

Abstract
The United States Food and Drug Administration (FDA) issued the dietary supplement (DS) current good manufacturing practice (GMP) regulations in compliance with the mandate from the Dietary Supplements Health and Education Act (DSHEA), with the intention of protecting public health by ensuring the quality of DS. The GMP regulations require manufacturers to establish their own quality specifications for identity, purity, strength, composition, and absence of contaminants. Numerous FDA‐conducted GMP inspections found that the private specifications set by these manufacturers are often insufficient to ensure adequate quality of dietary ingredients and DS. Wider use of the public standards developed by the United States Pharmacopeial Convention (USP), in conjunction with GMP compliance, can help ensure quality and consistency of DS as they do for medicines. Public health protection could be enhanced by strengthening the GMP provisions to require conformance with relevant United States Pharmacopeia–National Formulary (USP–NF) standards, or in the absence of USP standards, other public compendial standards. Another serious concern is the presence of synthetic drugs and drug analogues in products marketed as DS. Use of the new USP General Chapter Adulteration of Dietary Supplements with Drugs and Drug Analogs <2251> may reduce the exposure of consumers to dangerous drugs disguised as DS. © 2016 The Authors. Drug Testing and Analysis published by John Wiley & Sons Ltd.
Keywords: Dietary Supplements, DSHEA, Good Manufacturing Practices, USP–NF, Adulteration, PDE5
Dietary Supplements Health and Education Act (DSHEA), good manufacturing practice (GMP), and unresolved issues with quality of dietary supplements (DS)
In the United States, DSHEA (Public Law 103‐417, October 25, 1994) provides a regulatory framework for manufacturing and marketing DS that are intended to supplement the diet and contain dietary ingredients (vitamin, mineral, herb or other botanical, amino acid, or a concentrate, metabolite, constituent, or extract, or combination of any of these ingredients). DSHEA enabled consumer access to supplementation with the idea of promoting and maintaining health, and at the same time, it enabled the industry in the USA to grow from an initial base of about $4 billion in 1994 to an estimated $35 billion in 2015.1 Under DSHEA, DS manufacturers are responsible for establishing the safety and quality of a product, but they are not required to share that information with the Food and Drug Administration (FDA) before the product enters the market, unless it contains a new dietary ingredient (NDI), i.e., introduced to the market after October 15, 1994. Manufacturers are required to file a 75‐day pre‐market notification to the FDA for any NDIs, with the information based on which the manufacturers reached the conclusion of reasonable expectation of safety. The FDA has the authority to remove a product from the marketplace if it presents ‘significant or unreasonable risk of illness or injury’. However, the FDA is charged with the responsibility of proving that the product presents such a risk to public health, which is a resource‐intensive activity. It took a concerted effort pooling resources from multiple agencies to initiate legal action with the Department of Justice leading the charge.2 It remains to be seen if similar actions will be regularly in place to monitor compliance. Because the FDA has limited resources to ascertain harm, products of dubious quality can stay in the market without consequence. Moreover, by taking advantage of DSHEA's minimum requirements of pre‐marketing oversight, prescription drugs and their analogues, masqueraded as DS, have been illegally introduced.3 In addition, deficiencies in the current surveillance system have been reported.4, 5, 6
To address quality issues, DSHEA gave the FDA authority to prescribe and implement current GMPs for DS. Thirteen years after the passing of DSHEA, GMP became a rule in the Code of Federal Regulations (CFR) in 21 CFR Part 111 [Current Good Manufacturing Practice in Manufacturing, Packaging, Labeling, or Holding Operations for Dietary Supplements].7 Regarding the quality of DS, GMPs require manufacturers to establish their own specifications for dietary ingredients, other components, in‐process materials, and finished dietary supplements. Implementation of GMP requirements through manufacturer's private specifications has led to a lack of uniformity across the industry.8 Two products from different manufacturers may both carry an identical label, yet be formulated to very different quality specifications. GMP regulations also allow end‐product testing by just one of the established specifications to serve as a proxy for all quality attributes of the finished DS (21 CFR 111.75). Therefore, manufacturers may choose different tests to evaluate the quality of DS, even if they contain the same ingredient or are labelled with the same generic name. With regard to the analytical methods used as part of these specifications, GMP regulations require manufacturers to verify that the laboratory examination and testing methodologies are appropriate for their intended use, and to identify and use appropriate scientifically valid method(s) for each established specification for which testing or examination is required (21 CFR 111.320), but the rule does not require a complete formal validation for the analytical procedures.
Even with these limited requirements, there were numerous non‐compliance observations noted by the FDA during GMP inspections, where the analytical methods employed were either non‐specific or not fit for purpose.9 As an example of the inadequacy of analytical procedures within private manufacturer specifications, the FDA has noted in one of the warning letters that colour, particle size, pH, and comparison of the certificate of analysis supplied by the ingredient manufacturer with the specifications are not suitable tests for identity, because these tests neither uniquely identify an ingredient nor discriminate it from other ingredients.10, 11 Regulatory investigators also may have a hard time in determining what test should be used in cases of suspected adulteration, given the wide variety of dietary ingredients. Recent regulatory investigations, which used a DNA‐based method as the sole basis for determining adulteration of several botanical DS products, including ginkgo, St John's Wort, valerian, echinacea, and garlic are an example of this.12 While sensitivity of detection is a major attribute of DNA‐based methods, there are limitations to the use of these methods. False negatives may be reported as a result of the DNA being damaged during processing of the botanical ingredients (extraction, heat, and other manufacturing processes) or due to clearance of the DNA resulting from purification steps intended to enrich the content of constituents with bioactivity. In these cases, DNA may be no longer recoverable, even when the bioactive constituents are present in the formulation at the intended level. A negative test result may also be due to interferences from the matrix, which may complicate the recovery of a sufficient amount of DNA.13 On the other hand, false positive identification is also possible due to organic matter naturally occurring in the plant material at low but allowable levels (no more than 2%). Due to their high sensitivity, DNA‐based methods may be fooled by the deliberate addition of minute amounts of material rich in DNA from the plant stated on the label, even when the constituents desired for bioactivity are absent from the formulation. It is for these reasons that, although DNA techniques are powerful tools for authentication of plant materials at the early stages of processing, unless complemented with other orthogonal techniques, the presence or absence of DNA in more processed botanicals (i.e., extracts or finished dietary supplements) should not be used as the sole basis to evaluate quality in processed botanicals.
The second serious concern arises from several reports of adulteration of products marketed as dietary supplements with synthetic drugs and drug analogues. The addition of these synthetic substances (such as recent DMAA or BMPEA)14, 15, 16 is illegal since they do not meet the legal definition of a DS under the Food & Drugs Control Administration (FDCA), are not declared on the product label, and present a significant threat to consumer health, considering that these products are consumed without medical supervision, may contain toxic constituents or substances whose safety has never been examined, and whose interaction with medications may be unpredictable or lethal. The significant public health problem posed by products that are marketed as DS but contain undeclared substances was recognized as a major concern in a letter from the FDA Commissioner, Dr Margaret Hamburg.3 These synthetic substances could be prescription drugs, their unapproved analogues, or other compounds, such as novel synthetic steroids, that do not qualify as dietary ingredients. Use of these substances can pose considerable dangers to consumers who may take these products without knowing that the ingredients are present, since these undeclared ingredients may be associated with serious side effects or may interact with other products consumers may be taking. Dr Hamburg's letter noted that the FDA received numerous reports of serious adverse events associated with consumer use of these tainted products including strokes, acute liver injury, kidney failure, pulmonary embolisms (artery blockage in the lung), and even death. Adulteration of finished DS products in the following categories has been recognized as a major concern in the letter from Dr Hamburg:
Sexual enhancement: Also referred to as the Erectile Dysfunction category, this encompasses a functionally coherent group of adulterants, including several approved drugs (e.g. sildenafil), their numerous approved and unapproved analogues, synthetic intermediates, and derivatives.16, 17 Their functionality is manifested by selective inhibition of phosphodiesterase type 5 enzyme (PDE5), which hydrolyzes cyclic guanosine 3,5‐monophosphate (GMP).
Weight loss: This category comprises a functionally and chemically diverse collection of compounds that include stimulants, laxatives, diuretics, anorexiants, and psychoactive drugs.18 Although stimulants constitute an important segment of weight loss adulterants, the oral anorexiant sibutramine dominates this category, frequently in combination with phenolphthalein, a banned laxative.
Sports performance enhancement: Professional and amateur athletes are targeted with designer anabolic steroids and stimulants, many of which are banned by the World Anti‐Doping Agency (WADA).19 Functional and structural diversity, synthetic proclivity of the adulterators, and the generally small amounts of the infringing substances required to elicit a therapeutic effect make this category especially challenging to address. These DS are customarily formulated in protein‐ and fat‐rich matrices, thereby further complicating detection.
The nature of intentional adulteration is inherently not predictable and variable, since the adulterators are not guided, let alone bound, by GMP controls.
USP dietary supplement standards
For nearly 200 years, the United States Pharmacopeial Convention (USP), an independent, non‐profit, scientific‐based organization, has worked with volunteer experts from a wide cross‐section of stakeholders to develop and continuously revise and update science‐based quality standards for medicines, including their test methods and other tools that help protect public health. Standardization of botanicals and minerals dates back to the first edition of the USP in 1820, when physicians concerned with the quality of medicinal products developed a formulary, which later became an official compendium.20 Vitamins were admitted into the USP during the early 1900s. Since 1992, after the passing of the Nutritional Labeling and Education Act, the USP has developed the same kind of science‐based quality standards for nutritional and DS following an open and transparent public consultation process, whereby input from manufacturers, regulators, suppliers, and any other interested party is considered and evaluated by volunteer experts organized in Expert Committees. The USP prioritizes DS standards development based on considerations that include the extent of use, evidence of benefit, interest from a governmental body, and safety risk associated with its use. The USP admission evaluation process involves consideration of safety information from multiple sources, including adverse event reports from FDA MedWatch.21, 22 This assessment is conducted for the sole purpose of determining whether or not to develop a compendial monograph that is admitted in the United States Pharmacopeia–National Formulary (USP–NF) and is not intended as a determination of the intrinsic safety or efficacy of the DS ingredient or product under review. While estimates vary, the number of DS products in the market are estimated to be over 55 000, with a majority of the market value covered by about 100 ingredients (e.g. fish oil, calcium, glucosamine/chondroitin, CoQ10, and ginkgo), and major product categories (e.g. multivitamins, sports nutrition, and probiotics).1, 23, 24 The current revision of the USP–NF includes almost 500 monographs for DS ingredients and finished DS that cover most of the commonly used DS in commerce. USP–NF standards are used in about 140 countries worldwide, and are often referred as the basis for the specifications agreed in contractual agreements between buyers and sellers in international trade. Within the USA, the FDCA and its subsequent amendments recognize the USP and NF as ‘official compendia of the United States’.25 Federal regulations governing drugs require mandatory compliance with the USP–NF. However, compliance with the official compendia is only optional for DS.
Under DSHEA, a DS may be deemed ‘misbranded’ if the manufacturer claims conformance with specifications of an official compendium (USP–NF) and fails to comply. Because the enforceability of compliance with USP standards is conditional to the claim, DS manufacturers typically avoid claiming USP quality on labels in order to avoid the risk of being deemed misbranding because of an eventual lack of compliance. Despite the good intentions to include compendial standards in the law as a resource for manufacturers, by incorporating them under the misbranding provisions rather than as a minimum requirement for quality, DSHEA has effectively created a disincentive for manufacturers to claim compendial standards on their labels in detriment of transparency for the consumers.
Given the complexity of the DS matrices, attributes of an analytical method in USP monographs that are fit for the intended purpose depend on the nature of the analyte (ingredient or a product), as well as the analytical objectives (qualitative, quantitative, or others). Accordingly, methods for identification, composition, or strength, and limits for contamination require the consideration of the types of DS or dietary ingredients (e.g. botanicals or non‐botanicals), and whether the DS is administered in solid oral dosage forms, solutions, or suspensions. For example, in addition to quality standards for the dietary ingredients – the raw material (Ginkgo leaves) and the extract (Powdered Ginkgo Extract), the USP‐NF also provides standards for the final dosage forms, such as Ginkgo Capsules and Ginkgo Tablets.26 In addition, the USP–NF provides guidelines and general chapters applicable to DS related to methods and limits for pesticide residues, elemental contaminants (such as arsenic, lead, mercury, and cadmium), residual solvents, microbial contamination, and detection of irradiated botanical ingredients. Compendial identification tests for dietary ingredients include use of macroscopic/microscopic, chemical, spectroscopic and chromatographic methods. Since the objective of an identification test method is to be able to discriminate between related species and/or potential adulterants or substitutes, which are likely to be present, the specific tests for a botanical ingredient usually include a combination of two or three procedures.
Suitability of an analytical method depends on the matrix of the analyte as well as the objectives of the analysis. While an intact botanical plant material or its powdered form may be identified by macroscopic or microscopic features, the identifying features are lost when such material is extracted or processed. In these cases, chromatographic procedures, such as high‐performance thin‐layer chromatography (HPTLC) or high‐performance liquid chromatography (HPLC), are used for qualitative and quantitative assessment of identity, composition, and detection of adulterants. Continuing with the example of ginkgo, adulteration is known to occur with less expensive flavonol aglycones to achieve the market desired 24% flavonol glycoside content or by using other parts of the plant (root bark) to achieve the standard of 6% of ginkgo terpene lactones.27, 28 USP standards for Powdered Ginkgo Extract include the following orthogonal test methods and acceptance criteria that define the essential quality attributes and help detect adulteration26:
Identification by two complementary methods (HPTLC and HPLC). These qualitative methods compare chromatographic patterns of the sample with that of the reference standard, and define the acceptance criteria in terms of the ratio and relative abundance of flavonol glycosides and ginkgo terpene lactones (Figures 1a–1c).
Composition: These quantitative methods specify the acceptance criteria for the content of flavonol glycosides (22–27%), and the terpene lactones (5.4–12.0%), with specific content of ginkgolides A, B, and C and bilobalide that are unique to ginkgo (Figures 1c and 1d).
Limits for contaminants: pesticide residues (USP General Chapter <561>); elemental impurities (USP General Chapter <561>); microbial load (USP General Chapters <2021> and <2022>).
Specific tests: These tests specify limits for the content of rutin (not more than 4%) and quercetin (not more than 0.5%) which may be added in adulterated products; limits for the ginkgolic acid29 at not more than 5 µg/g (Figure 1e); and limit of residual solvents (USP General Chapter <565>).
Figure 1.
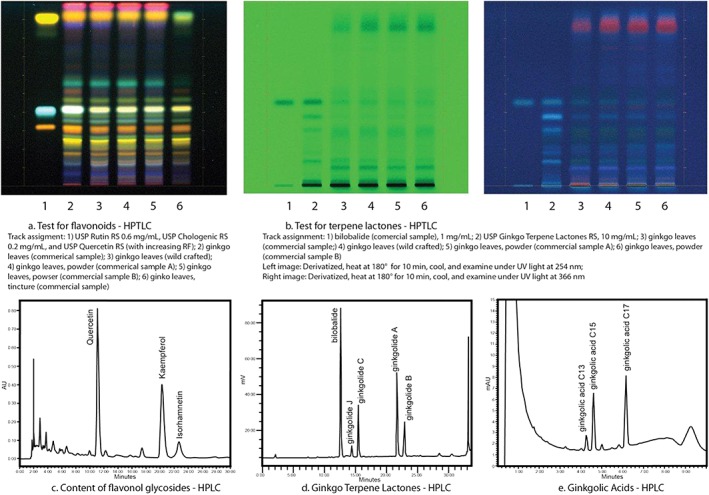
Tests of Monograph Standards: Ginkgo. Examples of the testing standards used with USP monographs26
While these science‐based methods and acceptance criteria from USP–NF public standards are available to define the quality of ginkgo and to prevent adulteration, incidence of ginkgo adulteration may occur when these are ignored.30 The most common form of ginkgo adulteration is spiking with pure flavonoids (rutin and quercetin), hydrolyzed extracts or extracts from other flavonoid‐rich material such as Japanese sophora (Styphnolobium japonicum)31 in order to comply with the compendial requirement for not less than 22% of flavonol glycosides. The limits for the content of rutin (not more than 4%) and quercetin (not more than 0.5%) was introduced in the compendia to detect these adulterated products.26 Further, the compendial requirement for terpene lactones (not less than 5.4%) can prevent ginkgo adulteration with botanicals containing flavonol glycosides but lack the unique terpene lactones.
USP–NF standards for ginkgo illustrate the approach to use orthogonal methods to define the identity and quality of a botanical ingredient since any one test method cannot be a surrogate for each of the diverse quality attributes. DNA testing is arising as another useful orthogonal analytical procedure to ensure plant authentication and became an official USP method in General Chapter <563> Identification of the Articles of Botanical Origin in December 2014. DNA methods are not yet referenced in any DS monograph in USP–NF, though when the validation data is available, USP may incorporate DNA tests into specific monographs, but even then the test is not expected to be utilized as a standalone procedure, but as a complement to chromatographic, spectroscopic, and botanical (microscopic or macroscopic) procedures.32
The Office of Dietary Supplements at National Institutes of Health runs Analytical Methods and Reference Materials Program33 which funds AOAC International to develop analytical methods for select DS. The USP participates in the AOAC method development process as a stakeholder. Some of the resulting analytical methods developed through public funding could be adopted into the USP. The Office of Dietary Supplements also supports development of National Institute of Standards and Technology reference materials. These methods and reference materials complement the USP DS quality standards (analytical methods, acceptance criteria and reference standards).
Potential solutions to lack of uniformity in product quality
For generic and over‐the‐counter (OTC) drugs, minimum quality is ensured by mandatory compliance with the minimum requirements set in the official compendia. Adherence to public standards set by USP for DS in the same way that works so well for generics and OTCs would alleviate the problem of quality disparity; if all products comply with the minimum standard required in the compendia, then the public will know that two products labelled Echinacea Tablets share at least the minimum standards for quality.
The FDA has recognized the value of USP–NF‐validated analytical methods in the preamble of the GMP as it noted: ‘We [FDA] explicitly stated that you may use validated methods that can be found in official references, such as AOAC International [an analytical methods development organization], USP, and others’ and that ‘compendial standards may be appropriate reference materials for use in conducting tests or examinations’.34 Unfortunately, validation of analytical methods is not required, nor is the use of validated method such as those in USP or AOAC. Here again, the USP can help to alleviate the problem should industry follow General Chapter <1225> Validation of Compendial Methods, which defines the parameters to be used to determine fitness for purpose.35
Given the dialogue concerning the availability of methods that are suitable for intended purposes, public quality standards provide a uniform point of reference for regulators and manufacturers, and promote consumer confidence. If DS manufacturers and government regulators adopt USP–NF public standards, they acquire a transparent means to help ensure the quality of DS products through the supply chain, and allow consumers to have confidence in the quality of the products on the market.
Currently, manufacturers may self‐determine the quality of their products indicating compliance with USP public standards by listing the monograph title of the article along with the letters U‐S‐P on the product label. Participation in the USP's voluntary third‐party verification program is another way to demonstrate the quality of DS. USP DS verification services include (1) an on‐site facility audit for compliance with FDA GMPs and USP's more rigorous GMPs in General Chapter <2750> Manufacturing Practices for Dietary Supplements; (2) a thorough review of manufacturing and quality control product documentation; (3) comprehensive laboratory testing for conformance to dietary supplement standards found in the USP–NF; (4) continuous change control monitoring; and (5) off‐the‐shelf surveillance testing of randomly selected samples of products to confirm that USP‐verified products continue to meet the USP's stringent standards. It is primarily the combination of the GMP audit and the product documentation review that forms the basis of product quality and (batch‐to‐batch) consistency. This approach confirms the principle that quality needs to be built into the product, not tested into the product.36
USP tools to detect intentional adulteration of dietary supplements
In order to address the serious concerns arising from the adulteration of products marketed as DS with synthetic drugs and drug analogues, a case could be made for public standards and reference materials for targeted common adulterants considering the repeat offences in specific product categories. In 2013, the USP convened an Expert Panel to investigate and recommend analytical methodologies capable of detection of pharmaceutically adulterated DS. In May 2015, the work of the Expert Panel led to a proposed new guidance document in the form of General Chapter <2251> Adulteration of Dietary Supplements with Drugs and Drug Analogs.37 Presently, the chapter targets supplements adulterated with PDE5 inhibitors; subsequent revisions will include methodologies specific to analysis of adulterated weight loss and sports performance enhancement products. The proposed chapter suggests multiple analytical methods, including HPLC with photodiode array and mass‐spectrometric (MS) detection, HPTLC with visual, UV, and MS detection, ambient ionization mass spectrometry, NMR spectroscopy (both low‐ and high‐field), and a bioluminescent phosphodiesterase inhibition method. It is advisable to use several screening techniques to maximize the potential for adulteration detection, because no single methodology is universally applicable. Supplementary material includes MS and UV absorbance data, relative retention time values for common adulterants, and chemical structures. Relevant USP–NFReference Standards for adulterant screening are included; however, considering the rate of propagation of structural analogues and proliferation of newly developed ‘designer’ molecules, establishing and maintaining an all‐inclusive catalog of reference materials would be challenging and impractical. USP public standards, including the monographs and General Chapter <2251>, are intended to provide the analytical tools that are necessary for detecting DS adulteration thereby enabling diligent manufacturers and regulators to assess the quality of their DS ingredients and products all through the supply chain. Availability of these tools is not sufficient to prevent unscrupulous criminal supply of adulterated DS in the US market, Therefore, the USP is developing a Dietary Supplements Adulteration Database of the incidences of DS adulteration to provide an easily searchable public database of the risks of adulteration and the available detection methods, similar to how the USP's Food Fraud Database has analyzed the economically motivated adulteration of food ingredients.38, 39 The DS adulteration database is also intended to highlight the gaps and needs for public standards to counteract adulteration. The analytical challenges in the detection of unintentional or deliberate adulterants are varied. USP–NF monographs utilize targeted analytical methods to assess quality of an ingredient or dosage form in terms of its identification, composition or strength, performance attributes, and limits of contaminants. Multiple tests are typically used to impart orthogonal assessments of the unique quality attributes of the test substance, and to increase confidence in the analysis. Compendial standards for ginkgo, ginseng, bilberry, and chondroitin sulfate, for example, include tests for common adulterants. However, detection of unlabeled adulterants demands the use of non‐targeted methods.
Targeted techniques are warranted when the analytes are known or can be reasonably anticipated. An example of a targeted approach is the monitoring a chromatographic run at a particular wavelength (or mass‐to‐charge ratios), and quantifying the analyte that appears within a pre‐defined retention‐time window. Targeted analysis is conceptually straightforward, because it relies on pre‐existing knowledge of the analyte and allows optimization of test methodology for its reliable detection. Targeted screening may be sometimes informed by functional categories as in case of PDE‐5 inhibitor analogues that are adulterants in products marketed as sexual enhancement DS. Bioassay based screening methods may be used to detect these class of compounds.
In contrast, non‐targeted methods are necessary because the nature of the analyte may be difficult to predict, and variable amounts of multiple adulterants, belonging to several functional categories, are commonplace. Non‐targeted screening trades precise knowledge of the analyte identity, along with specificity and accuracy, for a wider detection scope. Examples of non‐targeted chromatographic screening include acquisition of photodiode array data and full mass‐spectral scanning following a chromatographic separation. Adulteration paradigms favor utilization of detection techniques in a non‐targeted mode, thereby facilitating detection of a suspect adulterant even in the absence of a matching reference compound.
Conclusion
The lack of uniformity of product quality and adulteration of DS should be a concern for manufacturers, regulators and consumers alike. The provision of GMPs to allow manufacturers to set their own private standards contributes to a lack of transparency that makes it difficult for different parties to agree on what quality means for a given product. Therefore, the current GMP requirements provide limited assurance that the dietary ingredients and DS are of adequate and consistent quality across different manufacturers. The presence of products spiked with synthetic drugs marketed as DS demands the use of innovative tools to protect public health; USP has responded to the challenge by developing General Chapter <2251>. Stronger adoption of science‐based public quality standards by the industry or in the regulations would provide a solution to these issues. Compliance with USP–NF standards help ensure the consistency and quality of medicines in the USA and could do the same for DS. Public health is best served when public standards for quality are required as a minimum, as it is the case with drugs in the USA. We believe that the universal adoption of the USP–NF science‐based public standards would serve regulators (e.g. the FDA), manufacturers and consumers by improving the consistency and quality of DS marketed in the USA. This may be accomplished by strengthening GMP provisions to require conformance with standards established by USP–NF or other compendia when a monograph title is used as the name of an ingredient or product.
Sarma, N. , Giancaspro, G. , and Venema, J. (2016) Dietary supplements quality analysis tools from the United States Pharmacopeia. Drug Test. Analysis, 8: 418–423. doi: 10.1002/dta.1940.
The copyright line for this article was changed on June 8, 2016 after original online publication.
References
- 1. Nutrition Business Journal, Penton Media Inc. NBJ's Global Supplement & Nutrition Industry Report, New Hope Natural Media, Boulder, Colorado, USA, 2014, pp. 39. [Google Scholar]
- 2. DOJ . Justice department and federal partners announce enforcement actions of dietary supplement cases. Available at: http://www.justice.gov/opa/pr/justice‐department‐and‐federal‐partners‐announce‐enforcement‐actions‐dietary‐supplement‐cases [23 November 2015].
- 3. FDA . Letter to manufacturers of dietary supplements. Dr. Margaret Hamburg. Dec 15, 2010. Available at: http://www.fda.gov/downloads/Drugs/ResourcesForYou/Consumers/BuyingUsingMedicineSafely/MedicationHealthFraud/UCM236985.pdf [06 August 2015].
- 4. Klontz K. C., DeBeck H. J., LeBlanc P., Mogen K. M., Wolpert B. J., Sabo J. L., Salter M., Seelman S. L., Lance S. E., Monahan C., Steigman D. S., Gensheimer K.. The role of adverse event reporting in the FDA response to a multistate outbreak of liver disease associated with a dietary supplement. Public Health Rep. 2015, 130, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohen P. A.. Hazards of hindsight—monitoring the safety of nutritional supplements. N. Engl. J. Med. 2014, 370, 1277. [DOI] [PubMed] [Google Scholar]
- 6. Gardiner P., Sarma D. N., Low Dog T., Barrett M. L., Chavez M. L., Ko R., Mahady G. B., Marles R. J., Pellicore L. S., Giancaspro G. I.. The state of dietary supplement adverse event reporting in the United States. Pharmacoepidemiol. Drug Saf. 2008, 17, 962. [DOI] [PubMed] [Google Scholar]
- 7. FDA . Current good manufacturing practice in manufacturing, packaging, labeling, or holding operations for dietary supplements. 21 CFR 111. [Docket No. 1996N–0417]. Available at: http://www.fda.gov/ohrms/dockets/98fr/cf0441.pdf p. 400 [6 August 2015].
- 8. Miller R. K., Celestino C., Giancaspro G. I., Williams R. L.. FDA's dietary supplement GMPs: standards without standardization. Food Drug Law J. 2008, 63, 929. [PubMed] [Google Scholar]
- 9. Long J.. More supplement manufacturers complying with FDA GMP regulations, but room for improvement. Available at: http://www.naturalproductsinsider.com/blogs/supplement‐law/2015/03/more‐supplement‐manufacturers‐complying‐with‐fda.aspx [13 November 2015].
- 10. FDA Warning Letter. Available at: http://www.fda.gov/iceci/enforcementactions/warningletters/2014/ucm406488.htm [6 August 2015].
- 11. FDA lab testing finds N‐acetyl‐leucine in samples of recalled Medisca product labeled as L‐citrulline. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm385964.htm [6 August 2015].
- 12. O'Conner A.. Retailers are warned over herbal supplements. The New York Times, 2015, February 3. Available at: http://www.nytimes.com/interactive/2015/02/02/health/herbal_supplement_letters.html [6 August 2015].
- 13. Little D. P.. Authentication of Ginkgo biloba herbal dietary supplements using DNA barcoding. Genome. 2014, 57, 513. [DOI] [PubMed] [Google Scholar]
- 14. DMAA in Dietary Supplements. Available at: http://www.fda.gov/Food/DietarySupplements/QADietarySupplements/ucm346576.htm [6 August 2015].
- 15. Cohen P.A., Bloszies C., Yee C., Gerona R.. An amphetamine isomer whose efficacy and safety in humans has never been studied, β‐methylphenylethylamine (BMPEA), is found in multiple dietary supplements. Drug Test. Anal. 2015. . DOI: 10.1002/dta.1793. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 16. Venhuis B. J., de Kaste D.. Towards a decade of detecting new analogues of sildenafil, tadalafil and vardenafil in food supplements: a history, analytical aspects and health risks. J. Pharm. Biomed. Anal. 2012, 69, 196. [DOI] [PubMed] [Google Scholar]
- 17. Patel D. N., Li L., Kee C. L., Ge X., Low M. Y., Koh H. L.. Screening of synthetic PDE‐5 inhibitors and their analogues as adulterants: analytical techniques and challenges. J. Pharm. Biomed. Anal. 2014, 87, 176. [DOI] [PubMed] [Google Scholar]
- 18. Venhuis B.J., Zwaagstra M.E., van den Berg J.D.J., Wagenaar H.W.G., van Riel A.J.H.P., Barends D. M., de Kaste D.. Trends in drug substances detected in illegal weight‐loss medicines and dietary supplements. A 2002‐2007 survey and health risk analysis. RIVM Report 370030002/2009. Available at: http://vorige.nrc.nl/multimedia/archive/00213/370030002_213599a.pdf [6 August 2015].
- 19. Thevis M., Kuuranne T., Geyer H., Schänzer W.. Annual banned‐substance review: analytical approaches in human sports drug testing. Drug Test. Anal. 2015, 7, 1. [DOI] [PubMed] [Google Scholar]
- 20. Schiff P. L. Jr., Srinivasan V. S., Giancaspro G. I., Roll D. B., Salguero J., Sharaf M. H.. The development of USP botanical dietary supplement monographs, 1995‐2005. J. Nat. Prod. 2006, 69, 464. [DOI] [PubMed] [Google Scholar]
- 21. USP Admission Criteria and Safety Classification for Dietary Supplements Guideline. Available at: http://www.usp.org/sites/default/files/usp_pdf/EN/dietarySupp/admissiondsguideline_vers_1.1.pdf [6 August 2015].
- 22. FDA MedWatch. Available at: http://www.fda.gov/Safety/MedWatch/default.htm [6 August 2015].
- 23. Dietary Supplements Label Database – Preamble. Available at: http://ods.od.nih.gov/Research/Dietary_Supplement_Label_Database.aspx [6 August 2015].
- 24. Dietary Supplements Label Database – Ginkgo products. Available at: http://www.dsld.nlm.nih.gov/dsld/rptQSearch.jsp?item=ginkgo&db=adsld [6 August 2015].
- 25. Federal Food Drug Cosmetic Act. § 201(j); 21 U.S.C. § 321(j). Available at: http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/FDCActChaptersIandIIShortTitleandDefinitions/ucm086297.htm [6 August 2015].
- 26. USP Ginkgo monographs . USP 38, United States Pharmacopeial Convention, Rockville, MD, 2015, pp. 6061. [Google Scholar]
- 27. Harnly J. M., Luthria D., Chen P.. Detection of adulterated Ginkgo biloba supplements using chromatographic and spectral fingerprints. J. AOAC Int. 2012, 95, 1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bzhelyansky A., Sarma N. D., Giancaspro G. I.. Letter to the Editor in response to ‘Adulteration of Ginkgo biloba products and a simple method to improve its detection’ by Wohlmuth et al . Phytomedicine. 2014, 21, 1497. [DOI] [PubMed] [Google Scholar]
- 29. van Beek T. A., Montoro P.. Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J. Chromatogr. A. 2009, 1216, 2002. [DOI] [PubMed] [Google Scholar]
- 30. FDA . Warning letter to Iowa Select Herbs 4/18/14. Available at: http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/2014/ucm394259.htm [06 August 2015.
- 31. Avula B., Sagi S., Gafner S., Upton R., Wang Y. H., Wang M., Khan I. A.. Identification of Ginkgo biloba supplements adulteration using high performance thin layer chromatography and ultra high performance liquid chromatography‐diode array detector‐quadrupole time of flight‐mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 7733. [DOI] [PubMed] [Google Scholar]
- 32. USP . The United States Pharmacopeial Convention urges scientific validation of DNA test methods for regulating the quality of herbal supplements. Available at: http://www.usp.org/news/united‐states‐pharmacopeial‐convention‐urges‐scientific‐validation‐dna‐test‐methods‐regulating‐quality‐herbal‐supplements. [6 August 2015].
- 33. NIH . Office of Dietary Supplements – Analytical methods and reference materials program. Available at: https://ods.od.nih.gov/Research/AMRMProgramWebsite.aspx [23 November 2015].
- 34. FDA . Current good manufacturing practice in manufacturing, packaging, labeling, or holding operations for dietary supplements. 21 CFR 111. [Docket No. 1996N–0417]. Available at: http://www.fda.gov/ohrms/dockets/98fr/cf0441.pdf; pp 402 and 563 [6 August 2015].
- 35. USP . <1225> Validation of Compendial Procedures. USP 38 . In: USP 38–NF 33. United States Pharmacopeial Convention, Rockville, MD, 2015, pp. 1445.
- 36. USP Dietary Supplements Verification Program. Available at: http://www.usp.org/usp‐verification‐services [6 August 2015].
- 37. USP . <2251> Adulteration of dietary supplements with drugs and drug analogs. Pharmacopeial Forum, 2014, 41(3). United States Pharmacopeial Convention, Rockville, MD.
- 38. USP . Food Fraud Database. Available at: http://www.usp.org/food‐ingredients/food‐fraud‐database [6 August 2015].
- 39. Moore J. C., Spink J., Lipp M.. Development and application of a database of food ingredient fraud and economically motivated adulteration from 1980 to 2010. J. Food Sci. 2012, 77, R118. [DOI] [PubMed] [Google Scholar]