

Summary
American Foulbrood, caused by Paenibacillus larvae, is the most severe bacterial disease of honey bees (Apis mellifera). To perform genotyping of P. larvae in an epidemiological context, there is a need of a fast and cheap method with a high resolution. Here, we propose Multiple Locus Variable number of tandem repeat Analysis (MLVA). MLVA has been used for typing a collection of 209 P. larvae strains from which 23 different MLVA types could be identified. Moreover, the developed methodology not only permits the identification of the four Enterobacterial Repetitive Intergenic Consensus (ERIC) genotypes, but allows also a discriminatory subdivision of the most dominant ERIC type I and ERIC type II genotypes. A biogeographical study has been conducted showing a significant correlation between MLVA genotype and the geographical region where it was isolated.
Introduction
The gram‐positive, rod‐shaped, endospore‐forming bacterium Paenibacillus larvae is the ethiological agent of American foulbrood (AFB) (Genersch et al., 2006), a deadly brood disease of the honey bee (Apis mellifera). Very young larvae, below 36 h of age, are most susceptible. Transmission occurs by the oral uptake of the highly resistant spores that are spread inside the colony or between colonies by adult bees that carry them or by interventions of the beekeeper (Lindstrom, 2008; Lindstrom et al., 2008). AFB is classified as a notifiable disease in most countries and depending on the local control strategy, sick colonies are destroyed by burning, decontaminated by the shaking method or treated with antibiotics. Hence, P. larvae is responsible for considerable economic losses in the beekeeping sector worldwide.
Historically, it was thought that AFB and ‘powdery scale disease’ were caused by different species, Bacillus larvae and Bacillus pulvifaciens respectively (Katznelson, 1950). After different rounds of reclassification (Ash et al., 1991,1993), they were classified as the same species, but split at subspecies level (Heyndrickx et al., 1996). In 2006 was demonstrated that both should be classified as the species P. larvae without separation at the subspecies level (Genersch et al., 2006).
Over the years, different techniques have been developed for genotyping of P. larvae, as there are Restriction Endonuclease Fragment Patterns (Djordjevic et al., 1994; Alippi et al., 2002), rep‐PCR (Alippi and Aguilar, 1998; Genersch and Otten, 2003), Pulsed‐Field Gel Electrophoresis (Wu et al., 2005; Genersch et al., 2006), Amplified Fragment Length Polymorphism (de Graaf et al., 2006), Denaturing Gradient Gel Electrophoresis (Antunez et al., 2007) and most recently Multi Locus Sequence Typing (MLST) (Morrissey et al., 2015). Of these techniques, only rep‐PCR has commonly been used for genotyping P. larvae. Enterobacterial Repetitive Intergenic Consensus (ERIC) rep‐PCR of the bacteria revealed four genotypes (Genersch and Otten, 2003; Genersch et al., 2006). Both genotypes ERIC I and ERIC II have a worldwide distribution (Schafer et al., 2014; Morrissey et al., 2015). Genotypes ERIC III and IV have not only been identified in field for decades, but exist as few isolates in culture collections (Genersch, 2010). ERIC genotyping has the advantage that it splits the species into biological relevant groups which differ in colony morphology and virulence at individual and colony level (Neuendorf et al., 2004; Genersch et al., 2005, 2006). The sequencing of the genomes of P. larvae ERIC I and ERIC II strains (Qin et al., 2006; Chan et al., 2011; Djukic et al., 2014) provided an important step towards the development of better molecular typing methods. The recently developed MLST method (Morrissey et al., 2015) ratifies and extends the ERIC typing scheme. Although this MLST is a very useful method for epidemiology and source tracking, its use in diagnostic labs is hampered by its expensive and labour‐intensive sequencing step.
We developed an alternative, equivalent technique called Multi Locus Variable number of tandem repeat Analysis (MLVA) (van Belkum, 1999) for genotyping P. larvae. MLVA is a proven highly discriminatory subtyping method used for many pathogens, such as Mycobacterium tuberculosis (Mazars et al., 2001), Yersinia pestis (Klevytska et al., 2001), Staphylococcus aureus (Malachowa et al., 2005), Erwinia amylovora (Buhlmann et al., 2014) and Campylobacter jejuni (Techaruvichit et al., 2015). The method has also been used to link genotype information with geographical information to study how bacteria (Y. pestis, Burkholderia pseudomallei, Xanthomonas citri pv. citri, Bacillus anthracis and E. amylovora) behave within smaller geographical areas or single outbreaks (Girard et al., 2004; U'Ren et al., 2007; Bui et al., 2009; Stratilo and Bader, 2012; Buhlmann et al., 2014). The typing method uses the Variation in Number of Tandem Repeats (VNTR) on different loci among the genome to classify the strains (van Belkum, 1999). These variations are caused by slipped strand mispairing (Torres‐Cruz and van der Woude, 2003). VNTRs have been described as fast molecular clocks (van Belkum, 1999). Indeed, since dynamics of VNTRs depend on repeat copy number (Vogler et al., 2006), different VNTRs show different clock speeds (Lindstedt, 2005; van Belkum, 2007). This makes the method suitable for phylogenetic and evolutionary studies. In MLVA, different VNTR loci are combined, allowing inspection of phylogenetic relationships among different bacterial strains. Typically, 5 to 6 loci are combined in one multiplex PCR, which can be analysed using electrophoresis. In this paper, we describe the development of a MLVA‐based genotyping protocol for P. larvae, and the subsequent typing of a collection of 209 P. larvae strains, demonstrating that MLVA has great potential for genotyping P. larvae in an epidemiological context.
Results and discussion
Identification of tandem repeat regions
We screened three publically available P. larvae genomes [BRL 230010 (Qin et al., 2006) and its updated sequence B3650 (Chan et al., 2011), DSM 25430 and DSM 25719 (Djukic et al., 2014)] for tandem repeats using an online software package (Tandem Repeats Database at https://tandem.bu.edu/cgi-bin/trdb/trdb.exe; Gelfand et al., 2006). BRL 230010 and DSM 25719 belong to the ERIC I genotype, whereas DSM 25430 is an ERIC type II. To permit VNTR analysis on an agarose gel, the search criteria for tandem repeats were set at a size between 15 and 120 bp with 80% sequence match. This resulted in the finding of 40 different tandem repeat loci (Table S1).
The search for suitable VNTRs was continued by a two‐step procedure. Candidate loci were selected based on different criteria in silico, and their respective primers were designed. Three criteria were used: (i) the locus had a different copy number in at least two genomes or (ii) it had a pattern size between 15 and 30 or (iii) it had a copy number of more than five units. In a second step the differentiating power of these loci was tested on 13 different isolates. These isolates included the four different ERIC genoytpe and were isolated at different locations. ERIC types III and IV were represented once and ERIC II twice. The first selection criterium resulted in eight candidate loci, of which four were differentiating enough to be retained for implementation in the multiplex PCR, i.e. VNTR A, VNTR C, VNTR F and VNTR G (Table 1). An optimal MLVA procedure includes 5–6 VNTRs and therefore the search for more VTNRs was needed. The second selection criterium rendered six additional loci. Of these six loci, two were retained after testing in the 13 strains: VNTR B and VNTR D (Table 1). Finally, loci with more than five repeat units were screened. This gave two loci to be tested, one of which was retained: VNTR E (Table 1). Thus, in total seven loci were found with enough differentiating power for implementation in the multiplex PCR.
Table 1.
VNTR primer sequences
| VNTR | Length TR | Forward primer | Length 5′ | Reverse primer | Length 3′ |
|---|---|---|---|---|---|
| VNTR A* | 19 | GAGGGATATACCCCACCTCTTT | 5 | GGGGAAGTATGATCCCGAAG | 17 |
| VNTR B* | 21 | CCGGAATAATCCGCTTATGA | 22 | ATCACCAGAGTTGGCGATTC | 3 |
| VNTR C* | 24 | TGGTTTAGGAACCGGTGTTG | 47 | CACATTAAAGCCTGTGCAGGTA | 38 |
| VNTR D* | 24 | ATCATGGCGGTTGGGATG | 2 | CACAGGCTCGACAACCACTA | 13 |
| VNTR E* | 68 | TGTTCAATTTTGATTGTTTTGTTCA | 73 | TATATGGCGGTCGGCTTAAT | 2 |
| VNTR F | 48 | TACCCCAATCTGCCTTGTTG | 70 | CATGCTCCTGCGTGGTATAA | 41 |
| VNTR G | 18 | GTCATTACGGCCCAGGTG | 20 | TGAGGCTGCAAAGACAGATG | 22 |
Five VNTR loci (depicted with *) were used to combine in the multiplex PCR. Of each VNTR locus the length, forward primer and reverse primer are given. The distance (in base pairs) between the primer annealing site and the tandem repeat was mentioned as length 5′ and length 3′.
Multiplex PCR
Optimization of the multiplex PCR was realized by establishing the optimal concentration of MgCl2, primers and template DNA. In the final PCR, two loci were omitted because they created non‐target amplicons (VNTR F) or no amplicons during multiplexing (VNTR G). The obtained MLVA profiles were analysed using a 3% High Resolution Agarose Gel (Sigma‐Aldrich, St. Louis, MO, USA) (Fig. 1). The screening of the 13 strains created a total of nine different MLVA patterns. The screening was repeated three times and gave reproducable results.
Figure 1.
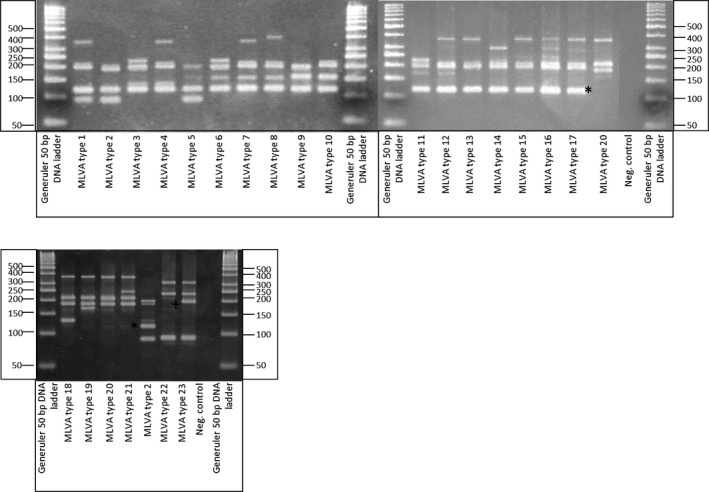
MLVA types. The tested collection of Paenibacillus larvae strains contained 23 different MLVA types. The ERIC I types (MLVA type 1–17) all have a band of 120 bp (*), which is absent in all other ERIC types. ERIC III (MLVA type 22) could be differentiated from ERIC type IV (MLVA type 23) by the presence of a 190 bp band (+). ERIC types II and IV do not show an VNTR B amplicon, ERIC type III does not show an VNTR E amplicon.
MLVA genotyping
The Laboratory of Molecular Entomology and Bee Pathology have access to a large collection of P. larvae strains available from the bacteria collection of the Belgian Co‐ordinated Collections of Micro‐organisms (BCCM/LMG) and from its own working collection. The strains that were retained for this study originated from a nationwide honey screening in Belgium (de Graaf et al., 2001) (116 isolates) or from clinical outbreaks of AFB in Belgian apiaries between 2013 and 2015 (74 isolates), and were extended with three additional reference strains (BRL 230010; Qin et al., 2006; LMG 16252, LMG 16247; both obtained from BCCM/LMG), six Austrian ERIC II isolates (Loncaric et al., 2009) and 10 Italian ERIC II strains (Bassi et al., 2015). Using these 209 different isolates, 14 new MLVA types were discovered resulting in 23 MLVA patterns.
A representative isolate for each pattern was chosen and its amplicons were sequenced. The resulting sequences confirmed that the primers targeted their respective tandem repeat in each MLVA pattern and that no off‐targets were amplified. The copy size of the tandem repeats could be established for each locus. These were consistent with the size of the amplicon on the gel (after substracting the flanking sequences) (Table 1 and Fig. 1). In Table 2, the typical VNTR‐code is given for the different MLVA patterns. To facilitate the analysis, we attributed a MLVA type number (MLVA 1 – 23) to each unique VNTR combination (Table 2).
Table 2.
MLVA types
| MLVA | VNTR‐code | ERIC genotype | Prevalence (%) |
|---|---|---|---|
| 1 | 2‐6‐4‐3‐5 | I | 1.0 |
| 2 | 4‐6‐3‐3‐1 | I | 30.1 |
| 3 | 4‐6‐3‐3‐3 | I | 1.0 |
| 4 | 4‐6‐4‐3‐5 | I | 1.4 |
| 5 | 5‐6‐3‐3‐1 | I | 1.0 |
| 6 | 5‐6‐3‐3‐3 | I | 3.8 |
| 7 | 5‐6‐4‐3‐5 | I | 4.3 |
| 8 | 5‐6‐4‐3‐6 | I | 1.0 |
| 9 | 6‐6‐3‐3‐2 | I | 2.3 |
| 10 | 6‐6‐4‐3‐2 | I | 11.0 |
| 11 | 6‐6‐4‐3‐3 | I | 1.4 |
| 12 | 6‐6‐4‐3‐5 | I | 1.9 |
| 13 | 7‐6‐4‐3‐5 | I | 17.7 |
| 14 | 10‐6‐4‐3‐4 | I | 3.8 |
| 15 | 10‐6‐4‐3‐5 | I | 4.8 |
| 16 | 13‐6‐4‐3‐4 | I | 0.5 |
| 17 | 13‐6‐4‐3‐5 | I | 3.3 |
| 18 | 4‐0‐3‐7‐5 | II | 0.5 |
| 19 | 6‐0‐3‐7‐5 | II | 1.0 |
| 20 | 7‐0‐3‐7‐5 | II | 6.2 |
| 21 | 8‐0‐3‐7‐5 | II | 0.5 |
| 22 | 2‐6‐5‐11‐0 | III | 0.5 |
| 23 | 2‐0‐5‐11‐0 | IV | 0.5 |
Of each MLVA type the VNTR‐code, ERIC genotype and prevalence in our dataset is given. Seventeen MLVA types belonged to ERIC type I strains, four belong to ERIC type II. ERIC type III and IV were represented by only one MLVA type. Most MLVA types show a very low prevalence, only three types have a prevalence higher than 10%.
VNTR characteristics
VNTR A was found to be the most diverse locus, splitting up in 8 different lengths of tandem repeats. The number of tandem repeats ranged from 2 to 13. The second most divers VNTR was VNTR E, followed by VNTR C, D and B (Table 3 and Fig. 2). The discriminatory power of the MLVA is due to the complementary discriminating power of each VNTR locus. The MLVA typing method based on these VNTR loci is powerful enough to find new MLVA types for strains that are not present in the tested dataset.
Table 3.
VNTR characteristics of the full dataset (193 isolates)
| VNTR | Length TR | No. alleles | No. repeat copies | Min rep | Max rep | Shannon | Simpson |
|---|---|---|---|---|---|---|---|
| VNTR A | 19 | 8 | 2, 4, 5, 6, 7, 8, 10, 13 | 2 | 13 | 1.68 | 0.78 |
| VNTR B | 21 | 1 | 6 | 6 | 6 | 0.29 | 0.16 |
| VNTR C | 24 | 3 | 3, 4, 5 | 3 | 5 | 0.74 | 0.51 |
| VNTR D | 24 | 3 | 3, 7, 11 | 3 | 11 | 0.34 | 0.17 |
| VNTR E | 68 | 6 | 1, 2, 3, 4, 5, 6 | 1 | 6 | 1.39 | 0.70 |
The length of the tandem repeat, the number of different alleles (No. alleles), the number of tandem repeats in each allele (No. repeat copies), the maximum and the minimum number of repeats (max rep, min rep) are given. The Shannon Index and Simpson Index were both used to measure the diversity.
Figure 2.
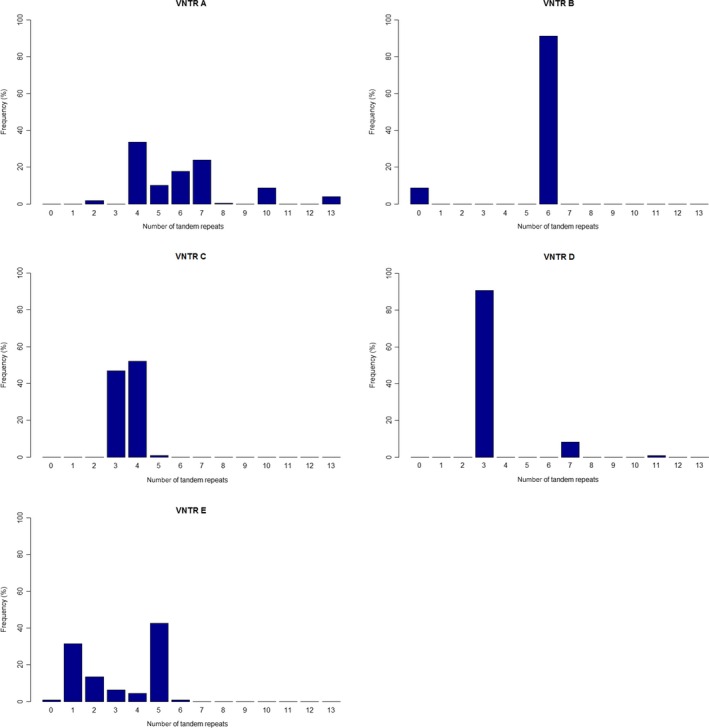
Diversity of VNTR loci. For each VNTR locus the tandem repeat (TR) copy numbers are given with their respective prevalence in our dataset. VNTR A: 8 alleles (2, 4, 5, 6, 7, 8, 10, 13 TR), VNTR B: 1 allele (6 TR) and no amplicon in 8.2% of the strains (the ERIC II and IV strains), VNTR C: 3 alleles (3, 4, 5 TR), VNTR D: 3 alleles (3, 7, 11 TR), VNTR E: 6 alleles (1, 2, 3, 4, 5, 6) and no amplicon in 0.5% of the strains (ERIC III strains).
Evolutionary history
The five loci had all different molecular clock speeds, which makes it possible to look at the evolutionary history of the MLVA types (Fig. 3). The first node in the tree clustered the types 22 and 23 apart. To these MLVA types belong the ERIC type III and type IV strains respectively. These are the only strains of the tested collection that correspond to the former P. larvae pulvifaciens subspecies. The second node clustered all MLVA types that contained ERIC type I strains apart from the types that contained the ERIC type II strains.
Figure 3.
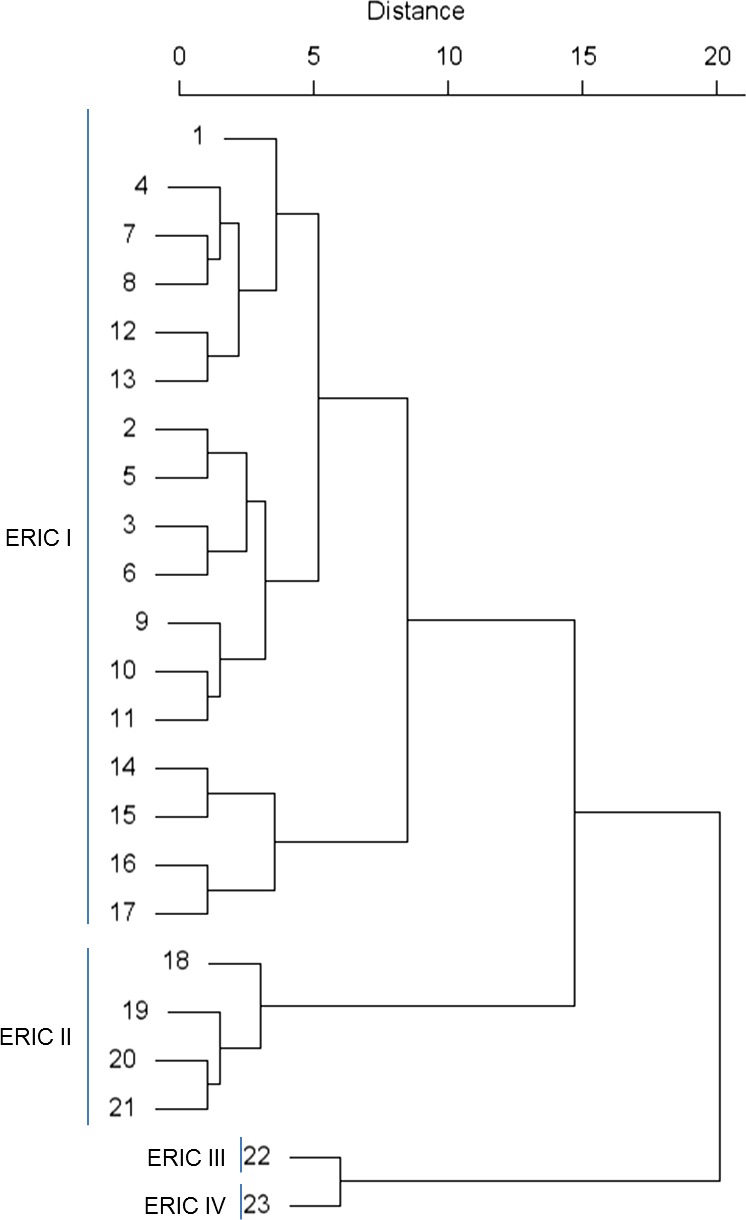
Evolutionary history of MLVA types. The evolutionary history of the MLVA types is based on hierarchical clustering. The distances were calculated using the sum of absolute differences in VNTR repeat number. The grouping reflected the ERIC‐typing method.
ERIC genotyping
All strains were also typed using ERIC rep‐PCR, which confirmed the presence of 189 ERIC type I strains, 18 ERIC type II strains, 1 ERIC type III and 1 ERIC type IV strain. Within ERIC type I, 17 MLVA genotypes could be discriminated and ERIC type II 4 MLVA genotypes were identified. Within ERIC type III and ERIC type IV, each time a single MLVA type could be attributed (Table 2). The subdivision of ERIC type I and ERIC type II in multiple subtypes is in the line of the findings of Morrissey (Morrissey et al., 2015) who found by MLST 16 subtypes in 173 ERIC type I strains and three subtypes in 92 ERIC II strains. Both typing schemes have thus on average the same resolution.
The present MLVA genotyping protocol also permits to differentiate between the ERIC genotypes by the presence of some discriminatory bands. Indeed, a band of 120 base pairs is always present in ERIC type I, but not in the ERIC type II. ERIC type III and type IV have a similar pattern, however, ERIC type III has always a band of 200 base pairs, which is absent in ERIC type IV (Fig. 1). The VNTR B primers do not create an amplicon in ERIC II and IV strains, while the VNTR E primers do not create an amplicon in ERIC II strains.
Prevalence and biogeography
The prevalence of the MLVA types within the complete collection differed significantly (Table 2). Only three types had a prevalence of more than 10.0%: MLVA 2 (30.1%), MLVA 13 (17.7%) and MLVA 10 (11.0%). Within the ERIC II strains, MLVA 20 was predominant (14/18 isolates), this type contained isolates from Belgium, Italy and Austria. Almost half of the MLVA types (10 of 23) were represented by 1 or 2 isolates, corresponding to a prevalence of 0.5–1%.
As the geographical spread of the apiaries where the isolates originated from (honey or brood samples) were mostly known (169 strains), we were able to provide the distribution per province (Fig. 4). A chi‐squared test using the geographical region (province) of the apiaries and the MLVA type was conducted and gave a very significant P‐value of < 0.001 (χ2 = 153.33, df = 144). We saw a much greater diversity in the subset of strains that originated from the honey screen compared with those from the clinical outbreaks (Shannon diversity index of 2.11 and 1.36, respectively). In fact, in the latter mostly 1 or 2 MLVA types were found per province, this could be due to the rapid spread of the spores in the vicinity of a clinical outbreak as described in de Graaf et al. (2001).
Figure 4.
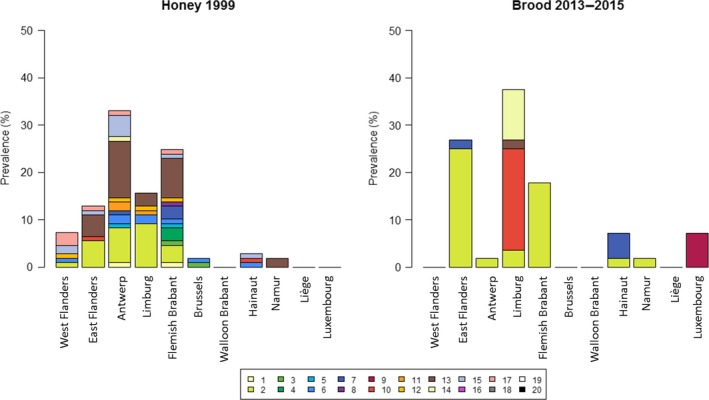
Distribution of MLVA‐types. The geographical location of each isolate from honey in 1999 and diseased brood in 2013–2015 was recorded. A significant difference in distribution over the provinces was observed. A much higher diversity in MLVA‐types was found in the honey samples than in the brood samples.
Remarkably, the MLVA type 2 which was most abundant in clinical cases in East Flanders, Antwerp, Limburg and Flemish Brabant and was likewise abundant in honey samples taken almost two decades earlier. It is not unravelled yet whether this is due to failure to eliminate the contamination with this MLVA type completely or to its biological characteristics (virulence). Another MLVA type (MLVA 13) that was abundant in honey samples of the same provinces, was almost entirely absent in the clinical cases of the period 2015–2013. In the brood samples of the province Limburg, MLVA 10 represents more than half of the isolates, while it is not present in any of the clinical cases of the other provinces.
Finally, it was investigated if classification of the isolates using a dissimilarity matrix resulted in the same MLVA types classified in the same group. Agglomerative hierarchical clustering was used as the algorithm. From the resulting tree (Fig. 5), we can conclude that indeed strains with the same MLVA type are on average more similar to each other based on non‐genetic traits (regional code, province, beekeeper, isolation year, isolation source) than to other MLVA types. The significant correlation with the region is also visual.
Figure 5.
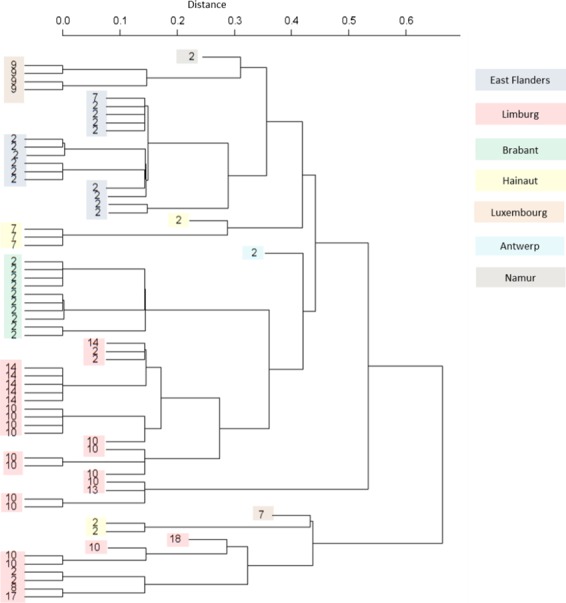
Agnes hierarchical clustering. Agnes hierarchical clustering based on the dissimilarities of non‐genetic traits clustered the strains by MLVA type and province. The correlation between these two parameters was visual.
Conclusions
In this paper, the development of a new genotyping method for P. larvae is presented. Using this method, we could subdivide ERIC type I into 17 different genotypes and ERIC type II into four different genotypes. ERIC type III and IV were represented each time by only a single MLVA genotype. Moreover, in the present MLVA genotyping protocol, amplicons indicative for the ERIC genotype were included, making it possible to extract information of the ERIC genotype from the MLVA pattern. This study clearly shows the usefulness of the MLVA method in epidemiological and biogeographical studies. MLVA genotyping has the unique combination of desirable properties for a genotyping method. It is very fast, highly specific, cheap and not labour intensive, which makes it an almost perfect method to implement in bee health extension laboratories. The method is usable for epidemiological, phylogenetic and evolutionary studies.
Experimental procedures
Dataset
In the library, 116 isolates were collected in the context of the honey screening in 1999 (de Graaf et al., 2001). This study was conducted anonymously throughout Belgium, with the postal code of the area where the apiary was located as geographical reference. One honey sample was collected per apiary. The library also consisted of isolates collected from diagnostic AFB cases in Belgium, including a set of isolates from clinical outbreaks from 2013 to 2015. From the latter, the postal code and beekeeper was recorded. Finally, the set of field isolates was completed with six Austrian ERIC type II isolates (Loncaric et al., 2009), 10 Italian ERIC type II isolates (Bassi et al., 2015), ERIC type III (LMG 16525), and ERIC type IV (LMG 16247) (both from the BCCM/LMG bacteria collection) reference strains, and a sequenced ERIC type I strain (BRL 230010) (Qin et al., 2006; Chan et al., 2011).
Preparation dataset
Bacterial genomic DNA was prepared using InstaGene matrix (Bio‐Rad, Hercules, CA, USA) according to the protocol described by Genersch and Otten (Genersch and Otten, 2003). To confirm that the isolates belong to P. larvae a 16S rDNA PCR was performed as described by Dobbelaere (Dobbelaere et al., 2001). Of each strain, the ERIC genotype has been determined according to the protocol of Genersch and Otten (2003).
VNTR prediction
The publically available genomes of BRL 230010 (Qin et al., 2006; updated by Chan et al., 2011), DSM25430 (Djukic et al., 2014) and DSM 25719 (Djukic et al., 2014) were used as input in the Tandem Repeats database (Gelfand et al., 2006). As selection criteria, we set the alignment parameters to match = 2, mismatch = 5, indels = 7, the minimal alignment score = 80, the pattern size ≤ 120 bp. From the obtained list of possible targets (Table S1), primers (IDT, Leuven, Belgium) were designed for loci that (i) had a different copy number in at least two genomes or (ii) had a pattern size between 15 and 30 or (iii) had a copy number of more than five units. The primers were constructed targeting the flanking regions of the tandem repeat locus and to have an annealing temperature of 52°C. The differentiating power of the tandem repeat loci was tested using 13 P. larvae isolates. Ten of the 13 isolates were randomly selected from the dataset, and were extended with R‐20833 (ERIC type II), LMG 16252 (ERIC type III) and LMG 16247 (ERIC type IV). A locus suitable to include in the multiplex PCR should generate a specific amplicon for each isolate and had to make discriminate between strains possible using agarose gel electrophoresis.
Construction multiplex PCR
Seven loci were initially combined in a multiplex PCR. Two loci were omitted, because the first one generated off target amplicons and the second failed to generate amplicons. The multiplex PCR was optimized by testing a DNA‐concentration gradient (20–120 ng DNA), MgCl2 gradient (1–3 nM) and variable combinations of primer concentrations (0.2–4 μM).
The final multiplex PCR used the HotStarTaq Plus DNA Polymerase kit (Qiagen, Hilden, Germany) and consisted of 1× PCR buffer, 2.5 mM MgCl2, 1× Q‐solution, 400 μM dNTPs, 0.8 μM of each VNTR A primer (IDT), 0.4 μM of each VNTR B – D primer and 4 μM of each VNTR E primer (Table 1). The total reaction volume was 25 μl and 100 ng total DNA is used as template. The PCR‐programme was as followed: 5′ 95°C, 30× (1′ 94°C, 1′ 52°C, 1′ 72°C) and 10′ 72°C. The multiplex PCR was repeated three times on the 13 isolates and proved to give reproducible results.
Data scoring and description profiles
The length and number of tandem repeats was determined by analysing the agarose gel and confirmed by sequencing the differing amplicons of the 23 MLVA types. Sequencing was done by Sanger sequencing performed by GATC‐Biotech (Constance, Germany). The number of repeat units was rounded to the next highest integer number. If more than one amplicon for a specific VNTR was detected and the difference in length between these amplicons was higher than the repeats length (stutter peaks), the amplicon with the most intense band was used to assign the repeat number. The Shannon and Simpson diversity index was calculated using R (R Core Team, 2015). Using agglomerative hierarchical clustering (Agnes), evolutionary analysis was performed. The clustering method used the sum of the absolute distances to calculate the dissimilarities between observations.
Analysis correlations
All statistical analysis is performed using R (R Core Team, 2015). Correlations were analysed using a Pearson's Chi‐square test with the geographical location (province) and MLVA type as variables. To perform a reliable test, MLVA types with less than five isolates were removed, provinces containing less than five isolates were joined with neighbouring provinces. A Pearson's chi‐squared test was performed on the full dataset.
Agnes classification
In a final stage of the epidemiological analysis, an Agnes classification was performed. This is a hierarchical classification and can be used to show how the different MLVA classes cluster together when the MLVA type is not taken into consideration. Since our variables are almost exclusively categorical variables, a dissimilarity matrix had to be constructed first. This numerical dissimilarity matrix was then be used as input in Agnes.
Supporting information
Table S1. Tandem repeats of published genomes.
Microbial Biotechnology (2016) 9(6), 772–781
Funding Information
The study was funded by the Research Foundation of Flanders (FWO‐Vlaanderen G.0163.11). The funder had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The authors thank Dr. Stefano Bassi (IZSLER ‘Bruno Ubertini’) and Dr. Irmgard Derakhshifar (Institute for apiculture AGES) for their kind donation of ERIC II isolates of Paenibacillus larvae. The practical assistance of Klaas Van De Loock was greatly acknowledged.
References
- Alippi, A.M. , and Aguilar, O.M. (1998) Characterization of isolates of Paenibacillus larvae subsp. larvae from diverse geographical origin by the polymerase chain reaction and BOX primers. J Invertebr Pathol 72: 21–27. [DOI] [PubMed] [Google Scholar]
- Alippi, A.M. , Lopez, A.C. , and Aguilar, O.M. (2002) Differentiation of Paenibacillus larvae subsp larvae, the cause of American foulbrood of honeybees, by using PCR and restriction fragment analysis of genes encoding 16S rRNA. Appl Environ Microbiol 68: 3655–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunez, K. , Piccini, C. , Castro‐Sowinski, S. , Rosado, A.S. , Seldin, L. , and Zunino, P. (2007) Phenotypic and genotypic characterization of Paenibacillus larvae isolates. Vet Microbiol 124: 178–183. [DOI] [PubMed] [Google Scholar]
- Ash, C. , Farrow, J.A.E. , Wallbanks, S. , and Collins, M.D. (1991) Phylogenetic heterogeneity of the genus Bacillus revealed by comparative‐analysis of small‐subunit‐ribosomal RNA sequences. Lett Appl Microbiol 13: 202–206. [Google Scholar]
- Ash, C. , Priest, F.G. , and Collins, M.D. (1993) Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Proposal for the creation of a new genus Paenibacillus . Antonie Van Leeuwenhoek 64: 253–260. [DOI] [PubMed] [Google Scholar]
- Bassi, S. , Formato, G. , Milito, M. , Trevisiol, K. , Salogni, C. , and Carra, E. (2015) Phenotypic characterization and ERIC–PCR based genotyping of Paenibacillus larvae isolates recovered from American foulbrood outbreaks in honey bees from Italy. Vet Quarterly 35: 27–32. [DOI] [PubMed] [Google Scholar]
- van Belkum, A. (1999) The role of short sequence repeats in epidemiologic typing. Curr Opin Microbiol 2: 306–311. [DOI] [PubMed] [Google Scholar]
- van Belkum, A. (2007) Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA). FEMS Immunol Med Microbiol 49: 22–27. [DOI] [PubMed] [Google Scholar]
- Buhlmann, A. , Dreo, T. , Rezzonico, F. , Pothier, J.F. , Smits, T.H. , Ravnikar, M. , et al (2014) Phylogeography and population structure of the biologically invasive phytopathogen Erwinia amylovora inferred using minisatellites. Environ Microbiol 16: 2112–2125. [DOI] [PubMed] [Google Scholar]
- Bui, T.N. , Verniere, C. , Jarne, P. , Brisse, S. , Guerin, F. , Boutry, S. , et al (2009) From local surveys to global surveillance: three high‐throughput genotyping methods for epidemiological monitoring of Xanthomonas citri pv. citri pathotypes. Appl Environ Microbiol 75: 1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, Q.W. , Cornman, R.S. , Birol, I. , Liao, N.Y. , Chan, S.K. , Docking, T.R. , et al (2011) Updated genome assembly and annotation of Paenibacillus larvae, the agent of American foulbrood disease of honey bees. BMC Genom 12: 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djordjevic, S. , Hoshon, M. , and Hornitzky, M. (1994) DNA restriction‐endonuclease profiles and typing of geographically diverse isolates of Bacillus‐Larvae . J Apic Res 33: 95–103. [Google Scholar]
- Djukic, M. , Brzuszkiewicz, E. , Funfhaus, A. , Voss, J. , Gollnow, K. , Poppinga, L. , et al (2014) How to kill the honey bee larva: genomic potential and virulence mechanisms of Paenibacillus larvae . PLoS ONE 9: e90914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbelaere, W. , de Graaf, D.C. , Peeters, J.E. , and Jacobs, F.J. (2001) Development of a fast and reliable diagnostic method for American foulbrood disease (Paenibacillus larvae subsp larvae) using a 16S rRNA gene based PCR. Apidology 32: 363–370. [Google Scholar]
- Gelfand, J. , Rodriguez, A. , and Benson, G. (2006) TRDB‐The tandem repeats database. Nucleic Acids Res 00: D1–D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genersch, E. (2010) American Foulbrood in honeybees and its causative agent, Paenibacillus larvae . J Invertebr Pathol 103(Suppl. 1): S10–S19. [DOI] [PubMed] [Google Scholar]
- Genersch, E. , and Otten, C. (2003) The use of repetitive element PCR fingerprinting (rep‐PCR) for genetic subtyping of German field isolates of Paenibacillus larvae subsp larvae . Apidologie 34: 195–206. [Google Scholar]
- Genersch, E. , Ashiralieva, A. , and Fries, I. (2005) Strain‐ and genotype‐specific differences in virulence of Paenibacillus larvae subsp. larvae, a bacterial pathogen causing American foulbrood disease in honeybees. Appl Environ Microbiol 71: 7551–7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genersch, E. , Forsgren, E. , Pentikainen, J. , Ashiralieva, A. , Rauch, S. , Kilwinski, J. , and Fries, I. (2006) Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without subspecies differentiation. Int J Syst Evol Microbiol 56: 501–511. [DOI] [PubMed] [Google Scholar]
- Girard, J.M. , Wagner, D.M. , Vogler, A.J. , Keys, C. , Allender, C.J. , Drickamer, L.C. , and Keim, P. (2004) Differential plague‐transmission dynamics determine Yersinia pestis population genetic structure on local, regional, and global scales. PNAS 101: 8408–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf, D.C. , Vanderkerckhove, D. , Dobbelaere, W. , Peeters, J. , and Jacobs, F.J. (2001) Influence of the proximity of American foulbrood cases and apiculture management on the prevalence of Paenibacillus larvae spores in Belgian honey. Apidologie 32: 587–599. [Google Scholar]
- de Graaf, D.C. , De Vos, P. , Heyndrickx, M. , Van Trappen, S. , Peiren, N. , and Jacobs, F.J. (2006) Identification of Paenibacillus larvae to the subspecies level: an obstacle for AFB diagnosis. J Invertebr Pathol 91: 115–123. [DOI] [PubMed] [Google Scholar]
- Heyndrickx, M. , Vandemeulebroecke, K. , Hoste, B. , Janssen, P. , Kersters, K. , De Vos, P. , et al (1996) Reclassification of Paenibacillus (formerly Bacillus) pulvifaciens (Nakamura 1984) Ash et al. 1994, a later subjective synonym of Paenibacillus (formerly Bacillus) larvae (White 1906) Ash et al. 1994, as a subspecies of P. larvae, with emended descriptions of P. larvae as P. larvae subsp. larvae and P. larvae subsp. pulvifaciens. Int J Syst Bacteriol 46: 270–279. [DOI] [PubMed] [Google Scholar]
- Katznelson, H. (1950) Bacillus pulvifaciens (N. SP.), an organism associated with powdery scale of honeybee larvae. J Bacteriol 59: 153–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevytska, A.M. , Price, L.B. , Schupp, J.M. , Worsham, P.L. , Wong, J. , and Keim, P. (2001) Identification and characterization of variable‐number tandem repeats in the Yersinia pestis genome. J Clin Microbiol 39: 3179–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstedt, B.A. (2005) Multiple‐locus variable number tandem repeats analysis for genetic fingerprinting of pathogenic bacteria. Electrophoresis 26: 2567–2582. [DOI] [PubMed] [Google Scholar]
- Lindstrom, A. (2008) Distribution of Paenibacillus larvae spores among adult honey bees (Apis mellifera) and the relationship with clinical symptoms of American foulbrood. Microb Ecol 56: 253–259. [DOI] [PubMed] [Google Scholar]
- Lindstrom, A. , Korpela, S. , and Fries, I. (2008) The distribution of Paenibacillus larvae spores in adult bees and honey and larval mortality, following the addition of American foulbrood diseased brood or spore‐contaminated honey in honey bee (Apis mellifera) colonies. J Invertebr Pathol 99: 82–86. [DOI] [PubMed] [Google Scholar]
- Loncaric, I. , Derakhshifar, I. , Oberlerchner, J.T. , Köglberger, H. , and Moosbeckhofer, R. (2009) Genetic diversity among isolates of Paenibacillus larvae from Austria. J Inverebr Pathol 100: 44–46. [DOI] [PubMed] [Google Scholar]
- Malachowa, N. , Sabat, A. , Gniadkowski, M. , Krzyszton‐Russjan, J. , Empel, J. , Miedzobrodzki, J. , et al (2005) Comparison of multiple‐locus variable‐number tandem‐repeat analysis with pulsed‐field gel electrophoresis, spa typing, and multilocus sequence typing for clonal characterization of Staphylococcus aureus isolates. J Clin Microbiol 43: 3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazars, E. , Lesjean, S. , Banuls, A.L. , Gilbert, M. , Vincent, V. , Gicquel, B. , et al (2001) High‐resolution minisatellite‐based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. PNAS 98: 1901–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey, B.J. , Helgason, T. , Poppinga, L. , Funfhaus, A. , Genersch, E. , and Budge, G.E. (2015) Biogeography of Paenibacillus larvae, the causative agent of American foulbrood, using a new multilocus sequence typing scheme. Environ Microbiol 17: 1414–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuendorf, S. , Hedtke, K. , Tangen, G. , and Genersch, E. (2004) Biochemical characterization of different genotypes of Paenibacillus larvae subsp. larvae, a honey bee bacterial pathogen. Microbiology 150: 2381–2390. [DOI] [PubMed] [Google Scholar]
- Qin, X. , Evans, J.D. , Aronstein, K.A. , Murray, K.D. , and Weinstock, G.M. (2006) Genome sequences of the honey bee pathogens Paenibacillus larvae and Ascosphaera apis . Insect Mol Biol 15: 715–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2015). R: A language and environment for statistical computing. URL https://www.R-project.org/.
- Schafer, M.O. , Genersch, E. , Funfhaus, A. , Poppinga, L. , Formella, N. , Bettin, B. , and Karger, A. (2014) Rapid identification of differentially virulent genotypes of Paenibacillus larvae, the causative organism of American foulbrood of honey bees, by whole cell MALDI‐TOF mass spectrometry. Vet Microbiol 170: 291–297. [DOI] [PubMed] [Google Scholar]
- Stratilo, C.W. , and Bader, D.E. (2012) Genetic diversity among Bacillus anthracis soil isolates at fine geographic scales. Appl Environ Microbiol 78: 6433–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techaruvichit, P. , Takahashi, H. , Vesaratchavest, M. , Keeratipibul, S. , Kuda, T. , and Kimura, B. (2015) Development of multiple‐locus variable‐number tandem‐repeat analysis for molecular subtyping of Campylobacter jejuni by using capillary electrophoresis. Appl Environ Microbiol 81: 5318–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres‐Cruz, J. , and van der Woude, M.W. (2003) Slipped‐strand mispairing can function as a phase variation mechanism in Escherichia coli . J Bacteriol 185: 6990–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U'Ren, J.M. , Hornstra, H. , Pearson, T. , Schupp, J.M. , Leadem, B. , Georgia, S. , et al (2007) Fine‐scale genetic diversity among Burkholderia pseudomallei soil isolates in northeast Thailand. Appl Environ Microbiol 73: 6678–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler, A.J. , Keys, C. , Nemoto, Y. , Colman, R.E. , Jay, Z. , and Keim, P. (2006) Effect of repeat copy number on variable‐number tandem repeat mutations in Escherichia coli O157:H7. J Bacteriol 188: 4253–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X.Y. , Chin, J. , Ghalayini, A. , and Hornitzky, M. (2005) Pulsed‐field gel electrophoresis typing and oxytetracycline sensitivity of Paenibacillus larvae subsp larvae isolates of Australian origin and those recovered from honey imported from Argentina. J Apic Res 44: 87–92. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Tandem repeats of published genomes.