

Abstract
Although recent years have witnessed significant advances in the development of catalytic, enantioselective halofunctionalizations of alkenes, the related dihalogenation of olefins to afford enantioenriched vicinal dihalide products remains comparatively underdeveloped. However, the growing number of complex natural products bearing halogen atoms at stereogenic centers has underscored this critical gap in the synthetic chemist’s arsenal. This Review highlights the selectivity challenges inherent in the design of enantioselective dihalogenation processes, and formulates a mechanism-based classification of alkene dihalogenations, including those that may circumvent the “classical” haliranium (or alkene-dihalogen π-complex) intermediates. A variety of metal and main group halide reagents that have been used for the dichlorination or dibromination of alkenes are discussed, and the proposed mechanisms of these transformations are critically evaluated.
Keywords: dihalogenation, alkenes, enantioselective, catalysis, mechanism
Catalysis of alkene dihalogenation , under different activation modes, with control over both the absolute and relative stereochemical course of dihalogen addition, is one of the most vexing problems in stereoselective synthesis. Dihalogenations that circumvent the “classical” haliranium (or alkene-dihalogen π complex) intermediates provide new and exciting opportunities for catalysis, potentially having broader implications for the design of stereoselective alkene difunctionalizations.
1. Introduction: State of the Art
The oxidative difunctionalization of alkenes with electrophilic halogen sources is among the most direct and versatile of strategies for the installation of vicinal, heteroatombearing, stereogenic centers with highly predictable diastereoand constitutional selectivity. 1 However, despite spectacular successes in the development of highly enantioselective alkene dioxygenation protocols – principally epoxidation 2 and dihydroxylation 3 – the control of enantioselectivity in alkene halogenation has only recently begun to capture the imagination of the synthetic organic community. Whilst the field is undoubtedly still in its infancy, a variety of catalytic, enantioselective olefin difunctionalization reactions involving all four (common) halogens have now been realized, and these transformations fall under the broad umbrella of “alkene halofunctionalization” reactions.4
The majority of successful catalytic, enantioselective reactions in this class have involved the intramolecular capture of (putative) haliranium ion intermediates 5 by tethered nucleophiles, including carboxylic acids, alcohols, and (protected) amines, amongst others. However, much less attention has been paid to the development of catalytic, enantioselective variants of the prototypical alkene halogenation process – the addition of molecular dihalogens (X2) to alkenes to afford vicinal dihalides. A particularly striking illustration of this gap in enantioselective synthetic methodology is evident in the landmark total synthesis of the chlorosulfolipid mytilipin A 3 by Carreira and co-workers, in which the very first step of the synthesis – the vicinal dichlorination of ethyl sorbate 1 to give dichloride 2 – is carried out in racemic fashion, necessarily leading to the racemic natural product 3 (Scheme 1).6
Scheme 1.

A racemic alkene dichlorination as the first step of Carreira’s landmark chlorosulfolipid total synthesis.
In fact, despite the ever growing number of halogenated natural products on record, 7 only two examples of enantioselective alkene dihalogenations in total synthesis have been reported, one of which – the synthesis of (+)-bromochloromyrcene8 – was accomplished only recently (vide infra). The first instance of an enantioselective alkene dihalogenation in the preparation of a natural product was described by Snyder et al. during their synthesis of (−)-napyradiomycin 7, although the dichlorination employed a stoichiometric amount of a chiral modifier. Thus, using the chiral, non-racemic dialkoxyborane 5 as a (super)stoichiometric additive to form a chiral 2:1 complex with alkene 4, subsequent treatment with Cl2 delivered the dichloride 6 in 93.5:6.5 er (Scheme 2).9 The interaction of two equivalents of 5 with 4 was proposed to shield one enantioface of the alkene, leading to the preferential formation of one enantiomer of the dichloride.
Scheme 2.

Snyder’s stoichiometric, enantioselective alkene dichlorination en route to (–)-napyradiomycin 7. MOM = methoxymethyl.
This strategy bears some resemblance to an earlier enantioselective dihalogenation based on host-guest inclusion complexes of unsaturated acids in crystalline α- or β-cyclodextrin, in which methacrylic acid in particular could be dichlorinated with high enantioselectivity upon treatment with Cl2.10
In another effort to effect enantioselective dichlorinations of olefins, Snyder and co-workers examined chiral S-Cl sulfonium salts as stoichiometric reagents for enantioselective chlorenium ion transfer. However, the treatment of 1,2-dihydronaphthalene 8 with S-Cl sulfonium salt 9 in CH2Cl2 delivered the vicinal dichloride 10 in 57% yield but only 57:43 er (Scheme 3).11
Scheme 3.

Stoichiometric, enantioselective alkene dichlorination employing a chiral, non-racemic, S-Cl sulfonium salt 9 as a chlorenium ion transfer reagent.
The first, practical, catalytic, enantioselective dichlorination of alkenes was reported by Nicolaou and co-workers in 2011. In this process, 4-Ph(C6H4)ICl2 is employed as the chlorinating agent and (DHQ)2PHAL serves as the catalyst. A number of (E)-configured, 3-aryl 2-propenyl alcohols are dichlorinated in moderate to good er (71.5:28.5 to 90.5:9.5 er), although O-protected, (Z)-configured, or aliphatic allylic alcohols are generally less selective (<52.5:47.5 to 81:19 er) (Scheme 4).12
Scheme 4.

Catalytic, enantioselective alkene dichlorination.
Although detailed mechanistic investigations were not undertaken, the mode of catalysis was speculated to involve Lewis base activation13 of the iodine(III) dichlorinating agent by one of the quinuclidine nitrogens of (DHQ)2PHAL, with a potential hydrogen bonding interaction between a phthalazine nitrogen and the hydroxyl group of the substrate as a stereocontrolling element (Figure 1).
Figure 1.

Proposed stereoinduction model.
Another landmark achievement in this area is the catalytic, enantioselective dibromination of alkenes recently developed by Burns and co-workers, employing diethyl α,α-dibromomalonate 11 as a Br+ equivalent in conjunction with BrTi(Oi-Pr)3 as a Lewis acid bound source of Br−. The dibromination of a range of (E)-configured, 3-aryl 2-propenyl alcohols can be effected using 20 mol% of TADDOL14 additive 12 to provide the corresponding dibromides with good enantioselectivities (85.5:14.5 to 92.5:7.5 er) (Scheme 5). Slightly higher enantioselectivities (5–10% ee increase) can be achieved when the diol 12 is used as a stoichiometric agent (100 mol%). 15
Scheme 5.

Catalytic, enantioselective alkene dibromination.
A proposed catalytic cycle for this reaction is outlined in Scheme 6. Ligand exchange at titanium may lead to the coordinatively saturated complex I, loaded with substrate, bromide ion, diethyl α,α-dibromomalonate 11, and chiral diol 12. Bromenium ion delivery to the alkene, assumed to be intramolecular and reversible, would then give the bromiranium ion species II. An enantioselectivity-determining attack of bromide ion at the benzylic carbon of the substrate may then ensue, setting the absolute configuration of the vicinal dibromide motif, and it was speculated that the bromide may be transferred intramolecularly from the coordination sphere of Ti. Ligand exchange at Ti by i-PrOH – presumably reversible – would then release the dibromide product and enable turnover of the diol 12. Although this mechanistic scenario was described by the authors as a dynamic kinetic resolution of a reversibly formed, chiral bromiranium ion, the presence of the chiral diol ligand renders the two possible bromiranium ions diastereomeric rather than enantiomeric, and as such this mechanism would be classified as a Type 1 dynamic kinetic asymmetric transformation (DyKAT).16 However, the authors did stress that an enantiodetermining, irreversible bromiranium ion formation, or even a concerted dibromination step, could not be ruled out with the evidence available. On the basis of an appreciable racemic background reaction in the absence of the chiral diol 12, and on the fact that even substoichiometric amounts of 12 impart significant enantioselectivity, the authors suggest that this reaction constitutes an example of ligand-accelerated catalysis.17 However, the origin of the acceleration is currently unclear.
Scheme 6.

Proposed catalytic cycle. 2-Np = 2-naphthyl.
In a further development, Burns and co-workers have recently modified their enantioselective dihalogenation method to accomplish the first regio- and enantioselective chlorobromination reactions of alkenes.8 In this study, N-bromosuccinimide (NBS) is employed as the Br+ source, ClTi(Oi-Pr)3 serves as a Lewis acid bound source of Cl−, and tridentate Schiff base 13 is now used as a chiral, non-racemic catalyst (or possibly precatalyst). A variety of allylic alcohols are converted to the corresponding β-chlorobromides with generally high enantioselectivities (89:11 to 98.5:1.5 er) and site selectivities (6:1 to >20:1 rr) (Scheme 7). Perhaps the most remarkable aspect of this work is the fact that the catalyst is able to overturn the intrinsic (substrate-controlled) site selectivity of chloride ion addition to the putative bromiranium ion intermediates, as evidenced by control experiments conducted on a representative substrate. Although detailed mechanistic information is not yet available, the authors suggest that intramolecular chloride delivery to the bromiranium ion may occur from an alkoxy-ligated titanium center, with the enantioselectivity-determining step of the reaction yet to be established.
Scheme 7.

Catalytic, enantioselective alkene chlorobromination. NBS = N-bromosuccinimide.
To showcase the power of this new method in the synthesis of halogenated natural products, the reaction was also applied to a concise, gram-scale synthesis of (+)-bromochloromyrcene 14 (Scheme 8). As well as providing the first example of a catalytic, enantioselective alkene dihalogenation in total synthesis, the halogenation step itself comprises an impressive case of catalyst-controlled chemo-, site- and enantioselectivity.
Scheme 8.

Enantioselective synthesis of (+)-bromochloromyrcene 14. DMP = Dess-Martin periodinane. NBS = N-bromosuccinimide.
In the same report, Burns et al. also disclosed preliminary results for the catalytic, enantioselective dichlorination and dibromination of allylic alcohols bearing only alkyl substitution on the olefin.8 This stands in contrast to their earlier dibromination reaction (Scheme 5), for which only cinnamyl alcohol derivatives delivered high enantioselectivities.15 Thus, allylic alcohol 15 is dichlorinated in 95:5 er using tert-butyl hypochlorite (t-BuOCl) as the Cl+ source (i.e., replacing NBS in their chlorobromination protocol), and diastereomeric alkenes 16 and 17 are dibrominated in 96:4 and 97.5:2.5 er, respectively, using BrTi(Oi-Pr)3 in place of ClTi(Oi-Pr)3 (Scheme 9).
Scheme 9.

Preliminary examples of catalytic, enantioselective alkene dichlorination and dibromination. NBS = N-bromosuccinimide.
Notably, these results constitute the first examples of highly enantioselective dihalogenation of non-conjugated (i.e., non-aryl-substituted) alkenes, although these substrates do still impose an electronic bias on the site selectivity of the nucleophilic halide addition by virtue of their alkene substitution patterns, and/or the inductive effect of the hydroxyl group. Thus, at the time of writing, there are no examples of a catalytic, highly enantioselective dihalogenation of electronically-unbiased alkenes, or those that lack catalyst directing groups. In this Review, the selectivity issues underpinning this “grand challenge” of asymmetric synthesis are analyzed and potential solutions are offered to the problems identified.
In passing it should be mentioned that other reports of enantioselective dihalogenations can also be found in the literature. However, in several cases the enantioselectivities are not unambiguously determined, and only optical rotations for the dihalide products are provided, typically without comparison to the specific rotations of the enantiopure substances (which were unknown).18,19 In another (highly cited) case, remarkable levels of enantioselectivity have been claimed (up to 98.5:1.5 er) for an enantioselective dibromination of alkenes catalyzed by palladium complexes.20 However, this method has never been applied or reproduced, 21 and the Supporting Information is limited and incomplete (e.g., missing optical rotations for several products, ambiguity surrounding the palladium precatalysts employed). For these reasons, these particular contributions will not be discussed further.
In the remainder of this Review, various selectivity challenges will be examined that must be overcome to effect successful catalytic, enantioselective dihalogenations of alkenes, and several mechanistically distinct pathways by which such processes may occur are proposed. Additionally, a number of main group and transition metal halide reagents are presented that have been used to dihalogenate alkenes, and the available evidence regarding the mechanisms of these reactions is critically assessed. It is not the intention of this Review to provide a comprehensive overview of the dihalogenating agents currently available, 22 nor the additions of the molecular dihalogens themselves23 (i.e., chlorine24 or bromine25), but rather to focus on halogenating reagents that may provide opportunities for new catalytic (and potentially enantioselective) reactions. Because of the unique challenges specific to alkene difluorination reactions26 and the thermodynamic disadvantage and reversibility of alkene diiodinations,23 only dichlorinations and dibrominations will be considered. Halogenation reactions comprising the addition of two different halogen atoms across a C=C bond are also beyond the scope of this Review.
2. Challenges for Stereoselective Catalysis
2.1. Preamble: Dihalogenating Reagents
Much of the activity in (non-industrial) halogenation chemistry has comprised the development of new reagents which are easier to handle than the molecular dihalogens, as well as offering differential reactivity to the latter (i.e., being either more reactive or more selective). Moreover, such reagents often allow precise control of stoichiometry, which is a particular advantage for chlorination, given the difficulties in dispensing accurate quantities of chlorine gas on a laboratory scale. For example, ammonium polyhalide salts, of general formula [R4N]+[(X2)nX]−, incorporate one or more equivalents of molecular dihalogen and are (typically) solid, crystalline compounds that have proven popular for alkene dihalogenation. For dibromination, pyridinium tribromide 27 and several other ammonium tribromide salts are commercially available and have enjoyed widespread use, although similar reagents for dichlorination – most notably Et4NCl3 (Mioskowski’s reagent)28 – are not commercial due to the slow release of Cl2 on storage, and the laboratory synthesis of this compound requires the handling of elemental chlorine. Besides this approach of “dihalogen carrier” reagents, another strategy is to generate molecular dihalogens (or their formal equivalent) in situ via the oxidation of halide sources with strong oxidants, 29 and this method is especially attractive for dichlorination. A large number of these protocols, summarized elsewhere,29, 30 have been developed for dibromination, although notably fewer are evident for dichlorination, including H2O2-HCl, 31 KMnO4-Me3SiCl-BnEt3NCl,32 and Oxone®-NaCl.33 As a complement to “halide oxidation” (X− → X+ + 2e−), the reverse process of “halenium reduction” (X+ + 2e− → X−) has also been employed, in which two equivalents of an X+ reagent react with one equivalent of a suitable 2e− reductant, generating X− ions alongside the X+ source (which may combine to give the molecular dihalogen34,35). Yoshimitsu’s protocol for alkene dichlorination using a 2:1 NCS:PPh3 reagent system (NCS = N-chlorosuccinimide), in which PPh3 serves as the reducing agent, is a good example,36 and several “organocatalytic” alkene dibrominations also fall into this category.35
However, none of the above reagents or reagent systems are well-suited for most catalytic dihalogenations (i.e., in which enantio- or diastereoselectivity is controlled), as all of these halogenating agents (e.g., dihalogens) exhibit a fast background rate of addition to alkenes.37 An attractive alternative is to avoid molecular dihalogens altogether and employ either a combination of separate halenium (X+) and halide (X−) equivalents or a single dihalogen equivalent (SO2Cl2, PhICl2, etc.) (Figure 2). Provided that these are sufficiently slow to react with the alkene substrate in the absence of a catalyst, then one has an ideal platform for catalytic dihalogenation. Alternative strategies to avoid background reactivity are outlined in Section 2.2.5, whereby only halide (X−) ions or only halenium (X+) sources are used as the source of halogen atoms, and the necessary X+ or X− reaction partners are generated catalytically (by oxidation or reduction, respectively).
Figure 2.

Strategies for avoiding the use of molecular dihalogens.
2.2. The Catalysis Problem
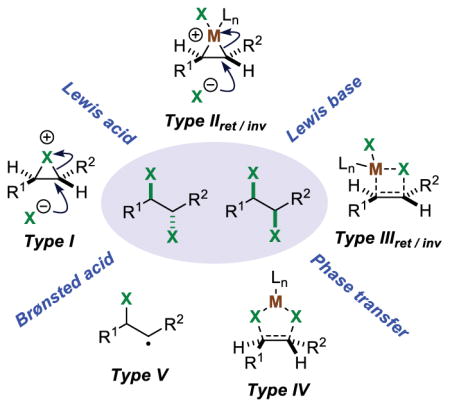
In our previous analysis of catalytic strategies to effect enantioselective halofunctionalizations of alkenes with oxygen-, nitrogen-, and carbon-centered nucleophiles, it was identified that most of the existing methods rely upon Brønsted acid, Lewis acid, or Lewis base catalysts to enhance the electrophilicity of an otherwise weakly reactive halenium ion (X+) source.4f In this way, an uncatalyzed background reaction of the alkene with the halogenating agent can be minimized. Another distinct strategy is phase transfer catalysis, in which the alkene substrate and reactive halogenating agent are physically separated in different phases until brought into contact by the catalyst. All of these general catalysis strategies are summarized in Scheme 10.
Scheme 10.

General strategies for catalysis of halofunctionalization.
In this Review, these strategies will be reexamined in the context of alkene dihalogenation. An additional catalysis mode, termed “redox catalysis”, will also be introduced, as this can offer conceptually and mechanistically distinct strategies for alkene dihalogenation (and perhaps halofunctionalization more broadly). It should be stressed that some of the catalytic cycles outlined in this section are currently only hypothetical for alkene dihalogenation, although known examples from the literature are highlighted where possible. The main objective is to provide the basic foundations on which to build new catalytic methods for alkene dihalogenation, and to preempt any obstacles that may arise in certain catalytic manifolds.
It should also be mentioned that there are several reports of alkene dihalogenations for which the generation of X2 (from X+ equivalents), rather than the addition step to the alkene, is “catalyzed” (or perhaps just initiated).35 Thus, any further reference to “catalytic” dihalogenations in this Review refers only to those reactions in which the alkene dihalogenation process itself is catalyzed, rather than the release of molecular dihalogen.
2.2.1. Brønsted Acid Catalysis
Brønsted acid catalysts may activate halenium ion (X+) sources III either by protonation or by hydrogen-bonding (these being the limiting cases of a continuum), and this can be represented by the activated species IV (Scheme 11). Moreover, protonation of the X+ equivalent (e.g., N-haloimide) may lead to halenium ion transfer to the conjugate base of the Brønsted acid, generating an alternative reactive electrophile V. Electrophilic attack of either IV or V on the alkene substrate may generate a haliranium ion intermediate VII, that can be attacked by a halide ion to generate the vicinal dihalide product. However, haliranium ions need not necessarily be intermediates, and other possibilities include direct attack of halide ion on alkene-“halogen” π-complexes, or the intermediacy of species featuring covalent bonds to metal or main group elements (see Sections 4 and 5). An undesired side reaction, which must be avoided in this mechanistic manifold, is the reaction of species IV or V with halide ion to release the molecular dihalogen (X2), which could contribute to an uncatalyzed background dihalogenation. However, the requirement for halide ion trapping of the haliranium ion presents a unique problem not encountered with neutral (protonated) trapping nucleophiles such as alcohols, carboxylic acids or amides. Specifically, the latter nucleophiles release a proton (H+) following nucleophilic attack on the haliranium ion, and this H+ ion serves to turnover the Brønsted acid catalyst. An anionic halide nucleophile (X−) on the other hand carries no such acidic proton, and the only proton source available is the conjugate acid VI of the leaving group VIII from the X+ reagent III. As the pKa of VI will be significantly higher than that of the Brønsted acid catalyst, turnover will not proceed, and the anionic leaving group VIII of the halenium source III will have deactivated the catalyst. Unfortunately, simply using the corresponding hydrohalic acid (HX) as a nucleophile is unlikely to resolve this issue, as the halogen atom is not nucleophilic in this form and the strongly acidic HX would promote a racemic Brønsted acid-catalyzed pathway.
Scheme 11.

A Brønsted acid-catalyzed alkene dihalogenation using separate
A possible solution to this turnover problem may be to use a complex anion IX of the halide (e.g., BCl4−, SbCl6−) as the nucleophile source – one which is sufficiently nucleophilic to trap the haliranium ion intermediate VII11, 38 and then afterward release a Lewis acid X to sequester the reagent-derived anion as a complex XI (Scheme 12). A potential danger, however, is that the Lewis acid X may itself catalyze a racemic background reaction if it is not rapidly “quenched” by complexation with VI.
Scheme 12.

A Brønsted acid-catalyzed alkene dihalogenation using an X+ reagent III in combination with a complex anion of the halide as an X− source.
Notably, the same turnover problem does not arise if one employs a dihalogen equivalent XII, because in this case only a neutral by-product XIV is generated that cannot sequester the proton of the Brønsted acid (Scheme 13). The mode of activation could involve hydrogen-bonding polarization of XII via a complex such as XIII, as exemplified by a (racemic) Brønsted acid-catalyzed alkene dichlorination with PhICl2 (see Section 4.5.1).39 Note that in this case there is no longer an exogenous source of halide ions required (i.e., the halide is incorporated into the reagent XII), so that any undesired molecular dihalogen formation would result from the decomposition of complex XIII.
Scheme 13.

A Brønsted acid-catalyzed alkene dihalogenation using a dihalogen equivalent XII as a single reagent.
2.2.2. Lewis Acid Catalysis
Lewis acid catalysis, which has been invoked in several catalytic, enantioselective halocyclizations, 40 provides another means of activating halogenating agents that is conceptually very similar to Brønsted acid catalysts (vide supra). Just as in that case, the use of separate halenium (X+) and halide (X−) equivalents leads to a catalytic turnover problem due to the anionic leaving group VIII of the halenium source III deactivating the acidic catalyst (Scheme 14). As for Brønsted acid catalysis, this problem could potentially be solved through the use of a complex anion IX of the halide (e.g., BCl4−, SbCl6−) as the nucleophile source, releasing a stoichiometric Lewis acid by-product X to facilitate turnover (vide supra).
Scheme 14.

A Lewis acid-catalyzed alkene dihalogenation using separate X+ and X− sources.
As for Brønsted acid catalysis (vide supra), no turnover problem exists if a dihalogen equivalent XII is employed, as the neutral by-product XIV does not strongly bind the Lewis acid catalyst (Scheme 15). Although no clear-cut examples are on record for alkene dihalogenation, Lewis acid catalysts have been used to effect the ionic chlorination of aromatics with SO2Cl2 via X–S–X bond polarization.41
Scheme 15.

A Lewis acid-catalyzed alkene dihalogenation using a dihalogen equivalent XII as a single reagent.
2.2.3. Lewis Base Catalysis
Following the general mechanism for Lewis base activation of Lewis acids,13 a Lewis base catalyst may activate a halenium source III by forming a polarized complex XV, possibly in equilibrium with a highly reactive ion pair XVI, as the active halenium transfer agent. As always, an undesired reaction of XV or XVI with halide ion could potentially generate the molecular dihalogen (X2) (Scheme 16).
Scheme 16.

A Lewis base-catalyzed alkene dihalogenation using separate X+ and X− sources.
A similar catalytic cycle can be constructed for a Lewis base-catalyzed alkene dihalogenation process using a dihalogen equivalent XII (Scheme 17). Although complex XVII and ion pair XVIII are depicted as the activated species (by analogy to Scheme 16), it should be borne in mind that the Lewis base might also interact at an electrophilic site other than the halogen atom (e.g., a metal or main group element center, represented here by the shaded block).
Scheme 17.

A Lewis base-catalyzed alkene dihalogenation using a dihalogen equivalent XII as a single reagent.
An example of the latter type of Lewis base-catalyzed process is Nicolaou and co-worker’s enantioselective dichlorination reaction, in which the dihalogen equivalent 4-Ph(C6H4)ICl2 is thought to be activated by substitution of a chloride ligand on the iodine(III) center with a neutral tertiary amine donor [i.e., a quinuclidine nitrogen on (DHQ)2PHAL], conferring a positive charge to the reagent which increases its electrophilicity (Figure 1).12 In contrast, Burns et al. have proposed a rather different role for the Lewis base (pre)catalyst (i.e., diol 12) in their enantioselective alkene dibromination protocol, suggesting that it may serve to accelerate the nucleophilic trapping of the bromiranium ion intermediate with (Ti-bound) bromide ion, rather than the formation of the bromiranium ion itself (Scheme 5).15 If this were true, it would constitute “nucleophile activation” as opposed to “electrophile activation” (as depicted in Scheme 6), which is another well-recognized facet of Lewis base catalysis.13 However, this mechanistic proposal is somewhat speculative, and an “electrophile activation” mode of catalysis, involving interaction of the diol moiety 12 with the Ti center to enhance its Lewis acidity (which would indirectly activate the Ti-bound halogen electrophile), could not be discounted.
2.2.4. Phase Transfer Catalysis
Another potential means of catalyzing alkene dihalogenation reactions is phase transfer (PT) catalysis, whereby a catalyst serves to chaperone a charged reactant or intermediate across a phase boundary (usually liquid-liquid or solid-liquid).42 If the catalyst-derived counterion present in the enantiodetermining step of the reaction is chiral and non-racemic, it may potentially impart enantioselectivity in this step via noncovalent interactions with one or both of the reacting partners.42 One attractive strategy may be to capitalize on the ability of molecular dihalogens such as Br2 and Cl2 to reversibly form trihalide anions, X3−, with their corresponding halide salts.43 In the presence of a PT catalyst featuring a chiral, lipophilic cation (e.g., NR4+, PR4+), an X3− anion could be transported from an aqueous (or solid) phase environment to the organic phase, at which point it could dihalogenate an alkene substrate and generate a halide ion as the by-product, which would return to the aqueous (or solid) phase along with the lipophilic cation. In non-polar reaction media, the dibromination of alkenes with Br3− anions (or Br2 in the presence of at least an equimolar amount of Br− ions) is thought to occur by rate- and product-determining attack of a Br− ion on a 1:1 alkene-Br2 π-complex.44 As a counter cation must necessarily be closely associated with the Br− ion during this step, the prospects for asymmetric induction with a chiral, non-racemic counter cation are promising, and similar considerations could apply to dichlorinations with Cl3− sources (Scheme 18).
Scheme 18.

Cationic phase transfer-catalyzed alkene dihalogenation.
It should be noted that an enantioselective, phase transfer-catalyzed alkene dichlorination has already been claimed,18 but as the product enantioselectivities were not actually measured (i.e., only optical rotations were provided), the feasibility of this approach for achieving meaningful levels of asymmetric induction has yet to be convincingly demonstrated.
In recent years, catalytic, enantioselective halofunctionalizations based on the concept of anionic phase transfer catalysis have begun to surface,45 and although initially applied solely to fluorination processes, 46 this concept has recently been extended to bromo- and iodocyclizations by Toste and co-workers. 47 By employing insoluble, DABCO-derived, tricationic “Br+” salts of the form 19, in combination with a chiral, lipophilic phosphoric acid 20 (as a precursor to the chiral anion PT catalyst), highly enantioselective bromocyclizations of amido alkenes such as 18 could be achieved by solid-liquid phase transfer catalysis (Scheme 19). Similar results were obtained in iodocyclizations using an “I+” salt analogous to 19.
Scheme 19.

Toste’s enantioselective halocyclization via chiral anion phase transfer catalysis.
Subsequently, Ma and co-workers showed that highly enantioselective bromocyclizations of tryptamine derivatives such as 21 could be achieved with the alternative “monocationic” “Br+” salt 22 which, in this reaction at least, gives more reproducible results than the “tricationic” “Br+” salts (e.g., 19) developed by Toste (Scheme 20).48 Later work by the same authors on a closely related bromocyclization found that mixtures of “monocationic” and “tricationic” DABCO-derived “Br+” salts closely related to 22 and 19 gives optimal performance.49
Scheme 20.

Ma’s enantioselective bromocyclization via chiral anion phase transfer catalysis.
Taking the “monocationic”, DABCO-derived “Br+” salts of type 22 for illustration, one could envisage a solid-liquid phase transfer catalysis process in which a chiral anion PT catalyst transports a monocationic, amine-bound X2 complex into the organic phase, where it may undergo reaction with an alkene substrate (Scheme 21). If the Lewis basic nitrogen of the amine remains associated with the electrophilic bromine atom during the alkene addition step, 50 enantiotopic face selection might occur by virtue of the chiral counter anion. Such a process is speculative, however, and it remains to be seen whether an anionic phase transfer-catalyzed alkene dihalogenation (enantioselective or not) can be reduced to practice.
Scheme 21.

Anionic phase transfer-catalyzed alkene dihalogenation.
2.2.5. Redox Catalysis
So far, all of the catalytic activation modes discussed have featured catalysts which undergo no formal change in oxidation state during the catalytic cycle. However, catalytic cycles are also possible with redox-active catalysts, based on transition metal or main group element centers, in which catalyst oxidation state changes do occur. A popular analogy might be the cross-coupling of a carbon nucleophile with a carbon electrophile under the agency of a transition metal catalyst – a transformation which is isohypsic but nevertheless involves both oxidation and reduction events at the catalytic metal center. This type of catalysis, which has previously been classified under the rather broad banner of “group-transfer catalysis”, 51 will be referred to as “redox catalysis” in this Review. Simple schematics of “isohypsic” alkene dihalogenations occurring under redox catalysis are shown in Scheme 22, both for: (a) separate halenium (X+) and halide (X−) equivalents and for (b) a single dihalogen equivalent. Although a two-electron oxidation state change at the “metal” is implied, a bimetallic mechanism involving one-electron changes at two “metals” could also be operative in principle. Moreover, this classification is silent regarding the mechanism of halogen transfer to the olefin from the “metal” halide, which could be polar, radical or concerted in nature. Clearly the involvement of metal or main group elements opens up new mechanistic possibilities beyond the typical haliranium ion (or alkene-“halogen” π-complex) intermediates, and this particular aspect is explored in detail in Sections 4 and 5.
Scheme 22.

“Isohypsic” redox catalysis for alkene dihalogenation. M = transition metal or main group element.
Keeping the analogy with metal-catalyzed cross-coupling reactions, one can also envisage “oxidative” or “reductive” variants of redox-catalyzed alkene dihalogenation reactions (Scheme 23). In the “oxidative” case, two equivalents of a halide source (X−) are employed in conjunction with a redox-active catalyst able to formally invert the reactivity of one X− ion (i.e., generate a formal X+ source) and an external (non-halogenating) oxidant to reoxidize the catalyst. An obvious requirement is that the reduced form of the catalyst is oxidized in preference to the stoichiometric halide source, in order to avoid the in situ generation of halogen electrophiles with high background reactivity (e.g., the molecular dihalogen). An example of an “oxidative” catalytic dihalogenation is Denmark and co-worker’s syn-stereospecific dichlorination of alkenes, which relies upon an N-fluoropyridinium reagent as the stoichiometric re-oxidant (see Section 4.4.2). 52 The third strategy of “reductive” dihalogenation is the polar opposite of the “oxidative” approach: two equivalents of a halenium source (X+) are employed in conjunction with a redox-active catalyst and an external reductant to regenerate the active catalyst. Similarly, the oxidized form of the catalyst must be reduced in preference to the X+ source. At the time of writing, there are no examples of this strategy yet reported for catalytic dihalogenation, providing an exciting opportunity for reaction invention.
Scheme 23.

“Oxidative” and “reductive” redox catalysis for alkene dihalogenation (note that all oxidation states are relative rather than absolute). M = transition metal or main group element.
2.3. The Enantioselectivity Problem
For alkene dihalogenation reactions proceeding via a two stage mechanism comprising haliranium ion (or alkene-dihalogen π-complex) intermediate formation and subsequent nucleophilic attack of halide ion, the absolute configuration of the vicinal dihalide product can potentially be decided at either, or both, of these stages. For the sake of simplicity, the following discussion will focus on haliranium ions as intermediates (setting aside the complication of “open” β-halo carbocations), although much of what follows is equally applicable to reactions involving direct nucleophilic attack on alkene-dihalogen π-complexes.44
The first line of analysis is to consider the symmetry properties of the alkene, which will in turn dictate the stereochemical features of the overall addition process (Scheme 24). For alkenes within the C2h point group, such as symmetrical, (E)-configured olefins, the haliranium ion is chiral but the dihalide products, arising from attack of a halide ion at one of two homotopic carbon termini, are necessarily achiral (category A). However, for a symmetrical, (Z)-alkene (i.e., C2v point group) the carbon termini within the (achiral) haliranium ion are enantiotopic, and so the enantiomer of the dihalide product formed depends solely on which of these two carbon atoms is attacked by the halide nucleophile (category B). In other words, nucleophilic trapping rather than haliranium ion formation is enantiodetermining for this class of alkene. In the case of unsymmetrical alkenes (Cs symmetry), of either (E)- or (Z)-configuration, the situation is more complicated: the haliranium ion intermediates are chiral and the two carbon termini are now constitutionally heterotopic (category C). In many alkene addition reactions via such chiral -iranium ions, the trapping nucleophile is different from the bridging group of the cyclic -onium species, and so the products of attack at the two different carbon termini are constitutional isomers. In such a scenario, the enantiomeric composition of both products is determined solely by the extent of enantiofacial discrimination in the -iranium ion formation step (assuming that no chiral catalyst-induced kinetic resolution occurs during trapping of the -iranium ions, which could lead to unequal enantiomeric ratios for the two constitutional isomers 53 ). However, in the case of alkene dihalogenation the two halogen atoms are indistinguishable, and the products of attack at the two different carbon termini are now related as enantiomers. Consequently, if the formation of a haliranium ion is irreversible, both its formation and nucleophilic trapping will determine the enantiomeric composition of the products. Accordingly, both of these processes must be highly selective to ensure significant enantioenrichment in the vicinal dihalide product.
Scheme 24.

Symmetry-based analysis of alkene dihalogenations (via haliranium ions), with enantiodetermining steps highlighted by bold arrows. X = halogen atom.
For alkenes falling into category C, situations may arise in which only one of the two stages, rather than both, is enantiodetermining, and this can significantly improve the prospects of achieving highly enantioselective reactions. These special cases are described in detail below, starting with enantiodetermining nucleophilic trapping (Section 2.3.1) and followed by enantiodetermining haliranium ion formation (Section 2.3.2).
2.3.1. Enantiodetermining Nucleophilic Trapping
If haliranium ion formation is fast and reversible,54 or if the chiral haliranium ions are able to rapidly interconvert (e.g., via alkene-to-alkene transfer, vide infra), then the nucleophilic trapping event becomes enantiodetermining. To selectively form one enantiomer of the dihalide product, the chiral catalyst must kinetically resolve the chiral haliranium ions, such that one enantiomer undergoes nucleophilic trapping faster than the other. Additionally, the catalyst must also control which of the two carbon atoms of the haliranium ion undergoes nucleophilic attack by halide ion, unless a substrate-controlled bias for the site of attack is already present (e.g., at a benzylic carbon).
Depending on whether the chiral haliranium ion intermediates are associated with the chiral catalyst during their interconversion, the situation is either a dynamic kinetic resolution (DKR) or a (Type 1) dynamic kinetic asymmetric transformation (DyKAT) [Scheme 25, arbitrarily depicting an (E)-alkene].16 Although challenging to distinguish experimentally, the distinction is important because the relative rates of nucleophilic trapping (and hence the enantioselectivity) depend not only on the rate constant ratio (kA/kB), but also on the concentrations of the stereoisomeric haliranium ions ([IA] and [IB]). In a DKR manifold, the haliranium ions are enantiomeric and their concentrations are equal (assuming racemization is much faster than trapping), and so a high enantioselectivity rests solely on a high kA/kB ratio (ideally >20). For a Type 1 DyKAT however, the haliranium ions are diastereomeric by virtue of their association with the chiral catalyst, and their concentrations can be different. In principle, a relatively low kA/kB ratio can be augmented if [IA] > [IB] (e.g., a kA/kB value of 5 in combination with [IA]/[IB] = 4 would still give 20:1 er in favor of A). For these reasons, it can be beneficial to design chiral catalysts able to associate with (stereomutating) haliranium ion intermediates,4f encouraging a DyKAT as opposed to a DKR process. One such tactic is to employ alkene substrates bearing catalyst-coordinating functionality,4f as typified by Nicolaou’s12 and Burns’s8,15 use of allylic alcohols in their respective dihalogenation protocols (see Section 1). Another possible strategy, recently showcased for enantioselective selenocyclizations (via seleniranium ion intermediates) is to use a chiral anion-binding catalyst, which essentially partners the -iranium ion intermediate with a chiral counter anion.55
Scheme 25.

Enantiodetermining nucleophilic attack of halide ion by kinetic resolution of haliranium ions. Cat = catalyst, X = halogen atom.
At least conceptually, an RRRM process (regiodivergent reaction of a racemic mixture) is possible if the catalyst can induce nucleophilic attack at opposite termini for each haliranium ion enantiomer, although such processes are difficult to rationally design [Scheme 26, arbitrarily depicting an (E)-alkene].56 Rather unusually, an RRRM process in the current context would generate the same enantiomer of dihalide product in both cases, as opposed to the constitutional isomers more typical of RRRM reactions. Consequently, such a process – if at all possible – would constitute a transformation that is mechanistically an RRRM but nominally an enantioconvergent reaction.
Scheme 26.

Enantiodetermining nucleophilic attack of halide ion by regiodivergent (enantioconvergent) reaction of a racemic mixture. Cat = catalyst, X = halogen atom.
Although the above discussion has centered on haliranium ions as intermediates, a similar analysis may conceivably apply in dihalogenation reactions involving direct halide ion attack on (reversibly-formed) alkene-dihalogen π-complexes.44
2.3.2. Enantiodetermining Haliranium Ion Formation
For haliranium ion formation to be enantiodetermining, two conditions must be met: (1) the halenium ion transfer to the olefin from the “X+” source must be irreversible, and (2) the haliranium ion thus produced must be configurationally stable prior to its nucleophilic trapping (i.e., it must not racemize). If either one of these criteria are not fulfilled, then the decision as to which enantiomer of the dihalide product is formed will occur at the (subsequent) nucleophilic trapping stage (see Section 2.3.1).
From the earlier symmetry-based analysis (Scheme 25), it is also clear that both haliranium ion formation and nucleophilic trapping with halide ion can be enantiodetermining. Even if a single enantiomer of haliranium ion is formed, the enantioselectivity can be eroded if nucleophilic trapping occurs without high site selectivity. Thus, for haliranium ion formation alone to be enantiodetermining, the nucleophilic attack of the halide ion must be completely biased toward one of the two carbon centers of the haliranium ion intermediate. In practice, this is most easily achieved by substrate control rather than catalyst control, and electronically-biased olefins such as aryl-conjugated alkenes will inherently direct halide attack toward the benzylic carbon center, for example.
The following analysis will not be concerned with the factors that lead to highly enantioselective haliranium ion formation (as this is largely a question of catalyst design on a case-by-case basis), but rather the general factors that mitigate against obtaining high enantioselectivity (other than the operation of a racemic background reaction, vide supra). A clear difficulty in enantioselective halenium ion transfer to an alkene is the distance between the alkene substituents and the catalyst which is covalently associated with the halogen atom. This arrangement arises because of the stereoelectronic requirement for the alkene to approach the σ* orbital of the X+–Cat* bond. A similar problem is encountered in gold catalysis because of the linear coordination geometry of Au(I) complexes, although chiral counter anions or specially designed chiral ligands have begun to provide solutions.57 By way of contrast, electrophilic species offering π* orbitals as a “docking” point for the catalyst (e.g., OsO4) enable more diverse geometries and a better stereochemical communication between the catalyst and the alkene (Figure 3).
Figure 3.

Challenges in stereochemical communication between the catalyst and the alkene substrate. Cat = catalyst, X = halogen atom.
Even if the haliranium ion can be formed with high enantioselectivity, intermolecular halenium ion transfer from one alkene to another can lead to rapid racemization, unless the nucleophilic trapping event is kinetically more competitive (Scheme 27). The occurrence of rapid, degenerate alkene-to-alkene halenium ion transfer processes was first demonstrated by Brown and co-workers, who studied adamantylidene adamantane and its corresponding (isolable) bromiranium or iodiranium ions.58,25d Computational studies support the notion that this process occurs via a low barrier, associative displacement at the halogen.58a, 59 Related alkene-to-alkene transfer processes have also been identified for thiiranium and seleniranium ions, similarly complicating the enantioselective additions of chalcogen electrophiles onto alkenes.60
Scheme 27.

Alkene-to-alkene transfer illustrated for ethylene and its corresponding bromiranium ion.
The operation of alkene-to-alkene transfer as a racemization mechanism under catalytically-relevant conditions has been probed by Denmark and co-workers. 61 Acetolysis experiments showed that the chiral bromiranium ion derived from enantioenriched bromo tosylate 24 can be captured with high enantiospecificity in the absence of alkenes.62 However, the inclusion of (E)-4-octene 26 as an additive severely eroded the selectivity (Scheme 28). The extent of erosion depends on the concentrations of the alkene and the nucleophile, as well as the identity of the counterion. Under similar reaction conditions, the analogous chloriranium ion from 25 is trapped with complete enantiospecificity, even in the presence of (E)-4-octene 26, implying that alkene-to-alkene transfer is inherently less facile with chloriranium ions.
Scheme 28.

Erosion of enantiospecificity in acetolysis from alkene-to-alkene transfer. HFIP = hexafluoroisopropanol, Tf = trifluoromethanesulfonyl, Ts = 4-toluenesulfonyl, es = (eeproduct/eestarting material) × 100%.
As alluded to in Section 2.3.1, fast racemization of chiral haliranium ions by alkene-to-alkene transfer is not necessarily an impediment to obtaining high enantioselectivities. In fact, it can actually be an advantage in cases for which the nucleophilic trapping step is enantiodetermining, as a concentration build up of the “undesired” (i.e., slower reacting) haliranium ion is avoided during the kinetic resolution process.
2.4. A Product Racemization Problem?
A less obvious source of imperfect enantioselectivity in alkene dihalogenation is that potentially arising from (partial) racemization of the vicinal dihalide products via a Type 1 dyotropic rearrangement.63 This process, first observed as the mutarotation of 5α,6β-dibromocholestane (the dibromination product of cholest-5-ene) by Grob and Winstein in 1952,64 is ascribed to a concerted pericyclic process in which the two halogen atoms migrate simultaneously and intramolecularly, with inversion of configuration at both carbon centers. In the first example of the racemization of an enantiopure, acyclic vicinal dibromide via a dyotropic rearrangement, Braddock, Schleyer and co-workers demonstrated that (R,R)- and (S,S)-1,2-dibromo-1,2-diphenylethane 27 racemize stereospecifically in refluxing benzene, without any crossover to the meso-diastereomer (Scheme 29).65
Scheme 29.

Racemization of an enantioenriched, acyclic vicinal dibromide via a Type 1 dyotropic rearrangement.
Although the elevated temperatures required for such dyotropic racemizations are not likely to render this a cause for concern under the conditions of an enantioselective dihalogenation process, strong heating of an enantioenriched vicinal dihalide post reaction (as during a hot recrystallization or drying process) should be carried out with caution. Particularly troubling is an observation that the equilibration of 1,2-dibromo-3-tert-butylcyclohexane diastereomers has been found to occur at the temperature of the injection block of a gas chromatograph66 – something that current practitioners should be aware of when relying upon chiral stationary phase GC methods to assess enantiomeric ratios!
2.5. The Diastereocontrol Problem
Unrelated to enantioselectivity, but nevertheless a challenging problem in stereoselective alkene dihalogenation, is the issue of control over the internal (or “simple”) diastereoselectivity of vicinal dihalide formation. As reactions proceeding via halide ion attack upon haliranium ion or alkene-dihalogen π complexes are mechanistically constrained to be anti-diastereospecific processes, access to diastereomeric dihalide products typically necessitates inversion of the relative configuration of the alkene starting material. For example, an anti-dihalogenation of a (Z)-alkene would furnish the same diastereomer as a syn-dihalogenation of the (E)-alkene (Scheme 30). However, the option of diastereodivergent dihalogenation from a single alkene geometry is clearly preferable and, at any rate, the (E)-isomers of cyclic alkenes are inaccessible for ring sizes below eight. Although Denmark, Cresswell and Eey have recently achieved the first catalytic, syn-stereospecific dichlorination of alkenes (see Section 4.4.2),52 an enantioselective variant of this reaction, as well as an analogous syn-dibromination process, remain elusive.67
Scheme 30.

Correlation between alkene geometry and vicinal dihalide relative configuration for anti- and syn-selective dihalogenations.
3. Mechanistic Classification of Alkene Dihalogenations
As should now be clear from the preceding discussion, dihalogenation reactions proceeding via halide ion attack upon haliranium ion or alkene-dihalogen π-complex intermediates present numerous challenges in terms of controlling the absolute configuration of the vicinal dihalide products. These include: (1) control of regioselectivity in the nucleophilic trapping with halide ion, (2) difficulties in transmitting stereochemical influence from a catalyst covalently bound to the halogen atom, and (3) potential for alkene-to-alkene halenium ion transfer processes causing racemization of enantioenriched haliranium ions. Though a separate issue, the control of relative configuration (i.e., internal diastereoselectivity) is also limited, as the reactions are stereoelectronically mandated to deliver anti-dihalogenated products, with no general means of overturning this selectivity.
In this Section, a variety of alternative mechanistic scenarios, previously suggested to be operative for main group element or metal-based halogenating reagents, are presented that may offer conceptually distinct strategies for stereoselective alkene dihalogenation. It must be emphasized, however, that a number of these mechanistic proposals are speculative, and some of them lack rigorous experimental or theoretical support. This is clearly an issue that needs to be addressed before one can rationally utilize these types of halogenating agents in enantioselective dihalogenation protocols.
3.1. Type I Dihalogenation
Type I dihalogenations are defined as those reactions involving halide ion attack upon haliranium ions (or possibly alkene-“halogen” π-complexes44) as the only electrophilic intermediates [Scheme 31, arbitrarily depicting an (E)-alkene]. This is of course the prototypical mechanistic manifold for (ionic) dihalogenations of olefins with the molecular dihalogens, and is so far the only pathway which has been exploited/invoked in existing (highly) enantioselective dihalogenations (see Section 1). Of course, the simplistic depiction in Scheme 31 (with generalized halenium-transfer reagents of the form X–Y) belies the mechanistic complexity encountered in ionic additions of the molecular dihalogens themselves; the reactions of olefins with Br2 in particular have been the subject of intense experimental and theoretical scrutiny since the late 1960s.25 Although beyond the scope of this Review, the detailed mechanisms of such processes have received extensive coverage elsewhere,23 including discussions of the role of the solvent and dihalogen/halide ion concentrations on the reaction pathway. The role of alkene-X2 π-complexes as obligatory intermediates has also been recognized.25c
Scheme 31.

A Type I dihalogenation process. Y = nucleofuge.
An additional complication in this type of dihalogenation is the extent of halogen atom bridging in the haliranium ion intermediate, and whether or not such intermediates are better formulated as β-halo carbocations. 68, 69 As a generalization, cation-stabilizing substituents (e.g., gem-dialkyl substitution, aryl groups) at one end of the halonium ion, high dielectric solvents, or chlorine as the halogen are all factors which conspire to reduce the degree of halogen bridging. 70 This has obvious implications for both reaction stereoselectivity and -specificity, with weakly bridged haliranium ions typically resulting in non-stereospecific dihalogenations to give anti/syn dihalide mixtures.23 In the dichlorinations of some conjugated alkene substrates, for example, imperfect anti-diastereoselectivity or even predominant syn-addition can occur,71 as in the reaction of (E)-stilbene 28 with molecular Cl2 (Scheme 32).71d However, the interpretation of such results as being the sole consequence of β-chloro carbocations should be made with caution, as the precise reaction conditions and concentrations have not always been specified, and the work of Poutsma has shown that certain alkenes at high concentrations can initiate radical chain additions of Cl2.24
Scheme 32.

Predominant syn-dichlorination in the reaction of (E)-stilbene 28 with molecular Cl2.
For the purposes of this Review, the generation of open β-halo carbocations as the first formed intermediates will also be referred to as a Type I dihalogenation reaction, regardless of the degree of anti-stereoselectivity or stereospecificity.
3.2. Type II Dihalogenation
In Type II dihalogenations, the alkene substrate is activated not by a halenium ion, X+, but by either an electrophilic metal or main group electrophile, via a π-complex or -iranium ion (note that “M” shall be used to denote either a metal or main group element). Outer sphere attack of a halide ion, X−, on such species results in anti-stereospecific addition of the elements of M–X across the alkene, this being the defining feature of a Type II dihalogenation. On the contrary, strained alkenes (e.g., norbornene) constitute potential exceptions to the anti-addition “rule”, favoring syn-addition pathways with certain metal salts that otherwise prefer anti-addition [e.g. mercury(II) or thallium(III) salts]; 72 these special cases fall outside of the Type II classification (see Type III mechanism, vide infra). Following an anti-addition of M–X across the olefin, the final stage of a Type II dihalogenation involves reductive elimination (with respect to M) to expel the reduced M species and deliver the vicinal dihalide product – a process which may proceed via a number of different pathways and with either retention or inversion of configuration (“Type IIret” or “Type IIinv”, respectively) [Scheme 33, arbitrarily depicting an (E)-alkene]. If the M center is not already high-valent prior to M–X addition to the alkene, an oxidative activation of M may be necessary before reductive elimination can occur. 73 Crucially, the stereochemical course of the reductive elimination dictates the overall stereochemical course of the dihalogenation, with Type IIret reactions resulting in anti-dihalogenation and Type IIinv reactions leading to syn-dihalogenation. Although a Type IIret process may potentially involve a haliranium ion intermediate “downstream” in the mechanism (by anchimeric assistance from the β-halogen74 to expel the M nucleofuge75,76), such halogenations would not be classified as Type I reactions because of the other electrophilic intermediates, featuring the metal or main group activator, which are actively involved. A third scenario (not depicted in Scheme 33) is also conceivable in which the final C–X bond formation occurs via a carbocation or carbon-centered radical – derived from C–M bond heterolysis or homolysis, respectively – which could potentially lead to non-stereospecific dihalogenation pathways.77
Scheme 33.

A Type II dihalogenation mechanism.
Examples of proposed Type IIret mechanisms include the anti-selective dichlorination of alkenes with PCl578 (Section 4.3.2) and the anti-selective dibrominations of alkenes with CuBr2 (Section 5.7.1).79 However, in neither of these processes is a Type IIret mechanism substantiated by experimental evidence (e.g., detection or isolation of intermediates), and so to the best of our knowledge there are currently no unambiguous cases of Type IIret alkene dihalogenation. There are however several other metal- or main group element-mediated alkene difunctionalization reactions, 80 including some halofunctionalizations,81 which share similar elementary steps to a Type II dihalogenation, such as the anti-addition of a metal/main group electrophile (M) and nucleophile (Nu) across an olefinic double bond. Furthermore, although rare, anti-chlorometalation reactions of alkenes are on record,82 and both stereoretentive76, 83 and invertive (SN2-like)76, 84 oxidatively-induced reductive eliminations of alkylmetal halide intermediates to furnish C(sp3)–halogen bonds are well precedented.85
A more clear-cut example of a Type IIinv process is the selenium-catalyzed, syn-dichlorination of olefins developed by Denmark et al.,52 for which the proposed catalytic cycle is comprised of steps which are all known stoichiometrically and are of well established stereochemical course (see Section 4.4.2).
3.3. Type III Dihalogenation
Type III dihalogenation is broadly similar to Type II dihalogenation, with the exception being that the initial addition of the elements of M–X across the alkene double bond proceeds with syn-stereospecificity. Such syn-additions can be formulated as migratory insertions of the olefin into the M–X bond of the metal (or metalloid) halide, proceeding in a concerted fashion via a 4-center transition state. Just as for Type II dihalogenations, the ensuing reductive elimination (with respect to M) may proceed via retentive or invertive pathways, leading to syn- or anti-dihalogenation, respectively [Scheme 34, arbitrarily depicting an (E)-alkene]. As before, a third type of reductive elimination may involve final C–X bond formation via a carbocation or carbon-centered radical, arising from C–M bond heterolysis or homolysis, respectively.77
Scheme 34.

A Type III dihalogenation mechanism. TS = transition state.
The proposal of syn-halometalation in Type III dihalogenations deserves further comment, particularly as migratory insertions of alkenes into M–X bonds are thermodynamically unfavorable for transition metals. It is in fact the microscopic reverse of this process – β-halide elimination – that is normally observed, accounting for the notorious instability of β-haloalkyl transition metal complexes.86 The driving force for β-halide elimination has largely been attributed to the differences between metal-carbon and metal-halide bond strengths,87 being most exothermic for “hard” early transition metals88 and relatively less so for “softer” late metals. 89 Of course, endergonic elementary steps embedded in multistep sequences can still lead to productive reactions if they are offset by subsequent, exergonic steps, and halometalations of alkenes have been proposed in several catalytic cycles. For instance, the chloropalladation of π-unsaturates 90 is well precedented, primarily for alkynes 91 but also with olefins,82, 92 with the stereochemical course of the insertion (syn or anti) being dependent on the precise reaction conditions (e.g., halide ion concentration). In contrast to transition metals, β-halometalations of alkenes with main group halides are well known (e.g., PhSeCl), and although many of these additions are anti-stereospecific processes, the (ionic) β-chlorotelluration of alkenes has been shown to proceed in a syn-stereospecific fashion.93
As one might gather from the above discussion regarding the instability of many β-haloalkyl transition metal complexes, which often precludes their isolation,86 direct experimental evidence for Type III dihalogenations is lacking, although such mechanisms have been invoked to account for both anti- and syn-selective dihalogenations of olefins mediated by high-valent metal or metalloid halides. For example, a Type IIIret mechanism involving syn-chlorometalation of an alkene followed by stereoretentive reductive elimination has been suggested by Sharpless and co-workers to account for the formation of syn-dichlorination by-products from the oxidation of alkenes with chromyl chloride (CrO2Cl2) at low temperature.94 Additionally, a Type IIIinv mechanism has been tentatively proposed for manganese(VII)-mediated alkene anti-dichlorinations, though it is not even clear in these cases that a manganese chloride species is the active chlorinating species (see Section 5.3.1).
3.4. Type IV Dihalogenation
Type IV dihalogenation involves a concerted delivery of two halogen atoms from a metal or main group element center to an alkene in a syn-stereospecific fashion, via a 5-membered transition state [Scheme 35, arbitrarily depicting an (E)-alkene].
Scheme 35.

A Type IV dihalogenation mechanism.
Originally invoked by Barton and Miller to account for the syn-selective dichlorination of cholesterol benzoate with PhICl2 under anhydrous conditions,95 this mechanism has since been discredited, despite receiving some theoretical support96 (see Section 4.5.1). A Type IV mechanism has also been suggested to be operative for syn-stereospecific dichlorinations of olefins by high-valent antimony and molybdenum chlorides (see Sections 4.3.3 and 5.2.2, respectively). Sharpless has also pointed out that a Type IV dichlorination process cannot be excluded as an explanation for the formation of syn-dichlorination products in the oxidation of alkenes by CrO2Cl2, although he favored a Type IIIret mechanism (vide supra).94 However, Nelson and co-workers have concluded that a Type IV dichlorination is indeed more likely for the latter transformation, based on linear correlations between log krel and either alkene ionization potentials or HOMO energies (i.e., showing that only electronic and not steric effects are important).97
In terms of product stereostructure, the Type IV mechanism is indistinguishable from a Type IIinv or Type IIIret dihalogenation for which obvious parallels can be drawn to the syn-dihydroxylation of alkenes by OsO4. In that case, both a [2+2] cycloaddition mechanism (analogous to the Type IIIret pathway) and a [3+2] cycloaddition (analogous to the Type IV pathway) have been advocated.98 However, compelling support for the [3+2] pathway from 12C/13C kinetic isotope effects99 and quantum chemical DFT and ab initio calculations100 has now firmly established that the [3+2], and not the [2+2], cycloaddition mechanism is in operation with OsO4.
3.5. Type V Dihalogenation
Type V dihalogenation involves halogen atom transfer to the alkene substrate to generate a β-halo radical, which can react further with a halogen atom donor (X–Y) to give a vicinal dihalide [Scheme 36, arbitrarily depicting an (E)-alkene]. If Y = X (or if Y can become X by extrusion of a neutral molecule), a radical chain reaction may ensue (e.g., as for Cl2, SO2Cl2, PhICl2, vide infra), although non-chain mechanisms may be operative in some metal-mediated or -catalyzed processes (e.g., for low-valent manganese, copper, and ruthenium halides, vide infra). The conditions under which molecular Cl2 undergoes radical chain additions to alkenes have been thoroughly studied by Poutsma, with high concentrations of certain alkene substrates able to initiate radical processes even in the dark under a nitrogen atmosphere.24 In terms of stereochemical course, the formation of vicinal dihalides by Type V mechanisms is generally non-stereospecific,24, 101 with the relative configurations of the products being determined by steric factors.
Scheme 36.

A Type V dihalogenation mechanism.
4. Alkene Dihalogenations with Main Group Halides as Reagents or Catalysts
To fully realize the largely untapped potential of main group or metal halides as reagents or catalysts for catalytic alkene dihalogenation protocols, some knowledge of the stoichiometric reactivity of these compounds with alkenes is required, and particularly the mechanistic and stereochemical features of such reactions. This aspect shall be the focus of the remainder of this Review, with main group halides covered in this Section and transition metal halides discussed in Section 5. Although most main group or metal halides have been used in stoichiometric amounts, there are clearly opportunities to render this chemistry catalytic in such species.52
4.1. Group 13 Halides
Uemura and co-workers have reported that the reaction of alkenes with molten, heterogeneous TlCl3•H2O in refluxing CCl4 gives vicinal dichlorides resulting from anti-addition in low yield, in addition to substantial amounts of hydrochlorinated products.102 For example, the reaction of excess (Z)-4-octene 31 with TlCl3•H2O gave an 80:20 mixture of anti- and syn-dichlorination products 32 and 33, respectively, in 5% combined yield (w.r.t. TlCl3), in addition to several hydrochlorination products in 33% yield (Scheme 37).
Scheme 37.

Low-yielding dichlorination reaction of (Z)-4-octene 31 with TlCl3•H2O.
However, the reaction could be rendered catalytic in TlCl3•H2O using CuCl2•2H2O as a co-catalyst and O2 as the terminal oxidant, with control experiments showing no reaction in the absence of TlCl3•H2O. Under these conditions, cyclohexene 34 gave anti-dichloride 35 in 50% yield (w.r.t. 34) and 98:2 dr, as well as cyclohexyl chloride 36 in 17% yield (Scheme 38). Although the catalyst loadings are very high – to such an extent that the reaction is not obviously catalytic based on the product yield – this process does constitute a very rare example of metal-catalyzed alkene dihalogenation.
Scheme 38.

A thallium-catalyzed alkene dichlorination?
Little is known regarding the mechanism of alkene dichlorinations with TlCl3•H2O, although product distributions from norbornene103 and norbornadiene104 are suggestive of Type I processes involving chloriranium ion intermediates.
4.2. Group 14 Halides
For Group 14 halides in the +4 oxidation state, only lead(IV) chloride (PbCl4) possesses sufficient oxidizing power to dichlorinate alkenes,105 although it’s high oxidation potential and Lewis acidity, coupled with the severe cumulative toxicity of lead compounds, renders this a particularly unsavory reagent. A yellow, oily liquid in its pure form, PbCl4 can be prepared in situ from Pb(OAc)4 and excess dry HCl, although it thermally decomposes to PbCl2 and Cl2 above 0 °C. However, at cryogenic temperatures, in situ generated PbCl4 reacts with an excess of several alkenes (cyclohexene, 4-t-butylcyclohexene, cycloheptene, or Δ2-cholestene) to afford the corresponding anti-dichlorides in quantitative yield w.r.t. PbCl4 (Scheme 39).106 Unfortunately, a Lewis acid-catalyzed hydrochlorination with the excess HCl is a major side reaction (hence the requirement for excess alkene), and this dominates with trisubstituted alkenes (e.g., 1-methyl-4-t-butyl-cyclohexene) or alkenes which are slower to undergo 1,2-addition [e.g., (Z)-cyclooctene]. Little mechanistic information for this dichlorination is available, although product distributions for the reaction of PbCl4 with norbornene,103 norbornadiene,104 and cyclooctadienes 107 as mechanistic probes are suggestive of an ionic mechanism that does not involve molecular Cl2.
Scheme 39.

The dichlorination of excess cyclohexene 34 with in situ generated PbCl4, accompanied by extensive hydrochlorination.
Although difluorinations are beyond the scope of this Review, it is interesting to note in passing that the difluorination of alkenes with “PbF4” has also been reported.108 However, an early claim of the difluorination of stilbene with “PbF4” [prepared in situ from Pb(OAc)4 and HF], 109 was later found to be erroneous, with the purported vicinal difluoride product reassigned as a geminal difluoride.110 Furthermore, the active fluorinating agent generated from Pb(OAc)4 and HF has been proposed as Pb(OAc)2F2 and not PbF4.110 Bowers and coworkers have reported the syn-selective difluorination of pregnenolone acetate 37 using Pb(OAc)4-HF to give the 5α,6α-difluoride 38 in 27% yield (Scheme 40).111 To account for the stereochemical course, a concerted transfer of both fluorine atoms from PbF4 to the alkene (i.e., a Type IV dihalogenation) was suggested, although a radical (or even carbocation) based mechanism might also be consistent with this outcome (see Section 4.5.1 for a similar syn-selective dichlorination of a steroidal alkene). A mechanistic study on the reaction of Pb(OAc)4-HF with norbornene favored a syn-fluorometalation process (i.e., a Type III addition) followed by heterolytic cleavage of the C–Pb bond to furnish a carbocation intermediate (or homolytic cleavage and single electron transfer oxidation).112
Scheme 40.

Difluorination of pregnenolone acetate 37 using Pb(OAc)4-HF.
4.3. Group 15 Halides
4.3.1. Nitrogen
Kovacic and co-workers have studied the use of nitrogen trichloride, (NCl3) as a reagent for the dichlorination of alkenes in detail.113 Although pure NCl3 is an explosive yellow oil, it can be freshly prepared as a solution in chlorinated organic solvents via the reaction of NH4Cl with Ca(ClO)2, as described in an Organic Syntheses procedure.114 When used in this form, it is stable for several days at 0–5 °C, and gives exceptionally clean dichlorinations of simple (unfunctionalized) alkenes such as cyclohexene 34, albeit with the alkene used in excess (Scheme 41). The reaction probably proceeds via a radical mechanism,113b,115 although a competing ionic pathway may be operative for certain alkene substrates,116 much as for reactions with Cl2.24
Scheme 41.

The dichlorination of excess cyclohexene 34 with NCl3.
Other alkene dihalogenations are reliant on more stable N–X reagents such as N-haloimides and related compounds, in which these X+ equivalents are combined with a suitable source of nucleophilic halide ion. The halide can either be generated in situ by reduction of one equivalent of the N–X reagent with an added reductant (Section 2.1),35,36 or it can be introduced via a different reagent entirely [e.g., R4NBr,34 LiBr,117 BrTi(Oi-Pr)315].
4.3.2. Phosphorus
Although the vicinal dichlorination of alkenes with PCl5 has long been known,118 this method has rarely been applied in synthesis and has been subjected to only limited mechanistic scrutiny.78 In a representative example of this reaction, (E)-stilbene 28 reacts with an excess of PCl5 in refluxing chlorobenzene to give anti-dichloride 30 in 85% yield and syn-dichloride 29 in 13% yield (Scheme 42).
Scheme 42.

The dichlorination of (E)-stilbene 28 with PCl5.
Based on the known reaction of PCl5 with alkenes at low temperature to furnish β-chloro phosphinylated adducts,119 it has been suggested that the dichlorination may proceed via initial addition of PCl5 (in the form [PCl4]+[PCl6]−) to the alkene via a bridged phosphonium intermediate, followed by a stereoretentive SNi-type conversion of the C–PCl3 bond to a C–Cl bond (i.e., a Type IIret mechanism).78 However, the intermediacy of β-chloro phosphinylated adducts at the elevated temperatures employed was not demonstrated, nor was the decomposition of such species to vicinal dichlorides. Although anti-selective dichlorination of cyclohexene was taken as support for the proposed (ionic) mechanism, this result could equally well be ascribed to a radical process.120 It was however suggested that the stereochemical course of the reaction with (E)-stilbene 28 could be the result of parallel ionic and radical processes.
To gain further mechanistic insight, Uemura and coworkers examined the reactions of norbornene and 1,5-cyclooctadiene with PCl5, as these substrates are known to give distinct product distributions in ionic versus radical dichlorinations.121 With 1,5-cyclooctadiene 39, for example, the reaction with PCl5 in CHCl3 (ε = 4.70) at 65 °C gave almost exclusively the “ionic” dichloride 40, whereas the reaction in non-polar CCl4 (ε = 2.23) at a slightly higher temperature (76 °C) delivered predominantly the “radical” dichloride 41 (Scheme 43). On the basis that PCl5 is a covalent monomer in non-polar media such as benzene and CCl4,122 it was concluded that any “ionic” dichlorides produced in these solvents are unlikely to arise via a Type IIret mechanism,78 and that a Type I process may be in operation. As PCl5 is known to decompose slightly to PCl3 and Cl2 near 100 °C, molecular Cl2 could not be excluded as a contributor to the “ionic” pathway.
Scheme 43.

The dichlorination of 1,5-cyclooctadiene 39 with PCl5.
4.3.3. Antimony
On the basis of precedent for the use of SbCl5 as a catalyst for the dichlorination of alkenes with Cl2, 123 as well as an example of its use as a stoichiometric reagent with 1,2-dibromoethylene as the substrate,124 Uemura and co-workers have performed a detailed study on the dichlorination of alkenes and dienes with SbCl5 in chlorocarbon solvents.125,126 Related studies have also been carried out by Heasley and coworkers, 127 in addition to other brief investigations on the use of liquid SO2 as a solvent128 and 1,3-butadiene as a substrate.129 One of the most remarkable features of this reaction is the production of vicinal dichlorides resulting from a stereospecific130 syn-dichlorination of the alkene. For example, addition of excess (E)-2-butene 42 (2.8 equiv) to a solution of SbCl5 in CCl4 at 73 °C gave an 82:18 mixture of dichlorides 43 and 44, resulting from syn- and anti-selective addition respectively, in 96% combined yield (w.r.t. SbCl5). The diastereomeric alkene (Z)-45 behaved almost identically (i.e., 84:16 syn:anti addition), confirming the stereospecific nature of the process (Scheme 44). Control experiments verified that the syn-selectivity was the result of kinetic control. However, no reaction occurred with electron-deficient alkenes such as acrylonitrile, ethyl maleate, ethyl fumarate or tetrachloroethylene, and with styrene or ethyl vinyl ether, vigorous reactions ensued to give polymeric material.
Scheme 44.

Syn-stereospecific dichlorination of alkenes using SbCl5.
The syn-selectivity was found to be sensitive to both the polarity of the chlorocarbon solvent and the reaction temperature, with higher syn-selectivities obtained in more polar media at elevated temperatures. Thus, the ratios of syn:anti addition for the dichlorination of cyclohexene in 1,2-dichloroethane (ε = 10.37) at 83 °C and in CCl4 (ε = 2.23) at 30 °C were 89:11 and 60:40, respectively. The speciation of SbCl5 in solution depends on the polarity of the solvent: in MeCN (ε = 37.5), conductivity data indicate that SbCl5 is in equilibrium with [SbCl4]+[SbCl6]− 48,131 whereas in CCl4 (ε = 2.23) the conductivity data132 and vibrational spectra133 are consistent with molecular SbCl5 46 (of D3h symmetry).134 Uemura et al. have suggested that solutions of SbCl5 in chlorocarbons may comprise an equilibrium mixture of SbCl5 46, Sb2Cl10 47,135 and [SbCl4]+[SbCl6]− 48 (Scheme 45).125
Scheme 45.

Proposed equilibria in chlorocarbon solutions of SbCl5.
To rationalize the syn-stereospecificity of the dichlorination, a Type IV mechanism involving concerted transfer of both chlorine atoms from SbCl5 46 (or possibly Sb2Cl10 47) to the alkene via a 5-membered cyclic transition state XIX has been proposed.125 A Type IIIret mechanism involving syn-chlorometalation followed by stereoretentive reductive elimination (w.r.t. Sb) was seemingly not considered and should not be ruled out, especially as TeCl4 (tellurium being the Group 16 neighbor of antimony) is known to β-chlorotellurate olefins in a syn-stereospecific fashion.93 The formation of dichlorides resulting from anti-addition was ascribed to ion pair [SbCl4]+[SbCl6]− 48 as the reactive chlorinating species,136 which may promote an ionic mechanism via β-chloro carbenium ion intermediates XX (Scheme 46). An interaction of the chlorine atom in intermediates XX with antimony, which prevents bridging to give a chloriranium ion, was invoked to explain the formation of 1,1- and 1,3-dichloride products from cyclopentene (vide infra).125,127 In support of the mechanistic proposal in Scheme 46, the syn:anti selectivity decreased considerably upon addition of a strong Lewis acid (AlCl3 or SbF3) to the SbCl5, possibly due to the formation of [SbCl4]+[MXnCl]− ion pair complexes. Additionally, as the ionization SbCl5 → [SbCl4]+[SbCl6]− is likely to be exothermic (as for PCl5122), the amount of [SbCl4]+[SbCl6]− 48 would be expected to decrease as the temperature is raised, and this might account for the fact that syn-selectivity increases at higher temperatures. The effect of solvent – in which syn-selectivity increases in more polar solvents – was rationalized in terms of the different Ingold-Hughes charge-type classifications137 of the syn- and anti-dichlorination pathways. Specifically, a more polar solvent should accelerate the syn-addition (a reaction between two uncharged species) because charge separation is greater in the transition state (TS) than the starting materials, and the former will be relatively more stabilized. Conversely, a polar solvent should retard the anti-addition (a reaction between an uncharged and a charged species) because the charge becomes more dispersed in the TS, relative to the starting species.
Scheme 46.

Proposed parallel mechanisms for the formation of syn- and anti-dichlorides, respectively, arbitrarily illustrated for an (E)-configured alkene.
For certain alkenes, such as cyclopentene,125,127 norbornene,103,125 or norbornadiene,104 rearrangement products consistent with carbocation intermediates of the form XX are produced. For instance, with cyclopentene, both 1,1- and 1,3-dichlorides (the products of 1,2-hydride shifts127) are obtained in addition to the 1,2-anti-dichloride product, with none of the 1,2-syn-dichloride detected.125 This, and other unusual results,103,127 have been explained in terms of carbocation stability (according to the Hammond postulate 138 ), in which alkenes able to generate more stable carbocations are more prone to ionic reaction pathways. An alternative explanation for the anomalous reactivity of cyclopentene and norbornene with SbCl5 is the unfavorable steric interaction expected between SbCl5 and the proximal methylene group in a concerted syn-addition to the alkene substrate (Figure 4).125,139
Figure 4.
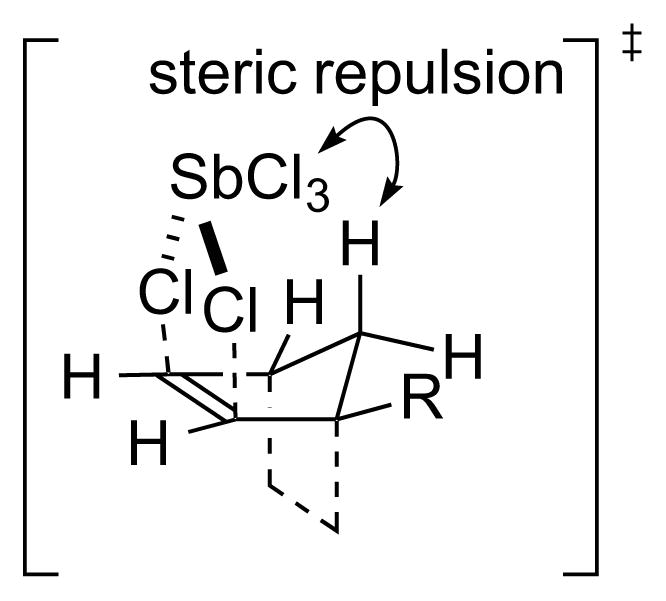
Anticipated destabilizing steric interaction in the concerted addition of SbCl5 to either cyclopentene or norbornene.
Uemura and co-workers have also extended the use of SbCl5 to the syn-selective dichlorination of alkynes, albeit with low yields (20–30%).140
4.4. Group 16 Halides
4.4.1. Sulfur
The reaction of sulfuryl chloride (SO2Cl2) with alkenes to afford vicinal dichlorides has long been known,113a,118, 141 and Kharasch and Brown have shown that these reactions can be initiated with organic peroxides.142 On this basis, it was proposed that the dichlorinations proceed via a radical chain mechanism (i.e., Type V dichlorination), in which the initiation step involves the generation of chlorosulfuryl radicals (·SO2Cl) from SO2Cl2. These radicals may lose SO2 to release Cl· atoms, which will add to olefins to generate β-chloro radical intermediates XXI, although ·SO2Cl could potentially transfer chlorine to the olefin directly. With regards the latter possibility, the ·SO2Cl radical is thought to be at least partly responsible (along with Cl·) for hydrogen-abstraction in photoinitiated C–H chlorinations of hydrocarbons with SO2Cl2.143 In either case, chlorine transfer from another molecule of SO2Cl2 to the β-chloro radical XXI could then furnish the vicinal dichloride directly and regenerate the ·SO2Cl radical.142 Alternatively XXI may react with SO2Cl2 to furnish chlorosulfite esters XXII as primary products (which are isolable at lower temperatures) and these may decompose on heating to give vicinal dichlorides (Scheme 47).144 Typical side products of SO2Cl2 radical dichlorinations include higher chlorinated compounds from chlorine substitution along the alkyl chain,142 as well as symmetrical β-chloro-n-alkyl sulfones arising from direct attack of ·SO2Cl on the olefin.144 The vicinal dichlorination of alkynes with SO2Cl2 is also possible and is thought to follow a similar radical chain pathway.145
Scheme 47.

Dichlorination of alkenes with SO2Cl2 via a radical chain mechanism (with Cl· depicted as the chain-carrier).
Uemura and co-workers have also found that reactions of SO2Cl2 with alkenes via an ionic (possibly Type I) pathway are possible, as evidenced by the change in product distribution for the dichlorination of 1,5-cyclooctadiene 39 in the presence versus absence of 1,3-dinitrobenzene as a radical inhibitor.121 Furthermore, on the basis such that silica gel promotes electrophilic aromatic chlorinations with SO2Cl2, 146 a similar effect was noted for alkene dichlorinations, and the addition of silica gel strongly promotes the ionic pathway (Scheme 48). Comparable results are also obtained with norbornene as a mechanistic probe for ionic versus radical dichlorination. These particular results have interesting implications for the development of Brønsted/Lewis acid or Lewis base-catalyzed alkene dichlorinations with SO2Cl2 as a dihalogen equivalent, as opposed to the less stable and non-commercially available PhICl2 (which has already been applied in Brønsted acid-catalyzed39 and Lewis base-catalyzed12 dichlorinations). Notably, Lewis acid catalysts have been used to promote the ionic chlorination of aromatic compounds with SO2Cl2 via X–S–X bond polarization.41
Scheme 48.

The dichlorination of 1,5-cyclooctadiene 39 with SO2Cl2. 1,3-DNB = 1,3-dinitrobenzene.
4.4.2. Selenium
Although SeCl4 is known to react with alkenes to produce bis(2-chloroalkyl)selenium dichlorides, 147 Uemura and co-workers have shown that this reagent is capable of dichlorinating various alkenes in CCl4, including cyclohexene,103 norbornene,103 norbornadiene,104 (Z)-cyclooctene,126a and cyclooctadienes.107 Control experiments indicated that organoselenium compounds were not involved as intermediates, and product distributions from the aforementioned alkenes were most consistent with a Type I ionic mechanism, as opposed to a radical (Type V) dichlorination process.
More recently, Denmark and co-workers reported a selenium-catalyzed alkene dichlorination reaction that also constitutes the first catalytic, syn-stereospecific dichlorination of alkenes.52 On the basis of known anti-stereospecific β-chloroselenylation of alkenes with PhSeCl3,148 as well as the invertive nucleophilic displacement of the high-valent selenium(IV) moiety149 by chloride ions in such adducts to afford syn-dichlorides, 150 it was surmised that a direct alkene syn-dichlorination that is catalytic in selenium may be possible. The key challenge in formulating such a catalytic cycle was the identification of a stoichiometric oxidant to selectively reoxidize Se(II) to Se(IV), but does not interfere with the alkene substrate, nor oxidize chloride ions to Cl2. Thus, with diphenyl diselenide (PhSeSePh) as the precatalyst, benzyltriethylammonium chloride (BnEt3NCl) as the chloride source, and an N-fluoropyridinium salt 49 as the oxidant, a wide variety of functionalized 1,2-disubstituted (E)- and (Z)-configured alkenes, including simple allylic alcohols, deliver syn-dichlorides with exquisite stereocontrol (Scheme 49).
Scheme 49.

Selenium-catalyzed syn-dichlorination of alkenes.
Chlorotrimethylsilane (Me3SiCl) proved crucial as an additive to facilitate turnover, presumably serving as a scavenger for fluoride ions generated from oxidant 49. Additionally, the inclusion of 2,6-lutidine N-oxide 50 as an (optional) additive was found to enhance the rate of the reactions (but not the yields or selectivities), although it actually proved deleterious in some cases and was omitted (e.g., for allylic alcohol substrates).
A proposed catalytic cycle for these reactions – which are believed to follow a Type IIinv dihalogenation mechanism – is illustrated in Scheme 50 (without the 2,6-lutidine N-oxide 50). Following precatalyst activation under the oxidative conditions (i.e., PhSeSePh → PhSeCl3), the first step is thought to be addition of PhSeCl3 to the alkene substrate to generate a seleniranium ion intermediate XXIII. Nucleophilic ring-opening by Cl− ion then gives a (β-chloroalkyl)phenylselenium dichloride XXIV,148 following which stereoinvertive displacement of the Se(IV) nucleofuge by Cl− ion delivers the vicinal dichloride product, in an overall syn-stereospecific addition process. Oxidation of PhSeCl to PhSeCl3 in the presence of oxidant 49 and Me3SiCl completes the catalytic cycle.
Scheme 50.

Proposed catalytic cycle for the selenium-catalyzed syn-dichlorination of alkenes.
4.5. Group 17 Halides (Except Dihalogens)
4.5.1. Iodine
Dichloroiodobenzene (PhICl2) – a yellow crystalline solid originally isolated in 1886 by Willgerodt,151 and the first example of a hypervalent iodine(III) reagent152 – is renowned for its ability to cleanly dichlorinate olefins under mild conditions. Although not commercially available due to its instability (being storable at −20 °C for about two weeks), it can be prepared by the treatment of iodobenzene with chlorine gas (via an Organic Syntheses procedure153) or, more conveniently, by the in situ generation of Cl2 using strong oxidants with chloride sources.154 However, PhICl2 and other ArylICl2 reagents undergo rapid and reversible dissociation to Cl2 and the corresponding aryl iodide in polar solvents (e.g., AcOH, MeNO2), 155 and even do so in non-polar solvents (e.g., CCl4) if a strong acid catalyst is present (e.g., CF3CO2H). 156 Moreover, certain Lewis basic ortho-substituents on the aryl ring of ArylICl2 reagents greatly accelerate this decomposition process (in AcOH), with 2-nitro and 2-carboxymethyl groups exerting the strongest effect.157
In general, dichlorinations of alkenes with PhICl2 in chlorocarbon solvents follow either a radical chain mechanism (Type V) or an ionic mechanism (Type I or Type II), depending on the reaction conditions.115,158,159 The radical chain mechanism (Scheme 51), originally proposed by Bloomfield, 160 can be initiated either thermally or photochemically (in the absence of radical inhibitors such as oxygen), and support for this mechanism comes from studies of certain alkenes as mechanistic probes for ionic versus radical dichlorination, including norbornene,24b,121,158,159 cyclooctene,126 cyclooctadienes,107,121 cyclopentadiene and 1,3-cyclohexadiene,115 vinylcyclopropanes,116 and others.159 By analogy to radical dichlorinations with SO2Cl2 (vide supra),142 it has been proposed that Cl· is the chain-carrying radical,158,159 although Ph(Cl)I· could also be the chain-carrier.159,171, 161 In possible support of the latter scenario, Ph(Cl)I· (as opposed to Cl·) is thought to be a competent hydrogen-abstractor in photoinitiated C–H chlorinations of hydrocarbons with PhICl2, based on comparison of product distributions to similar reactions with Cl2.162 A related question has arisen in ionic mechanisms with PhICl2 as to whether Cl− or Ph(Cl)I− is the active nucleophile (vide infra).115,159,163
Scheme 51.

Dichlorination of alkenes with PhICl2 via a radical chain mechanism (with Cl· depicted as the chain-carrier).
Regarding the stereochemical course of these radical dichlorinations, mixtures of anti- and syn-dichlorination products are frequently obtained, which for cyclic alkenes typically (though not always) favor the anti-diastereomer.121,126,158,164 High stereoselectivity can even be obtained with acyclic alkenes: β-methylstyrenes 51 and 52, for example, give 92:8 and 91:9 ratios of anti-53/syn-54 dichlorides, respectively, upon photoinitiated reaction with PhICl2 (Scheme 52).165 In all of these cases, the simple (internal) diastereoselectivity is explained on the basis of steric interactions between the β-chloro radical intermediate and PhICl2 as a very bulky chlorine atom donor.115,161,164,165
Scheme 52.

The stereoconvergent dichlorination of β-methylstyrenes (E)-51 and (Z)-52 with PhICl2.
For some cyclic alkenes, however, including 1,5-cyclooctadiene107,121 substituted norbornenes,166 and various steroidal alkenes (e.g., cholesterol benzoate),95,161, 167 syn-selective dichlorination can occur with PhICl2 under typical radical conditions. Barton and Miller originally rationalized this syn-addition pathway as being the result of a concerted, molecular addition mechanism involving synchronous transfer of both chlorine atoms from PhICl2 onto the double bond (i.e., a Type IV dihalogenation)95 – a process which has been calculated to be theoretically feasible.96 However, this proposal has since been discredited on the basis that the “anomalous” syn-selectivity is readily accommodated by a Type V radical process.24b,158,159 Specifically, in the case of cholesterol benzoate 55, attack of a Ph(Cl)I· radical may occur at the less hindered α-face of the alkene to afford two constitutionally isomeric β-chloro radical intermediates XXV and XXVI. As the axial methyl group in XXVI disfavors chlorine atom transfer by PhICl2 to the β-face of the C(5) radical, attack should preferentially take place on the α-face, affording syn-dichloride 57 as the major product. Conversely, chlorine transfer to the C(6) radical of XXV might be expected to occur with similar facility from both the α- and β-faces, delivering a mixture of syn-57 and anti-56 dichlorides (Scheme 53).
Scheme 53.

Mechanistic rationale for the syn-selective dichlorination of cholesterol benzoate 55 via a Type V radical chain pathway.
The ionic pathway for alkene dichlorination with PhICl2 proceeds when reactions are performed thermally in the presence of radical inhibitors (e.g., molecular oxygen), although these reactions tend to be very slow in (dry) chlorocarbon solvents in the absence of catalysts (vide infra), and can take several days to reach completion.115,158,159, 168 Typically these additions are highly anti-selective (at least in chlorocarbon media), and often more selective than Cl2 when direct comparisons have been made.115, 169, 170 Consequently, it has been suggested that Ph(Cl)I− rather than Cl− may be the active nucleophile that traps a chloriranium ion (or β-chloro carbocation) intermediate (i.e., Type I mechanism).115,159,163 However, a Type IIret mechanism proceeding via an iodiranium ion intermediate could also be envisioned, and this would be consistent with the observed anti-selectivity (Scheme 54). A Type IIinv mechanism has even been suggested to be (at least partly) operative in Lewis basic solvents like THF, or in chlorocarbon solvents in the present of DMSO as an additive, to account for unusually high proportions of syn-dichlorination [particularly from (Z)-alkenes] under these conditions (Scheme 54). 171 It is conceivable that THF or DMSO may serve to catalyze the SN2 displacement from the intermediate alkene-PhICl2 adduct by generating a cationic iodine(III) nucleofuge and free chloride ion.
Scheme 54.

Possible reaction pathways for the ionic dichlorination of alkenes with PhICl2.
Of potential relevance to catalysis is the fact that various additives greatly accelerate anti-selective ionic dichlorinations of olefins with PhICl2. For example, Cotter et al. have shown that trifluoroacetic acid (CF3CO2H) can catalyze the ionic dichlorination of alkenes by PhICl2, probably via hydrogen-bond-induced polarization of the Cl–I–Cl unit (Scheme 55).39,159 Provided that the alkene concentration is sufficiently high, this direct reaction of the olefin with the (activated) PhICl2 outpaces the acid-catalyzed decomposition of PhICl2 to molecular chlorine and iodobenzene.156 A Type I mechanism has been suggested to be operative in this case, but an (ionic) Type IIret process, in which the alkene attacks the activated PhICl2 moiety at iodine to give an intermediate iodiranium ion species, should not be excluded. In a related phenomenon, the addition of up to 1.0 equivalent of H2O promotes the ionic addition of PhICl2 to cholesterol benzoate in refluxing CHCl3, at the expense of the radical pathway.95 Although this rate enhancement was originally ascribed to a H2O-catalyzed dissociation of PhICl2 into PhI and Cl2, this was later disproved by other authors,155b and the generation of trace HCl (perhaps from the hydrolysis of PhICl2) has instead been suggested to catalyze the direct attack of the alkene onto PhICl2 (activated by Cl–I–Cl polarization as in Scheme 55).159
Scheme 55.

Trifluoroacetic acid-catalyzed alkene dichlorination with PhICl2 in CCl4.
In a potential example of Lewis base catalysis,13 the addition of pyridine is also found to greatly accelerate the ionic dichlorination of alkenes in chlorocarbon solvents such as CCl4,172 although the origin of the rate enhancement is unclear. Either N-chloropyridinium salt XXVII or PhICl2•py complex XXVIII could plausibly be the active chlorinating species in these reactions, or the pyridine may even be serving to catalyze the formation of molecular Cl2 (Scheme 56). An obvious parallel can be drawn to Nicolaou and co-worker’s enantioselective dichlorination protocol, which uses 4-Ph(C6H4)ICl2 as the chlorinating agent and (DHQ)2PHAL as a chiral amine Lewis base catalyst (Section 1).12 The fact that the enantioselectivity of their dichlorination shows a dependence on the nature of the aryl group of the iodoarene dichloride reagent supports the notion that PhICl2•amine complexes such as XXVIII may be competent chlorinating species.
Scheme 56.

Plausible active chlorinating agents in the pyridine-promoted dichlorination of alkenes with PhICl2.
The in situ generation of iodine(III)-based halogenating agents derived from PhI(OAc)2 and pyridinium halide salts has been reported by Lupton and co-workers, and alkene dichlorinations and dibrominations were briefly explored. 173 Whilst dibromination of (E)-β-methylstyrene 51 gave the anti-dibromide 58 exclusively, the analogous dichlorination afforded the anti-dichloride 53 in only 2:1 dr. An enantioselective variant of the dibromination of (E)-51 was also attempted using the chiral, non-racemic iodoxyarene 59, but only negligible enantioselectivity was obtained (51.5:48.5 er) (Scheme 57).
Scheme 57.

Dihalogenation of (E)-β-methylstyrene 51 with PhI(OAc)2 and Py•HX (X = Br or Cl), and attempted enantioselective dibromination with chiral, non-racemic iodoxyarene 59.
In contrast to alkene dichlorination reactions mediated by PhICl2, the corresponding difluorination reactions with iodoarene difluorides (ArIF2) are far less general. However, Hara, Yoneda and co-workers have reported that the combination of 4-iodotoluene difluoride (4-TolIF2) in CH2Cl2 with Et3N•5HF as a co-solvent enables the vicinal difluorination of (mostly terminal) alkenes in moderate to good yields.174 As the sole example of an internal alkene, cyclohexene derivative 60 gave syn-difluoride 61 in 55% yield as a single diastereomer, providing some insight into the reaction mechanism. It is proposed that HF serves to activate the 4-TolIF2 reagent via hydrogen-bonding (i.e., X–I–X polarization39), such that reaction with the alkene leads to iodiranium ion XXIX. Invertive ring-opening of XXIX by fluoride ion would give the anti-configured β-fluoro iodonium adduct XXX (examples of which have been isolated 175 ), and then SN2 displacement of the iodine(III) nucleofuge by a second fluoride ion would deliver the syn-difluorinated product (i.e., a Type IIinv dihalogenation pathway) (Scheme 58).
Scheme 58.

Vicinal difluorination of alkene 60 with 4-TolIF2 and Et3N•5HF and the proposed Type IIinv mechanism.
5. Alkene Dihalogenations with Transition Metal Halides as Reagents or Catalysts
5.1. Group 5 Metal Halides
5.1.1. Vanadium
On the basis a precedent for the use of vanadium(IV) chloride (VCl4) as a chlorinating agent for arenes,176 Uemura et al. have briefly investigated its competency as a dichlorinating agent for alkenes, reacting it with norbornene,103 norbornadiene,104 and (Z)-cyclooctene.125,126 With norbornene 62, a mixture of chlorinated products 63–69 is obtained, although syn-dichloride 63 predominates, and control experiments indicate that the product ratio is the result of kinetic control (Scheme 59). In contrast, almost no syn-dichloride is formed in similar reactions with cyclohexene and norbornadiene. Although a radical mechanism was excluded on the basis that no change in the isomer distribution occurs when the reactions are conducted in the presence of 1,3-dinitrobenzene or oxygen, the presence of 63 and 64 as major products is uncharacteristic of a purely ionic pathway. The results with norbornadiene are similarly inconclusive104 and although some 1,4-dichlorination is observed with (Z)-cyclooctene (indicative of an ionic pathway), the yield and selectivity are low.126
Scheme 59.

Dichlorination of norbornene 62 using VCl4.
5.1.2. Niobium and Tantalum
Both niobium(V) chloride (NbCl5) and tantalum(V) chloride (TaCl5) are unreactive with cyclohexene and norbornene upon heating in CCl4,103 and there are no reports of their successful use as dichlorinating agents.
5.2. Group 6 Metal Halides
5.2.1. Chromium
Chromium halides have not been used preparatively for alkene dihalogenation, but Sharpless and co-workers have reported that syn-dichlorides are formed as minor products (in up to 13% yield) from the oxidation of alkenes with chromyl chloride (CrO2Cl2) at low temperature.94 Although a Type IIIret mechanism was proposed by Sharpless et al., Nelson and co-workers suggest that a concerted Type IV dichlorination is in operation based on linear correlations between log krel and either alkene ionization potentials or HOMO energies.97
5.2.2. Molybdenum
The dichlorination of alkenes with stoichiometric amounts of MoCl5 shares many similarities with the corresponding reactions using SbCl5 as the chlorinating agent (Section 4.3.3); syn-stereospecific dichlorination occurs, and typically with lower yields but higher diastereoselectivities than the SbCl5-mediated processes. Following an early report on the dichlorination of tetrachloroethylene with MoCl5 to give hexachloroethane,177 the groups of Uemura178 and San Filippo,179 generalized the reaction and revealed its unusual syn-stereospecificity.180 Thus, addition of excess (E)-2-butene 42 (35 equiv) to a pale red-brown solution of MoCl5 in CCl4 at 74 °C provides a 92:8 mixture of syn/anti dichlorination products 43:44 in 85% combined yield (w.r.t. MoCl5). An analogous dichlorination of (Z)-2-butene 45 confirmed the stereospecific nature of the process (Scheme 60). As a large excess of the alkene component is generally required due to competing polymerization, all reported yields are based on MoCl5 as the limiting reagent. 1,2-Disubstituted alkenes typically give reasonable quantities of syn-dichlorinated products (63–68%), but the use of terminal, tri- and tetrasubstituted alkenes did not lead to synthetically useful results (≤10% product).179 Monochlorides were also formed as by-products in many cases which is ascribed to the in situ generation of HCl by hydride abstraction from the allylic position of the alkenes by MoCl5.178 No reaction occurs with electron-deficient alkenes such as ethyl fumarate and ethyl maleate, and only a trace (<5%) of dichloride is obtained from styrene.178
Scheme 60.

Syn-stereospecific dichlorination of alkenes using MoCl5.
By analogy to their earlier proposal for SbCl5-mediated syn-dichlorination,125 Uemura and co-workers proposed that a concerted transfer of both chlorine atoms may occur from monomeric181 MoCl5 70 (or possibly dimeric Mo2Cl10 71182) to the alkene via a 5-membered cyclic transition state XXXI (i.e., a Type IV mechanism).178 A Type IIIret mechanism involving syn-chlorometalation followed by stereoretentive reductive elimination (w.r.t. Mo) was seemingly not considered and should not be ruled out. The formation of dichlorides resulting from anti-addition, as well as certain rearranged products from norbornene and norbornadiene, is ascribed to ion pair [MoCl4]+[MoCl6]− 72 as the reactive chlorinating species,176 which may promote an ionic mechanism via β-chloro carbocation XXXII (or chloriranium ion) intermediates (Scheme 61). Comparison of the results with those for SbCl5 as a chlorinating agent125 indicates that MoCl5 may exist primarily in neutral forms 70 or 71 in CCl4, in contrast to SbCl5 whereby an ion pair analogous to 72 (i.e., [SbCl4]+[SbCl6]−) appears to make a larger contribution, probably as a consequence of the higher Lewis acidity of SbCl5.
Scheme 61.

Proposed parallel mechanisms for the formation of syn- and anti-dichlorination products, respectively, arbitrarily illustrated for an (E)-configured alkene.
Considering the synthetic limitations of the aforementioned alkene dichlorinations with MoCl5, Nugent has developed a milder and more selective variant of this reaction using tetrabutylammonium octamolybdate [(Bu4N)4Mo8O26] in combination with acetyl chloride (AcCl) as the chloride source, in which a polychloromolybdenum(VI) chlorinating agent is thought to be generated in situ. 183 This method enables the clean syn-dichlorination of a range of alkenes in excellent yield (mostly 83–94% based on alkene), and all with >98:2 syn:anti addition selectivity, with the reactions being run at ambient temperature in CH2Cl2 (Scheme 62). In view of the isolation of Mo(V) complexes Bu4NMoOCl4 184 and MoOCl3 (as its OPPh3 complex185) as by-products, the stoichiometry of the reaction is thought to be: alkene + 0.25 Mo8O26 4− + 9 AcCl → dichloride + MoOCl4 − + 4 MoOCl3 + 4.5 Ac2O In contrast to Uemura et al.,178 Nugent suggests a Type IIIret mechanism to account for the syn-stereospecificity.
Scheme 62.

Nugent’s protocol for molybdenum-mediated syn-dichlorination of alkenes.
5.2.3. Tungsten
Tungsten hexachloride (WCl6) was found to be a competent chlorinating agent for (excess) cyclohexene, affording 41% of the corresponding syn-dichloride and <1% of the anti-dichloride, 179 although it is unreactive with norbornene.103 Moreover, in situ prepared tungsten hexabromide (WBr6) reacts with an excess of cyclohexene to give a mixture of syn-dibromide (40–45%) and anti-dibromide (5–10%).179
5.3. Group 7 Metal Halides
5.3.1. Manganese(VII)
The combination of organic soluble benzyltriethyl-ammonium permanganate (BnNEt3MnO4) with chloride-based promoters has been developed by Markó and co-workers as a reagent system for the highly anti-stereospecific dichlorination of alkenes.32 Although the initial reports of this method employed oxalyl chloride [(COCl)2] as the promoter and chloride source,32b,c the use of chlorotrimethylsilane (Me3SiCl) was later found to be more practical,32a allowing the reactions to be run at ambient temperature as opposed to under cryogenic (−45 °C) conditions. Thus, the reaction of 5-decenes (E)-73 or (Z)-75 with a pre-formed mixture of KMnO4:BnNEt3Cl:Me3SiCl (1:1:4) in CH2Cl2 affords dichlorides anti-74 or syn-76, respectively, as single diastereomers in excellent yield (Scheme 63). A similar protocol has also been reported by Hazra and Pore, using tetradecyltrimethylammonium permanganate (H29C14NMe3MnO4) as a “safer” alternative to (isolated) BnNEt3MnO4 (the former does not detonate upon drying of the solid reagent at elevated temperatures).186 With Me3SiBr in place of Me3SiCl, the latter reagent system has also been employed for the anti-selective dibromination of alkenes. 187 The use of BnNEt3MnO4 with (COCl)2 or Me3SiCl as promoters has also been extended to the vicinal dichlorination of allenes.188
Scheme 63.

Anti-stereospecific dichlorination of alkenes with Markó‘s reagent.
Markó et al. have suggested that chloriranium ions (arising either from molecular Cl2 or some other Cl+ equivalents) are unlikely to be intermediates in these manganese(VII)-mediated dichlorinations. Specifically, different product distributions were observed for the dichlorination of cyclooctadiene monoepoxide 77 with either Cl2 (in CH2Cl2 at 20 °C) or Markó’s reagent [using either (COCl)2 at −40 °C32b or Me3SiCl at 0→20 °C32a]. Whereas Cl2 leads to extensive transannular participation by the oxirane oxygen to give dichlorinated bicyclic ethers 79 and 80 (via a putative chloriranium ion XXXIII), the manganese(VII)-mediated reactions give predominantly 1,2-dichloride 78, and only traces of bicyclic products 79 and 80 (Scheme 64). Although this was cited as evidence for the lack of a chloriranium ion intermediate in the latter case, there are alternative explanations which are compatible with chloriranium ion (or alkene-Cl2 π-complex) intermediacy in both cases.189 Moreover, it is conceivable that the other components present in the manganese(VII)-mediated reactions may simply have suppressed the epoxide participation (e.g., Lewis acidic Mn salts complexing the oxirane), or that the chloride ion concentration is higher than in the reaction with Cl2.36 The difference in reaction temperatures also calls into question the validity of direct comparisons.
Scheme 64.

Experiments to probe the intermediacy of chloriranium ions as intermediates.
Other observations, such as a lack of chlorination activity on pre-warming the active reagent to room temperature [with (COCl)2 as promoter]32b,c and the differing product distributions for dichlorinations of certain dienes with either “excess” Cl2 or “excess” manganese-based reagent186 have also been invoked as evidence that molecular Cl2 is not the active chlorinating agent, although again the assumption that “all else is equal” may be invalid. Although further studies are needed to exclude chloriranium ions as intermediates (i.e., a Type I mechanism), both Markó32 and Hazra186 have proposed that the manganese(VII)-mediated reactions proceed via a Type IIIinv mechanism, with an initial syn-chlorometalation of the alkene to give a β-chloro alkylmanganese species XXXIV, followed by an invertive SN2-type reductive elimination of [Mn] by chloride ion (Scheme 65). An argument against a Type IIIinv process has been made by Vanderwal and co-workers, who noted that the Markó reagent is also capable of cleanly effecting the anti-dichlorination of alkynes, in which case a similar syn-chlorometalation would then necessitate an unlikely SN2 invertive displacement by chloride ion at a C(sp2) center.190
Scheme 65.

Mechanistic proposal for the anti-dichlorination of alkenes by Markó‘s reagent.
With Me3SiCl as the promoter, Hazra and Pore have probed the in situ generated reagent by both EPR and UV-vis spectroscopy and have cautiously assigned the active species as manganese(V) oxide trichloride (Cl3MnO).186 Their assignment was based upon a UV-vis absorbance at 400 nm, which matched that reported for an independently prepared sample of Cl3MnO.191 An EPR spectrum was also obtained that consisted of “six sharp lines”, and this was stated as being consistent with a “lower valent manganese species with unpaired electrons”.186 However, only transition metal complexes possessing an odd number of unpaired electrons (i.e., d1, d3, d5, d7, or d9 configurations) are EPR active at the X-band frequencies which are typically employed,192 so Cl3MnO as a Mn(V) (d2) species cannot be responsible for the observed EPR signal. The signal would be consistent however with either Mn(IV)193 (d3) or Mn(II) (d5).
The presence of lower valent manganese species in solution following the addition of Me3SiCl, but prior to the addition of the alkene, proves that a redox reaction has occurred between Me3SiCl and the MnO4 − ion. By direct analogy to the oxidation of Cl− by permanganate ions in acidic media, this reaction may be: 2 [R4N+][MnO4 −] + 16 Me3SiCl → 2 [R4N+][MnCl3 −] + 5 Cl2 + 8 (Me3Si)2O. However, the stoichiometry required by this equation (i.e., 1:8 MnO4 −:Me3SiCl) implies that only half of the MnO4 − employed in the Markó protocol (1:4 MnO4 −:Me3SiCl) should be consumed by this process (generating 1.25 equiv of Cl2 w.r.t. the alkene). The excess MnO4 − may react with the initial Mn(II) product in a process such as Mn(VII) + Mn(II) → Mn(V) + Mn(IV), which could account for the EPR activity [i.e. Mn(IV)193] and the assignment of the Mn(V) species Cl3MnO from UV-vis data (vide supra). A final complication arises from the fact that M(V) oxo compounds of the form O=Mn(salen)Cl are known to abstract chlorine atoms from CH2Cl2 to generate Mn(IV) hypochlorite complexes [i.e., Mn(salen)(OCl)Cl] which are able to dichlorinate alkenes.193a
To conclude, if Cl2 is indeed being generated in this redox process,194 it is presumably responsible for dichlorinating the alkene, and there are even potential explanations for the loss of chlorination activity on warming.195 Furthermore, Vanderwal and co-workers have reported that the diastereoselectivities of the dichlorination of chiral allylic alcohols with the Markó reagent and Mioskowski reagent (Et4NCl3)28 are virtually identical, providing circumstantial evidence that the former reagent system may simply involve an in situ generation of an R4NCl3-type species, with Cl3 − as the active chlorinating species.190 Clearly, the overall picture for manganese(VII)-mediated alkene dichlorinations is complex, and further work is necessary to fully deconvolute the process.
5.3.2. Manganese(III) and (IV)
In addition to manganese(VII)-mediated alkene dichlorinations, a variety of other protocols employing lower valent manganese reagents [i.e., Mn(III) or Mn(IV)] have also been described. For example, isolated Mn(III) chlorides such as MnCl3•HOAc or salts of the form M2[MnCl5] (e.g., M+ = NH4 +, NMe4 +, PyH+, PhCH2NMe3 +) are competent dichlorinating agents for alkenes, and Mn(OAc)3 in AcOH combined with either CaCl2 or AcCl can be used to generate “MnCl3” in situ.196 A related reagent system for alkene dichlorination is MnO2-MnCl2-AcCl (1.5:1:6) in DMF, which is thought to generate Mn(III) chlorides by the disproportionation of Mn(II) and Mn(IV) [i.e., Mn(II) + Mn(IV) → 2 Mn(III)].197 Thus, the dichlorination of (E)-2-hexene 81 under the latter conditions affords a 64:36 mixture of anti/syn dichlorination products 82 in 95% combined yield. On the basis of the relatively low diastereoselectivity and the absence of ionic pathway-derived by-products from certain alkenes (e.g., t-butylethylene, norbornene), as well as positive tests for radical cyclizations, a Type V dichlorination via a non-chain radical mechanism has been proposed for these Mn(III)-mediated reactions (Scheme 66).196,197
Scheme 66.

Alkene dichlorination mediated by Mn(III) chlorides.
The vicinal dichlorination of alkenes has even been reported using a substoichiometric amount of Mn(acac)3 (i.e., 1 mol%) with sodium chlorite (NaClO2) as the re-oxidant. For example, reaction of (E)-2-octene 83 under these conditions gives dichloride(s) 84 in 80% yield (Scheme 67).198 However, the authors declined to report the relative configurations or diastereomeric compositions of the dichloride products, and they also did not report a control experiment in the absence of Mn(acac)3. Furthermore, based on the reactivity of Mn(III)(salen)Cl 87 with sodium hypochlorite (NaOCl) in CH2Cl2 (vide infra), the possibility that the CH2Cl2 solvent may be a source of chlorine atoms cannot be excluded.193a
Scheme 67.

Mn(III)-catalyzed dichlorination of alkenes.
Manganese(IV) chlorides have also been implicated as dichlorinating agents for alkenes. Despite the fact that MnO2 is well known to oxidize aqueous HCl to molecular chlorine,194 the reagent combination of MnO2-Me3SiCl in THF reacts with alkenes at 40–60 °C to give vicinal dichlorides without the distinctive by-products characteristic of Cl2 involvement.199 For example, and similarly to the Mn(III)-mediated dichlorinations above, no skeletal rearrangements take place with t-butylethylene or norbornene, and no allylic chlorination occurs with trisubstituted alkenes. In the reaction with norbornene 62, for example, clean conversion to vicinal dichlorides 64 and 63 is observed, and a Type V non-chain radical mechanism has been proposed for these reactions (Scheme 68).
Scheme 68.

Alkene dichlorination mediated by Mn(IV) chlorides.
Pecoraro and co-workers have reported Mn(IV)(salpn)Cl2 85 as the first structurally well-characterized manganese chloride species capable of dichlorinating alkenes. 200 For example, the treatment of norbornene 62 with 2.0 equiv of 85 gave dichlorides 64 and 63 in yields of 81% and 16%, respectively. The Mn product of the reaction was identified as Mn(III)(salpn)Cl 86 (Scheme 69).
Scheme 69.

Alkene dichlorination with a structurally well-characterized Mn(IV) halide.
During a study on the possible involvement of Mn(IV) species in the Jacobsen-Katsuki epoxidation, Mock-Knoblauch and co-workers showed that the treatment of Jacobsen’s catalyst Mn(III)(salen)Cl 87 with either PhIO or NaOCl in CH2Cl2 gave, inter alia, Mn(IV) hypochlorite complex 88.193a On the basis of EPR and mass spectrometry data, a mechanism for the formation of 88 involving chlorine abstraction from CH2Cl2 by a transient O=Mn(V)(salen)Cl intermediate XXXV is proposed (Scheme 70).
Scheme 70.

Chlorine atom abstraction from CH2Cl2 to generate a Mn(IV) hypochlorite complex 88.
This Mn(IV) hypochlorite complex 88 is capable of dichlorinating alkenes. For example, the reaction of 88 with 1,2-dihydronaphthalene 8 in CH2Cl2 affords anti-dichloride 10 as the major product, in addition to some chlorohydrin 89 and epoxide 90 (Scheme 71). Although the chlorinating agent 88 is chiral and non-racemic, 10 is formed with only negligible enantioenrichment (<52.5:47.5 er).
Scheme 71.

Ability of Mn(IV) hypochlorite complex 88 to dichlorinate alkenes.
5.3.3. Rhenium
Rhenium(V) chloride (ReCl5) is known to react stoichiometrically with tetrachloroethylene to generate hexachloroethane, although the targeted compound in this particular reaction is actually the reduced metal salt, rhenium(IV) chloride (ReCl4).201
5.4. Group 8 Metal Halides
5.4.1. Iron
Although iron(III) chloride (FeCl3) has been used as a stoichiometric reagent to effect the chlorination of arenes (i.e., Ar–H + 2 FeCl3 → Ar–Cl + 2 FeCl2 + HCl),202 it is unreactive toward norbornene,103 and there are no examples of alkene dichlorination with this reagent.
5.4.2. Ruthenium
Sakai and co-workers have shown that the dichlorination of a variety of alkenes can be effected using hexachloroethane as the chlorinating agent in the presence of 1 mol% RuCl2(PPh3)2 in refluxing toluene (Scheme 72).203 This reaction is closely related to metal-catalyzed variants of the Kharasch addition of polyhaloalkanes to alkenes,204 with the exception here that the intermediate carbon radical derived from the polyhaloalkane undergoes β-scission to give a chlorine atom radical (and tetrachloroethylene) before it engages the olefin. Unfortunately the stereochemical course of the reaction, which might help to support or refute the proposed Type V mechanism, was not evaluated.
Scheme 72.

Ruthenium-catalyzed alkene dichlorination by a Type V mechanism.
5.5. Group 9 Metal Halides
To the best of our knowledge, no reports of the use of cobalt, rhodium, or iridium halides as stoichiometric reagents for the dichlorination or dibromination of alkenes are extant, nor is the use of these elements as redox catalysts for such transformations. Cobalt(III) fluoride (CoF3) has been used however to difluorinate simple halogenated olefins.205
5.6. Group 10 Metal Halides
5.6.1. Palladium
Despite the fact that chloropalladations of alkenes with Pd(II)–Cl complexes are known,82,92 as well as oxidatively-induced stereoretentive76,83a–f,i and invertive (SN2-like)76,84 reductive eliminations of alkyl–Pd(II) intermediates to form C(sp3)–Cl bonds, no Pd-catalyzed alkene dichlorinations combining these elementary steps are on record (Scheme 73).206
Scheme 73.

A palladium-catalyzed dichlorination of alkenes?
However, the vicinal dichlorination of allenes with a stoichiometric amount of PdCl2(PhCN)2 has been achieved by Bäckvall and Jonasson, in which these two elementary steps are performed in a stepwise manner. 207 Thus, the reaction of 1,2-heptadiene 91 with stoichiometric PdCl2(PhCN)2 gave 2-chloro π-allyl complex 92 in 58% yield, and subsequent treatment with p-benzoquinone and LiCl in AcOH gave the corresponding vicinal dichloride 93 (of unspecified dr and in unspecified yield) (Scheme 74). Attempts to run the dichlorination under catalytic conditions were unsuccessful due to a competing reaction of the π-allyl complex 92 with unreacted allene 91.
Scheme 74.

Stepwise dichlorination of an allene using stoichiometric palladium.
Interestingly, a catalytic dibromination of allenes could be achieved by replacing LiCl with LiBr, and this was applied to a range of allenes (Scheme 75).207 The success may be ascribed to the greater nucleophilicity of the bromide ion (w.r.t. chloride ion), which enables the halide to outcompete the allene starting material in intercepting the π-allyl intermediate.
Scheme 75.

Palladium-catalyzed dibrominations of allenes. Cy = cyclohexyl.
Of course the crucial difference between allenes versus alkenes as substrates for this transformation is the thermodynamic favorability of the halopalladation step in the former case – a consequence of the formation of a stable Pd(II) π-allyl complex. On the other hand, halometalations of alkenes in general are endergonic, and the reverse process of β-halo elimination renders the β-halo metalated intermediates highly unstable.86 This suggests that successful Pd-catalyzed alkene dihalogenations may benefit from stabilization of the alkyl–Pd(II) intermediate, perhaps by π-benzyl formation (i.e., by employing aryl alkene substrates) or by chelation with a proximal functional group (Scheme 76).
Scheme 76.

Potential strategies for stabilizing the alkyl–Pd(II) intermediate against unproductive β-halo elimination. LB = Lewis base.
Another strategy to avoid unproductive β-halo elimination from a chloropalladated intermediate might be to circumvent the alkyl–Pd(II) species altogether, and design a catalytic system in which oxidation of Pd(II) → Pd(IV) precedes interaction with the alkene substrate. With this in mind, Kraft and co-workers have shown that the isolated LPd(IV)Cl4 complex 95 [prepared by oxidation of the corresponding LPd(II)Cl2 complex 94 with Cl2] is capable of dichlorinating alkenes in an anti-selective fashion.208 For example, the reaction of 1-hexene 96 with LPd(IV)Cl4 95 and Bu4NCl in CDCl3 at rt affords dichloride 97 in 89% NMR yield (w.r.t. 96). Mechanistic studies support a Type I mechanism involving the slow formation of cationic LPd(IV)Cl3 + XXXVI, followed by chlorenium ion transfer to the alkene (in a ligand-mediated process devoid of π-coordination) to give a chloriranium ion species XXXVII, which is then trapped by chloride ion (Scheme 77). In subsequent work, Kraft et al. showed that pyridine significantly enhances the rate of the reaction of LPd(IV)Cl4 95 with alkenes, and that a “catalytic” dichlorination of styrene is possible with LPd(IV)Cl3(py)+ as an in situ generated catalyst and LPd(IV)Cl4 95 as the stoichiometric oxidant.209
Scheme 77.

Pd(IV)-mediated dichlorination of alkenes.
5.7. Group 11 Metal Halides
5.7.1. Copper
It has long been known that the gas-phase reaction of alkenes with solid-supported CuCl2 at high temperatures (220–330 °C) results in the formation of vicinal dichlorides, along with reduction of CuCl2 to CuCl. As the so-called “oxychlorination” process, this reaction is used on industrial scale for the synthesis of 1,2-dichloroethane, with the CuCl being reoxidized to CuCl2 with HCl and O2 (Scheme 78).210
Scheme 78.

The “oxychlorination” process for the dichlorination of ethylene.
The mechanism of the (stoichiometric) reaction of alkenes with pumice-supported CuCl2 has been studied,211 as has the chemisorption of ethylene with CuCl2 and CuCl,212 and the initial step of the process is thought to involve π-complex formation between the olefin and surface Cu(II) ions.211a,212 The mechanistic course from this point depends on the nature of the olefin, with (E)-d2-ethylene 98 giving poorly stereoselective dichlorination and 2-butenes 42 and 45 undergoing anti-dichlorination with moderately high stereospecificity (Scheme 79).211a Control experiments indicated that neither alkene nor dichloride isomerization was significant for the reaction of (E)-d2-98, but both occurred to some extent for 42 and 45, eroding the stereospecificity. It was surmised that the reaction partitions between a radical and ionic pathway, depending on the substitution of the alkene. A similar phenomenon has been observed by Poutsma in the dark chlorination of several olefins using Cl2 under nitrogen, with the ionic route being favored for more highly substituted alkenes at low alkene concentrations, and the radical addition being far less stereoselective than the former.24 Control experiments indicated that Cl2 is not the active chlorine source in these reactions,213 although it was assumed that the rate of disproportionation of CuCl2 is unaffected by [ethylene].
Scheme 79.

Gas-phase dichlorination of alkenes on pumice-supported CuCl2.
For solution-phase dichlorinations of olefins with CuCl2, early work involving photochemical activation of CuCl2 by Kochi214 was followed by the development of a thermal reaction in aqueous solution by Miller and co-workers. 215 The dichlorination of alkenes with CuCl2 in organic solvents (AcOH or alcohols) was later studied by Uemura et al., including the reaction scope, 216 kinetics, 217 effect of additives (NaOAc, NaCl),218 and the use of butadiene as a substrate,219 all of which are summarized in a subsequent overview article.220 Further detailed investigations by Koyano and co-workers,221 including establishing the stereochemical course of the reaction,222 the influence of additives (e.g., LiCl),223 and the kinetics.223
In general, these dichlorination reactions are conducted using stoichiometric amounts of CuCl2, typically in AcOH or alcohol solvents, at temperatures ranging from 70 to 150 °C (though often >100 °C).220,221 The use of somewhat lower temperatures (70–80 °C) is possible in MeCN as the solvent,224 but LiCl is usually added in these cases to solubilize the CuCl2 and to further enhance the rate.220,225 Chlorine has been deemed unlikely to be the active chlorinating agent in these reactions221 as CuCl2 is known not to decompose to CuCl and Cl2 at these temperatures in the solvents employed226 (albeit in the absence of olefins). Dichlorinations conducted in alcohol solvents are often complicated by competing chloroalkoxylation processes220,221 although the addition of LiCl promotes the formation of dichloride products.223 In contrast to the gas-phase chemistry,211 the reactions of 2-butenes 42 and 45 in alcohols, AcOH or MeNO2 as the solvent are not stereospecific, yielding a common ~60:40 mixture of anti/syn dichlorination products 44:43 (Scheme 80, data shown for MeOH). Control experiments probing isomerization of either the alkene substrates or dichloride products indicated that this ratio is the result of kinetic control.
Scheme 80.

Non-stereospecific dichlorination of 2-butenes with CuCl2 in MeOH.
The reaction kinetics in alcohol solvents have been studied by the groups of Uemura220 (with styrene in n-PrOH) and Koyano (with 1-octene in MeOH),223 with both sets of authors arriving at a similar (initial) rate law of the form: (where n = 1.8 for Uemura and n = 1.6 for Koyano). Additionally, a reactivity order (based on conversion of CuCl2 in independent experiments) of ethylene > propylene > 1-butene > 2-butenes was also established. This parallels the stability of Ag(I)-olefin π-complexes,227 and is taken as support for Cu(II)-alkene π-complex intermediates. On the basis of all of the above findings, Koyano et al. have proposed the following mechanism (Scheme 81).221 Initial formation of a Cu(II)-alkene π-complex XXXVIII is followed by transfer of Cl+ from the Cu to generate a β-chlorocarbenium ion intermediate XXXIX, in which association of the chlorine atom with the Cu prevents its efficient bridging to give a chloriranium ion species. 228 Rapid interconversion of these carbocations XXXIXA and XXXIXB prior to nucleophilic trapping would account for the lack of stereospecificity. From the experimental rate law, the rate-determining step (RDS) is proposed to be the nucleophilic trapping of carbocations XXXIX. Considering an order in CuCl2 of 1.8 (assumed to be 2 within error), Uemura and co-workers proposed that CuCl2 may be the active nucleophile in this process,220 but Koyano et al. showed that an order of 1.5 in CuCl2 would be expected for (CuCl2-derived) Cl− ion as the active nucleophile – in good agreement with their order of 1.6.223 Both the CuCl by-product and H2O are potent inhibitors, probably due to the former sequestering free Cl− ions223 and the latter forming stable aqua complexes of CuCl2. In contrast, the RDS in MeCN is believed to be formation of either the alkene-CuCl2 π-complex XXXVIII or the β-chlorocarbenium ion XXXIX, based on a first-order dependence on both alkene and CuCl2.220
Scheme 81.

Mechanistic proposal for the dichlorination of alkenes with stoichiometric amounts of CuCl2 in alcohol solvents.
However, the proposed intermediacy of β-chlorocarbenium ions XXXIX in these reactions is difficult to reconcile with the results of other studies conducted with CuCl2-LiCl in MeCN. Thus, a variety of alkenes as mechanistic probes for ionic versus radical dichlorination have been examined with CuCl2-LiCl in MeCN, including norbornene,103 norbornadiene,104 cyclooctadienes,107 and (Z)-cyclooctene.126 In most cases the product distributions were more representative of radical rather than ionic processes, but the insensitivity of the reaction to radical inhibitors argued against a radical chain mechanism.
An alternative mechanism that is consistent with all of the experimental observations (i.e., kinetics, stereoconvergency, lack of carbocation-derived products) is outlined in Scheme 82. Thus, chloride ion attack on a Cu(II)-alkene π-complex XL (i.e., a Type II addition), followed by homolysis of the C–Cu bond in XLI would furnish a β-chloroalkyl radical XLII. This carbon radical could then intercept another molecule of CuCl2 to give a transient Cu(III) alkyl species XLIII, which will reductively eliminate to give the dichloride product. Similar mechanisms involving nucleocupration of alkenes, C–Cu bond homolysis, and reductive elimination from Cu(III) intermediates are well established for other copper-mediated alkene difunctionalizations.229
Scheme 82.

A plausible mechanism for alkene dichlorinations with CuCl2 that is consistent with all of the experimental observations.
In contrast to alkene dichlorinations with CuCl2, the analogous dibrominations with CuBr2 proceed readily at ambient temperature,79,224,226b,230 and are also highly anti-stereospecific in both alcohol230 and MeCN solvents.224 For example, the reactions of 2-butenes (E)-42 and (Z)-45 with CuBr2 in MeCN at rt gives dibromides anti-99 and syn-100, respectively, in 91% yield (w.r.t. CuBr2) (Scheme 83).
Scheme 83.

Stereospecific dibromination of 2-butenes with CuBr2.
Baird and co-workers have found that certain “soft” or π-acidic ligands can substantially increase the chemical yield of alkene dibrominations with CuBr2, and that in some cases these ligands are effective in only substoichiometric amounts [e.g., MeCN, Ph3P, dppe [1,2-bis(diphenyl-phosphino)ethane], (t-BuO)3P].224 In fact, the effectiveness of MeCN as a solvent for CuBr2-mediated dibrominations is attributed to its π-acidic character as a ligand. These results are interpreted as being a consequence of the disproportionation of CuBr2 to CuBr and molecular bromine (i.e., CuBr2 → CuBr + ½ Br2), with “soft” or π-acidic ligands stabilizing the Cu(I) product and driving the equilibrium to the right. An earlier kinetic study of these dibrominations by Castro et al., which found a second-order dependence on CuBr2,226b was cited as support for this disproportionation as the rate-determining step, although other interpretations of this rate law are viable (c.f. mechanism of CuCl2-mediated dichlorinations, vide supra). However, several other pieces of evidence, including the product distribution from the reaction of norbornadiene, the relative ordering of olefin reactivity (R2C=CR2 > R2C=CHR ≫ RCH=CHR > RCH=CH2), and spectrophotometric evidence231 for the generation of Br2 and CuBr3 − in MeCN solutions of CuBr2 are all supportive of Br2 as the active brominating agent.224
However, for the dibromination of n-hexene with CuBr2 in MeOH at 40 or 50 °C, a mechanism similar to that in Scheme 81 for CuCl2 (albeit with fully bridged bromiranium ions) has been proposed by Koyano et al., on the basis of approximate third-order kinetics (~second-order in CuBr2 and first-order in alkene).230 A different mechanism altogether, from DFT calculations, has been advanced by Fraser-Reid, Snyder and co-workers for alkene dibrominations using a combination of CuBr2 and LiBr in 3:1 MeCN:THF.79 In what could be described as a Type IIret mechanism (calculated for ethylene), attack of bromide ion on a Cu(II)-alkene π-complex XLIV gives a square planar Cu(II) anion XLV, followed by single electron transfer to CuBr2 to generate a neutral Cu(III) species XLVI. Reductive elimination then delivers the dibromide product and CuBr(MeCN) (Scheme 84). Ma and Wu proposed a similar mechanism for CuX2-mediated (X = Cl, Br) cyclization reactions of 2,3-allenoic acids.232
Scheme 84.

Calculated Type IIret mechanism for alkene dibromination with CuBr2 and LiCl in MeCN:THF.
The dihalogenation of alkynes with CuCl2 233 and CuBr2 234 has also been reported, and is usually (though not always) anti-selective.
5.7.2. Silver
With the exception of silver(II) fluoride (AgF2), which has been shown to difluorinate alkenes,205 silver does not form halides in oxidation states higher than +1, and neither AgCl nor AgBr are sufficiently oxidizing to effect halogenation reactions.
5.7.3. Gold
Unlike silver, gold is capable of forming halides in the +3 oxidation state, and gold(III) chloride (AuCl3) is known to react with alkenes to afford the corresponding vicinal dichlorides.235 Exposure of various alkenes (in excess) to AuCl3 in cyclohexane at 20 °C over several hours provides vicinal dichlorides in excellent yields (w.r.t. AuCl3) along with Au metal, but the reactions of 2-butenes 42 and 45 are non-stereospecific. Considering the isolation of intermediates, the net reactions are believed to be a composite of three sequential reactions (1)–(3), in which A is the alkene and B is the vicinal dichloride (Scheme 85).
Scheme 85.

Sequential reactions in the dichlorination of alkenes with AuCl3.
The (alkene)3•Au2Cl4 complexes implicated in this reaction manifold could actually be isolated at low temperature (−60 °C) in CHCl3. The decomposition of these isolated (alkene)3•Au2Cl4 complexes in CHCl3 at −30 °C (a model of reaction (2), Scheme 85), followed by sequestration of gold salts with added pyridine, affords the corresponding vicinal dichlorides. Curiously, under these conditions, the reactions of (alkene)3•Au2Cl4 complexes derived from 2-butenes (E)-42 and (Z)-45 are moderately stereospecific (Scheme 86). Furthermore, the decomposition of an authentic sample of alkene•AuCl [derived from (Z)-2-butene 45] at 20 °C in CHCl3 (a model of reaction (3), Scheme 85) is similarly stereospecific, giving 43 in 75:25 dr.
Scheme 86.

Stereospecific dichlorination of 2-butenes with AuCl3.
6. Closing Remarks
In the intervening years since the publication of our review on enantioselective halofunctionalization of alkenes,4f the pace of research activity in this burgeoning field has continued to increase. Impressive advances have been recorded for the functionalization of different types of alkenes engaging all of the common halogens in combination with a wide variety of nucleophilic heteroatoms. Although the vast majority of these halofunctionalization reactions are intramolecular and form small ring heterocycles, intermolecular process have also been described with increasingly better scope and stereoselectivity. Moreover, the development of novel reactivity concepts for the generation of electrophilic halenium sources has been parlayed with novel stereocontrol mechanisms as was presaged in our treatise.
Although this field is still in its infancy, by comparison the number of enantioselective dihalogenations hardly constitutes a field with fewer than a handful of reports to date. Yet, as is hopefully apparent from the foregoing disquisition, ample opportunities exist for a wide variety of reagents and reaction types. Moreover, given the remarkable range of mechanistic manifolds that are available for dihalogenation, opportunities for creative engineering of stereochemically robust and controllable processes abound. It is our hope that the possibilities revealed by this essay will stimulate inspired and intrepid incursions into the unexplored territories in the landscape of alkene difunctionalization.
Acknowledgments
We are grateful to the National Institutes of Health (GM R01-085235) for generous financial support. S. T.-C. E. thanks the Agency for Science, Technology, and Research of Singapore (A*STAR) for a postdoctoral fellowship. We thank Dr. Carl Poree and Dr. Dietrich Böse for their assistance with the translation of several papers.
Biographies

Alex Cresswell was born in Bradford, West Yorkshire, in 1985. He received his D.Phil. from the University of Oxford in 2012, under the supervision of Professor Stephen G. Davies. Following a two year postdoctoral stay with Professor Scott E. Denmark at the University of Illinois, he is currently undertaking a second postdoctoral appointment with Professor Guy C. Lloyd-Jones FRS at the University of Edinburgh.

Stanley T.-C. Eey was born in Singapore in 1981. He received his Ph.D. at the National University of Singapore in 2012, under the supervision of Dr. Martin J. Lear. He was working as R&D scientist for a biotech spin-off company from NUS before joining Professor Scott E. Denmark at the University of Illinois for a two year postdoctoral stay, funded by the A*STAR International Fellowship. He is currently a scientist at the Institute of Chemical and Engineering Sciences, A*STAR.

Scott E. Denmark obtained an S.B. degree from MIT in 1975 and his D.Sc.Tech. (under the direction of Albert Eschenmoser) from the ETH Zürich in 1980. That same year he began his career at the University of Illinois and since 1991 he has been the Reynold C. Fuson Professor of Chemistry. His research interests include the invention of new synthetic reactions, exploratory organoelement chemistry, and the origin of stereocontrol in fundamental carbon-carbon bond-forming processes.
Footnotes
Dedicated to Prof. Dr. Albert Eschenmoser on the festive occasion of his 90th birthday
References
- 1.a) French AN, Bissmire S, Wirth T. Chem Soc Rev. 2004;33:354–362. doi: 10.1039/b310389g. [DOI] [PubMed] [Google Scholar]; b) Ranganathan S, Muraleedharan KM, Vaish NK, Jayaraman N. Tetrahedron. 2004;60:5273–5308. [Google Scholar]; c) Cardillo G, Orena M. Tetrahedron. 1990;46:3321–3408. [Google Scholar]; d) Trost BM, Fleming I, Semmelheck MF, editors. Comprehensive Organic Synthesis. Vol. 4. Pergamon Press; Oxford: 1991. [Google Scholar]; e) Mulzer J. In: Organic Synthesis Highlights. Mulzer J, Altenbach H-J, Braun M, Krohn K, Reißig H-U, editors. VCH; Weinheim: 1991. pp. 158–164. [Google Scholar]; f) Bartlett PA. In: Asymmetric Synthesis. Morrison J, editor. Vol. 3. Academic Press; Orlando: 1983. pp. 411–454. [Google Scholar]
- 2.a) Zhu Y, Wang Q, Cornwall RG, Shi Y. Chem Rev. 2014;114:8199–8256. doi: 10.1021/cr500064w. [DOI] [PubMed] [Google Scholar]; b) Xia QH, Ge HQ, Ye CP, Liu ZM, Su KX. Chem Rev. 2005;105:1603–1662. doi: 10.1021/cr0406458. [DOI] [PubMed] [Google Scholar]
- 3.Kolb HC, Vannieuwenhze MS, Sharpless KB. Chem Rev. 1994;94:2483–2547. [Google Scholar]
- 4.a) Hennecke U, Wilking M. Synlett. 2014;25:1633–1637. [Google Scholar]; b) Cheng YA, Yu WZ, Yeung YY. Org Biomol Chem. 2014;12:2333–2343. doi: 10.1039/c3ob42335b. [DOI] [PubMed] [Google Scholar]; c) Chen J, Zhou L. Synthesis. 2014;46:586–595. [Google Scholar]; d) Wang SH, Li BS, Tu YQ. Chem Commun. 2014;50:2393–2408. doi: 10.1039/c3cc48547a. [DOI] [PubMed] [Google Scholar]; e) Snyder SA, Brooks AP. In: Asymmetric Synthesis II: More Methods and Applications. Christmann M, Bräse S, editors. Chapter 20 Wiley–VCH; Weinheim: 2012. [Google Scholar]; f) Denmark SE, Kuester WE, Burk MT. Angew Chem Int Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Hennecke U. Chem Asian J. 2012;7:456–465. doi: 10.1002/asia.201100856. [DOI] [PubMed] [Google Scholar]; h) Tan C, Zhou L, Yeung YY. Synlett. 2011:1335–1339. [Google Scholar]; i) Castellanos A, Fletcher SP. Chem Eur J. 2011;17:5766–5776. doi: 10.1002/chem.201100105. [DOI] [PubMed] [Google Scholar]; j) Snyder SA, Treitler DS, Brucks AP. Aldrichim Acta. 2011;44:35–48. [Google Scholar]
- 5.Haliranium (bridged halonium) ion intermediates were first proposed in 1937 to explain the anti-stereospecific dihalogenation of alkenes with dihalogens; see: Roberts I, Kimball GE. J Am Chem Soc. 1937;59:947–948.
- 6.Nilewski C, Geisser RW, Carreira EM. Nature. 2009;547:573–576. doi: 10.1038/nature07734. [DOI] [PubMed] [Google Scholar]
- 7.a) Kladi M, Vagias C, Roussis V. Phytochem Rev. 2004;3:337–366. [Google Scholar]; b) Gribble GW. Chemosphere. 2003;52:289–297. doi: 10.1016/S0045-6535(03)00207-8. [DOI] [PubMed] [Google Scholar]; c) Gribble GW. Acc Chem Res. 1998;31:141–152. [Google Scholar]; d) Gribble GW. Progress in the Chemistry of Organic Natural Products. 1996;68:1–498. doi: 10.1007/978-3-031-26629-4_1. [DOI] [PubMed] [Google Scholar]
- 8.Hu DX, Seidl FJ, Bucher C, Burns NZ. J Am Chem Soc. 2015;137:3795–3798. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]
- 9.Snyder SA, Tang Z-Y, Gupta R. J Am Chem Soc. 2009;131:5744–5745. doi: 10.1021/ja9014716. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka Y, Sakuraba H, Nakanishi H. J Chem Soc, Chem Commun. 1983:947–948. [Google Scholar]
- 11.a) Brooks AP, Treitler DS, Liu SA, Snyder SA. Synthesis. 2013;45:1886–1898. [Google Scholar]; b) Snyder SA, Treitler DS, Brooks AP. J Am Chem Soc. 2010;132:14303–14314. doi: 10.1021/ja106813s. [DOI] [PubMed] [Google Scholar]
- 12.Nicolaou KC, Simmons NL, Ying Y, Heretsch PM, Chen JS. J Am Chem Soc. 2011;133:8134–8137. doi: 10.1021/ja202555m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denmark SE, Beutner GL. Angew Chem Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 14.Seebach D, Beck AK, Heckel A. Angew Chem Int Ed. 2001;40:92–138. [PubMed] [Google Scholar]
- 15.Hu DX, Shibuya GM, Burns NZ. J Am Chem Soc. 2013;135:12960–12963. doi: 10.1021/ja4083182. [DOI] [PubMed] [Google Scholar]
- 16.Steinreiber J, Faber K, Griengl H. Chem Eur J. 2008;14:8060–8072. doi: 10.1002/chem.200701643. [DOI] [PubMed] [Google Scholar]
- 17.Berrisford DJ, Bolm C, Sharpless KB. Angew Chem Int Ed Engl. 1995;34:1059–1070. [Google Scholar]
- 18.Juliá S, Ginebreda A. Tetrahedron Lett. 1979;20:2171–2174. [Google Scholar]
- 19.a) Bellucci G, Berti G, Marioni F, Marsili A. Tetrahedron. 1970;26:4627–4633. [Google Scholar]; b) Bellucci G, Giordano C, Marsili A, Berti G. Tetrahedron. 1969;25:4515–4522. [Google Scholar]; c) Bellucci G, Marioni F, Marsili A. Tetrahedron. 1969;25:4167–4172. [Google Scholar]; d) Berti G, Marsili A. Tetrahedron. 1966;22:2977–2988. doi: 10.1016/s0040-4020(01)82275-4. [DOI] [PubMed] [Google Scholar]
- 20.El-Qisairi AK, Qaseer HA, Katsigras G, Lorenzi P, Trivedi U, Tracz S, Hartman A, Miller JA, Henry PM. Org Lett. 2003;5:439–441. doi: 10.1021/ol0273093. [DOI] [PubMed] [Google Scholar]
- 21.We have attempted unsuccessfully to reproduce the results described in these reports. Although we were able to obtain the dibrominated products from four different alkenes using three different catalyst systems, all products were racemic as determined by CSP-HPLC. The original authors employed lanthanide chiral shift reagents to determine the enantiomeric composition of the products, unfortunately without providing the spectra in the Supporting Information.
- 22.a) Schaumann E, editor. Science of Synthesis, Vol. 34: Chlorine, Bromine, and Iodine. Thieme; Stuttgart: 2007. [Google Scholar]; b) Larock RC. Comprehensive Organic Transformations. VCH; New York: 1989. [Google Scholar]
- 23.a) Kočovskí P. In: Addition Reactions: Polar Addition in Organic Reaction Mechanisms. Knipe AC, editor. Wiley; Chichester, UK: 2007. [Google Scholar]; b) De la Mare PBD, Bolton R. Electrophilic Additions to Unsaturated Systems. 2. Elsevier; New York: 1982. pp. 136–197. [Google Scholar]; c) V’yunov KA, Ginak AI. Russ Chem Rev (Engl Transl) 1981;50:151–163. [Google Scholar]; d) Schmid GH, Garratt DG. In: The Chemistry of Doubly Bonded Functional Groups. Patai S, editor. Wiley-Interscience; New York: 1977. Supplement A, Part 2, Chap. 9. [Google Scholar]; e) De la Mare PBD. Electrophilic Halogenation. Cambridge University Press; Cambridge: 1976. [Google Scholar]; f) Freeman F. Chem Rev. 1975;75:439–490. [Google Scholar]; g) Fahey RC. In: Topics in Stereochemistry. Eliel EL, Allinger NL, editors. Vol. 3. Wiley; New York: 1968. p. 237. [Google Scholar]
- 24.a) Poutsma ML. Science. 1967;157:997–1005. doi: 10.1126/science.157.3792.997. [DOI] [PubMed] [Google Scholar]; b) Poutsma ML. J Am Chem Soc. 1965;87:4285–4293. [Google Scholar]; c) Poutsma ML. J Am Chem Soc. 1965;87:2172–2183. [Google Scholar]; d) Poutsma ML. J Am Chem Soc. 1965;87:2161–2171. [Google Scholar]
- 25.a) Islam SM, Poirier RA. J Phys Chem A. 2008;112:152–159. doi: 10.1021/jp077306d. [DOI] [PubMed] [Google Scholar]; b) Islam SM, Poirier RA. J Phys Chem A. 2007;111:13218–13232. doi: 10.1021/jp075674b. [DOI] [PubMed] [Google Scholar]; c) Lenoir D, Chappe C. Chem Eur J. 2003;9:1036–1044. doi: 10.1002/chem.200390097. [DOI] [PubMed] [Google Scholar]; d) Brown RS. Acc Chem Res. 1997;30:131–137. [Google Scholar]; e) Ruasse MF. Adv Phys Org Chem. 1993;28:207–291. [Google Scholar]; f) Ruasse MF. Acc Chem Res. 1990;23:87–93. [Google Scholar]
- 26.a) Sandford G. J Fluorine Chem. 2007;128:90–104. [Google Scholar]; b) Hutchinson J, Sandford G. Top Curr Chem. 1997;193:1–43. [Google Scholar]; c) Rozen S. Acc Chem Res. 1988;21:307–312. [Google Scholar]
- 27.Rosemund KW, Kuhnhenn W. Ber. 1923;56:1262–1269. [Google Scholar]
- 28.Schlama T, Gabriel K, Gouverneur V, Mioskowski C. Angew Chem Int Ed Engl. 1997;36:2342–2344. [Google Scholar]
- 29.Eissen M, Lenoir D. Chem Eur J. 2008;14:9830–9841. doi: 10.1002/chem.200800462. [DOI] [PubMed] [Google Scholar]
- 30.Karki M, Magolan J. J Org Chem. 2015;80:3701–3707. doi: 10.1021/acs.joc.5b00211. [DOI] [PubMed] [Google Scholar]
- 31.Ho T-L, Gupta BGB, Olah GA. Synthesis. 1977:676–677. [Google Scholar]
- 32.a) Markó IE, Richardson PF, Bailey M, Maguire AR, Coughlan N. Tetrahedron Lett. 1997;38:2339–2342. [Google Scholar]; b) Markó IE, Richardson PF. Synlett. 1991:733–736. [Google Scholar]; c) Markó IE, Richardson PF. Tetrahedron Lett. 1991;32:1831–1834. [Google Scholar]
- 33.Ren J, Tong R. Org Biomol Chem. 2013;11:4312–4315. doi: 10.1039/c3ob40670a. [DOI] [PubMed] [Google Scholar]
- 34.Finkelstein M, Hart SA, Moore WM, Ross SD, Eberson L. J Org Chem. 1986;51:3548–3551. [Google Scholar]
- 35.a) Stodulski M, Goetzinger A, Kohlhepp SV, Gulder T. Chem Commun. 2014;50:3435–3438. doi: 10.1039/c3cc49850f. [DOI] [PubMed] [Google Scholar]; b) Xue H, Tan H, Wei D, Wei Y, Lin S, Liang F, Zhao B. RSC Advances. 2013;3:5382–5385. [Google Scholar]; c) Hernández-Torres G, Tan B, Barbas CF., III Org Lett. 2012;14:1858–1861. doi: 10.1021/ol300456x. [DOI] [PubMed] [Google Scholar]; d) Zhu M, Lin S, Zhao GL, Sun J, Córdova A. Tetrahedron Lett. 2010;51:2708–2712. [Google Scholar]
- 36.Kamada Y, Kitamura Y, Tanaka T, Yoshimitsu T. Org Biomol Chem. 2013;11:1598–1601. doi: 10.1039/c3ob27345h. [DOI] [PubMed] [Google Scholar]
- 37.For a remarkable case of “negative catalysis”, in which the catalyzed reaction is actually slower or comparable to the “background” reaction in the absence of the catalyst, see: Denmark SE, Chi HM. J Am Chem Soc. 2014;136:3655–3663. doi: 10.1021/ja413270h.
- 38.For example, tetrafluoroborate (BF4−) ions are known to be able to transfer fluoride ion (F−) to haliranium ion intermediates; see: Cresswell AJ, Davies SG, Roberts PM, Thomson JE. Chem Rev. 2015;115:566–611. doi: 10.1021/cr5001805.
- 39.Cotter JL, Andrews LJ, Keefer RM. J Am Chem Soc. 1962;84:793–797. [Google Scholar]
- 40.a) Ning Z, Jin R, Ding J, Gao L. Synlett. 2009:2291–2294. [Google Scholar]; b) Kwon HY, Park CM, Lee SB, Youn JH, Kang SH. Chem Eur J. 2008;14:1023–1028. doi: 10.1002/chem.200701199. [DOI] [PubMed] [Google Scholar]; c) Kang SH, Lee SB, Park CM. J Am Chem Soc. 2003;125:15748–15749. doi: 10.1021/ja0369921. [DOI] [PubMed] [Google Scholar]
- 41.a) Watson WD. J Org Chem. 1985;50:2145–2148. [Google Scholar]; b) Watson WD. Tetrahedron Lett. 1976;17:2591–2594. [Google Scholar]
- 42.Maruoka K, editor. Asymmetric Phase Transfer Catalysis. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 43.Nelson IV, Iwamoto RT. J Electroanal Chem. 1964;7:218–221. [Google Scholar]
- 44.a) Bellucci G, Chiappe C, Moro Giacomo Lo. J Org Chem. 1997;62:3176–3182. doi: 10.1021/jo9620526. [DOI] [PubMed] [Google Scholar]; b) Bellucci G, Berti G, Bianchini R, Ingrosso G, Yates K. J Org Chem. 1981;46:2315–2323. [Google Scholar]; c) DeYoung S, Berliner E. J Org Chem. 1979;44:1088–1092. and references cited therein. [Google Scholar]
- 45.Phipps RJ, Hamilton DL, Toste FD. Nature Chem. 2012;4:603–614. doi: 10.1038/nchem.1405. [DOI] [PubMed] [Google Scholar]
- 46.Yang X, Wu T, Phipps RJ, Toste FD. Chem Rev. 2015;115:826–870. doi: 10.1021/cr500277b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y-M, Wu J, Hoong C, Rauniyar V, Toste FD. J Am Chem Soc. 2012;134:12928–12931. doi: 10.1021/ja305795x. [DOI] [PubMed] [Google Scholar]
- 48.Xie W, Jiang G, Liu H, Hu J, Pan X, Zhang H, Wan X, Lai Y, Ma D. Angew Chem Int Ed. 2013;52:12924–12927. doi: 10.1002/anie.201306774. [DOI] [PubMed] [Google Scholar]
- 49.Liu H, Jiang G, Pan X, Wan X, Lai Y, Ma D, Xie W. Org Lett. 2014;16:1908–1911. doi: 10.1021/ol5004109. [DOI] [PubMed] [Google Scholar]
- 50.Neverov AA, Brown RS. J Org Chem. 1998;63:5977–5982. doi: 10.1021/jo980627o. [DOI] [PubMed] [Google Scholar]
- 51.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito, CA: 2009. [Google Scholar]
- 52.Cresswell AJ, Eey ST-C, Denmark SE. Nature Chem. 2015;7:146–152. doi: 10.1038/nchem.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Denmark SE, Burk MT. Org Lett. 2012;14:256–259. doi: 10.1021/ol203033k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng CY, Tilk-Slebocka H, Nagorski RW, Alvarado L, Brown RS. J Org Chem. 1993;58:2122–2127. [Google Scholar]
- 55.Zhang H, Lin S, Jacobsen EN. J Am Chem Soc. 2014;136:16485–16488. doi: 10.1021/ja510113s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller LC, Sarpong R. Chem Soc Rev. 2011;40:4550–4562. doi: 10.1039/c1cs15069c. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y-M, Lackner AD, Toste FD. Acc Chem Res. 2013;47:889–901. doi: 10.1021/ar400188g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.a) Neverov AA, Brown RS. J Org Chem. 1996;61:962–968. [Google Scholar]; b) Brown RS, Nagorski RW, Bennet AJ, McClung RED, Aarts GHM, Klobukowski M, McDonald R, Santarsiero BD. J Am Chem Soc. 1994;116:2448–2456. [Google Scholar]
- 59.Sølling TI, Radom L. Int J Mass Spectrom. 1999;185–187:263–270. [Google Scholar]
- 60.Denmark SE, Collins WR, Cullen MD. J Am Chem Soc. 2009;131:3490–3492. doi: 10.1021/ja900187y. [DOI] [PubMed] [Google Scholar]
- 61.Denmark SE, Burk MT, Hoover AJ. J Am Chem Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]
- 62.For related studies on the generation and enantiospecific trapping of enantioenriched bromiranium ions see: Braddock DC, Marklew JS, Thomas AJF. Chem Commun. 2011;47:9051–9053. doi: 10.1039/c1cc13619d.Braddock DC, Hermitage SA, Kwok L, Pouwer R, Redmond JM, White AJP. Chem Commun. 2009:1082–1084. doi: 10.1039/b816914d.Braddock DC, Marklew JS, Foote KM, White AJP. Chirality. 2013;25:692–700. doi: 10.1002/chir.22194.
- 63.a) Fernández I, Cossío FP, Sierra MA. Chem Rev. 2009;109:6687–6711. doi: 10.1021/cr900209c. [DOI] [PubMed] [Google Scholar]; b) Reetz M. Angew Chem Int Ed Engl. 1972;11:129–130. [Google Scholar]
- 64.Grob CA, Winstein S. Helv Chim Acta. 1952;99:782–802. [Google Scholar]
- 65.Braddock DC, Roy D, Lenoir D, Moore E, Rzepa HS, Wu JI-C, von Ragué Schleyer P. Chem Commun. 2012;48:8943–8945. doi: 10.1039/c2cc33676f. [DOI] [PubMed] [Google Scholar]
- 66.Barilli PL, Bellucci G, Berti G, Marioni F, Marsili A, Morelli I. Chem Commun. 1970:1437–1438. [Google Scholar]
- 67.A catalyticsyn-dihalogenation of alkenes using molecular dihalogens in the presence of iron(III) complexes as catalysts has been claimed, but no product characterization data was reported, and the method has never been applied or reproduced; see: Tsipis CA, Katsoulos GA, Vakoulis FD. J Chem Soc, Chem Commun. 1985:1404–1405.
- 68.Based on the isotopic perturbation of degenerate equilibrium method, Ohta and co-workers have suggested that the tetramethylbromonium ion consists of rapidly equilibrating β-bromo carbocation structures, rather than a symmetrical, bridged bromiranium ion; see: Ohta BK, Hough RE, Schubert JW. Org Lett. 2007;9:2317–2320. doi: 10.1021/ol070673n.. Subsequently, Singleton and Bogle presented a reinterpretation of this data that refutes Ohta’s conclusion, supporting the notion of a symmetrical bromiranium ion; see: Bogle XS, Singleton DA. J Am Chem Soc. 2011;133:17172–17175. doi: 10.1021/ja2084288.
- 69.For evidence of open β-chloro carbocations as intermediates in a catalytic, enantioselective chlorolactonization reaction see: Yousefi R, Ashtekar KD, Whitehead DC, Jackson JE, Borhan B. J Am Chem Soc. 2013;135:14524–14527. doi: 10.1021/ja4072145.
- 70.a) Haubenstock H, Sauers RR. Tetrahedron. 2005;61:8358–8365. [Google Scholar]; b) Yamabe S, Tsuji T, Hirao K. Chem Phys Lett. 1988;146:236–242. [Google Scholar]; c) Berman DW, Anicich V, Beauchamp JL. J Am Chem Soc. 1979;101:1239–1248. [Google Scholar]; d) Olah GA, Westermann PW, Melby EG, Mo YK. J Am Chem Soc. 1974;96:3565–3573. [Google Scholar]; e) Olah GA, Bollinger JM, Mo YK, Brinich JM. J Am Chem Soc. 1972;94:1164–1168. [Google Scholar]; f) Olah GA, Bollinger JM. J Am Chem Soc. 1968;90:947–953. [Google Scholar]
- 71.Syn-addition of Cl2 to acenaphthalene [ Cristol SJ, Stermitz FR, Ramey PS. J Am Chem Soc. 1956;78:4939–4941.] and phenanthrene [ de la Mare PBD, Klassen NV, Koenigsberger R. J Chem Soc. 1961:5285–5293.] has been noted. Syn-addition was also suspected in the cases of 1-phenylpropene [ Fahey RC, Schubert C. J Am Chem Soc. 1965;87:5172–5179.] and stilbene [ Cristol SJ, Bly RS. J Am Chem Soc. 1960;82:142–145.]. The addition of Cl2 to cyclopentadiene has been observed to produce, inter alia, a 24% yield of the dichloride syn-3,4-dichlorocyclopentene; see: Heasley VL, Davis PD, Ingle DM, Rold KD, Heasley GE. J Org Chem. 1973;39:736–737.
- 72.Traylor TG. J Am Chem Soc. 1969;2:152–160. [Google Scholar]
- 73.For an example of oxidatively-induced reductive elimination from an β-chloroalkyl tellurium species to form a vicinal dichloride, see: Uemura S, Fukuzawa SI. J Organomet Chem. 1984;268:223–234.Uemura S, Fukuzawa S-I. J Chem Soc, Chem Commun. 1980:1033–1034.
- 74.Although β-chloro substituents are comparatively reluctant to engage in anchimeric assistance, β-bromo substituents do so more readily; see: Grunwald E. J Am Chem Soc. 1951;73:5458–5459.
- 75.For a conceptually similiar process involving anti-selective oxythallation of an olefin followed by expulsion of the Tl(III) nucleofuge by neighboring group participation from the β-acetoxy group, see: Anderson CB, Winstein S. J Org Chem. 1963;28:605–606.
- 76.For neighboring group participation by a β-phenyl group in the expulsion of a high-valent palladium nucleofuge, leading to stereoretentive C(sp3)–Cl bond formation, see: Bäckvall J-E, Nordberg RE. J Am Chem Soc. 1980;102:393–395.
- 77.Although not dihalogenations, oxidative 1,2-difunctionalizations of alkenes by thallium(III) salts are thought to proceed via heterolysis of the C–Tl bond in the thallated adducts to generate carbocation intermediates; see: Uemura S. In: Main Group Metals in Organic Synthesis. Yamamoto H, Oshima K, editors. Chapter 9 Wiley-VCH; Weinheim: 2004.
- 78.Wyman DP, Wang JYC, Freeman WR. J Org Chem. 1963;28:3173–3177. [Google Scholar]
- 79.Rodebaugh R, Debenham JS, Fraser-Reid B, Snyder JP. J Org Chem. 1999;64:1758–1761. doi: 10.1021/jo9718509. [DOI] [PubMed] [Google Scholar]
- 80.For reviews of palladium-catalyzed alkene difunctionalizations see: McDonald RI, Liu G, Stahl SS. Chem Rev. 2011;111:2981–3019. doi: 10.1021/cr100371y.Sehnal P, Taylor RJK, Fairlamb IJS. Chem Rev. 2010;110:824–889. doi: 10.1021/cr9003242.Xu LM, Li BJ, Yang Z, Shi ZJ. Chem Soc Rev. 2010;39:712–733. doi: 10.1039/b809912j.Jacques B, Muñiz K. In: Catalyzed Carbon-Heteroatom Bond Formation. Yudin AK, editor. Chapter 4 Wiley-VCH; Weinheim: 2010. Muñiz K. Angew Chem Int Ed. 2009;48:9412–9423. doi: 10.1002/anie.200903671.Jensen KH, Sigman MS. Org Biomol Chem. 2008;6:4083–4088. doi: 10.1039/b813246a.For a representative gold-catalyzed alkene difunctionalization see: de Haro T, Nevado C. Angew Chem Int Ed. 2011;50:906–10. doi: 10.1002/anie.201005763.For reviews of iodine-mediated/catalyzed alkene difunctionalizations see: Brown M, Farid U, Wirth T. Synlett. 2013;24:424–431.Finkbeiner P, Nachtsheim B. Synthesis. 2013;45:979–999.
- 81.For a review of catalytic aminohalogenations see: Chemler SR, Bovino MT. ACS Catal. 2013;3:1076–1091. doi: 10.1021/cs4001249.
- 82.a) Wipke WT, Goeke GL. J Am Chem Soc. 1974;96:4244–4249. [Google Scholar]; b) Wiger G, Albelo G, Rettig MF. J Chem Soc, Dalton Trans. 1974:2242–2247. [Google Scholar]
- 83.a) Yin G, Wu T, Liu G. Chem Eur J. 2012;18:451–455. doi: 10.1002/chem.201102776. [DOI] [PubMed] [Google Scholar]; b) Kalyani D, Satterfield AD, Sanford MS. J Am Chem Soc. 2010;132:8419–8427. doi: 10.1021/ja101851v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kalyani D, Sanford MS. J Am Chem Soc. 2008;130:2150–2151. doi: 10.1021/ja0782798. [DOI] [PubMed] [Google Scholar]; d) Yin G, Liu G. Angew Chem, Int Ed. 2008;47:5442–5445. doi: 10.1002/anie.200801438. [DOI] [PubMed] [Google Scholar]; e) Zhu G, Lu X. J Organomet Chem. 1996;508:83–90. [Google Scholar]; f) Zhu G, Ma S, Lu X, Huang Q. J Chem Soc, Chem Commun. 1995:271–273. [Google Scholar]; g) Pearson RG, Muir WR. J Am Chem Soc. 1970;92:5519–5520. [Google Scholar]; h) Johnson RW, Pearson RG. Chem Commun. 1970:986–987. [Google Scholar]; i) Coulson DR. J Am Chem Soc. 1969;91:200–202. [Google Scholar]
- 84.Bäckvall J-E. Tetrahedron Lett. 1977;18:467–468. [Google Scholar]
- 85.For an example of either stereoretention or -inversion as a function of reaction conditions, see: Wong PK, Stille JK. J Organomet Chem. 1974;70:121–132.
- 86.Friedrich HB, Moss JR. Adv Organomet Chem. 1991;33:235–290. [Google Scholar]
- 87.Luo YR. Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton, FL: 2007. [Google Scholar]
- 88.a) Cundari TR, Taylor CD. Organometallics. 2003;22:4047–4059. [Google Scholar]; b) Stockland RA, Foley SR, Jordan RF. J Am Chem Soc. 2003;125:796–809. doi: 10.1021/ja028530d. [DOI] [PubMed] [Google Scholar]; c) Strazisar SA, Wolczanski PT. J Am Chem Soc. 2001;123:4728–4740. doi: 10.1021/ja003315n. [DOI] [PubMed] [Google Scholar]; d) Stockland RA, Jordan RF. J Am Chem Soc. 2000;122:6315–6316. [Google Scholar]
- 89.a) Carpenter AE, McNeece AJ, Barnett BR, Estrada AL, Mokhtarzadeh CC, Moore CE, Rheingold AL, Perrin CL, Figueroa JS. J Am Chem Soc. 2014;136:15481–15484. doi: 10.1021/ja508956q. [DOI] [PubMed] [Google Scholar]; b) Foley SR, Stockland RA, Shen H, Jordan RF. J Am Chem Soc. 2003;125:4350–4361. doi: 10.1021/ja029823+. [DOI] [PubMed] [Google Scholar]
- 90.a) Lu X. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-i., editor. Chapter V.3.5 Wiley; New York: 2002. [Google Scholar]; b) Ogawa A. In: Handbook of Organopalladium Chemistry for Organic Synthesis. Negishi E-i., editor. Chapter VII.6 Wiley; New York: 2002. [Google Scholar]
- 91.Bäckvall J-E, Nilsson YIM, Gatti RGP. Organometallics. 1995;14:4242–4246. [Google Scholar]
- 92.For the proposed syn-chloropalladation of a vinyl chloride see: Henry PM. J Org Chem. 1972;37:2443–2447.
- 93.Bäckvall JE, Bergaman J, Engman L. J Org Chem. 1983;48:3918–3923. [Google Scholar]
- 94.Sharpless KB, Teranishi AY, Bäckvall J-E. J Am Chem Soc. 1977;99:3120–3128. [Google Scholar]
- 95.Barton DHR, Miller E. J Am Chem Soc. 1950;72:370–374. [Google Scholar]
- 96.Mylonas VE, Sigalas MP, Katsoulos GA, Tsipis CA, Varvoglis AG. J Chem Soc, Perkin Trans 2. 1994:1691–1696. [Google Scholar]
- 97.Nelson DJ, Li R, Brammer C. J Phys Org Chem. 2004;17:1033–1038. [Google Scholar]
- 98.Norrby P-O, Gable KP. J Chem Soc, Perkin Trans 2. 1996:171–178. and references cited therein. [Google Scholar]
- 99.DelMonte AJ, Haller J, Houk KN, Sharpless KB, Singleton DA, Strassner T, Thomas AA. J Am Chem Soc. 1997;119:9907–9908. [Google Scholar]
- 100.a) Haunschild R, Loschen C, Tüllmann S, Cappel D, Hölscher M, Holthausen MC, Frenking G. J Phys Org Chem. 2007;20:11–18. [Google Scholar]; b) Deubel DV, Loschen C, Frenking G. Top Organomet Chem. 2005;12:109–144. [Google Scholar]; c) Hölscher M, Leitner W, Holthausen MC, Frenking G. Chem Eur J. 2005;11:4700–4708. doi: 10.1002/chem.200500217. [DOI] [PubMed] [Google Scholar]
- 101.Radical additions to alkenes can in principle be stereospecific if the intermediate carbon radical can be captured stereoselectively prior to C–C bond rotation. For example, the radical addition of HBr to (E)- and (Z)-2-bromo-2-butene is highly anti-stereospecific at low temperature (−78 °C), becoming almost non-stereospecific at room temperature; see: Goering HL, Larsen DW. J Am Chem Soc. 1959;81:5937–5942.
- 102.Uemura S, Sasaki O, Okano M. Bull Chem Soc Jpn. 1972;45:1482–1484. [Google Scholar]
- 103.Uemura S, Onoe A, Okano M. Bull Chem Soc Jpn. 1975;48:3702–3705. [Google Scholar]
- 104.Onoe A, Uemura S, Okano M. Bull Chem Soc Jpn. 1976;49:345–346. [Google Scholar]
- 105.For example, tin(IV) chloride (SnCl4) was found to be unreactive with norbornene; see: ref. 103.
- 106.Henniger PW, Wapenaar E, Havinga E. Rec Trav Chim. 1966;85:1177–1187. [Google Scholar]
- 107.Uemura S, Onoe A, Okazaki H, Okano M, Ichikawa K. Bull Chem Soc Jpn. 1976;49:1437–1438. [Google Scholar]
- 108.For the difluorination of perhalogenated alkenes with lead(IV) fluorides see: Bissell ER, Fields DB. J Org Chem. 1964;29:1591–1593.Henne AL, Newby TH. J Am Chem Soc. 1948;70:130–132.Henne AL, Waalkes TP. J Am Chem Soc. 1945;67:1639–1640.
- 109.Dimroth O, Bockemüller W. Chem Ber. 1931;64:516–522. [Google Scholar]
- 110.Bornstein J, Skarlos L. J Am Chem Soc. 1968;90:5044. [Google Scholar]
- 111.Bowers A, Holton PG, Denot E, Loza MC, Urquiza R. J Am Chem Soc. 1962;84:1050–1053.Bowers A, Denot E, Urquiza R. Tetrahedron Lett. 1960;1:34–37.Also see: Bruckner K, Mannhardt HJ. 1167828. German Patent. 1964
- 112.Tanner DD, Van Bostelen P. J Am Chem Soc. 1972;94:3187–3195. [Google Scholar]
- 113.a) Strand JW, Kovacic P. Synth Commun. 1972;2:129–137. [Google Scholar]; b) Field KW, Kovacic P. J Org Chem. 1971;36:3566–3571. [Google Scholar]; c) Field KW, Kovacic P. Synthesis. 1969:135. [Google Scholar]
- 114.Kovacic P, Chaudhary SS. Org Synth. 1968;48:4. [Google Scholar]
- 115.Heasley VL, Rold KD, McKee DB. J Org Chem. 1976;41:1287–1289. [Google Scholar]
- 116.Shellhamer DF, McKee DB, Leach CT. J Org Chem. 1976;41:1972–1976. [Google Scholar]
- 117.Shao L-X, Shi M. Synlett. 2006:1269–1271. [Google Scholar]
- 118.For an early example of alkene dichlorination with PCl5, see: Spiegler L, Tinker JM. J Am Chem Soc. 1939;61:940–942.
- 119.Pudovik NA, Khairullin VK. Russ Chem Rev. 1968;37:317–332. [Google Scholar]
- 120.Russell GA, Ito A, Konaka R. J Am Chem Soc. 1963;85:2988–2991. [Google Scholar]
- 121.Uemura S, Okazaki H, Onoe A, Okano M. Bull Chem Soc Jpn. 1978;51:3568–3570. [Google Scholar]
- 122.Suter RW, Knachel HC, Petro VP, Howatson JH, Shore SG. J Am Chem Soc. 1973;95:1474–1479. [Google Scholar]
- 123.a) Buckles RF, Knaack DF. J Org Chem. 1960;25:20–24. [Google Scholar]; b) Hoff MC, Greenlee KW, Boord CE. J Am Chem Soc. 1951;73:3329–3336. [Google Scholar]; c) Radcliffe MR, Best CE. 2445729. US Patent. 1948; d) Nieuwland JA. Ind Eng Chem. 1935;27:850–854. [Google Scholar]
- 124.Van de Walle H. Bull Soc Chim Belg. 1919;28:304. [Google Scholar]
- 125.a) Uemura S, Onoe A, Okano M. Bull Chem Soc Jpn. 1974;47:692–697. [Google Scholar]; b) Uemura S, Sasaki O, Okano M. Chem Commun. 1971:1064–1065. [Google Scholar]
- 126.For the chlorination of (Z,Z)-cycloocta-1,5-diene and (Z)-cyclooctene with SbCl4 to give dichlorides resulting from transannular reactions see: Uemura S, Onoe A, Okano M. Bull Chem Soc Jpn. 1977;50:1078–1081.Uemura S, Onoe A, Okano M. J Chem Soc, Chem Commun. 1975:210–211.
- 127.Heasley VL, Rold KD, Titterington DR, Leach CT, Gipe BT, McKee DB, Heasley GE. J Org Chem. 1976;41:3997–4001. [Google Scholar]
- 128.Akiyama F, Horie T, Matsuda M. Bull Chem Soc Jpn. 1973;46:1888–1890. [Google Scholar]
- 129.Vignes RP, Hamer J. J Org Chem. 1974;39:849–851. [Google Scholar]
- 130.The term “stereospecific” is used throughout this Review to refer to reactions in which two different stereoisomeric starting materials (enantiomers or diastereoisomers) react to give two different stereoisomeric products. For reactions exhibiting imperfect stereospecificity, the degree of stereospecificity can be expressed as a percentage; see: Eliel E, Wilen S. Stereochemistry of Organic Compounds. Wiley; New York: 1994. p. 837.Fleming I. Molecular Orbitals and Organic Chemical Reactions: Reference Edition. Wiley; Chichester, U.K: 2010. pp. 205–207.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; 2006. pp. 319–320.
- 131.Kolditz L, Preiss H. Z Anorg Allg Chem. 1961;310:242–247. [Google Scholar]
- 132.Vignes R, Hamer J. J Org Chem. 1974;39:849–851. [Google Scholar]
- 133.a) Beattie IR, Gilson T, Livingston K, Fawcett V, Ozin GA. J Chem Soc, A. 1967:712–718. [Google Scholar]; b) Carlson GL. Spectrochim Acta. 1963;19:1291–1307. [Google Scholar]
- 134.The molecular SbCl5 unit persists in the solid state structure; see: Ohlberg SM. J Am Chem Soc. 1959;81:811–813.
- 135.Brockner W, Cyvin SJ, Hovdan H. Inorg Nucl Chem Lett. 1979;11:171–176. [Google Scholar]
- 136.It has been suggested that SbCl4 + is the active chlorinating species in the reaction of benzene, toluene, and chlorobenzene with SbCl5 (below 40 °C), where an excess aromatic component (ε = 2.27–5.61) is used as the solvent; see: Kovacic P, Sparks AK. J Am Chem Soc. 1960;82:5740–5743.
- 137.Hughes ED, Ingold CK. J Chem Soc. 1935:244–255. [Google Scholar]
- 138.Hammond GS. J Am Chem Soc. 1955;77:334–338. [Google Scholar]
- 139.It is known that cyclopentene prefers electrophilic addition on the same face of the alkene as the C(4) methylene group due to minimization of torsional strain in the transition state; see: Poli G. Tetrahedron Lett. 1989;30:7385–7388.
- 140.a) Uemura S, Okazaki H, Onoe A, Okano M. J Chem Soc, Perkin Trans 1. 1979:548–552. [Google Scholar]; b) Uemura S, Onoe A, Okano M. J Chem Soc, Chem Commun. 1976:145. [Google Scholar]
- 141.For early (pre-1940) examples of alkene dichlorination with SO2Cl2 see: Friese H, Djiang D. Ber Dtsch Chem Ges. 1938;71:667–670.Durrans TH. J Chem Soc, Trans. 1923;123:1424–1429.Norris JF, Thomas R, Brown BM. Ber Dtsch Chem Ges. 1910;43:2940–2959.
- 142.Kharasch MS, Brown HC. J Am Chem Soc. 1939;61:3432–3434. [Google Scholar]
- 143.Russell GA. J Am Chem Soc. 1958;80:5002–5003. [Google Scholar]
- 144.Kharasch MS, Zavist AF. J Am Chem Soc. 1951;73:964–967. doi: 10.1021/ja01190a518. [DOI] [PubMed] [Google Scholar]
- 145.Uemura S, Masaki G, Toshimitsu A, Sawada S. Bull Chem Soc Jpn. 1981;54:2843–2844. [Google Scholar]
- 146.Hojo M, Matsuda R. Synth Commun. 1975;5:169–171. [Google Scholar]
- 147.Riley RF, Flato J, Bengels D. J Org Chem. 1962;27:2651–2653. [Google Scholar]
- 148.a) Engman L. J Org Chem. 1987;52:4086–4094. [Google Scholar]; b) Garratt DG, Schmid GH. Can J Chem. 1974;52:3599–3606. [Google Scholar]
- 149.Paulmier C. Phosphorus Sulfur Silicon Relat Elem. 2001;172:25–54. [Google Scholar]
- 150.a) Morella AM, Ward DA. Tetrahedron Lett. 1985;26:2899–2900. [Google Scholar]; b) Morella AM, Ward DA. Tetrahedron Lett. 1984;25:1197–1200. [Google Scholar]
- 151.Willgerodt O. J Prakt Chem. 1886;33:154–160. [Google Scholar]
- 152.For reviews see: Zhdankin VV, Stang PJ. Chem Rev. 2008;108:5299–5358. doi: 10.1021/cr800332c.Zhdankin V, Stang P. Chem Rev. 2002;102:2523–2584. doi: 10.1021/cr010003+.Varvoglis A. Hypervalent Iodine in Organic Synthesis. Academic Press; London: 1997. Varvoglis A. Tetrahedron. 1997;53:1179–1255.Stang PJ, Zhdankin VV. Chem Rev. 1996;96:1123–1178. doi: 10.1021/cr940424+.Varvoglis A. Synthesis. 1984:709–726.Banks DF. Chem Rev. 1966;66:243–266.
- 153.Lucas HJ, Kennedy ER. Org Synth. 1942;22:69. [Google Scholar]
- 154.Podgoršek A, Iskra J. Molecules. 2010;15:2857–2871. doi: 10.3390/molecules15042857. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.a) Keefer RM, Andrews LJ. J Am Chem Soc. 1958;80:5350–5355. [Google Scholar]; b) Andrews LJ, Keefer RM. J Am Chem Soc. 1958;80:1723–1728. [Google Scholar]; c) Keefer RM, Andrews LJ. J Am Chem Soc. 1958;80:277–281. [Google Scholar]
- 156.Andrews LJ, Keefer RM. J Am Chem Soc. 1960;82:3059–3063. [Google Scholar]
- 157.a) Jeffery EA, Andrews LJ, Keefer RM. J Org Chem. 1965;30:617–620. [Google Scholar]; b) Andrews LJ, Spears LJ, Keefer RM. J Am Chem Soc. 1964;86:687–690. [Google Scholar]; c) Keefer RM, Andrews LJ. J Am Chem Soc. 1959;81:5329–5333. [Google Scholar]; d) Andrews LJ, Keefer RM. J Am Chem Soc. 1959;81:4218–4223. [Google Scholar]; e) Keefer RM, Andrews LJ. J Am Chem Soc. 1959;81:2374–2379. [Google Scholar]
- 158.Tanner DT, Gidley GC. J Org Chem. 1968;33:38–43. [Google Scholar]
- 159.Masson S, Thuillier A. Bull Soc Chim Fr. 1969:4368–4377. [Google Scholar]
- 160.Bloomfield GF. J Chem Soc. 1944:114–120. [Google Scholar]
- 161.Zarecki A, Wicha J, Kocor M. Tetrahedron. 1976;32:559–563. [Google Scholar]
- 162.a) Tanner DD, Van Bostelen PB. J Org Chem. 1967;32:1517–1521. [Google Scholar]; b) Banks DF, Huyser ES, Kleinberg J. J Org Chem. 1964;29:3692–3693. [Google Scholar]
- 163.Dewar MJS, Fahey RC. J Am Chem Soc. 1963;85:2248–2252. [Google Scholar]
- 164.Heasley GE, Bower TR, Dougharty KW, Easdon JC, Heasley VL, Arnold S, Carter TL, Yaeger DB, Gipe BT, Shellhamer DP. J Org Chem. 1980;45:5150–5155. [Google Scholar]
- 165.Shellhamer DP, Ragains ML, Gipe BT, Heasley VL. J Fluorine Chem. 1982;20:13–18. [Google Scholar]
- 166.Masson S, Thuillier A. C R Hebd Seances Acad Sci, Ser C. 1967;264:1189–1192. [Google Scholar]
- 167.Berg CJ, Wallis ES. J Biol Chem. 1946;162:683–693. [PubMed] [Google Scholar]
- 168.However, Tanner and Gidley have noted that carrying out the dichlorination of norbornene in CCl4 at reflux (80 °C) in an open vessel proceeds mainly by a (fast) radical addition process even when air is bubbled through the solution, and this is attributed to the low solubility of dioxygen (as a radical inhibitor) in CCl4 at this temperature (see ref. 158). This observation makes it difficult to judge whether some reports of very highly anti-selective dichlorinations of cyclic alkenes (which were not specified as being performed under oxygen-free conditions) are actually proceeding via radical or ionic mechanisms.
- 169.Summerbell RK, Lunk HE. J Am Chem Soc. 1957;79:4802–4805. [Google Scholar]
- 170.Campbell JR, Jones JKN, Wolfe S. Can J Chem. 1966;44:2339–2342. [Google Scholar]
- 171.Devillier M, Bodot H. Bull Soc Chim Fr. 1972:227–232. [Google Scholar]
- 172.Wicha J, Zarecki A, Kocór M. Tetrahedron Lett. 1973;14:3635–3638. [Google Scholar]
- 173.Ngatimin M, Gartshore CJ, Kindler JP, Naidu S, Lupton DW. Tetrahedron Lett. 2009;50:6008–6011. [Google Scholar]
- 174.Hara S, Nakahigashi J, Ishi-i K, Sawaguchi M, Sakai H, Fukuhara T, Yoneda N. Synlett. 1998:495–496. [Google Scholar]
- 175.Holton PG, Cross AD, Bowers A. Steroids. 1963;2:71–79. [Google Scholar]
- 176.Kovacic P, Lange RM. J Org Chem. 1965;30:4251–4254. [Google Scholar]
- 177.a) Brown TM, McCann EL. Inorg Synth. 1970;12:181–186. [Google Scholar]; b) Kepert DL, Mandyczewsky R. Inorg Chem. 1968;7:2091–2093. [Google Scholar]; c) Brown TM, McCann EL. Inorg Chem. 1968;7:1227–1229. [Google Scholar]
- 178.Uemura S, Onoe A, Okano M. Bull Chem Soc Jpn. 1974;47:3121–3124. [Google Scholar]
- 179.San Filippo JS, Jr, Sowinski AF, Romano LJ. J Am Chem Soc. 1975;97:1599–1600. [Google Scholar]
- 180.For the syn-dichlorination of alkynes with MoCl5, see ref. 179.
- 181.Cotton FA, Wilkinson G. Advanced Inorganic Chemistry. 3. Interscience; New York: 1972. p. 958. [Google Scholar]
- 182.Larson ML. J Am Chem Soc. 1960;82:1223–1226. [Google Scholar]
- 183.Nugent WA. Tetrahedron Lett. 1978;19:3427–3430. [Google Scholar]
- 184.Piovesana O, Furlani C. Inorg Nucl Chem Lett. 1967;3:535–538. [Google Scholar]
- 185.Horner SM, Tyree SY. Inorg Chem. 1962;1:122–127. [Google Scholar]
- 186.a) Hazra BG, Pore VS. J Indian Chem Soc. 2003;80:1065–1071. [Google Scholar]; b) Hazra BG, Chordia MD, Basu S, Bahule BB, Pore VS, Naskar DB. J Chem Res (S) 1998:8–9. [Google Scholar]; c) Hazra BG, Chordia MD, Basu S, Bahule BB, Pore VS, Naskar DB. J Chem Res (M) 1998:143–150. [Google Scholar]
- 187.Hazra BG, Chordia MD, Bahule BB, Pore VS, Basu S. J Chem Soc, Perkin Trans 1. 1994:1667–1669. [Google Scholar]
- 188.Boyes AL, Wild M. Tetrahedron Lett. 1998;39:6725–6728. [Google Scholar]
- 189.It is possible for instance that the selectivity-determining steps are different under the two sets of conditions. Thus, if chloriranium ion formation is irreversible under Markó’s conditions and kinetically in favour of attack on the olefin face syn to the oxirane, then transannular participation by the epoxide becomes stereochemically unfeasible. On the other hand, if chloriranium ion (or alkene-Cl2 π-complex) formation is reversible under the conditions with Cl2, then the nucleophilic opening (or direct attack on the π-complex) becomes selectivity-determining, and transannular trapping by the oxirane might be expected to outpace the attack of chloride ion.
- 190.Shibuya GM, Kanady JS, Vanderwal CD. J Am Chem Soc. 2008;130:12514–12518. doi: 10.1021/ja804167v. [DOI] [PubMed] [Google Scholar]
- 191.Brigg TS. Inorg Nucl Chem. 1968;30:2866–2869. [Google Scholar]
- 192.Eaton GR, Eaton SS, Barr DP, Weber RT. Quantitative EPR. Springer; New York: 2010. pp. 32–34. [Google Scholar]
- 193.a) Adam W, Mock-Knoblauch C, Saha-Möller CR, Herderich M. J Am Chem Soc. 2000;122:9685–9691. [Google Scholar]; b) Chandra SK, Basu P, Ray D, Pal S, Chakravorty A. Inorg Chem. 1990;29:2423–2428. [Google Scholar]; c) Kessissoglou DP, Li X, Butler WM, Pecoraro VL. Inorg Chem. 1987;26:2487–2492. [Google Scholar]; d) Burriel R, O’Connor CJ, Carlin RL. Inorg Chem. 1985;24:3706–3708. [Google Scholar]; e) Camenzind MJ, Hollander FJ, Hill CL. Inorg Chem. 1983;22:3776–3784. [Google Scholar]
- 194.On a historical note, Scheele’s discovery of molecular Cl2 actually involved the oxidation of chloride ion by MnO2; see: Weeks ME. Discovery of the Elements. 3. Journal of Chemical Education; Easton, PA: 1935. pp. 253–257.. This process has also been used industrially for the manufacture of Cl2 (i.e., MnO2 + 4 HCl → MnCl2 + 2 H2O + Cl2); see: Greenwood NN, Earnshaw A. Chemistry of the Elements. 2. Butterworth-Heinemann; Oxford: 1997. p. 1048.
- 195.On this basis, the reported loss of chlorination activity on warming could simply be the result of the Cl2 steadily reacting with the CH2Cl2 solvent to give CHCl3 and HCl – a process which has been used to prepare CHCl3 on an industrial scale under mild conditions (35–50 °C) (see 2316534. Russian Patent. 2008). The fact that the reagent system must be kept below around −40 °C when using (COCl)2 as the promoter may be a consequence of Cl2 reacting at more elevated temperatures with dissolved CO [an expected by-product from (COCl)2].
- 196.Donnelly KD, Fristad WE, Gellerman BJ, Peterson JR, Selle BJ. Tetrahedron Lett. 1984;25:607–610. [Google Scholar]
- 197.Bellesia F, Ghelfi F, Pagnoni UM, Pinetti A. Synth Commun. 1991;21:489–494. [Google Scholar]
- 198.Yakabe S, Hirano M, Morimoto T. Synth Commun. 1998;28:1871–1878. [Google Scholar]
- 199.a) Bellesia F, Ghelfi F, Pagnoni UM, Pinetti A. J Chem Res (S) 1989:360–361. [Google Scholar]; b) Bellesia F, Ghelfi F, Pagnoni UM, Pinetti A. J Chem Res (S) 1989:108–109. [Google Scholar]
- 200.Law NA, Machonkin TE, McGorman JP, Larson EJ, Kampf JW, Pecoraro VL. J Chem Soc, Chem Commun. 1995:2015–2016. [Google Scholar]
- 201.Brignole A, Cotton FA. Chem Commun. 1971:706a. [Google Scholar]
- 202.Kovacic P, Brace NO. J Am Chem Soc. 1954;76:5491–5494. [Google Scholar]
- 203.Sakai K, Sugimoto K, Shigeizumi S, Kondo K. Tetrahedron Lett. 1994;35:737–740. [Google Scholar]
- 204.Kharasch MS, Jensen EV, Urry WH. Science. 1945;102:128. doi: 10.1126/science.102.2640.128. [DOI] [PubMed] [Google Scholar]
- 205.Rausch DA, Davis RA, Osbourne DW. J Org Chem. 1963;28:494–497. [Google Scholar]
- 206.Although Henry and co-workers have reported a palladium-catalyzed dibromination of alkenes (see ref. 20), this work is not discussed in this Review due to concerns over its reproducibility (see endnote 21) and incomplete Supporting Information.
- 207.Bäckvall J-E, Jonasson C. Tetrahedron Lett. 1997;38:291–294. [Google Scholar]
- 208.McCall AS, Wang H, Desper JM, Kraft S. J Am Chem Soc. 2011;133:1832–1848. doi: 10.1021/ja107342b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.McCall AS, Kraft S. Organometallics. 2012;31:3527–3538. [Google Scholar]
- 210.Magistro AJ, Cowfer JA. J Chem Educ. 1986;63:1056–1058. [Google Scholar]
- 211.a) Nicholas PP, Carroll RT. J Org Chem. 1968;33:2345–2349. [Google Scholar]; b) Arganbright RP, Yates WF. J Org Chem. 1962;27:1205–1208. [Google Scholar]
- 212.Hall PG, Parsley M, Rosseinsky DR, Mann RA, Waugh KC. J Chem Soc, Faraday Trans 1. 1983;79:343–361. [Google Scholar]
- 213.Although some chlorine production was detected at 320 °C by flowing nitrogen (rather than ethylene gas) over the solid-supported CuCl2, the measured rate of Cl2 generation was only around 1.8% that expected if CuCl2 was rapidly and reversibly undergoing disproportionation to CuCl2 and Cl2 (based upon the expected partial pressure of Cl2 at equilibrium and taking into account the nitrogen gas flow rate). Moreover, the rate of 1,2-dichloroethane production was 167 times faster than the (separately measured) rate of Cl2 generation in the absence of ethylene. See ref. 211a.
- 214.Kochi JK. J Am Chem Soc. 1962;84:2121–2127. [Google Scholar]
- 215.Spector ML, Heinemann H, Miller KD. Ind Eng Chem Process Des Develop. 1967;6:327–331. [Google Scholar]
- 216.Ichikawa K, Uemura S, Hiramoto T, Takagaki Y. Kogyo Kagaku Zasshi. 1968;71:1657–1663. [Google Scholar]
- 217.Ichikawa K, Uemura S, Hiramoto T, Takagaki Y. Kogyo Kagaku Zasshi. 1969;72:2577–2580. [Google Scholar]
- 218.Ichikawa K, Uemura S, Hiramoto T, Takagaki Y. Kogyo Kagaku Zasshi. 1969;72:2390–2392. [Google Scholar]
- 219.Ichikawa K, Uemura S, Hiramoto T, Takagaki Y. Kogyo Kagaku Zasshi. 1969;72:1096–1098. [Google Scholar]
- 220.Ichikawa K, Uemura S, Hiramoto T, Takagaki Y. Bull Japan Petrol Inst. 1970;12:77–83. [Google Scholar]
- 221.Koyano T. Bull Chem Soc Jpn. 1970;43:1439–1443. [Google Scholar]
- 222.Koyano T. Bull Chem Soc Jpn. 1970;43:3501–3504. [Google Scholar]
- 223.Koyano T, Watanabe O. Bull Chem Soc Jpn. 1971;44:1378–1381. [Google Scholar]
- 224.Baird WC, Jr, Surridge JH, Buza M. J Org Chem. 1971;36:3324–3330. [Google Scholar]
- 225.There is conflicting information on the effect of added chloride salts on dichlorinations with CuCl2 in MeCN. Baird and co-workers have noted that the addition of 1.0 equiv of KCl (w.r.t. to CuCl2) led to a 5-fold reduction in the yield of dichlorides, which was attributed to the formation of inactive polychloro copper(II) complexes; see ref. 224.
- 226.a) Ball MC, Coultard RFM. J Chem Soc A. 1968:1417–1419. [Google Scholar]; b) Castro CE, Gaughan EJ, Owsley DC. J Org Chem. 1965;30:587–592. [Google Scholar]
- 227.Bennet MA. Chem Rev. 1962;62:611–652. [Google Scholar]
- 228.Uemura and co-workers proposed a very similar sequence of events but were less explicit in their description of how the CuCl2 may activate the olefin, depicting a partially-formed, bridged chloriranium ion still bound to CuCl, which does not account for the stereochemical results of Koyano et al.. See ref. 220.
- 229.a) Sequeira FC, Chemler SR. Org Lett. 2012;14:4482–4485. doi: 10.1021/ol301984b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miller Y, Miao L, Hosseini A, Chemler SR. J Am Chem Soc. 2012;134:12149–12156. doi: 10.1021/ja3034075. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Paderes MC, Belding L, Fanovic B, Dudding T, Keister JB. Chem Eur J. 2012;18:1711–1726. doi: 10.1002/chem.201101703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Koyano T. Bull Chem Soc Jpn. 1971;44:1158–1160. [Google Scholar]
- 231.Schneider W, Zelewsky AV. Helv Chim Acta. 1963;46:1848–1863. [Google Scholar]
- 232.Ma S, Wu S. J Org Chem. 1999;64:9314–9317. [Google Scholar]
- 233.a) Uemura S, Okazaki H, Onoe A, Okano M. J Chem Soc, Perkin Trans 1. 1977:676–680. [Google Scholar]; b) Uemura S, Onoe A, Okano M. J Chem Soc, Chem Commun. 1975:925–926. [Google Scholar]
- 234.a) Xiang J, Yuan R, Wang R, Yi N, Lu L, Zou H, He W. J Org Chem. 2014;79:11378–11382. doi: 10.1021/jo501776b. [DOI] [PubMed] [Google Scholar]; b) Uemura S, Okazaki H, Okano M. J Chem Soc, Perkin Trans 1. 1978:1278–1282. [Google Scholar]
- 235.Hüttel R, Reinheimer H, Nowak K. Chem Ber. 1968;101:3761–3776. [Google Scholar]