

Abstract
We have developed a new class of cinchonium betaine catalysts bearing both a base moiety and an aromatic moiety as an N-substituent of the quinuclidine motif. These cinchonium betains were found to promote proton transfer catalysis with 1000–5000 turnovers per 24 hours, thereby enabling us to realize highly efficient enantioselective isomerization of trifluoromethyl imines to provide a practical access to optically active trifluoromethylated amines.
Graphical Abstract
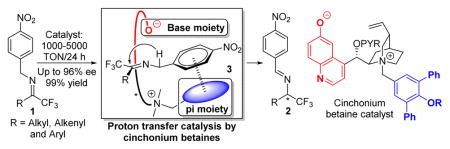
1. Introduction
We1 and others2 demonstrated that chiral organic catalysts containing both hydrogen bond donor and acceptor could facilitate biomimetic 1,3-proton transfer catalysis to promote highly enantioselective olefin and imine isomerizations. These enantioselective isomerizations provide new access to valuable chiral building blocks such as α,β-unsaturated butenolides,1a,3 α-amino acids,2c α,β-unsaturated cyclohexenones1c and trifluoromethylated amines.1b,2e Among these studies, the realization of the first highly enantioselective isomerization1b of trifluoromethyl imines with DHQ-3 stands as a conceptually significant progress as DHQ-3 achieved efficient catalytic chiral recognition of non-enolate carbanions for asymmetric reactions (Figure 1).4,5
Figure 1.

Drawbacks of asymmetric isomerization of trifluoromethyl imines with acid-base bifunctional catalysts.
However, the required high catalyst loading (10 mol%) and long reaction time (48–72 hours) severely hampered the application of this reaction in asymmetric synthesis. Moreover, the isomerizations with the pseudoenantiomeric catalyst DHQD-3 proceeded with significantly lower enantioselectivity under optimized conditions (Figure 1). Consequently, this method was not able to provide useful access to the S enantiomers of chiral trifluoromethylated amines. These problems also apply to another cinchona alkaloid-derived bifunctional catalyst reported by Shi and coworkers for the same isomerizations.2e These limitations associated with existing methods highlighted both the urgent need and challenges for the development of significantly more active and selective chiral catalysts for this promising asymmetric transformation. We wish to report in this article the development of new chiral cinchonium betaine catalysts that afford powerful enantioselective proton transfer catalysis, which overcomes the limitations associated with existing chiral catalysts. The general scope, simple protocol for reaction execution and product isolation and an extraordinarily low catalyst loading render this unprecedented, phase transfer catalysis-mediated enantioselective imine isomerization a practical method for the asymmetric synthesis of trifluoromethylated amines.6–9
2. Results and discussion
During our recent studies of C-C bond forming asymmetric umpolung reaction of imines10, we observed that certain cinchona alkaloid-derived chiral phase transfer catalysts promoted isomerization of trifluoromethyl imines. This observation raised the prospect of developing a new class of chiral catalysts for the promotion of asymmetric isomerizations of imines via a fundamentally different mechanism from those by acid-base bifunctional catalysts. Although highly enantioselective protonations of enol derivatives were reported,11 a literature search revealed that phase transfer catalyst-promoted proton transfer reactions are still rare and only with modest enantioselectivity. 12
Motivated by the possible discovery of new and powerful catalysts for enantioselective proton transfer catalysis, we began to explore cinchonium salts as catalysts for the isomerization of trifluoromethyl imine 1. At room temperature in the presence of aqueous KOH and 0.5 mol% cinchonium salts C-5 and C-6, the model isomerization was found to proceed in very low enantioselectivity (Entries 1–2, Table 1). We next examined catalysts C-7, which presented an electron-rich N-terphenyl group that was designed to engage in π-π interaction with the 2-azaallylanions.10 To our disappointment, C-7 afforded even worse enantioselectivity (Entry 3, Table 1). Moreover, imine 1a was found to be transformed into not only the chiral amine 2a but also the N-(1,1-difluoropropenyl) imine 4a. The latter, presumably formed by the loss of fluoride from 2-azaallylanion 10a (Figure 2a), was often the dominant product.13
Table 1.
Asymmetric isomerization of 1a with cinchonium betaine catalystsa
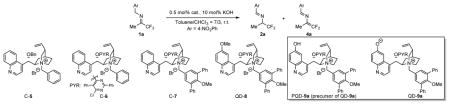
| |||||
|---|---|---|---|---|---|
| Entry | cat. | t (h) | conv. (%)b | ee (%)b | 2a/4ab |
| 1 | C-5 | 4 | 16 | −22 | 61/39 |
| 2 | C-6 | 4 | 3 | −26 | 45/55 |
| 3 | C-7 | 4 | 10 | −12 | 40/60 |
| 4 | QD-9ac | 1 | 100 | 91 | 97/3 |
| 5 | QD-8 | 4 | 10 | −33 | 34/66 |
Reactions were run with 1a (0.025 mmol), aqueous KOH (0.3 μL, 50 wt%, 10 mol%) and catalyst in toluene/CHCl3 (7/3 v/v, 0.25 mL) at room temperature.
Determined by HPLC analysis.
The Betaine catalyst QD-9a was generated in situ from PQD-9a.
Figure 2.

Working hypotheses.
These discouraging results indicated that, with cinchonium-based phase transfer catalysts, the enantioselective protonation of the 2-azaallyl anion 10a proceeded not only with low enantioselectivity but also too slow to establish the desired isomerization as the major reaction pathway (Figure 2a). Moreover, we suspected that the low enantioselectivity could be attributed to the difficulty in effectively biasing the two enantiotopic faces of the 2-azaallylanion 10a towards the intermolecular protonation by water, a small proton donor. Following these considerations, we concluded that the intermolecular nature of the protonation step in these cinchonium salts-mediated isomerizations was largely responsible for the poor catalytic activity and enantioselectivity.
We postulated that we might be able to address this problem by installing an alkoxide functionality into the cinchonium salt C-7, which bear a π-π interaction moiety. As illustrated in Figure 2b, we hypothesized that such a betaine catalyst11c,14–18 could, upon the binding of imine 1a to the catalyst via π-π interaction, promote both the deprotonation and the protonation steps in an intramolecular-like setting. The protonation step should be greatly accelerated as it involved proton transfer between the alkoxide and the 2-azaallyanion intermediate located close to each other. More importantly, the spatial relationship between the phenol as the proton donor and the 2-azaallyanion interacting with the catalyst via π-π and ion-pair interactions should be confined by the chiral backbone of the catalyst, thereby providing a more favorable setting for achieving fast and highly enantioselective protonation.
Guided by these considerations, we employed PQD-9a as a precursor of the cinchonium betaine catalyst QD-9a (Table 1). Initially, betaine QD-9a was examined for the isomerizations of 1a under the same conditions as those applied to catalysts C5–C7. We expected that QD-9a could be formed in situ via deprotonation of the PQD-9a by KOH. To our delight, the isomerization of 1a went to completion in one hour with 0.5 mol% of QD-9a to afford the desired chiral amine 2a in 91% ee as virtually the only detectable product (entry 4, Table 1). In contrast, the quinidine-derived cinchonium salt QD-8 afforded poor chemoselectivity and enantioselectivity, thereby showing the critical role played by the phenoxide of QD-9a for the efficient promotion of the enantioselective isomerization. The opposite sense of asymmetric induction by QD-9a versus that by C5-C7 and QD-8 indicated that QD-9a exercised its enantioselectivity via a distinct mechanism. This notion is consistent with the working hypothesis of our catalyst design as outlined in Figure 2.
To further test whether the betaine QD-9 was indeed responsible for the dramatically improved catalytic activity and selectivity, we carried out an isomerization with the preformed betaine QD-9a under base free conditions. Specifically, we first treated PQD-9a with solid KOH in toluene/CHCl3 for 15 minutes, when PQD-9a was shown by 1H NMR analysis to be converted into the betaine QD-9a.19 Then the desired amount of the QD-9a solution in toluene/CHCl3 was added to a solution of imine 1a in toluene/CHCl3. After four hours the isomerization was shown to proceed to 76% conversion to afford R-2a as the only detectable product in 92% ee (entry 1, Table 2). Upon further optimization by catalyst tuning (entries 2–3, Table 2), we were able to accomplish the isomerization in 98% conversion and 95% ee by employing betaine QD-9c.
Table 2.
Optimizations of catalyst and reaction conditions for enantioselective isomerization of 1aa
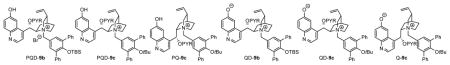
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | cat. | mol% of cat. | Base presentedin the reaction | Solvent | t (h) | conv. (%)b | ee (%)b |
| 1 | QD-9a | 0.5 | No | Toluene/CHCl3 = 7/3 | 4 | 76 | 92 |
| 2 | QD-9b | 0.5 | No | Toluene/CHCl3 = 7/3 | 4 | 97 | 93 |
| 3 | QD-9c | 0.5 | No | Toluene/CHCl3 = 7/3 | 4 | 98 | 95 |
| 4 | QD-9c | 0.2 | No | Toluene/CHCl3 = 7/3 | 24 | 77 | 95 |
| 5 | OD-9c | 0.2 | solid K2CO3 | Toluene/CHCl3 = 7/3 | 12 | 100 | 95 |
| 6 | QD-9c | 0.02 | solid K2CO3 | Toluene | 24 | 100 | 96c |
| 7 | QD-9c | 0.01 | solid K2CO3 | Toluene | 24 | 97 | 96 |
| 8 | Q-9c | 0.05 | solid K2CO3 | Toluene | 24 | 100 | −93 |
Betaine catalysts QD-9 and Q-9c were preformed from treatment of the corresponding precursors PQD-9 and PQ-9c with base (see supporting information for details). Reactions were run with 1a (0.025 mmol) in solvent (0.25 mL) indicated. The ratio of 2a/4a was determined to be greater than 99/1 by HPLC analysis.
Determined by HPLC analysis.
Absolute configuration was determined to be R. See Supporting Information for details.
We attempted to further reduce catalyst loading, but failed to accomplish a completed reaction with 0.2 mol% of QD-9c (entry 4, Table 2). Subsequently, we found that the presence of a solid base such as K2CO3 was highly beneficial for maintaining the activity of the betaine catalyst (entry 5 vs. 4, Table 2). Upon optimizations of solvent, a highly enantioselective and complete isomerization of 1a could be consistently accomplished in 24 hours in toluene with only 0.02 mol% of QD-9c (entry 6, Table 2). Notably, the conversion only suffered very slightly with 0.01 mol% of QD-9c without compromising enantioselectivity; reaching 97% in 24 hours. These results showed that the cinchonium betaine catalyst-mediated asymmetric proton transfer catalysis proceeded with an extraordinarily high catalyst turnover rate (entry 7, Table 2). Gratifyingly, a highly enantioselective isomerization of 1a into S-2a could be accomplished with 0.05 mol% of quinine-derived betaine Q-9c (entry 8, Table 2). Thus, the cinchonium betaine catalysts not only are far superior to existing organocatalysts in terms of catalyst efficiency but also bring a decisive synthetic advantage by offering useful access to either R or S enantiomer of the trifluoromethylated amines 2.
We next investigated the substrate scope. Excellent enantioselectivity could be readily accomplished for a variety of aliphatic trifluoromethyl imines of varying length (1a–1c) with QD-9c in 0.02–0.10 mol% loading (entries 1–3, Table 3). For the sterically more hindered cyclohexyl trifluoromethyl imine 1d, a catalyst loading of 0.50 mol% was required in order to achieve a highly enantioselective and complete isomerization in 24 hours (entry 4, Table 3). Interestingly, we found that QD-9c promoted an unprecedented isomerization of α,β-unsaturated imine 1e in excellent enantioselectivity and yield. It is noteworthy that the reaction proceeded without the formation of the 1,3-proton transfer product. The scope of the reaction was readily extended to a wide range of aryl trifluoromethyl imines (entries 6–12, Table 3). In most cases, a loading of 0.10–0.20 mol% of QD-9c was sufficient in affording a complete and highly enantioselective isomerization of imines 1 within 24 hours to generate the corresponding optically active chiral aryl trifluoromethylated amine 2 in close to quantitative yields. Aryl trifluoromethyl imines bearing a strongly electron-withdrawing substituent such as 1m and 1n proved to be more challenging substrates, nonetheless a quantitative reaction with good enantioselectivity could be mediated by 0.20 mol% of QD-9c. Importantly, isomerizations with betaine Q-9c in similarly low loading afforded the opposite enantiomer of the chiral trifluoromethylated amine in excellent yield and in only slightly lower optical purities (Table 3). The isomerization typically proceeded so cleanly that a simple filtration of the reaction mixture through a plug of deactivated silica gel followed by solvent removal furnished amines 2 in pure form as determined by NMR analysis. The NMR-pure isomerization product 2a, without further purification, could be hydrolyzed to give the desired chiral trifluoromethylated amine 11a in 86% yield and 96% ee.1b
Table 3.
Isomerization of 1 to 2 catalyzed by QD-9c and Q-9ca.
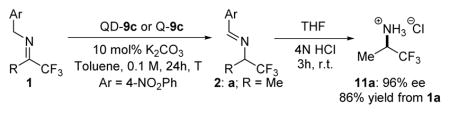
| ||||||
|---|---|---|---|---|---|---|
| Entry | 1 | R | T(°C) | mol% of cat. | Yield (%)b | ee (%)c |
| 1 | 1a | Me | r.t. | 0.02 (0.05) | 96 (96) | 96 (−93) |
| 2 | 1b | Et | r.t. | 0.08 (0.10) | 98 (97) | 95 (−91) |
| 3 | 1c | n-Bu | r.t. | 0.10 (0.20) | 97 (96) | 96 (−90) |
| 4 | 1dd | Cyclohexyl | r.t. | 0.50 (0.50) | 97 (74) | 94 (−86) |
| 5 | 1e | trans-Styrl | 0 | 0.40 (0.40) | 97 (96) | 95 (−85) |
| 6 | 1f | Ph | 0 | 0.10 (0.10) | 96 (96) | 90 (−81) |
| 7 | 1g | 4-Me-C6H4 | 0 | 0.10 (0.10) | 99 (97) | 93 (−85) |
| 8 | 1h | 3-Me-C6H4 | 0 | 0.10 (0.10) | 98 (98) | 93 (−84) |
| 9 | 1i | 4-OMe-C6H4 | 0 | 0.10 (0.10) | 99 (96) | 93 (−84) |
| 10 | 1j | 4-F-C6H4 | −20 | 0.20 (0.40) | 97 (95) | 90 (−84) |
| 11 | 1k | 4-Cl-C6H4 | −20 | 0.20 (0.40) | 95 (98) | 88 (−81) |
| 12 | 1l | 4-Br-C6H4 | −20 | 0.20 (0.40) | 98 (96) | 88 (−82) |
| 13 | 1m | 4-CF3-C6H4 | −20 | 0.20 (0.40) | 96 (98) | 83 (−76) |
| 14 | 1n | 4-COOtBu-C6H4 | −20 | 0.20 (0.40) | 95 (94) | 82 (−77) |
Betaine catalysts QD-9c and Q-9c were preformed from treatment of the corresponding precursors PQD-9c and PQ-9 with base(see supporting information for details). Reactions were run with 1 (0.2 mmol) in toluene (2.0 mL). Results in parentheses were obtained with Q-9c.
Isolated yield.
Determined by HPLC analysis.
An E/Z mixture of imine stereoisomers (E/Z = 3.2/1) was used.
To validate that the methine hydrogen in 2a came from one of the benzylic hydrogen in 1a, we first carried out the QD-9c catalyzed isomerization of 1a in toluene-d8 using the standard reaction conditions. We next carried out the same isomerization with dry K2CO3 in anhydrous toluene-d8 in dry box.19 As shown by 1H NMR analyses (Figure 3 and S-Figure 6 in Supporting Information), we found the chiral amine 2a was formed without incorporation of deuterium. We also investigated a QD-9c catalyzed isomerization of 1a in toluene-d8 with 40% KOD in D2O, instead of K2CO3, as the base. We found that 2a was formed in 92% ee and, once again, without incorporation of deuterium. 19 These results indicated that the methine hydrogen in 2a indeed came from the benzylic hydrogen in 1a, which was consistent with our hypothesis of how a cinchonium betain like QD-9c mediated this highly enantioselective 1,3-proton transfer (Figure 2b). To verify that the π-π interaction between imines 1 and catalyst 9c played an important role to the catalysis by 9c (Figure 2b), we investigated the isomerizations of both imines 1a′ and 1a″, which were derived by replacing the 4-NO2 benzyl group in 1a with a benzyl group and 4-carboxylatebenzyl group, respectively (Figure 4). Catalyst 9c was found to be inactive toward the isomerization of imine 1a′ and afforded significantly worse enantioselectivity (84% ee vs. 96% ee) and lower conversion (43% vs 100%) for the isomerization of 1a″. These results indicated that the 4-NO2 benzyl group not only rendered imine 1a more active toward the QD-9c catalyzed isomerization but also played an important role in the substrate-catalyst chiral recognition.
Figure 3.

1H NMR spectra of 2a isolated from labeling experiment in toluene-d8 at 298 K using the standard reaction conditions.
Figure 4.

Isomerization of imines 1a′ and 1a″ with QD-9c.
3. Conclusion
In conclusion, our exploratory studies of cinchonium betaines have led to the discovery of a new class of catalysts for enantioselective proton transfer catalysis. These new catalysts afforded remarkably high catalyst turnover rate for the promotion of asymmetric isomerizations of trifluoromethyl imines.20 Consequently, a broad range of alkyl, alkenyl and aryl trifluoromethyl imines could be converted in a highly enantioselective manner into either enantiomer of the corresponding optically active trifluoromethylated amines with typically 0.02 to 0.10 mol% of the cinchonium betaines. With a mechanistically distinct mode of catalysis, this reaction should provide a complementary approach to existing methods for the asymmetric synthesis of trifluoromethylated amines.
4. Experimental Section
4.1 General procedure for the asymmetric isomerization of alkyl trifluomethyl imines 1a to 1d with catalyst QD-9c
To a solution of catalyst PQD-9c (3.1 μmol, 3.0 mg) in toluene (180 μL) was added grounded K2CO3 (0.065 mmol, 9.0 mg). The suspension was then stirred vigorously at room temperature for 2 hours. After standing for 10 minutes, a portion of the clear solution (90 μL) was collected and diluted with toluene (810 μL). A portion of this diluted solution of QD-9c in toluene (0.040 μmol to 1.0 μmol, 23 μL to 580 μL) was placed into a vial (3.7 mL), to which was added sequentially toluene (1.977 mL to 1.42 mL), grounded K2CO3 (0.020 mmol, 2.8 mg) and trifluoromethyl imine 1 (0.20 mmol). Then the mixture was allowed to stir at room temperature for 24 hours. The reaction mixture was allowed to pass through a plug of deactivated silica gel21 to remove the catalyst. The deactivated silica gel plug was then washed with diethyl ether (2.0–4.0 mL). The filtrate was concentrated in vacuo to give trifluoromethylated amines 2.
4.2 General procedure for the asymmetric isomerization of alkenyl or aryl trifluomethyl imines 1e to 1n with catalyst QD-9c
To a solution of catalyst PQD-9c (3.1 μmol, 3.0 mg) in toluene (180 μL) was added grounded K2CO3 (0.065 mmol, 9.0 mg). The suspension was then stirred vigorously at room temperature for 2 hours. After standing for 10 minutes, a portion of the clear solution (90 μL) was collected and diluted with toluene (810 μL). A portion of this diluted solution of QD-9c in toluene (0.20 μmol to 0.80 μmol, 120 μL to 460 μL) was placed into a vial (3.7 mL), to which was added sequentially toluene (680 μL to 340 μL) and grounded K2CO3 (0.020 mmol, 2.8 mg). After the mixture was stirred at the designated temperature in Table 3 for 20 minutes, the solution of imine 1 (0.20 mmol) in toluene (1.2 mL) was added in one portion (the imine solution was also stirred at the specified temperature for 20 minutes). Then the mixture was allowed to stir at the specified temperature for 24 hours. The reaction mixture was allowed to pass through a plug of deactivated silica gel to remove the catalyst. The deactivated silica gel plug was then washed with diethyl ether (2.0–4.0 mL). The filtrate was concentrated in vacuo to give trifluoromethylated amines 2.
Supplementary Material
Acknowledgments
We are grateful for financial support from National Institutes of Health (Grant GM-61591) and the Keck Foundation.
Footnotes
The authors declare no competing financial interest.
Detailed experimental procedures; characterization of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Wu Y, Singh RP, Deng L. J Am Chem Soc. 2011;133:12458. doi: 10.1021/ja205674x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu Y, Deng L. J Am Chem Soc. 2012;134:14334. doi: 10.1021/ja306771n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee JH, Deng L. J Am Chem Soc. 2012;134:18209. doi: 10.1021/ja308623n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Liu HJ, Leow DS, Huang KW, Tan CH. J Am Chem Soc. 2009;131:7212. doi: 10.1021/ja901528b. [DOI] [PubMed] [Google Scholar]; (b) Liu H, Feng W, Kee CW, Leow D, Loh WT, Tan CH. Adv Synth Catal. 2010;352:3373. [Google Scholar]; (c) Xiao X, Xie Y, Su C, Liu M, Shi Y. J Am Chem Soc. 2011;133:12914. doi: 10.1021/ja203138q. [DOI] [PubMed] [Google Scholar]; (d) Inokuma T, Furukawa M, Suzuki Y, Kimachi T, Kobayashi Y, Takemoto Y. ChemCatChem. 2012;4:983. [Google Scholar]; (e) Liu M, Li J, Xiao X, Xie Y, Shi Y. Chem Commun. 2013;49:1404. doi: 10.1039/c2cc37423d. [DOI] [PubMed] [Google Scholar]
- 3.Hugelshofer CL, Magauer T. J Am Chem Soc. 2015;137:3807. doi: 10.1021/jacs.5b02021. [DOI] [PubMed] [Google Scholar]
- 4.For early attempts to develop catalytic enantioselective isomerizations of trifluoromethyl imines, see: Soloshonok VA, Kirilenko AG, Galushko SV, Kukhar VP. Tetrahedron Lett. 1994;35:5063.Soloshonok VA, Ono T. J Org Chem. 1997;62:3030. doi: 10.1021/jo970425c.Soloshonok VA, Yasumoto M. J Fluorine Chem. 2007;128:170.Michaut V, Metz F, Paris JM, Plaquevent JC. J Fluorine Chem. 2007;128:500.
- 5.Fehr C. Angew Chem, Int Ed Engl. 1996;35:2567. [Google Scholar]
- 6.For selected reviews on asymmetric synthesis of fluorinated molecules, see: Ma JA, Cahard D. Chem Rev. 2004;104:6119. doi: 10.1021/cr030143e.Nie J, Guo HC, Cahard D, Ma JA. Chem Rev. 2011;111:455. doi: 10.1021/cr100166a.
- 7.For selected examples of catalytic asymmetric hydrogenation of trifluoromethyl imines or enamides, see: Abe H, Amii H, Uneyama K. Org Lett. 2001;3:313. doi: 10.1021/ol0002471.Chen MW, Duan Y, Chen QA, Wang DS, Yu CB, Zhou YG. Org Lett. 2010;12:5075. doi: 10.1021/ol1020256.Jiang J, Lu W, Zhang X. Org Lett. 2015;17:1154. doi: 10.1021/acs.orglett.5b00087.
- 8.For selected examples of catalytic asymmetric transfer hydrogenation with trifluoromethyl imines, see: Henseler A, Kato M, Mori K, Akiyama T. Angew Chem, Int Ed. 2011;50:8180. doi: 10.1002/anie.201103240.Genoni A, Benaglia M, Massolo E, Rossi S. Chem Commun. 2013;49:8365. doi: 10.1039/c3cc43821j.Dai XY, Cahard D. Adv Synth Catal. 2014;356:1317.
- 9.For selected examples of catalytic asymmetric nucleophilic addition to trifluomethyl aldimines or aldimines, see: Funabiki K, Nagamori M, Goushi S, Matsui M. Chem Commun. 2004:1928. doi: 10.1039/b406000h.Gosselin F, O’Shea PD, Roy S, Reamer RA, Chen CY, Volante RP. Org Lett. 2005;7:355. doi: 10.1021/ol047431x.Kawai H, Kusuda A, Nakamura S, Shiro M, Shibata N. Angew Chem, Int Ed. 2009;48:6324. doi: 10.1002/anie.200902457.
- 10.Wu Y, Hu L, Li Z, Deng L. Nature. 2015;523:445. doi: 10.1038/nature14617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For examples of phase transfer catalyzed protonation reactions, see: Yamamoto E, Nagai A, Hamasaki A, Tokunaga M. Chem Eur J. 2011;17:7178. doi: 10.1002/chem.201100833.Yamamoto E, Gokuden D, Nagai A, Kamachi T, Yoshizawa K, Hamasaki A, Ishida T, Tokunaga M. Org Lett. 2012;14:6178. doi: 10.1021/ol3027363.Claraz A, Landelle G, Oudeyer S, Levacher V. Eur J Org Chem. 2013:7693.
- 12.For examples of phase transfer catalyzed proton transfer reactions, see: Oku M, Arai S, Katayama K, Shioiri T. Synlett. 2000:493.Shirakawa S, Tokuda T, Kan SBJ, Maruoka K. Org Chem Front. 2015;2:336.Roy A, Bhat BA, Lepore SD. Org Lett. 2016;18:1230. doi: 10.1021/acs.orglett.5b03681.
- 13.HF eliminations were previously reported, see: Perosa A, Selva M, Tundo P. J Chem Soc, Perkin Trans. 2002;2:1033.
- 14.For pioneering studies of chiral ammonium betaines, see: Uraguchi D, Koshimoto K, Ooi T. J Am Chem Soc. 2008;130:10878. doi: 10.1021/ja8041004.
- 15.For chiral BINOL-derived ammonium betaines as base catalysts, see: Uraguchi D, Koshimoto K, Sanada C, Ooi T. Tetrahedron: Asymmetry. 2010;21:1189.Uraguchi D, Koshimoto K, Ooi T. Chem Commun. 2010;46:300. doi: 10.1039/b916627k.Uraguchi D, Oyaizu K, Ooi T. Chem Eur J. 2012;18:8306. doi: 10.1002/chem.201201259.Uraguchi D, Oyaizu K, Noguchi H, Ooi T. Chem Asian J. 2015;10:334. doi: 10.1002/asia.201402943.Oyaizu K, Uraguchia D, Ooi T. Chem Commun. 2015;51:4437. doi: 10.1039/c4cc10261d.
- 16.For chiral BINOL-derived ammonium betaines as nucleophilic catalysts, see: Uraguchi D, Koshimoto K, Miyake S, Ooi T. Angew Chem Int Ed. 2010;49:5567. doi: 10.1002/anie.201002315.Uraguchi D, Koshimoto K, Ooi T. J Am Chem Soc. 2012;134:6972. doi: 10.1021/ja3022939.
- 17.For studies of 6′-OH cinchona alkaloid-derived ammonium salts, see: Liu Y, Provencher BA, Bartelson KJ, Deng L. Chem Sci. 2011;2:1301. doi: 10.1039/c1sc00137j.Provencher BA, Bartelson KJ, Liu Y, Foxman BM, Deng L. Angew Chem Int Ed. 2011;50:10565. doi: 10.1002/anie.201105536.
- 18.For cinchonium BINOL-derived betaines as base catalysts, see: Zhang W-Q, Cheng L-F, Yu J, Gong L-Z. Angew Chem, Int Ed. 2012;51:4085. doi: 10.1002/anie.201107741.
- 19.See Supporting Information for details.
- 20.For a review on low loading organocatalysis, see: Giacalone F, Gruttadauria M, Agrigento P, Noto R. Chem Soc Rev. 2012;41:2406. doi: 10.1039/c1cs15206h.
- 21.The deactivated silica gel was prepared from treating normal silica gel with NEt3. See supporting information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.