

Abstract
Methylation is a fundamental mechanism used in Nature to modify the structure and function of biomolecules, including proteins, DNA, RNA, and metabolites. Methyl groups are predominantly installed into biomolecules by a large and diverse class of S-adenosyl methionine (SAM)-dependent methyltransferases (MTs), of which there are ~200 known or putative members in the human proteome. Deregulated MT activity contributes to numerous diseases, including cancer, and several MT inhibitors are in clinical development. Nonetheless, a large fraction of the human MT family remains poorly characterized, underscoring the need for new technologies to characterize MTs and their inhibitors in native biological systems. Here, we describe a suite of S-adenosyl homocysteine (SAH) photoreactive probes and their application in chemical proteomic experiments to profile and enrich a large number of MTs (> 50) from human cancer cell lysates with remarkable specificity over other classes of proteins. We further demonstrate that the SAH probes can enrich MT-associated proteins and be used to screen for and assess the selectivity of MT inhibitors, leading to the discovery of a covalent inhibitor of nicotinamide N-methyltransferase (NNMT), an enzyme implicated in cancer and metabolic disorders. The chemical proteomics probes and methods for their utilization reported herein should prove of value for the functional characterization of MTs, MT complexes, and MT inhibitors in mammalian biology and disease.
Graphical abstract
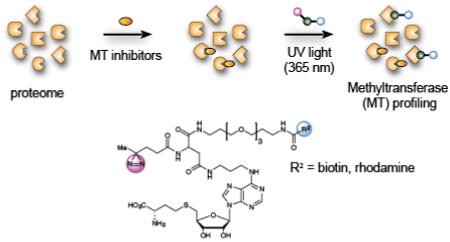
Introduction
Biological macromolecules, including proteins, RNA, and DNA, as well as metabolites, are structurally and functionally diversified by chemical modification. One such fundamental mechanism is methylation, or the replacement of a hydrogen atom on a biomolecule with a methyl group, CH3.1 Methylation can control DNA transcription (both via direct DNA methylation2 and indirectly via histone methylation3), RNA stability and localization,4 and protein activity, structure, localization, and protein/protein interactions.5-7 Endogenous metabolites and exogenous drugs are also methylated to alter their physicochemical properties and biological activities.8,9 Methyl groups, in a biological context, are predominantly installed by S-adenosyl methionine (SAM)-dependent methyltransferases (MTs), which transfer the sulfonium methyl group of SAM to carbon, nitrogen, oxygen, and other heteroatom centers on biomolecules.10 The metabolic product of catalysis – S-adenosyl-L-homocysteine (SAH) – can then be further catabolized to homocysteine, converted to methionine, and reincorporated into SAM,11 and the SAM:SAH ratio is considered to set the methylation potential of biological systems.12,13
A growing body of evidence suggests that deregulated methylation is an important feature of cancer,14 as well as other diseases, such as metabolic15 and neurological disorders,16 and that, in certain cases, inhibiting MTs can be therapeutic.17 Indeed, in addition to the clinical success of DNMT (DNA methyltransferase) inhibitors for treating cancer,18 inhibitors targeting additional MTs like DOTL1 and EZH2 are under active clinical investigation.19,20
MTs are one of the largest enzyme classes in Nature; however, many of the 200+ MTs in humans remain uncharacterized with regards to endogenous substrates and functions, and selective inhibitors have been developed for only a modest subset of these enzymes.10,17 By comparison, small-molecule inhibitors of other cofactor-dependent enzymes, such as kinases have shown broad utility in cancer and other diseases.21 Most kinase inhibitors compete for binding with the cofactor ATP to kinase actives sites, and advanced chemical proteomic technologies have exploited the centrality of the ATP-binding pocket of kinases to create platforms for profiling the activity and selectivity of kinase inhibitors, as well as to functionally characterize kinases and their substrates, in complex biological systems.22-24 Similar strategies have begun to emerge for MTs, where, for instance, SAM analogues that transfer an alkyne have been used to identify MT substrates.25 Likewise, first-generation photoreactive- and fluorescent/biotin-coupled probes based on the general MT inhibitors DzNep26 and SAH,27,28 as well as more selective DNMT29 and DOT1L30 inhibitors, have been introduced with the potential to enrich and identify MTs from native proteomes. While these previous efforts have succeeded in labeling and enriching a handful of MTs, the broader potential for chemical proteomic methods to target endogenous human MTs in complex biological systems with good scope and selectivity remains unclear.
Here we describe the synthesis of a suite of photoreactive SAH-based probes and their implementation in quantitative chemical proteomic experiments in human systems. We show through both UV light-dependent enrichment and competition experiments with SAH that the photoreactive SAH-based probes display good generality and remarkable specificity for MTs across the human proteome. We further provide evidence that SAH-based probes can, in certain instances, enrich MT-associated proteins and be used to screen for and assess the proteomic selectivity of MT inhibitors.
Results and Discussion
SAH-based probes for profiling MTs
We elected to synthesize a series of MT-directed photoaffinity probes based on the S-adenosyl-L-homocysteine (SAH) core, which is a product of MT-catalyzed reactions that binds with high nM to low-mid µM affinity to many MTs.31 We hoped that the ubiquitous nature of the SAH-binding motif would enable the photoaffinity probes to bind both broadly and specifically to MTs. Based on an analysis of available crystallographic data for SAM- and SAH-binding enzymes, and the work of Dalhoff and colleagues,27 we chose the N-6 position of SAH to attach photoreactive and enrichment/detection elements, as the site was expected to position the linker containing these elements toward solvent. Starting from the acetonide of 6-chloropurine riboside, we prepared probes 1-4 in eight synthetic steps (Figure 1; see Scheme S1 for details). Considering several recent successful examples of probe development utilizing aliphatic diazirines32-37 and benzophenones38-41 as photoreactive elements, we chose these groups (1, 2: diazirine; b: benzophenone), as well as an aryl azide (4) for incorporation into the MT-directed probes. By generating probes with three different photoreactive elements located in generally the same position, we hoped to gain perspective on the influence that photocrosslinker identity had on proteome-wide coverage of MTs. We created a fluorescent probe for visualizing protein targets by SDS-PAGE (probe 1 containing rhodamine) and biotinylated probes for enriching and identifying probe targets by quantitative mass spectrometry-based proteomics (probes 2-4). We also synthesized an alkyne SAH analogue with the expectation of performing proteomic studies using copper-catalyzed azide-alkyne cycloaddition (CuAAC),42 but we observed substantial non-specific (UV light-independent) proteomic reactivity with the SAH-based probes under standard CuAAC conditions (Figure S1).
Figure 1.

S-adenosylhomocysteine (SAH)-based photoreactive probes for chemical proteomic profiling of methyltransferases (MTs). R1 and R2 designate varying photoreactive elements and reporter tags (fluorophore, biotin), respectively.
Chemical proteomic profiling of human MTs with SAH-based probes
We first treated soluble proteomic lysates from three human cancer cell lines with probe 1 (25 µM), followed by photocrosslinking at 365 nm, SDS-PAGE, and in-gel fluorescence scanning, which revealed substantial UV light-dependent protein labeling (Fig. 2). Treatment of lysates with excess SAH blocked a subset of the probe 1 labeling events in each cell type (Fig. 2). We next employed quantitative LC-MS to enrich and identify proteins that reacted with biotinylated probe 2. For our initial studies, we used 769P cells (a human renal cell adenocarcinoma cell line) engineered to overexpress the metabolic MT N-nicotinamide N-methyltransferase (NNMT) (769P_NNMT cells8), anticipating that the overexpression of NNMT in these cells would provide a positive control for enrichment studies. Comparison of 769P and 769P_NNMT cell lysates showed a strong, SAH-competed probe 1-labeled protein in the latter cells that migrated at the expected ~30 KDa molecular weight for NNMT (Figure S2).
Figure 2.
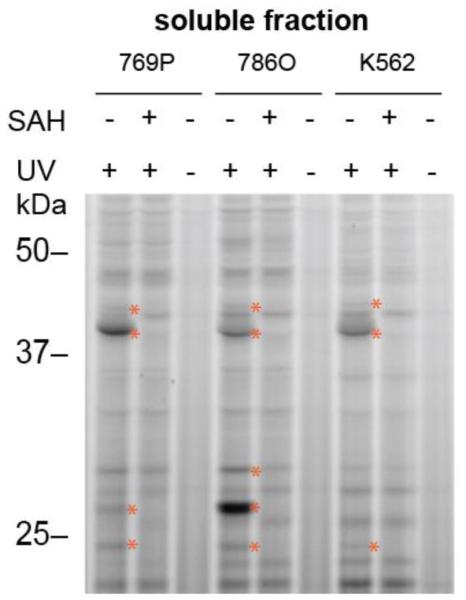
Gel-based profiling with fluorescent SAH-based probe 1. Probe 1 (25 µM) shows UV-dependent and SAH-competed reactivity with proteins in human cancer cell lysates (1 mg protein/mL). Proteins competed by excess SAH (100 µM) are marked with red asterisks. 769P and 786O are human renal cancer cell lines, and K562 is a human leukemia cell line.
To quantify 2-enriched proteins by LC-MS, we utilized stable-isotope labeling by amino acids in cell culture (SILAC) following established protocols.34,43 We performed two types of experiments: i) enrichment experiments, where soluble proteomic lysates from isotopically heavy amino acid-labeled cells were treated with 2 (25 µM) and exposed to UV light-induced protein crosslinking, and lysates from isotopically light amino acid-labeled cells were either treated with 2 without photocrosslinking (no-UV control) or without probe (no-probe control); and ii), competition experiments, where lysates from isotopically heavy cells were again treated with 2 (25 µM) and exposed to UV light, and lysates from isotopically light cells were treated with excess SAH (100 µM) and 2, followed by exposure to UV light.44 Protein targets were then categorized as enriched (heavy:light ratio > 4 in enrichment experiments), competed (heavy:light ratio > 4 in enrichment experiments), or both. Initial studies with both 769P and 769P_NNMT cell lysates showed that NNMT and several other MTs were strongly enriched by probe 2 (Figures 3A, 3B, and S3 and Table 1) and these enrichments were competed by SAH (Figure 3C). MTs also showed the expected 1:1 ratio in probe-vrs-probe control experiments where heavy and light cell lysates were both treated with probe and exposed to UV light (Figures 3A and S4, and Table S1). That NNMT could be enriched and identified not only from 769P-NNMT cells, but also at its lower endogenous expression levels in parental 769P cells (Figure 3A and Table S1) indicated the potential of probe 2 to profile MTs across a relatively broad range of expression levels. We therefore extended our analysis of targets of probe 2 to include two additional human cancer cell lines – 786O (an additional renal adenocarcinoma line) and K562 cells (a chronic myelogenous leukemia line) – and, for K562 cells, we profiled both soluble and particulate fractions.
Figure 3.

Quantitative MS-based profiling with biotinylated SAH probe 2. (A) Parent ion mass (MS1) traces for representative tryptic peptides for the indicated MTs that were specific (enriched and SAH-competed) targets of probe 2. Blue and red traces represent heavy (+ UV, no SAH) and light (− UV or + UV, SAH-competed) fractions, respectively. For +UV/+UV control group, both heavy and light cell lysates were treated with probe 2 and exposed to UV light. Note that a maximum ratio value of 20 was assigned to all ratios with 20 or greater values. 769P_NNMT cells are a variant of the human renal cancer cell line 769P that has been genetically engineered to overexpress NNMT.8 (B) SILAC ratio plot for total proteins identified in human cancer cell lysates treated with probe 2 (heavy: 25 µM, + UV; light: 25 µM, − UV). Results are average enrichment values (+ UV/− UV) for 13 independent experiments performed with 769P cells (+/− NNMT expression,), 786O cells, and K562 cells. (C) SILAC ratios plot for total proteins identified in human cancer cell lysates treated with probe 2 (25 µM, + UV) with or without excess SAH (500 µM) competitor. Results are average competition values (− SAH/+ SAH) for 24 independent experiments performed with 769P cells (+/− NNMT expression), 786O cells and K562 cells. Inset shows expanded image of SAH-competed proteins. For (B) and (C), red dashed lines mark four-fold ratio change used to designate UV-dependent and/or SAH-competed proteins enriched by probe 2, and red dots mark MT proteins in the profiles. (D) Categorization of MTs designated as specific (UV-enriched and SAH-competed) targets of probe 2 based on substrate class. Numbers in parentheses refer to the number of probe 2 targets identified in each MT category. MTs without a characterized substrate class are designated as “uncharacterized”.
Table 1.
List of specific targets of probe 2 and their substrate class (for MTs and kinases). “Enrich” category represents average SILAC ratio measurements for proteins in enrichment experiments (+UV/−UV); “Comp” category represents average SILAC ratio measurements for proteins in competition experiments (−SAH/+SAH).
| Uniprot | Gene | Substrate | Enrich | Comp | Uniprot | Gene | Substrate class | Enrich | Comp | |
|---|---|---|---|---|---|---|---|---|---|---|
| High-Confidence Targets | ||||||||||
| P26358 | DNMT1 | DNA | 16 | 20 | Q8N1G2 | CMTR1 | RNA | 20 | 20 | |
| Q8N4J0 | CARNMT | Metabolic | 20 | 20 | Q86U44 | METTL3 | RNA | 20 | 20 | |
| P21964 | COMT | Metabolic | 20 | 20 | P46087 | NOP2 | RNA | 20 | 17 | |
| Q5HYK3 | COQ5 | Metabolic | 20 | 20 | Q08J23 | NSUN2 | RNA | 20 | 20 | |
| Q14353 | GAMT | Metabolic | 20 | 20 | O43148 | RNMT | RNA | 20 | 20 | |
| P50135 | HNMT | Metabolic | 20 | 20 | Q9NXH9 | TRMT1 | RNA | 18 | 20 | |
| P40261 | NNMT | Metabolic | 20 | 20 | Q32P41 | TRMT5 | RNA | 20 | 20 | |
| P51580 | TPMT | Metabolic | 19 | 20 | O95671 | ASMTL | Uncharacterized | 20 | 20 | |
| Q9H2P9 | DPH5 | Other | 20 | 20 | Q96S19 | C16orf1 | Uncharacterized | 20 | 20 | |
| Q9UIC8 | LCMT1 | Other | 20 | 20 | O60344 | ECE2 | Uncharacterized | 20 | 20 | |
| Q9BV86 | NTMT1 | Other | 20 | 20 | Q8N7N1 | FAM86 | Uncharacterized | 20 | 20 | |
| P22061 | PCMT1 | Other | 20 | 20 | P0C5J1 | FAM86 | Uncharacterized | 20 | 20 | |
| Q86X55 | CARM1 | Protein | 15 | 20 | Q8N6R0 | METTL1 | Uncharacterized | 20 | 20 | |
| Q99873 | PRMT1 | Protein | 20 | 20 | Q9NRN9 | METTL5 | Uncharacterized | 20 | 12 | |
| O60678 | PRMT3 | Prortein | 15 | 20 | Q9H8H3 | METTL7 | Uncharacterized | 20 | 20 | |
| Q96LA8 | PRMT6 | Protein | 20 | 20 | Q9H1A3 | METTL9 | Uncharacterized | 20 | 20 | |
| Q96G04 | EEF2KM | Protein | 20 | 20 | Q7Z4G4 | TRMT1 | Uncharacterized | 20 | 20 | |
| Q5JPI9 | METTL1 | Protein | 20 | 20 | Non-MTs | |||||
| Q8WXB | METTL2 | Protein | 20 | 20 | Q2M2I8 | AAK1 | Protein (kinase) | 20 | 16 | |
| Q9NRG | SMYD2 | Protein | 20 | 20 | P55263 | ADK | Metabolic (kinase) | 20 | 16 | |
| Q9H7B4 | SMYD3 | Protein | 20 | 20 | Q9NSY1 | BMP2K | Protein (kinase) | 20 | 16 | |
| Q6GMV | SMYD5 | Protein | 20 | 20 | Q9UI30 | TRMT12 | None | 20 | 20 | |
| Q8IYT2 | CMTR2 | RNA | 20 | 20 | Q9H2G2 | SLK | Protein ( kinase) | 20 | 12 | |
| Medium-Confidence Targets | ||||||||||
| Q9NR22 | PRMT8 | Protein | N.D. | 20 | Q96FX7 | TRMT6 | RNA | 20 | N.D. | |
| O14744 | PRMT5 | Protein | 20 | N.D. | Q9BVS5 | TRMT6 | RNA | 20 | N.D. | |
| Q7Z624 | CAMKM | Protein | 20 | N.D. | O95568 | METTL1 | Uncharacterize | 20 | N.D. | |
| Q8TEK3 | DOT1L | Protein | N.D. | 20 | Q9BUU2 | METTL2 | Uncharacterize | 20 | N.D. | |
| Q15910 | EZTH2 | Protein | N.D. | 20 | Q96IZ6 | METTL2 | Uncharacterize | 20 | N.D. | |
| Q9NQR | SETD8 | Protein | N.D. | 20 | ||||||
In aggregate, we identified more than 350 proteins that were enriched by probe 2 in human cancer cell lysates (Table S1). Only 45 of these proteins, however, were also competed by SAH, and notably, 40 of these specific (enriched and competed) targets of probe 2 were known or predicted MTs (based on Uniprot designations) (Table 1).45 These MTs could be reliably detected across multiple experiments (≥2 quantified peptides in ≥2 datasets) and were therefore considered high-confidence targets of probe 2 (Table 1). Another 11 MTs were more sparsely detected across enrichment and competition data sets (medium-confidence; Table 1), but, regardless, all enriched MTs that were also detected and quantified in competition experiments showed clear competition by SAH (heavy:light ratios > 4). In contrast, the vast majority of non-MTs (249 out of 255 proteins) enriched by probe 2 were not competed by SAH, suggesting that they mostly reflect very low affinity or non-specific interactions with the probe. Based on these findings, we inferred that the handful of enriched MTs that were not detected in competition data sets (or vice versa) were also likely specific targets of probe 2, and, therefore, we also included these MTs in the final list shown in Table 1, bringing the total number of specific targets of probe 2 to 51 MTs and five non-MTs (Table 1).
The MTs enriched by probe 2 distributed across all major sub-classes, including protein (both Arg and Lys), nucleotide (both DNA and RNA), and metabolic MTs, as well as uncharacterized MTs that lack known substrates (Figure 3D and Table S1). Examples of biologically and biomedically important MTs profiled by probe 2 include: i) DNMT1, which methylates CpG sites in the genome and is targeted by anti-cancer drugs, such as azacitidine and decitabine;46 ii) COMT, which methylates and inactivates catecholamine neurotransmitters and is a potential target for neurological disorders, such as Parkinson’s disease;47 iii) several protein arginine (CARM1, PRMT1, PRMT3, PRMT5, PRMT6) and lysine (SYMD3) MTs implicated in cancer and inflammation;48-50 and iv) METTL3, which catalyzes N6-methyladenosine (m6A) of mRNA and long non-coding RNA.51 Among the uncharacterized MTs, ECE2 is an unusual protein predicted to have an N-terminal cytosolic MT domain followed by a transmembrane domain and C-terminal peptidase domain.52 Various proposed splice variants of ECE2 have been described some of which would generate truncated proteins,52 and it remains unclear which of these protein forms predominate in cells. In our proteomic data sets, we exclusively observed peptides for the MT domain of ECE2 (Figure S5), indicating that, for the cancer cell types under investigation, the MT domain of ECE2 likely exists as a separate protein (generated either by alternative splicing or proteolytic processing of a full-length form). In summary, from an analysis of three human cancer cell lines, we discovered that probe 2 enriched ~25% of the 200+ known and predicted human MTs. We suspect that some MTs were not detected because they have highly restricted cell and tissue distributions (e.g., GNMT is mainly expressed in liver). We also note that some of the sparsely detected MTs (e.g., DOT1L, EZH2) were found in the particulate fractions of cancer cell lysates, which were not extensively profiled in this study. This fraction may represent an enriched source for chromatin-associated MTs.
The enrichment of 50+ MTs from cancer cell lysates by probe 2 initially seemed to contradict the profile observed with fluorescent probe 1 by SDS-PAGE, where only a handful of SAH-competed bands were observed (Figure 2). We recombinantly expressed several of the specific MT targets of probe 2 in HEK293T cells and found that the proteins could be detected by SDS-PAGE with probe 1 and that their probe labeling exhibited UV-dependence and SAH competition (Figure 4 and Figure S6). These data indicate that many of the MTs enriched by probe 2 can also be labeled and detected by probe 1 when recombinantly expressed in human cells, but their endogenous expression levels may be too low in cancer cell lysates to be detected over non-specifically labeled (SAH-independent) background proteins by SDS-PAGE.
Figure 4.
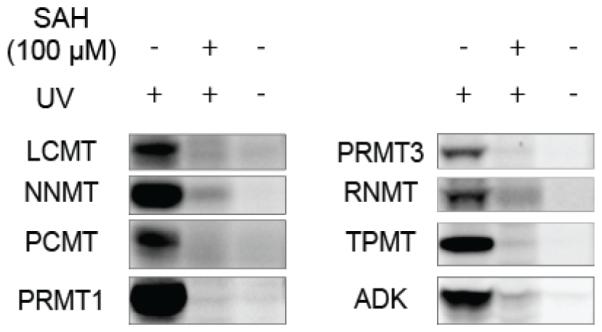
Probe 1 labeling of recombinantly expressed MTs and SAH-binding protein ADK. Proteins were expressed in HEK-293T cells by transient transfection and assayed in cell lysates, or expressed and purified from bacteria and doped into HEK293T lysates. Samples were treated with probe 1 (25 µM) ± UV light and with or without excess SAH (100 µM). For complete gels, including mock-transfected HEK-293T cells, see supporting Figures S6.
Considering that the vast majority of specific targets of probe 2 were confirmed or predicted MTs, we were curious about the rare exceptions and their mode of interaction with probe 2. Literature searches revealed that TRMT112 is the human orthologue of a yeast protein Trm112p that acts as a scaffolding protein to promote the activity of several MTs (e.g., Bud23, Mtq2, Trm9) involved in modifying tRNA, rRNA, and protein components of the cellular translational machinery.53-55 Two of the probe 2-enriched kinases – AAK1 and BMP2K – are sequence-related homologues of the yeast ARK kinases AKL1 and ARK1. A review of large-scale protein-protein interaction studies revealed that AKL1 associates with Trm9 (human orthologue ALKBH8) in yeast two-hybrid assays.56 Trm9 and ALKBH8 have been shown to generate the modified base 5-methoxycarbonylmethyluridine found at wobble positions of tRNAs.57-59 These findings suggest that at least a subset of the non-MT proteins enriched by probe 2 are components of larger protein complexes that contain MTs, which could explain their reactivity with probe 2, if, for instance, this probe bound to the MT component of the complexes, but crosslinked to these non-MT associated proteins (as has been observed for other photoreactive probes, such as those that target histone deacetylases60,61). Of note, we did not detect ALKBH8 (or human orthologues of Bud23 and Mtq2) as targets of probe 2, which could indicate that more efficient photocrosslinking occurs with other non-MT proteins (e.g., TRMT112, AAK1, BMP2K) within some MT protein complexes.
Another probe 2-enriched kinase – ADK, or adenosine kinase – is responsible for converting adenosine into adenosine monophosphate.62 Literature searches revealed that ADK activity is inhibited by SAH,63 providing some evidence for a direct interaction between SAH and this kinase. Consistent with this past work, we found that recombinantly expressed ADK reacted with probe 1 and this interaction was blocked by excess SAH (Figure 4). We also considered that probe 2 (and SAH) might be catabolized in cell lysates to an adenosine derivative, which then reacted with the adenosine binding site on ADK. However, we found that probe 2 reactivity with recombinant ADK was also blocked by the non-hydrolyzable natural product SAH analogue sinefungin64 (Figure S7). Adenosine, but not ATP, also competed probe 2 labeling of ADK (but not other endogenous MTs assessed in this experiment) (Figure S7), suggesting that SAH may bind to one of the two adenosine binding sites in this kinase.62
While the most common type of SAM-dependent enzyme in the human proteome catalyzes methyl-donating reactions, other SAM-utilizing enzymes exist, such as those involved in polyamine biosynthesis65 and the radical SAM superfamily.66 While we did not observe radical SAM enzymes as targets of probe 2, spermine synthase (SMS) was enriched by this probe (Table S1). However, SMS showed inconsistent competition by SAH and therefore did not meet the criteria required for designation as a high-confidence target of probe 2. Adenosyl homocysteinase (AHCY) showed a similar profile of enrichment, but inconsistent competition by excess SAH. Further studies may clarify whether these proteins are legitimate targets of probe 2.
Taken together, these findings demonstrate that the specific targets of probe 2 in human cell proteomes, as defined by exhibiting the dual properties of enrichment and SAH competition, are remarkably restricted to MTs, along with a handful of proteins that are part of MT complexes or independently bind SAH.
Evaluating the impact of photoreactive groups on MT profiles of SAH-based probes
We next asked whether the identity of the photoreactive group affected the target profile of SAH-based probes. Profiling the soluble proteomes from K562 cells with benzophenone and aryl azide probes 3 and 4, respectively, in both enrichment (probe +/− UV) and competition (probe +/− excess SAH) experiments identified 22 methyltransferases, only one of which (CMTR2) was not identified with diazirine probe 2 (Figure 5, Figure S8, and Table S1). Several other MT targets of probe 2 were either not enriched by probes 3 and 4 or enriched by probe4, but not competed by excess SAH. We are unsure why some MTs are enriched by probe 4, but not competed in their interaction by SAH. One possibility is that these enrichments reflect non-specific (i.e., non-active site) labeling of MTs. Alternatively, the differential SAH competition of some MTs in reactions with probes 2 versus 4 could reflect the varying reactive intermediates formed upon UV irradiation. Proteins labeled by probe 4 likely arise from direct crosslinking between the nitrene species initially formed upon UV irradiation and/or reaction of nucleophilic residues with the dehydroazepane formed upon nitrene rearrangement. Thus, poor SAH competition could reflect a persistent covalent mechanism for probe 4, which, instead of providing a transient snapshot of probe-protein interactions, can cumulatively label proteins over time thereby overcoming reversible competition by SAH. Regardless of the precise mechanistic differences, our data indicate that, for the SAH probes tested herein, the aliphatic diazirine group exhibited the broadest and most specific coverage of MTs in human cell proteomes.
Figure 5.

Distribution of MTs enriched by different photoreactive probes. Data are from K562 soluble lysates; 7-8 experiments for each probe that represent a combination of enrichment (UV-vs-no-UV) and SAH competition experiments. See Supporting Table S1, tab “Figure 5 data” for further details.
Discovery of a covalent inhibitor of NNMT using SAH-based probes
Cofactor-based probes have proven useful for the discovery and characterization of enzyme inhibitors,67-70 Building on previous studies showing that fluorescent SAH probes can be used to measure the binding affinity of SAM and SAH to MTs by competitive fluorescence polarization (FP),28,71 we tested whether probe1 could serve as a screening assay for the discovery of MT inhibitors. We selected NNMT for analysis because this MT has been shown to play important roles in cancer72,73 and metabolic disorders,15 but lacks selective inhibitors.
A review of the human NNMT crystal structure74 revealed the presence of a cysteine (C165) within the SAM-binding pocket that is not found in other MTs (Figure 6A). Considering that covalent ligands targeting non-catalytic cysteines in cofactor pockets have been developed for other enzymes, including kinases75 and MTs,76 we wondered if a similar approach could be applied to NNMT. Fragment-based methods offer a powerful way to discover protein ligands,77,78 and our lab recently reported a fragment electrophile library and its proteome-wide reactivity in human cancer cells.79 Within this large data set, we noted that one fragment (compound 8, Figure 6B) reacted strongly with C165 of NNMT. This cysteine, however, exhibited sparse coverage across the global proteomic data sets, and it was therefore unclear whether it also reacted with other fragments and, if so, with what potency. We therefore established a competitive FP screen where compounds could be tested for their ability to displace probe 1 from binding to recombinant, purified human NNMT. We first confirmed that probe 1 binding to NNMT in this FP assay was blocked by SAH in concentration-dependent manner with good Z’ (0.73) and signal:background (> 10:1) values. (Figure S9). We then screened ~20 electrophilic fragments (Figure S10) and identified several compounds that blocked the probe 1-NNMT interaction (Figure 6C) when tested at 500 µM, but only fragment 8 also showed activity at 25 µM. Fragment 8 also blocked in a concentration-dependent manner probe 1 labeling of an ~30 kDa protein in 786O cell lysates presumed to be endogenous NNMT (Figure 6B) (NNMT is 29.6 kDa in size and 786O cells express high levels of this protein8).
Figure 6.

Discovery of an inhibitor of NNMT that reacts with active-site cysteine C165. (A) Crystal structure (PDB 2IIP) of SAH bound to human NNMT; an active-site cysteine C165 is highlighted. (B) Structure of hit fragment 8 and concentration-dependent blockade of probe1 labeling of endogenous NNMT in 786O soluble lysates (2 mg protein/mL) by 8. (C) Fluorescence polarization screen of a fragment electrophile library73 for inhibition of probe 1 binding to recombinant, purified human NNMT. See Figure S10 for structures of fragment electrophiles screened. (D) Concentration-dependent effects of 26-30 on probe 1 labeling of NNMT. (E) Structure and characterization of the more potent NNMT inhibitor RS004 (31) and concentration-dependent blockade of probe1 labeling of NNMT by 31. (F) RS004 (indicated concentrations, 60 min) blocks binding of probe 1 to NNMT, but not the C165A-NNMT mutant as measured by fluorescence polarization. Data represent mean values +/− standard deviation; n = 4 per group. (G) RS004 (25 µM, 30 min) blocks probe 2 labeling of NNMT, but not other MTs in cancer cell lysates as measured by quantitative MS-based proteomics. Reported SILAC ratios are average competition values (DMSO/RS004) for three independent experiments performed with 786O cells. Representative MS1 traces for NNMT and other RS004-insensitive MTs are shown.
We synthesized and tested a series of amide analogues of fragment 8, which revealed that NNMT inhibition, as measured by probe 1 labeling in 786O cell lysates, tolerated replacement of one of the CF3 groups with a phenyl amide, but not other amide substitutions (Figure 6D). Further modifications furnished compound RS004 (31), which blocked probe 1 labeling of endogenous NNMT with an IC50 value of 10 µM (Figure 6E) and kobs/[I} value of 21 ± 4 M−1s−1 (Figure S11A). RS004 also inhibited the catalytic activity of recombinant, purified NNMT using a nicotinamide substrate assay in a concentration-dependent manner with an IC50 value of 1.0 µM (0.7-1.2 µM, 95% confidence intervals) (Figure S11B). Importantly, RS004 blocked probe 1 binding to and the catalytic activity of wild type, but not a C165A mutant of NNMT (Figure 6F andFigure S11B), supporting that this compound produces its inhibitory effect through reaction with C165. Finally, initial chemical proteomic studies confirmed that RS004 (25 µM, 30 min) blocked probe 2 reactivity with endogenous NNMT, but not other MTs in human cancer cell proteomes (Figure 6G), consistent with the unique positioning of C165 within the NNMT active site.
Conclusions
Despite previous reports of various chemical proteomic probes derived from SAH- and MT inhibitors,26-30 the utility of such probes for broadly profiling human MTs in complex biological systems has remained unclear. Probes 5 and 6, for instance, have been evaluated mainly in bacterial and plant systems, where they were found to enrich a handful of MTs.27,28,80,81 Inspired by recent studies demonstrating that dialkyl diazirines serve as useful photoreactive groups for chemical proteomic studies,33-35,37 we generated SAH-based probes bearing a diazirine photocrosslinker and found that these probes are capable of enriching a substantial fraction of human MTs from cancer cell proteomes. That these MTs originate from most of the major functional (protein, RNA, DNA, metabolite) and structural (seven-beta strand, SET, precorrin) subclasses10 reinforces the potential broad utility of probes 1 and 2 for functional characterization of this large and diverse enzyme family. On this subject, it was also exciting to find that several of the probe 2-enriched MTs represent poorly characterized enzymes for which knowledge of physiological substrates and functions is lacking. SAH-based probes thus provide an enabling assay format to profile these MTs in biological systems, as well as to screen for and develop inhibitors, as we showed for the cancer-related enzyme NNMT. Nonetheless, a full understanding of the scope of coverage of MTs provided by these SAH-based probes will require examination of more cell and tissue types beyond the handful of cancer cell lines examined herein. We also anticipate that placing the photoreactive and reporter groups at different positions on SAH probes may facilitate profiling of specific MTs that may exhibit poor binding or crosslinking to probes 1 and 2. In contrast, our results suggest that altering the identity of the photoreactive group itself may not improve coverage of MTs, as replacing the diazirine in 1 and 2 with benzophenone or aryl azide groups produced probes that mainly targeted subsets of the MTs enriched by the diazirine probes.
One of the most interesting findings to emerge from our studies is the remarkable specificity displayed by SAH-based probes for MTs. We found that, once proteomic data were filtered to identify proteins that were substantially enriched by probe 2 in a SAH-competed manner, virtually all (~90%) of the 50+ remaining targets were MTs, along with a handful of MT-associated proteins and proteins previously shown to bind SAH. Importantly, this remarkable selectivity was preserved even at the high concentrations of SAH probe and competitor tested herein, indicating that simultaneously profiling MTs that show a broad range of SAH-binding affinity should be possible. Thus, our data, combined with previous studies assessing the proteome-wide interactions of GTP82 and CoA83, support the value of cofactor-based photoreactive probes for enriching and profiling large classes of enzymes with good scope and specificity in native biological systems.
Projecting forward, it would be valuable to further modify SAH-based probes so that they display improved cell permeability for in situ profiling of MTs. Replacing the fluorophore and biotin tags with latent affinity handles compatible with bioorthogonal reactions is a clear strategy, but our initial attempts suggest that the classical CuAAC reaction may not be compatible with the SAH probes described herein. Fortunately, there are now many other bioorthogonal reactions that can be explored as alternatives.84-86 We also speculate that extending the length of the linker between the SAH and photoreactive groups could furnish probes with a greater capacity to label and enrich MT-associated proteins, albeit at the potential expense of reacting with MTs themselves. Regardless, one could envision that ultimately a suite of photoreactive SAH probes with varying structures and reporter tags would be used for the comprehensive characterization of MTs, MT-associated proteins, and MT inhibitors in native biological systems. These probes should offer a powerful tool set that complements other chemical biology methods to assess MT substrates25,87,88 toward the shared goal of improving our understanding of MTs and MT pathways in human biology and disease.
Supplementary Material
Acknowledgments
We thank members of the Cravatt lab for helpful discussions, and Steven Clarke and Jonathan Lowenson for a kind donation of human PCMT plasmid. This work was supported by the NIH (CA132630, CA186587).
Footnotes
Supporting Information. Detailed synthetic and experimental procedures, analytical (NMR, HRMS) characterization of compounds, supplementary figures, and supplementary tables containing proteomic data. This material is available free of charge via the internet at http://pubs.acs.org.
References
- (1).Fontecave M, Atta M, Mulliez E. Trends Biochem Sci. 2004;29:243. doi: 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- (2).Chen K, Zhao BS, He C. Cell Chem Biol. 2016;23:74. doi: 10.1016/j.chembiol.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Greer EL, Shi Y. Nat Rev Genet. 2012;13:343. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fu Y, Dominissini D, Rechavi G, He C. Nat Rev Genet. 2014;15:293. doi: 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- (5).Xing Y, Li Z, Chen Y, Stock JB, Jeffrey PD, Shi Y. Cell. 2008;133:154. doi: 10.1016/j.cell.2008.02.041. [DOI] [PubMed] [Google Scholar]
- (6).Biggar KK, Li SS. Nat Rev Mol Cell Biol. 2015;16:5. doi: 10.1038/nrm3915. [DOI] [PubMed] [Google Scholar]
- (7).Beaver JE, Waters ML. ACS Chem Biol. 2016;11:643. doi: 10.1021/acschembio.5b00996. [DOI] [PubMed] [Google Scholar]
- (8).Ulanovskaya OA, Zuhl AM, Cravatt BF. Nat Chem Biol. 2013;9:300. doi: 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jatana N, Apoorva N, Malik S, Sharma A, Latha N. Cent Nerv Syst Agents Med Chem. 2013;13:166. doi: 10.2174/1871524913666140109113341. [DOI] [PubMed] [Google Scholar]
- (10).Petrossian TC, Clarke SG. Mol Cell Proteomics. 2011;10:M110 000976. doi: 10.1074/mcp.M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lu SC. Int J Biochem Cell Biol. 2000;32:391. doi: 10.1016/s1357-2725(99)00139-9. [DOI] [PubMed] [Google Scholar]
- (12).Obeid R, Schadt A, Dillmann U, Kostopoulos P, Fassbender K, Herrmann W. Clin Chem. 2009;55:1852. doi: 10.1373/clinchem.2009.125021. [DOI] [PubMed] [Google Scholar]
- (13).Monteiro JP, Wise C, Morine MJ, Teitel C, Pence L, Williams A, McCabe-Sellers B, Champagne C, Turner J, Shelby B, Ning B, Oguntimein J, Taylor L, Toennessen T, Priami C, Beger RD, Bogle M, Kaput J. Genes Nutr. 2014;9:403. doi: 10.1007/s12263-014-0403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jones PA, Baylin SB. Cell. 2007;128:683. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, Cen Y, Sauve AA, Asara JM, Peroni OD, Monia BP, Bhanot S, Alhonen L, Puigserver P, Kahn BB. Nature. 2014;508:258. doi: 10.1038/nature13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jakovcevski M, Akbarian S. Nat Med. 2012;18:1194. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Copeland RA, Solomon ME, Richon VM. Nat Rev Drug Discov. 2009;8:724. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- (18).Gnyszka A, Jastrzebski Z, Flis S. Anticancer Res. 2013;33:2989. [PubMed] [Google Scholar]
- (19).Schapira M, Arrowsmith CH. Curr Opin Chem Biol. 2016;33:81. doi: 10.1016/j.cbpa.2016.05.030. [DOI] [PubMed] [Google Scholar]
- (20).Campbell RM, Tummino PJ. J Clin Invest. 2014;124:64. doi: 10.1172/JCI71605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wu P, Nielsen TE, Clausen MH. Drug Discov Today. 2016;21:5. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- (22).Rosenblum JS, Nomanbhoy TK, Kozarich JW. FEBS Lett. 2013;587:1870. doi: 10.1016/j.febslet.2013.05.008. [DOI] [PubMed] [Google Scholar]
- (23).Daub H. ACS Chem Biol. 2015;10:201. doi: 10.1021/cb5008794. [DOI] [PubMed] [Google Scholar]
- (24).Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM. Cell. 2013;154:737. doi: 10.1016/j.cell.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Blum G, Bothwell IR, Islam K, Luo M. Curr Protoc Chem Biol. 2013;5:67. doi: 10.1002/9780470559277.ch120241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Tam EK, Li Z, Goh YL, Cheng X, Wong SY, Santhanakrishnan S, Chai CL, Yao SQ. Chem Asian J. 2013;8:1818. doi: 10.1002/asia.201300303. [DOI] [PubMed] [Google Scholar]
- (27).Dalhoff C, Huben M, Lenz T, Poot P, Nordhoff E, Koster H, Weinhold E. Chembiochem. 2010;11:256. doi: 10.1002/cbic.200900349. [DOI] [PubMed] [Google Scholar]
- (28).Brown LJ, Baranowski M, Wang Y, Schrey AK, Lenz T, Taverna SD, Cole PA, Sefkow M. Anal Biochem. 2014;467:14. doi: 10.1016/j.ab.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhu B, Ge J, Yao SQ. Bioorg Med Chem. 2015;23:2917. doi: 10.1016/j.bmc.2015.03.006. [DOI] [PubMed] [Google Scholar]
- (30).Zhu B, Zhang H, Pan S, Wang C, Ge J, Lee JS, Yao SQ. Chemistry. 2016;22:7824. doi: 10.1002/chem.201600259. [DOI] [PubMed] [Google Scholar]
- (31).Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, Jerva LF, Scott MP, Copeland RA. Chem Biol Drug Des. 2011;78:199. doi: 10.1111/j.1747-0285.2011.01135.x. [DOI] [PubMed] [Google Scholar]
- (32).Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. Nat Methods. 2013;10:259. doi: 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kambe T, Correia BE, Niphakis MJ, Cravatt BF. J Am Chem Soc. 2014;136:10777. doi: 10.1021/ja505517t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Niphakis MJ, Lum KM, Cognetta AB, 3rd, Correia BE, Ichu TA, Olucha J, Brown SJ, Kundu S, Piscitelli F, Rosen H, Cravatt BF. Cell. 2015;161:1668. doi: 10.1016/j.cell.2015.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Li Z, Hao P, Li L, Tan CY, Cheng X, Chen GY, Sze SK, Shen HM, Yao SQ. Angew Chem Int Ed Engl. 2013;52:8551. doi: 10.1002/anie.201300683. [DOI] [PubMed] [Google Scholar]
- (36).Su Y, Pan S, Li Z, Li L, Wu X, Hao P, Sze SK, Yao SQ. Sci Rep. 2015;5:7724. doi: 10.1038/srep07724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bond MR, Zhang HC, Vu PD, Kohler JJ. Nat Protoc. 2009;4:1044. doi: 10.1038/nprot.2009.85. [DOI] [PubMed] [Google Scholar]
- (38).Cisar JS, Cravatt BF. J Am Chem Soc. 2012;134:10385. doi: 10.1021/ja304213w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Salisbury CM, Cravatt BF. P Natl Acad Sci USA. 2007;104:1171. doi: 10.1073/pnas.0608659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Khan AA, Kamena F, Timmer MS, Stocker BL. Org Biomol Chem. 2013;11:881. doi: 10.1039/c2ob27257a. [DOI] [PubMed] [Google Scholar]
- (41).Qvit N, Monderer-Rothkoff G, Ido A, Shalev DE, Amster-Choder O, Gilon C. Biopolymers. 2008;90:526. doi: 10.1002/bip.21010. [DOI] [PubMed] [Google Scholar]
- (42).Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed Engl. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- (43).Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- (44). In a subset of experiments, the treatment conditions for heavy and light fractions were reversed. See supporting discussion for further information.
- (45). See supporting information for a discussion of how proteins were categorized as methyltransferases.
- (46).Di Costanzo A, Del Gaudio N, Migliaccio A, Altucci L. Arch Toxicol. 2014;88:1651. doi: 10.1007/s00204-014-1315-6. [DOI] [PubMed] [Google Scholar]
- (47).Scheggia D, Sannino S, Scattoni ML, Papaleo F. CNS Neurol Disord Drug Targets. 2012;11:209. doi: 10.2174/187152712800672481. [DOI] [PubMed] [Google Scholar]
- (48).Hu H, Qian K, Ho MC, Zheng YG. Expert Opin Investig Drugs. 2016;25:335. doi: 10.1517/13543784.2016.1144747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kim JH, Yoo BC, Yang WS, Kim E, Hong S, Cho JY. Mediators Inflamm. 2016;2016:4028353. doi: 10.1155/2016/4028353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sarris ME, Moulos P, Haroniti A, Giakountis A, Talianidis I. Cancer Cell. 2016;29:354. doi: 10.1016/j.ccell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- (51).Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, Zou T, Yin P. Nature. 2016;534:575. doi: 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- (52).Tempel W, Wu H, Dombrovsky L, Zeng H, Loppnau P, Zhu H, Plotnikov AN, Bochkarev A. Proteins. 2009;74:789. doi: 10.1002/prot.22299. [DOI] [PubMed] [Google Scholar]
- (53).Letoquart J, van Tran N, Caroline V, Aleksandrov A, Lazar N, van Tilbeurgh H, Liger D, Graille M. Nucleic Acids Res. 2015;43:10989. doi: 10.1093/nar/gkv1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Letoquart J, Huvelle E, Wacheul L, Bourgeois G, Zorbas C, Graille M, Heurgue-Hamard V, Lafontaine DL. Proc Natl Acad Sci U S A. 2014;111:E5518. doi: 10.1073/pnas.1413089111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Liger D, Mora L, Lazar N, Figaro S, Henri J, Scrima N, Buckingham RH, van Tilbeurgh H, Heurgue-Hamard V, Graille M. Nucleic Acids Res. 2011;39:6249. doi: 10.1093/nar/gkr176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. Proc Natl Acad Sci U S A. 2001;98:4569. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Leihne V, Kirpekar F, Vagbo CB, van den Born E, Krokan HE, Grini PE, Meza TJ, Falnes PO. Nucleic Acids Res. 2011;39:7688. doi: 10.1093/nar/gkr406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).van den Born E, Vagbo CB, Songe-Moller L, Leihne V, Lien GF, Leszczynska G, Malkiewicz A, Krokan HE, Kirpekar F, Klungland A, Falnes PO. Nat Commun. 2011;2:172. doi: 10.1038/ncomms1173. [DOI] [PubMed] [Google Scholar]
- (59).Fu Y, Dai Q, Zhang W, Ren J, Pan T, He C. Angew Chem Int Ed Engl. 2010;49:8885. doi: 10.1002/anie.201001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Salisbury CM, Cravatt BF. J Am Chem Soc. 2008;130:2184. doi: 10.1021/ja074138u. [DOI] [PubMed] [Google Scholar]
- (61).Salisbury CM, Cravatt BF. Proc Natl Acad Sci U S A. 2007;104:1171. doi: 10.1073/pnas.0608659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Boison D. Pharmacological reviews. 2013;65:906. doi: 10.1124/pr.112.006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Palella TD, Andres CM, Fox IH. The Journal of biological chemistry. 1980;255:5264. [PubMed] [Google Scholar]
- (64).Zheng W, Ibanez G, Wu H, Blum G, Zeng H, Dong A, Li F, Hajian T, Allali-Hassani A, Amaya MF, Siarheyeva A, Yu W, Brown PJ, Schapira M, Vedadi M, Min J, Luo M. J Am Chem Soc. 2012;134:18004. doi: 10.1021/ja307060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Pegg AE. IUBMB Life. 2009;61:880. doi: 10.1002/iub.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Broderick JB, Duffus BR, Duschene KS, Shepard EM. Chem Rev. 2014;114:4229. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Biochemistry. 2007;46:350. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- (68).Patricelli MP, Nomanbhoy TK, Wu JY, Brown H, Zhou D, Zhang JM, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang QK, Lee JD, Gray NS, Kozarich JW. Chemistry & Biology. 2011;18:699. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Haystead TA. Curr Top Med Chem. 2006;6:1117. doi: 10.2174/156802606777812059. [DOI] [PubMed] [Google Scholar]
- (70).Carlson DA, Franke AS, Weitzel DH, Speer BL, Hughes PF, Hagerty L, Fortner CN, Veal JM, Barta TE, Zieba BJ, Somlyo AV, Sutherland C, Deng JT, Walsh MP, MacDonald JA, Haystead TA. Acs Chem Biol. 2013;8:2715. doi: 10.1021/cb400407c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Luan Y, Blazer LL, Hu H, Hajian T, Zhang J, Wu H, Houliston S, Arrowsmith CH, Vedadi M, Zheng YG. Org Biomol Chem. 2016;14:631. doi: 10.1039/c5ob01794g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Wu Y, Siadaty MS, Berens ME, Hampton GM, Theodorescu D. Oncogene. 2008;27:6679. doi: 10.1038/onc.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Kim J, Hong SJ, Lim EK, Yu YS, Kim SW, Roh JH, Do IG, Joh JW, Kim DS. Journal of experimental & clinical cancer research : CR. 2009;28:20. doi: 10.1186/1756-9966-28-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Peng Y, Sartini D, Pozzi V, Wilk D, Emanuelli M, Yee VC. Biochemistry. 2011;50:7800. doi: 10.1021/bi2007614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Chem Biol. 2013;20:146. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF. Nature. 2016;534:570. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Erlanson DA. Topics in current chemistry. 2012;317:1. doi: 10.1007/128_2011_180. [DOI] [PubMed] [Google Scholar]
- (78).Erlanson DA, McDowell RS, O'Brien T. J Med Chem. 2004;47:3463. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]
- (79).Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF. Nature. 2016 doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Lenz T, Poot P, Grabner O, Glinski M, Weinhold E, Dreger M, Koster H. Journal of visualized experiments : JoVE. 2010 doi: 10.3791/2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Wirsing L, Naumann K, Vogt T. Anal Biochem. 2011;408:220. doi: 10.1016/j.ab.2010.09.029. [DOI] [PubMed] [Google Scholar]
- (82).George Cisar EA, Nguyen N, Rosen H. J Am Chem Soc. 2013;135:4676. doi: 10.1021/ja400839e. [DOI] [PubMed] [Google Scholar]
- (83).Montgomery DC, Sorum AW, Meier JL. J Am Chem Soc. 2014;136:8669. doi: 10.1021/ja502372j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Sletten EM, Bertozzi CR. Angew Chem Int Ed Engl. 2009;48:6974. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Ramil CP, Lin Q. Chem Commun (Camb) 2013;49:11007. doi: 10.1039/c3cc44272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Patterson DM, Nazarova LA, Prescher JA. ACS Chem Biol. 2014;9:592. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- (87).Luo M. ACS Chem Biol. 2012;7:443. doi: 10.1021/cb200519y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Bicker KL, Obianyo O, Rust HL, Thompson PR. Mol Biosyst. 2011;7:48. doi: 10.1039/c0mb00015a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.