs that has dominated past research efforts. The initial models developed for the International Programme on Chemical Safety (IPCS) mode-of-action (MOA) framework (IPCS 2007), as well as many AOPs (Crofton and Zoeller 2005, Watanabe et al. 2011, Bushnell et al. 2010), describe a series of one-to-one relationships, in which the MIE initiates sequential KEs eventually leading to the AO. This linear sequence likely does not capture the whole complexity of molecular and cellular biology implicated in a neurotoxic response. Thus, systems approaches should be used to develop computational models of the networks of MIEs and KEs that collectively influence the AO. For example, Kleinstreuer et al. (2013) developed a novel multicellular agent-based model of vasculogenesis using the CompuCell3D (http://www.compucell3d.org/), which incorporates vascular endothelial growth factor signals, pro- and anti-angiogenic inflammatory chemokine signals, and the plasminogen activating system of enzymes and proteases to recapitulate disruption of vascular formation by environmental chemicals. Computational models of neurotoxicity that link networks of cellular and organ level systems provide one approach to reduce the complexity of the challenge of developing predictive models of neurotoxicity for use in regulatory decisions.
Identifying key events that lend themselves to high and medium throughput screening
One of the advantages of understanding an AOP is that the knowledge can be used in a predictive manner. Whether it be a simple “read across” approach using structural information about a chemical and its interactions with a molecular target (MIE) or a more quantitative approach, AOPs foster the ability to predict potential AOs for chemicals that have not been evaluated for toxicity in a test system.
A high priority for neurotoxicity AOPs is to identify KEs that represent points of convergence across multiple AOPs and that are biological responses, which can be easily incorporated into high or medium throughput screening assays. There are clear advantages and disadvantages to this approach. KEs occurring at more apical points in AOPs (i.e., closer to the AO) will by definition give rise to screening assays that detect broad classes of chemicals, but may not have the capability of distinguishing which “upstream” AOP has been activated by a chemical or class of chemicals (Woodruff et al. 2008). By contrast, assays based on early MIEs in an AOP will be test for direct interaction between chemicals and the biological target (e.g., receptor, enzyme) and thus be more specific to individual chemicals or chemical classes. This will facilitate SAR and QSAR model development. In any event, approaches that yield rapid, reliable, and high(er) throughput screening methods for detecting chemicals with the potential for neurotoxicity and developmental neurotoxicity are of high priority and data from screens that are based on AOPs will provide scientifically sound rationale to those making risk decisions about chemicals based on in vitro data.
Demonstrate the utility of the AOP approach to risk assessors using case studies
This workshop presents ten proposed AOPs related to neurotoxicity, but it was beyond the scope of the workshop to finalize comprehensive AOPs or demonstrate how to use for risk decision-making. Demonstration of the real-world applicability of neurotoxicity AOPs is paramount to having this concept more readily accepted and utilized by risk assessors. As such, an important goal for the neurotoxicology research community should be to identify a small number of case studies that demonstrate how applying an AOP approach to a risk-decision problem can improve the speed and/or confidence in a risk decision, or allows information from one chemical to be applied more broadly to an entire class of chemicals. Candidate AOPs for case studies may include the pyrethroids/sodium channels, thyroid hormone AOP, or the GABAA or NMDA-related AOPs; however, this has to be proven by empirical data.
Further development of the outlined AOPs to neurotoxicity
In this report, using the OECD template and information from literature searches, initial work has been conducted to identify and develop AOPs relevant to neurological outcomes. In all cases, the authors were asked to identify an MIE or a putative MIE, followed by responses at the cellular, tissue, organ, organism and population level, with each AOP summarized in a flow diagram. The presented AOPs are often based on a few well-studied model neurotoxicants and a summary of the qualitative understanding of each AOP has been briefly described. The KEs have been further evaluated in a correlative manner based on the available published data and the subjective interpretation of the strength of the scientific evidence.
The AOP descriptions are based on the OECD guidance following the rule that two anchors should be identified: one MIE linked in a causative manner to one AO. Indeed, in most of the described AOPs this principle has been followed. However, available scientific knowledge for some complex AOPs (e.g., AOP VIII: Neuroinflammation) suggests more than one putative MIE and AO. Such AOPs may in fact be seen as a network of AOPs where a number of different MIEs may lead to one particular AO, and where one MIE could also lead to a number of AOs. These draft AOPs will need further development following the OECD AOP Framework, identifying the most salient MIE to link with a specific AO and in so doing build a network of inter-related AOPs (Villeneuve et al. 2014a, 2014b).
The main aim of the workshop was to identify a set of putative AOPs related to neurotoxicity and developmental neurotoxicity that could be further elaborated. The next step for development of these AOPs will be application of the modified Bradford Hill considerations following the full OECD Template and Guidance on Developing and Assessing the Completeness of Adverse Outcome Pathway (OECD 2013). This implies evaluation of biological plausibility, concordance of dose-response, temporal concordance, consistency, and specificity of association between MIE and AO in a quantitative manner (Meek et al. 2014). These issues were outside the scope of this workshop and are thus not included in the proposed AOPs. In the near future, the ultimate goal for the listed AOPs is their submission to the OECD AOP Development Programme.
Supplementary Material
Appendix: Examples of the putative AOPs for neurotoxicity
Acknowledgments
The EU Reference Laboratory for Alternatives to Animal Testing (EURL ECVAM) together with the Safety Evaluation Ultimately Replacing Animal Testing (SEURAT-1) consortium organized a workshop (March 2013, Ispra) on Adverse Outcome Pathways (AOP) Relevant to Neurotoxicity, in context of the SEURAT-1 research initiative (Safety Evaluation Ultimately Replacing Animal Testing— see www.seurat-1.eu). The work of all workshop participants greatly contributed to this manuscript. In addition to the authors, the final version of this manuscript was reviewed following the internal procedures of the US EPA, the US National Institute of Environmental Health Sciences and the European Commission’s Joint Research Centre. We thank Sharon Munn (EURL ECVAM), William Mundy, and Mary Gilbert (US EPA) and Kristen Ryan (US NTP/NIEHS) for critically reading the manuscript and providing valuable comments.
Footnotes
Declaration of interest
The employment affiliation of the authors is shown on the cover page. The authors have sole responsibility for the writing and content of this paper. The contributing authors were participants of the workshop organized by the EURL ECVAM. The external workshop participants were invited on the basis of a survey performed by EURL ECVAM neurotoxicity experts of the latest literature to identify those with specific expertise in the relevant research fields. The workshop organization was financially supported by the European Commission, including the cost of travelling and per diem of all invited external experts. The strategy for preparing the report, the literature selected for review, the conclusions drawn and the recommendations made are exclusively the collective scientific output of the workshop participants and do not necessarily represent the views of the participants’ employers.
References
- Adeleye Y, Andersen M, Clewell R, Davies M, Dent M, Edwards S, et al. Implementing Toxicity Testing in the 21st Century (TT21C): Making safety decisions using toxicity pathways, and progress in a prototype risk assessment. Toxicology. 2014 doi: 10.1016/j.tox.2014.02.007. Epub ahead of print. [DOI] [PubMed]
- Aldridge WN. In: Mechanisms and concepts in toxicology. Aldridge WN, editor. University of Surrey; UK: Taylor & Francis; 1996. [Google Scholar]
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, et al. Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem. 2010;29:730–41. doi: 10.1002/etc.34. [DOI] [PubMed] [Google Scholar]
- Bal-Price AK, Coecke S, Costa L, Crofton KM, Fritsche E, Goldberg A, et al. Advancing the science of developmental neurotoxicity (DNT): testing for better safety evaluation. ALTEX. 2012;29:202–15. doi: 10.14573/altex.2012.2.202. [DOI] [PubMed] [Google Scholar]
- Bal-Price AK, Hogberg HT, Buzanska L, Lenas P, van Vliet E, Hartung T. In vitro developmental neurotoxicity (DNT) testing: Relevant models and endpoints. Neurotoxicology. 2010;31:545–54. doi: 10.1016/j.neuro.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Barone S, Jr, Das KP, Lassiter TL, White LD. Vulnerable processes of nervous system development: a review of markers and methods. Neurotoxicology. 2000;21:15–36. [PubMed] [Google Scholar]
- Barone S, Jr, Stanton ME, Mundy WR. Neurotoxic effects of neonatal triethyltin (TET) exposure are exacerbated with aging. Neurobiol Aging. 1995;16:723–35. doi: 10.1016/0197-4580(95)00089-w. [DOI] [PubMed] [Google Scholar]
- Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–35. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom B, Cohen RA, Freeman G. Summary health statistics for U.S. children: National Health Interview Survey, 2009. Vital Health Stat. 2010;247:1–82. [PubMed] [Google Scholar]
- Burbacher TM, Rodier PM, Weiss B. Methylmercury developmental neurotoxicity: a comparison of effects in humans and animals. Neurotoxicol Teratol. 1990;12:191–202. doi: 10.1016/0892-0362(90)90091-p. [DOI] [PubMed] [Google Scholar]
- Bushnell PJ, Kavlock RJ, Crofton KM, Weiss B, Rice DC. Behavioural toxicology in 21st century: challenges and opportunities for behavioural scientists. Summary of a symposium presented at the annual meeting of the neurobehavioral teratology society, June, 2009. Neurotox Teratol. 2010;32:313–28. doi: 10.1016/j.ntt.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Coecke S, Goldberg AM, Allen S, Buzanska L, Calamandrei G, Crofton K, et al. Workgroup report: incorporating in vitro alternative methods for developmental neurotoxicity into international hazard and risk assessment strategies. Environ Health Perspect. 2007;115:924–31. doi: 10.1289/ehp.9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Gray GM, Bucher JR. Toxicology. Transforming environmental health protection. Science. 2008;319:906–7. doi: 10.1126/science.1154619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofton KM, Mundy WR, Lein PJ, Bal-Price A, Coecke S, Seiler AE, et al. Developmental neurotoxicity testing: recommendations for developing alternative methods for the screening and prioritization of chemicals. ALTEX. 2011;28:9–15. [PubMed] [Google Scholar]
- Crofton KM, Zoeller RT. Mode of action: neurotoxicity induced by thyroid hormone disruption during development--hearing loss resulting from exposure to PHAHs. Crit Rev Toxicol. 2005;35:759–769. doi: 10.1080/10408440591007304. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell. 2001;105:521–32. doi: 10.1016/s0092-8674(01)00341-5. [DOI] [PubMed] [Google Scholar]
- Gassmann K, Abel J, Bothe H, Haarmann-Stemmann T, Merk HF, Quasthoff KN, et al. Species-specific differential AhR-expression protects human neural progenitor cells against developmental neurotoxicity of PAHs. Environ Health Perspect. 2010;118:1571–7. doi: 10.1289/ehp.0901545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma C, Vila J, Bachstetter A, Bickford PC. Oxidative stress and the aging brain: From theory to prevention. In: Riddle DR, editor. Brain Aging: Models, Methods, and Mechanisms. Chapter 15. Boca Raton (FL): CRC Press; 2007. [PubMed] [Google Scholar]
- Giedd JN. The teen brain: insights from neuroimaging. J Adolesc Health. 2008;42:335–43. doi: 10.1016/j.jadohealth.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Giedd JN. Structural magnetic resonance imaging of the adolescent brain. Ann N Y Acad Sci. 2004;1021:77–85. doi: 10.1196/annals.1308.009. [DOI] [PubMed] [Google Scholar]
- Graham DG, Amarnath V, Valentine WM, Pyle SJ, Anthony DC. Pathogenetic studies of hexane and carbon disulfide neurotoxicity. Crit Rev Toxicol. 1995;25:91–112. doi: 10.3109/10408449509021609. [DOI] [PubMed] [Google Scholar]
- Guerri C, Pascual M. Mechanisms involved in the neurotoxic, cognitive, and neurobehavioral effects of alcohol consumption during adolescence. Alcohol. 2010;44:15–26. doi: 10.1016/j.alcohol.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Harrill JA, Freudenrich TM, Robinette BL, Mundy WR. Comparative sensitivity of human and rat neural cultures to chemical-induced inhibition of neurite outgrowth. Toxicol Appl Pharmacol. 2011;256:268–80. doi: 10.1016/j.taap.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Yang CM. Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed Res Int. 2013;2013:484613. doi: 10.1155/2013/484613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IPCS. Harmonization project document No4. World Health Organisation; Geneva, Switzerland: 2007. IPCS Mode of Action Framework. [Google Scholar]
- Jenner P, Marsden CD. The actions of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in animals as a model of Parkinson’s disease. J Neural Transm Suppl. 1986;20:11–39. [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Absalom AR, Blomgren K, Brambrink A, Crosby G, Culley DJ, et al. Anaesthetic neurotoxicity and neuroplasticity: an expert group report and statement based on the BJA Salzburg Seminar. Br J Anaesth. 2013;111:143–51. doi: 10.1093/bja/aet177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T, Monk T, Rasmussen LS, Abildstrom H, Houx P, Korttila K, et al. Postoperative cognitive dysfunction in middle-aged patients. Anesthesiology. 2002;96:1351–7. doi: 10.1097/00000542-200206000-00014. [DOI] [PubMed] [Google Scholar]
- Kleinstreuer N, Dix D, Rountree M, Baker N, Sipes N, et al. A computational model predicting disruption of blood vessel development. PLoS Comput Biol. 2013;9:e1002996. doi: 10.1371/journal.pcbi.1002996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrigan PJ, Lambertini L, Birnbaum LS. A research strategy to discover the environmental causes of autism and neurodevelopmental disabilities. Environ Health Perspect. 2012;120:a258–60. doi: 10.1289/ehp.1104285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrigan PJ, Rauh VA, Galvez MP. Environmental justice and the health of children. Mt Sinai J Med. 2010;77:178–87. doi: 10.1002/msj.20173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein P, Locke P, Goldberg A. Meeting report: alternatives for developmental neurotoxicity testing – Test-Smart developmental neurotoxicology. Environ Health Perspect. 2007;115:764–8. doi: 10.1289/ehp.9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein P, Silbergeld E, Locke P, Goldberg AM. In vitro and other alternative approaches to developmental neurotoxicity testing (DNT) Environ Toxicol Pharmacol. 2005;19:735–44. doi: 10.1016/j.etap.2004.12.035. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, DeCaprio AP. Protein adduct formation as a molecular mechanism in neurotoxicity. Toxicol Sci. 2005;86:214–25. doi: 10.1093/toxsci/kfi197. [DOI] [PubMed] [Google Scholar]
- McDonald ME, Paul JF. Timing of increased autistic disorder cumulative incidence. Environ Sci Technol. 2010;44:2112–8. doi: 10.1021/es902057k. [DOI] [PubMed] [Google Scholar]
- MacKay C, Davies M, Summerfield V, Maxwell G. From pathways to people: applying the adverse outcome pathway (AOP) for skin sensitization to risk assessment. ALTEX. 2013;30:473–86. doi: 10.14573/altex.2013.4.473. [DOI] [PubMed] [Google Scholar]
- Meek ME, Klaunig JE. Proposed mode of action of benzene induced leukemia: interpreting available data and identifying critical data gaps for risk assessment. Chem Biol Interact. 2010;184:279–85. doi: 10.1016/j.cbi.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Meek ME, Boobis A, Cote I, Dellarco V, Fotakis G, Munn S, et al. New developments in the evolution and application of the WHO/IPCS framework on mode of action/species concordance analysis. J Appl Toxicol. 2014a;34:1–18. doi: 10.1002/jat.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek ME, Palermo CM, Bachman AN, North CM, Jeffrey Lewis R. Mode of action human relevance (species concordance) framework: Evolution of the Bradford Hill considerations and comparative analysis of weight of evidence. J Appl Toxicol. 2014;34:595–606. doi: 10.1002/jat.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller JT, Cluitmans P, Rasmussen LS, Houx P, Rasmussen H, Canet J, et al. Long-term postoperative cognitive dysfunction in the elderly ISPOCD1 study. ISPOCD investigators. International Study of Post-Operative Cognitive Dysfunction. Lancet. 1998;351:857–61. doi: 10.1016/s0140-6736(97)07382-0. [DOI] [PubMed] [Google Scholar]
- Mombelli E. Evaluation of the OECD (Q)SAR Application Toolbox for the profiling of estrogen receptor binding affinities. SAR QSAR Environ Res. 2012;23:37–57. doi: 10.1080/1062936X.2011.623325. [DOI] [PubMed] [Google Scholar]
- NRC. Toxicity Testing in the 21st Century: A Vision and a Strategy. Washington, DC: The National Academies Press; 2007. [Google Scholar]
- OECD. Guidance document on developing and assessing adverse outcome pathways. [Accessed on 29 November 2013];Series on Testing and Assessment No. 184 ENV/JM/MONO. 2013 :6. Available at: http://search.oecd.org/officialdocuments/displaydocumentpdf/?cote=env/jm/mono(2013)6anddoclanguage=en.
- Patlewicz GY, Lander DR. A step change towards risk assessment in the 21st century. Front Biosci (Elite Ed) 2013;5:418–34. doi: 10.2741/e625. [DOI] [PubMed] [Google Scholar]
- Paus T, Collins DL, Evans AC, Leonard G, Pike B, Zijdenbos A. Maturation of white matter in the human brain: a review of magnetic resonance studies. Brain Res Bull. 2001;54:255–66. doi: 10.1016/s0361-9230(00)00434-2. [DOI] [PubMed] [Google Scholar]
- Perluigi M, Swomley AM, Butterfield DA. Redox proteomics and the dynamic molecular landscape of the aging brain. Ageing Res Rev. 2014;13:75–89. doi: 10.1016/j.arr.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Rao DB, Jortner BS, Sills RC. Animal models of peripheral neuropathy due to environmental toxicants. ILAR J. 2014;54:315–23. doi: 10.1093/ilar/ilt058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108:511–33. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohan D, Buggy DJ, Crowley S, Ling FK, Gallagher H, Regan C, Moriarty DC. Increased incidence of post-operative cognitive dysfunction 24 hr after minor surgery in the elderly. Can J Anaesth. 2005;52:137–142. doi: 10.1007/BF03027718. [DOI] [PubMed] [Google Scholar]
- Sanders T, Liu Y, Buchner V, Tchounwou PB. Neurotoxic effects and biomarkers of lead exposure. Rev Environ Health. 2009;24:15–45. doi: 10.1515/reveh.2009.24.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder PK, Ankley G, Mekenyan O, Walker JD, Bradbury S. Quantitative structure-activity relationship models for prediction of estrogen receptor binding affinity of structurally diverse chemicals. Environ Toxicol Chem. 2003;22:1844–54. doi: 10.1897/01-345. [DOI] [PubMed] [Google Scholar]
- Schmieder PK, Tapper MA, Denny JS, Kolanczyk RC, Sheedy BR, Henry TR, Veith GD. Use of trout liver slices to enhance mechanistic interpretation of estrogen receptor binding for cost-effective prioritization of chemicals within large inventories. Environ Sci Technol. 2004;38:6333–42. doi: 10.1021/es0495314. [DOI] [PubMed] [Google Scholar]
- Shafer TJ, Meyer DA, Crofton KM. Developmental neurotoxicity of pyrethroid insecticides: critical review and future research needs. Environ Health Perspect. 2005;113:123–36. doi: 10.1289/ehp.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J, Jernigan TL. The basics of brain development. Neuropsychol Rev. 2010;20:327–48. doi: 10.1007/s11065-010-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartzwelder HS, Wilson WA, Tayyeb MI. Age-dependent inhibition of long-term potentiation by ethanol in immature versus mature hippocampus. Alcohol Clin Exp Res. 1995a;19:1480–5. doi: 10.1111/j.1530-0277.1995.tb01011.x. [DOI] [PubMed] [Google Scholar]
- Swartzwelder HS, Wilson WA, Tayyeb MI. Differential sensitivity of NMDA receptor-mediated synaptic potentials to ethanol in immature versus mature hippocampus. Alcohol Clin Exp Res. 1995b;19:320–3. doi: 10.1111/j.1530-0277.1995.tb01509.x. [DOI] [PubMed] [Google Scholar]
- Thomas RS, Philbert MA, Auerbach SS, Wetmore BA, Devito MJ, Cote I, et al. Incorporating new technologies into toxicity testing and risk assessment: moving from 21st century vision to a data-driven framework. Toxicol Sci. 2013;136:4–18. doi: 10.1093/toxsci/kft178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem. 1993;61:1191–206. doi: 10.1111/j.1471-4159.1993.tb13610.x. [DOI] [PubMed] [Google Scholar]
- van Wijngaarden P, Franklin RJ. Ageing stem and progenitor cells: implications for rejuvenation of the central nervous system. Development. 2013;140:2562–75. doi: 10.1242/dev.092262. [DOI] [PubMed] [Google Scholar]
- Villeneuve D, Crump D, Garcia-Reyero N, Hecker M, Hutchinson TH, LaLone CA, et al. Adverse outcome pathway (AOP) development I: strategies and principles. Toxicol Sci. 2014a doi: 10.1093/toxsci/kfu199. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia-Reyero N, Hecker M, Hutchinson TH, LaLone CA, et al. Adverse outcome pathway (AOP) development II: best practices. Toxicol Sci. 2014b doi: 10.1093/toxsci/kfu200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinken M. The adverse outcome pathway concept: a pragmatic tool in toxicology. Toxicology. 2013;312:158–65. doi: 10.1016/j.tox.2013.08.011. [DOI] [PubMed] [Google Scholar]
- Walhovd KB, Fjell AM, Espeseth T. Cognitive decline and brain pathology in aging - need for a dimensional, lifespan and systems vulnerability view. Scand J Psychol. 2014 doi: 10.1111/sjop.12120. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Watanabe KH, Andersen ME, Basu N, Carvan MJ, III, Crofton KM, King KA, et al. Defining and modeling known adverse outcome pathways: Domoic acid and neuronal signaling as a case study. Environ Toxicol Chem. 2011;30:9–21. doi: 10.1002/etc.373. [DOI] [PubMed] [Google Scholar]
- Westerholz S, de Lima AD, Voigt T. Regulation of early spontaneous network activity and GABAergic neurons development by thyroid hormone. Neuroscience. 2010;168:573–89. doi: 10.1016/j.neuroscience.2010.03.039. [DOI] [PubMed] [Google Scholar]
- Woodruff TJ, Zeise L, Axelrad DA, Guyton KZ, Janssen S, Miller M, et al. Meeting report: moving upstream-evaluating adverse upstream end points for improved risk assessment and decision-making. Environ Health Perspect. 2008;116:1568–75. doi: 10.1289/ehp.11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix: Examples of the putative AOPs for neurotoxicity