

Abstract
Esophageal adenocarcinoma (EAC) has a poor prognosis and is increasing in incidence in many Western populations. Neoadjuvant chemoradiation therapy (CRT) followed by surgery is increasingly the standard of care for locally advanced EAC; however, resistance to treatment is a significant clinical problem. The identification of both novel biomarkers predicting response to treatment and novel therapeutic targets to enhance the efficacy of CRT is key to improving survival rates in EAC. In this study, we performed global microRNA (miRNA) profiling of pretreatment EAC biopsies and identified 67 miRNAs significantly altered in patients who are resistant to CRT. One of these miRNAs, miR-187, was significantly decreased in pretreatment EAC tumors from patients having a poor response to neoadjuvant CRT, highlighting downregulation of miR-187 as a potential mediator of treatment resistance in EAC. In vitro, miR-187 was demonstrated to play a functional role in modulating sensitivity to X-ray radiation and cisplatin in EAC and its dysregulation was demonstrated to be due to chromosomal alterations. In vitro, miR-187 altered expression of a diverse array of pathways, including the immune regulator complement component 3 (C3), serum levels of which we have previously demonstrated to predict patient response to CRT. In vivo, expression of C3 was significantly increased in tumors from patients having a poor response to CRT. This study highlights for the first time a role for miR-187 as a novel biomarker of response to CRT and a potential therapeutic target for enhancing the efficacy of CRT in EAC.
INTRODUCTION
In recent decades, there has been a dramatic epidemiological shift in the incidence of esophageal adenocarcinoma (EAC), with rates rising by 600% over the last 30 years in Western populations (1). EAC is now the predominant histological subtype of esophageal cancer in Europe and the United States (2). This increase is linked to lifestyle factors, such as obesity, and incidence is expected to increase at a similar rate in the coming decades (3). Despite improvements in surveillance and diagnosis, the overall cure rate is <17%, resulting in an estimated 400,000 deaths worldwide each year (1). Consequently, a multimodal approach to treatment has been developed, with neoadjuvant chemoradiation therapy (CRT) followed by surgery increasingly becoming the standard of care in Ireland, North America and several European centers (4).
Tumor response to neoadjuvant CRT is the best predictor of overall and disease-free survival, with the attainment of a complete pathological response (pCR) associated with a 5-year survival rate of up to 60% (5). Unfortunately, a pCR is observed in only approximately 30% of patients receiving neoadjuvant CRT (6). The remaining 70% of patients are subject to the risk of toxicity and therapy-associated complications, and their prognosis is potentially worsened by the delay to surgery (7). There are currently no alternative treatment strategies for this majority of EAC patients.
The ability to identify, prior to neoadjuvant CRT, those patients likely to be resistant or sensitive to current treatment regimens may permit more appropriate stratification of treatment, which may ultimately improve survival rates. There are currently no routinely used predictive markers in the clinic. Consequently, there is an unmet clinical need to identify biomarkers, predicting response to neoadjuvant treatment and to develop novel treatment strategies to improve the tumor response to neoadjuvant CRT in EAC.
Patients with tumors of similar clinical characteristics can have vastly different responses to CRT, suggesting that the dichotomy in response is due to subtle differences in the tumor molecular genetic environment. This suggests that regulatory molecules, such as microRNAs (miRNAs), which are capable of regulating the expression of a large number of genes, could potentially play an important role in the tumor response to CRT and may therefore be useful biomarkers of response to treatment.
MiRNAs are single-stranded RNA molecules that regulate gene expression in cells by directly binding to and either degrading or translationally repressing targets (8). It is now well established that altered miRNA expression is intimately involved in tumorigenesis and cancer biology (8,9). Mounting evidence suggests that miRNA profiles may be more efficient than gene profiles in discriminating different disease states and diseased versus nondiseased states (9,10). A role for miRNAs has been implicated in the pathogenesis of EAC, with several potential diagnostic and prognostic miRNA markers identified (11,12). We have previously demonstrated that both miR-31 (13) and miR-330-5p (14) are significantly decreased in pretreatment tumor biopsies from esophageal cancer patients who subsequently have a poor response to neoadjuvant CRT, highlighting a potential role for these miRNAs in the tumor response to CRT. Furthermore, in vitro, in EAC cells, we demonstrated a functional role for both miR-31 and miR-330-5p in modulating radioresponse to a clinically relevant dose of X-ray radiation.
In this study, we demonstrate for the first time the dysregulated expression of miR-187 in EAC tumors from patients resistant to neoadjuvant CRT and importantly demonstrate a functional role for miR-187 in modulating the response of EAC to radiation and cisplatin. This highlights miR-187 as a novel biomarker of CRT response and a novel therapeutic target to enhance the efficacy of standard CRT treatment regimens in EAC.
MATERIALS AND METHODS
Patients, Treatment and Histology
Following ethical approval (Joint St James’s Hospital and AMNCH ethical review board) and written informed consent, diagnostic biopsy specimens were taken from patients with a diagnosis of operable EAC, prior to neoadjuvant therapy. Patients received a complete course of neoadjuvant CRT. Chemotherapy consisted of two courses of 5-fluorouracil (5-FU) and cisplatin, as described previously (15). Patients received 40.05 gray (Gy) in 15 daily fractions (2.67 Gy/fraction) over 3 wks, as described previously (15). Surgical resection involved transthoracic esophagectomy, including en-bloc lymphadenectomy of the abdominal and mediastinal nodes and was performed approximately 1 month following the completion of the CRT regimen. All resected esophagectomy specimens were assessed by an experienced pathologist. Tumor response to treatment was assigned one of five tumor regression grades (TRGs), TRG 1 (complete regression) to TRG 5 (no regression) based on the presence of residual cancer cells and the degree of fibrotic change, as described previously (16). Responders were classified as patients achieving a TRG of 1 or 2, while nonresponders were classified as patients having a TRG of 3, 4 or 5, as described previously (13).
Tissue Collection
Diagnostic endoscopic biopsies were obtained by a qualified endoscopist prior to neoadjuvant therapy. Immediately adjacent tissue was taken for histologic confirmation, which was performed using routine hematoxylin and eosin staining. Specimens were immediately placed in RNAlater (Ambion) and refrigerated for 24 h, before removal of RNAlater and storage at –80°C.
RNA Isolation
RNA from patient tumor tissue samples was isolated using an All-in-One purification kit (Norgen Biotek), as per the manufacturer’s instructions. Total RNA from cell lines was isolated using an RNeasy mini kit (Qiagen) or TriReagent RNA isolation reagent (Molecular Research Centre Inc.), as per the manufacturer’s instructions. For digital gene expression profiling, RNA was isolated using an RNeasy mini kit (Qiagen). In all cases, RNA was quantified spectrophotometrically using a Nanodrop 1000 spectrophotometer v3.3 (Thermo Scientific).
MiRNA Arrays
For pretreatment biopsy experiments, miRNA (40 ng) was converted to cDNA using Universal RT primers and miRNA reverse transcription kit (Exiqon), as per the manufacturer’s instructions. Global miRNA expression was assessed using human miRCURY LNA Universal RT miRNA arrays, panels I and II (Exiqon) using a 7900HT RT-PCR system (Applied Biosystems). Analysis was performed using GenEx 5.0 software (MultiD Analyses AB). Data were normalized using the global mean and one sample was set as the calibrator for relative quantification analysis.
Cell Lines
The human EAC cell lines OE33 P and OE33 R were generated and cultured in our laboratory, as described previously (17). The human EAC cell lines, OE33 and SK-GT-4, were obtained from the European Collection of Cell Cultures and cultured as monolayers in Roswell Park Memorial Institute (RPMI) 1640 medium, supplemented with 10% (v/v) heat-inactivated fetal bovine serum and 1% (v/v) penicillin–streptomycin (50 U/mL penicillin, 50 U/mL streptomycin). Cells were maintained at 37°C in 95% humidified air containing 5% CO2.
Quantitative PCR
For assessment of miRNA expression, total RNA (10 ng) was reverse transcribed to cDNA using a TaqMan MicroRNA reverse transcription kit (Life Technologies), as per the manufacturer’s instructions. qPCR was performed using a TaqMan miRNA assay kit (Applied Biosystems), and RNU6 was used as an endogenous control for data normalization. For validation of gene targets, gene expression analysis was performed using SYBR green or TaqMan qPCR. For the TaqMan PCR, total RNA (1 μg) was reverse transcribed to cDNA using random hexamers (Invitrogen) and Bioscript enzyme (Bioline). qPCR was performed using TaqMan primer probes and an ABI Prism 7900HT real-time thermal cycler (Applied Biosystems). For the SYBR green PCR, total RNA (1 μg) was reverse transcribed to cDNA using a QuantiTect RT kit (Qiagen). qPCR was performed using QuantiTect Primer Assays (Qiagen) and a Step One Plus Real-Time PCR System (Applied Biosystems). B2M or 18S was used as an endogenous control for data normalization. PCR data were analyzed by the 2–ΔΔCt Livak method (18).
Overexpression of miR-187
Transient overexpression of miR-187 was performed using miRNA precursor plasmids (System Biosciences). Cells were transfected with a pre-miR-187 vector (Catalogue number: PMIRH187PA-1) or scrambled vector control (Catalogue number: CD511B-1). Cells were transfected at a seeding density of ~7 × 105 cells in a T25 flask. Lipofectamine 2000 transfection reagent was diluted: 7.8 μL in 390 μL Opti-MEM medium (Invitrogen). The plasmid was diluted: 4.6 μg in 455 μL Opti-MEM. The DNA–lipid complexes were prepared by combining 390 μL of the diluted plasmid with the 390 μL of diluted transfection reagent. The solution was mixed well and incubated at room temperature for 5–10 min, and then 650 μL of the DNA–lipid solution was added to the T25 flask with an additional 5 mL of Opti-MEM.
Irradiation
Irradiation was performed using an RS-2000 Pro biological research irradiator (Rad Source Technologies) at a dose rate of 1.87 Gy/min. Dosimetry and mapping were optimized to United Kingdom National Physical Laboratory standards.
Clonogenic Assay
At 24 h post-transfection, cells were seeded at optimized cell seeding densities (7 × 102–1.5 × 103) in 6-well plates. Cells were allowed to adhere overnight and were irradiated or treated with cisplatin the following day. The cells were either irradiated with 2 Gy X-ray radiation and controls were mock irradiated or, treated for 24 h with 1 μmol/L cisplatin and controls were treated with PBS vehicle control. Cells were then incubated at 37°C in 5% CO2/95% humidified air for 8–14 d to allow colonies to form. Colonies were fixed and stained with crystal violet (70% methanol, 30% H2O, 0.1% w/v crystal violet) for 1 h at room temperature, followed by destaining in H2O. Air-dried plates were imaged and colonies were counted using a GelCount instrument (Oxford Optronics). The surviving fraction was calculated, as described previously (17).
Digital Gene Expression Profiling
Total RNA was extracted from OE33 R cells at 24 h following transfection with the pre-miR-187 vector or the scrambled control vector. Total RNA extracts (6 μg) were prepared for shipping as advised by LC Sciences. Whole transcriptome digital RNA-seq was performed using Illumina sequencing by synthesis technology. Analyzed data sets, including GO and KEGG analysis, were provided by LC Sciences.
DNA Extraction
Cells were seeded in 12-well plates at a density of 1.2 × 105 cells/well and allowed to adhere for 24 h. Cells were trypsinized, centrifuged at 180g for 3 min and aspirated leaving approximately 20 μL of residual fluid. Cell lysis solution (600 μL) was added to the samples, which were vortexed and incubated at 55°C for 1 h with 3 μL proteinase K solution. A 3 μL volume of RNase A solution was added. Samples were mixed, pulse centrifuged and incubated in a water bath at 37°C for 35 min before cooling on ice for 1 min. Protein precipitation solution (200 μL) was added and the samples were vortexed vigorously for 20 s, placed on ice for 5 min and centrifuged at 17,000g for 3 min. Supernatants were transferred to 1.5 mL tubes containing 600 μL isopropanol and inverted 50 times before centrifuging at 17,000g for 3 min. Samples were washed in 70% EtOH, air dried for 10 min, hydrated with 30 μL of DNA hydration mix and incubated overnight at room temperature.
Chromosomal Microarray Data Analysis
Chromosomal microarray analysis was performed using an Affymetrix CytoScan HD microarray. This microarray contains 2,696,550 oligonucleotide probes, including 1,953,246 unique nonpolymorphic probes and 743,304 single nucleotide polymorphism probes. Single nucleotide polymorphism arrays were conducted according to the manufacturers’ instructions (Affymetrix). Briefly, for each sample, DNA was digested with Affymetrix digestion kit (1 × Nsp1 buffer, 1 × BSA and Nsp1 enzyme) and ligated with Affymetrix ligation master mix (1 × T4 DNA ligase buffer, 1.5 μmol/L Nsp1 adaptor and T4 DNA ligase). PCR purification was performed after mixing with PCR Master Mix (1 × TITANIUM, TM Taq PCR buffer, dNTP mixture and PCR primer 002). DNA was quantified spectrophotometrically. DNA samples were fragmented, labeled, hybridized and washed, stained and scanned. Amplicon size was determined by running 8 μL of fragmentation samples on a 4% TBE (Tris/borate/EDTA) gel. Amplified DNA with an average size of 25–125 bp was used for Chromosomal Microarray Analysis (CGH). Patient hybridization parameters were compared with data derived from phenotypically normal individuals. Deletions smaller than 50 kb and duplications smaller than 400 kb were not reviewed. Detected copy number variations (CNVs) and copy neutral loss of heterozygosity (CN-LOH) were reported when found to have clear or suspected clinical relevance. CNVs devoid of relevant gene content or reported as common findings in the general population may not be reported. Genomic linear positions are given relative to NCBI build 37 (hg19).
Statistical Analysis
Statistical analysis was carried out using GraphPad InStat v3 (GraphPad software Inc). All data are expressed as mean ± the standard error of the mean (SEM). Statistical analysis was performed using a two-tailed Student t test, Mann–Whitney U test or Wilcoxin-matched pairs test. For all statistical analysis, differences were considered to be statistically significant at p ≤ 0.05.
All supplementary materials are available online at www.molmed.org.
RESULTS
MiR-187 Is Significantly Decreased in Pretreatment EAC Tumor Biopsies from Nonresponder Patients
To investigate the role of miRNAs in the response of EAC tumors to neoadjuvant CRT, the expression of 742 miRNAs was assessed in 18 pretreatment diagnostic EAC tumor biopsies taken prior to neoadjuvant CRT using miRNA arrays. Of the 18 patient cohort, 44% (n = 8) were classified as responders (TRG 1 and 2), whereas 56% (n = 10) were classified as nonresponders (TRG 4 and 5). Patient characteristics are outlined in Table 1.
Table 1.
Patient characteristics diagnostic tumor biopsy studies.
| miR-187 study (n = 18) | C3 study (n = 13) | |
|---|---|---|
| Male | 16 | 10 |
| Female | 2 | 3 |
| Age (years)a | 63 (37–75) | 64 (53–75) |
| Clinical TNMb stage | ||
| I | 0 | 0 |
| IIa | 6 | 2 |
| IIb | 2 | 2 |
| III | 10 | 9 |
| IV | 0 | 0 |
| TRGc | ||
| 1 | 3 | 4 |
| 2 | 5 | 4 |
| 4 | 8 | 4 |
| 5 | 2 | 1 |
| Pathological | ||
| Nodal status | 1 Patient NSd | 1 Patient NS |
| N0e | 7 | 6 |
| N1f | 10 | 5 |
Values given are mean (range).
TNM, Tumor node metastasis clinical staging classification.
TRG, tumor regression grade.
NS, not specified.
N0, lymph node negative.
N1, lymph node positive.
Sixty-seven miRNAs were demonstrated to be significantly altered in pretreatment biopsies from nonresponders, when compared with responders (Supplementary Table S1). Of these significantly altered miRNAs, 32 (48%) were significantly decreased, whereas 35 (52%) were significantly increased in nonresponders compared with responders, respectively. The altered expression of miRNAs in pretreatment EAC biopsies from patients having a subsequent response to neoadjuvant CRT supports a role for miRNAs in the tumor response to treatment in EAC.
One of these altered miRNAs, miR-187, was chosen for further investigation as it has been previously implicated in the tumorigenesis of a number of solid malignancies (19,20). MiR-187 was expressed in all tumor samples and expression was significantly lower (p = 0.005) in patients having a subsequent poor response to neoadjuvant CRT, when compared with responder patients (Figure 1A). The significantly higher expression of miR-187 in responders suggests a role for miR-187 in promoting tumor sensitivity in vivo. Supporting this, miR-187 expression was significantly lower (p < 0.01) in tumors of patients who were pathologically lymph node positive following neoadjuvant CRT, when compared with patients who were negative for lymph node involvement (Figure 1B).
Figure 1.

MiR-187 is lower in EAC tumors from nonresponders and patients with lymph node positivity. (A) MiR-187 expression was assessed in pretreatment diagnostic tumor biopsies from EAC patients (n = 18) by qPCR. MiR-187 expression was significantly lower in nonresponder patients (TRG 4 and 5) when compared with responders (TRG 1 and 2). Statistical analysis was performed using an unpaired, two-tailed Student t test, **p < 0.01. (B) MiR-187 was significantly lower in patients who were pathologically lymph node positive (N1) when compared with patients who were lymph node negative (N0). Statistical analysis was performed using a Mann–Whitney U test, *p < 0.05.
Taken together, these data suggest that reduced expression of miR-187 in EAC tumors may be important for the response to treatment of EAC, highlighting its potential as a predictive biomarker.
The Genomic Region Encoding miR-187 Is Deleted in EAC Cells
The role of miR-187 in the response to therapy in EAC was further investigated in vitro using a novel isogenic radioresistant EAC model OE33 P and OE33 R (17). The radioresistant OE33 R subline, which was generated by chronic irradiation with clinically relevant fractionated doses of 2 Gy X-ray radiation, displays enhanced resistance to radiation when compared with its radiosensitive OE33 parent (OE33 P) cell line (17). These two cell lines, of the same origin but with distinctly different radiosensitivities, provide a unique model to investigate the molecular determinants of response to radiation in EAC. Interestingly, endogenous expression of miR-187 was undetectable in OE33 P and OE33 R cell lines by qPCR (Supplementary Figure S1), suggesting its dysregulation in this cell line model. Multiple mechanisms of miRNA dysregulation have been demonstrated in cancer, including chromosomal alterations resulting in deletion, amplification and translocation of miRNA genomic loci (21). To investigate if chromosomal alterations may be involved in the altered expression of miR-187 in EAC cell lines, we performed Affymetrix CytoScan HD microarray analysis on both OE33 P and OE33 R cell lines. MiR-187 is encoded at 18q12.2 (Figure 2A). Cytoscan HD microarray analysis demonstrated copy number loss in the region encoding miR-187 in both OE33 P and OE33 R cell lines (Figure 2B), highlighting this as a mechanism of altered miR-187 expression in EAC cells.
Figure 2.
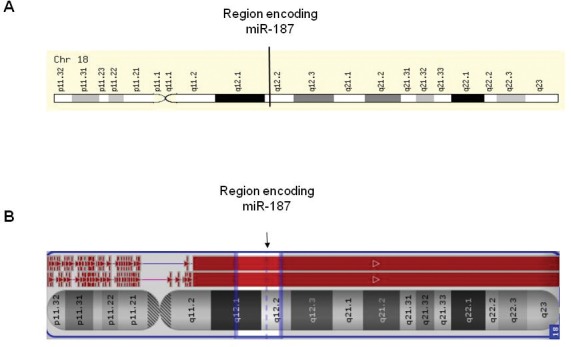
Chromosomal region encoding miR-187 in OE33 P and OE33 R is deleted. Affymetrix CytoScan HD microarray analysis was performed on OE33 P and OE33 R cells. (A) Depicts region encoding miR-187 on chromosome 18. (B) Cytoscan microarray analysis of chromosome 18 in OE33 P and OE33 R cell lines. OE33 P is represented by the pink line, and OE33 R is represented by the blue line. The red box corresponds to areas of copy number loss. A loss of copy number is demonstrated in both OE33 P and OE33 R at the region encoding miR-187.
MiR-187 Modulates Radiosensitivity and Chemosensitivity in EAC Cell Lines
To investigate the functional role of miR-187 in modulating the response to radiation in EAC, radioresistant OE33 R cells were transiently transfected with pre-miR–187 or a scrambled vector control and the sensitivity to a clinically relevant dose of 2 Gy X-ray radiation was assessed by clonogenic assay. Overexpression of miR-187 was confirmed by qPCR (Supplementary Figure S1). Supporting the in vivo data, overexpression of miR-187 significantly sensitized OE33 R cells to 2 Gy radiation (Figure 3A), supporting a functional role for miR-187 in modulating radiosensitivity. To investigate if this effect was specific to a radioresistant phenotype, miR-187 was transiently overexpressed in radiosensitive OE33 P cells and the effects on sensitivity to 2 Gy radiation was assessed. Interestingly, miR-187 overexpression significantly sensitized OE33 P cells to 2 Gy radiation when compared with a scrambled vector control (Figure 3A), further supporting a functional role for miR-187 in modulating radiosensitivity in EAC.
Figure 3.
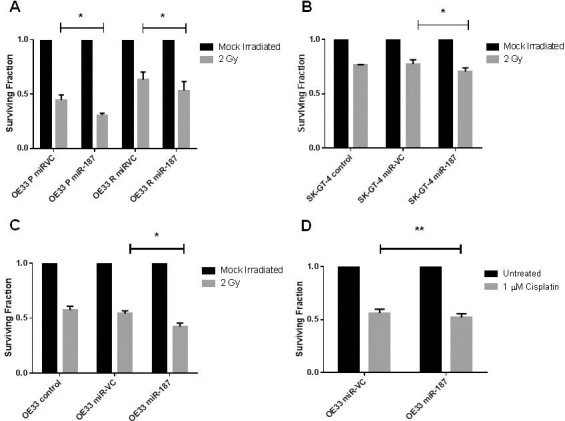
MiR-187 modulates radiosensitivity and cisplatin sensitivity in EAC cells. MiR-187 was transiently overexpressed in EAC cells, and the effect of 2 Gy X-ray radiation or 1 μM cisplatin was assessed by clonogenic assay. (A) Transient overexpression of miR-187 significantly sensitized OE33P and OE33 R cells to 2 Gy radiation when compared with scrambled vector control (miR-VC). (B) Transient overexpression of miR-187 significantly sensitized SK-GT-4 cells to 2 Gy radiation when compared with scrambled vector control (miR-VC). (C) Transient overexpression of miR-187 significantly sensitized OE33 cells to 2 Gy radiation when compared with a scrambled control. (D) Transient overexpression of miR-187 significantly sensitized OE33 cells to cisplatin when compared with a scrambled vector control. Statistical analysis was performed using (A) OE33 P paired t-test and OE33 R Wilcoxon matched pairs test (B and D) paired test and (C) unpaired t test; *p < 0.05, **p < 0.01.
This isogenic model is a model of acquired radioresistance as it was generated by the chronic irradiation of OE33 EAC cells. To investigate if miR-187 could functionally modulate radiosensitivity in EAC cell lines with inherently different radiosensitivities, miR-187 was overexpressed in the OE33 and SK-GT-4 cell lines. SK-GT-4 cells were significantly more resistant to radiation at 2 Gy, when compared with OE33 cells (surviving fraction; OE33 0.58 ± 0.03 and SK-GT-4 0.77 ± 0.005). Interestingly, endogenous miR-187 expression was also undetectable in the OE33 and SK-GT-4 cell lines (data not shown). The overexpression of miR-187 was demonstrated to significantly sensitize SK-GT-4 and OE33 cells to 2 Gy radiation when compared with a scrambled control (Figure 3B, C), further supporting a functional role for miR-187 in modulating radiosensitivity of EAC cells to a clinically relevant dose of X-ray radiation.
To investigate if the effects of miR-187 were radiation-specific, we transiently overexpressed miR-187 in OE33 EAC cells and assessed the sensitivity to the chemotherapeutic cisplatin (1 μmol/L), a chemotherapeutic commonly used to treat EAC, by clonogenic assay. Interestingly, miR-187 significantly sensitized OE33 cells to cisplatin when compared with a scrambled vector control (Figure 3D). Taken together, these data strongly support a role for miR-187 as a functional modulator of cellular sensitivity to X-ray radiation and cisplatin in EAC in vitro, and further supports a role for miR-187 in modulating the tumor response to neoadjuvant CRT in vivo.
Identification of miR-187-Regulated Gene Targets
Considering miR-187 is differentially expressed in EAC tumors in vivo and alters cellular sensitivity to radiation and cisplatin in vitro, miR-187 was transiently overexpressed in OE33 R cells and digital gene expression profiling was performed to identify potential gene targets in order to investigate the biological significance of miR-187 dysregulation. Overexpression of miR-187 significantly altered the expression of 303 genes when compared with a scrambled vector control. Of these altered genes, 162 were upregulated and 141 were downregulated as a result of miR-187 overexpression, suggesting miR-187-mediated regulation of these genes. Interestingly, 29 significantly altered genes (~9%) were novel non-annotated genes (Supplementary Table S2).
Gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of these altered genes was performed to further investigate the biological significance of miR-187 dysregulation. KEGG analysis highlighted that upregulated genes were associated with pathways in cancer and included the tumor suppressors APC and phosphatase and tensin homolog (PTEN), the transcription factor and proto-oncogene JUN and the transcriptional regulator HDAC2 (Table 2). Downregulated genes were enriched in RIG-I-like receptor signaling, cytosolic DNA-sensing, cytokine–cytokine receptor interaction, Toll-like receptor signaling, Jak-STAT signaling, antigen processing and presentation and chemokine signaling pathways (Table 2).
Table 2.
KEGG analysis of pathways regulated by miR-187.
| KEGG pathway | Fold enrichment | Genes |
|---|---|---|
| Pathways in cancer | +3.55 | HDAC2, JUN, KITLG, CDK6, APPL1, PTEN, FH, APC |
| Chemokine signaling | –3.09 | CCL22, CXCL11, CCL5, STAT2, CXCL10 |
| Cytokine–cytokine receptor interaction | –4.41 | CCL22, TNFSF10, IL22RA1, IL29, IFNB1, CXCL11, IL28B, CCL5, IL28A, CXCL10 |
| Jak-STAT signaling | –4.47 | IL22RA1, IL29, 1FNB1, IL28A, IL28B, STAT2 |
| Antigen processing and presentation | –5.57 | TAP1, HLA-B, CD74, HLA-F |
| Toll-like receptor signaling | –6.87 | IFNB1, IRF7, TLR3, CXCL11, CCL5, CXCL10 |
| Rig-1-like receptor signaling | –11.39 | DDX58, IFIH1, ISG15, IFNB1, IRF7, DHX58, CXCL10 |
| Cytosolic DNA-sensing | –12.61 | DDX58, IFNB1, IRF7, CCL5, ZBP1, CXCL10 |
Based on their involvement in pathways implicated in CRT resistance, we selected six upregulated target genes (PYROXD2, APC, PTEN, JUN, tumor necrosis factor [TNF] and CDK6) and nine downregulated target genes (C3, CXCL10, CFB, TRANK1, APOL2, OAS1, IL28A/B/C, interferon β1 [IFNβ1] and IL22RA1) from the transcriptome data set to further validate by qPCR. Two of the upregulated genes (PTEN and TNF) were validated to be significantly upregulated following overexpression of miR-187 (Figure 4A), and eight downregulated targets selected for validation were confirmed as downregulated following overexpression of miR-187 (Figure 4B), further supporting their regulation by miR-187. These data support miR-187-mediated regulation of these gene targets in EAC cells. Given the decreased expression of miR-187 in EAC tumors from poor responders, this suggests that the genes and/or pathways downregulated by miR-187 are likely to be increased and/or overactive in poor responder tumors, which may provide a mechanism for resistance to treatment in vivo.
Figure 4.
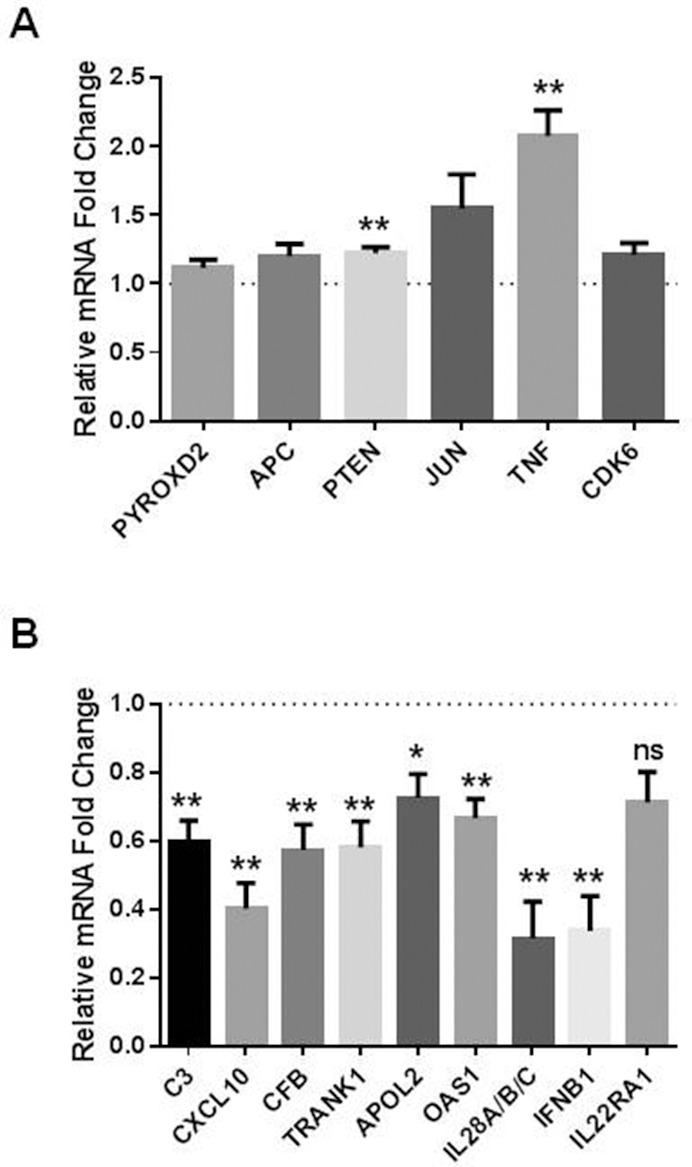
Identification and validation of miR-187-regulated target genes. Digital gene expression profiling was performed on OE33 R cells transfected with either pre-miR-187 vector or a scrambled vector control; miR-VC expression is normalized to 1 and is represented by the dashed line. (A) qPCR validation of genes upregulated following miR-187 overexpression. PTEN and TNF are significantly increased in cells overexpressing miR-187 when compared with a scrambled vector control (miR-VC). (B) qPCR validation of genes downregulated following miR-187 overexpression. C3, CXCL10, CFB, TRANK1, APOL2, OAS1, IL28A/B/C and IFNβ1 were significantly downregulated in cells overexpressing miR-187 when compared with a scrambled control. Data are presented as the mean ± SEM from five independent experiments. Statistical analysis was performed using the one sample t test; *p < 0.05, **p < 0.01, ns not significant.
C3 Is Altered in Pretreatment EAC Tumors from Nonresponders
Interestingly, one of the genes downregulated in miR-187-overexpressing cells, complement component 3 (C3), has been demonstrated previously by our group to be altered in the sera of poor responder esophageal cancer patients and predicts response to treatment with moderate sensitivity and specificity (22). Given that miR-187 expression is significantly lower in pretreatment tumor biopsies from poor responder EAC patients (Figure 1A), we hypothesized that C3 expression may also be altered in poor responder tumors. RNA was available for 13 EAC patients, 62% of whom were classified as responders (TRG 1 and 2), whereas 38% were classified as nonresponders (TRG 4 and 5). Patient characteristics are outlined in Table 1. C3 mRNA expression was increased significantly (p = 0.039) in patients having a subsequent nonresponse to neoadjuvant CRT when compared with responder patients (Figure 5). This demonstrates for the first time that C3 is expressed in EAC tumors and is altered in tumors from patients who respond poorly to neoadjuvant CRT when compared with responders. The increased expression of C3 in nonresponder tumors supports its negative regulation by miR-187 in vivo and further highlights its potential role in the tumor response to neoadjuvant CRT in EAC.
Figure 5.
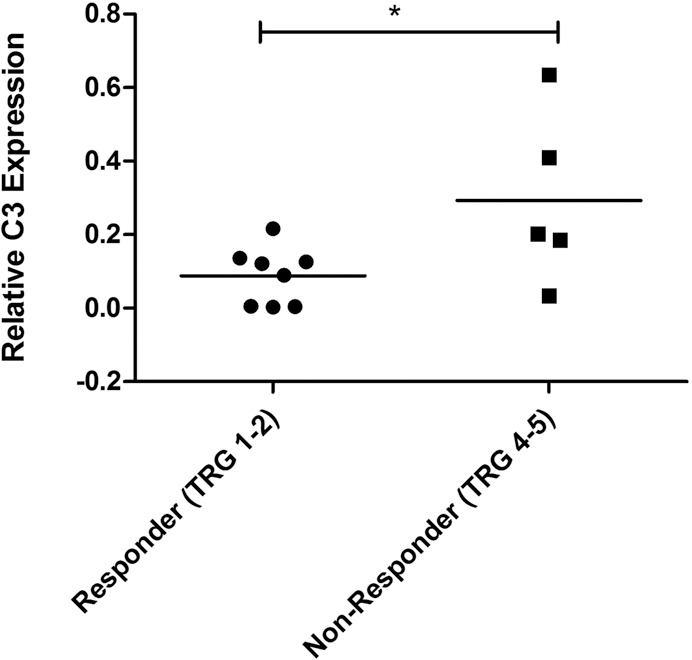
C3 is altered in EAC tumors from nonresponders. C3 mRNA expression was assessed in pretreatment diagnostic tumor biopsies from EAC patients (n = 13) by qPCR. C3 expression was significantly higher in nonresponders (TRG 4 and 5) when compared with responders (TRG 1 and 2). Statistical analysis was performed using an unpaired, two-tailed Student t test, *p < 0.05.
DISCUSSION
EAC is an aggressive cancer with a poor prognosis and is increasing at an alarming rate in Western populations (1) Although a good response to neoadjuvant CRT results in improved survival (5,7), resistance to CRT is a devastating clinical problem, with seven out of ten patients demonstrating no or moderate response to treatment. The clinical challenge is to identify, prior to treatment, those patients who will derive a benefit from neoadjuvant CRT, and conversely, those who will not and who may benefit from more timely surgery. For EAC patients whose tumors display resistance, the identification of novel therapeutic strategies to enhance the tumor response to CRT is vital for improving survival rates and quality of life for patients and for reducing the economic cost of failed treatment. Additionally, the identification of patients with resistant tumors also provides the opportunity to differentially stratify these patients to alternative treatments, which may yield a more favorable prognosis.
Global miRNA profiling of pretreatment EAC tumor biopsies identified 67 miRNAs that were significantly altered in patients who had a subsequent poor response to neoadjuvant CRT when compared with responders (Supplementary Table S1). Of the differentially expressed miRNAs identified here, miR-31 (13), miR-330-5p (14) and members of the let-7 family (23,24) have previously been implicated in the cellular response to radiation and chemotherapy, further supporting this 67 miRNA profile as a signature of response to CRT. Supporting miRNAs as predictive markers of response to treatment in EAC, a recent study by Skinner et al. demonstrated that dysregulation of four miRNAs (miR-505*, miR-99b, miR-451 and miR-145*) was significantly associated with a pCR (12). Three miRNAs (miR-145*, miR-886-5p and miR-720) that we identified as being altered between responders and nonresponders in this study were also highlighted by Skinner and colleagues in pCR versus non-pCR patients. However, it must be considered that in the Skinner study, patients were grouped as pCR versus non-pCR, which may explain the discrepancies between the two studies.
In this study, miR-187 was chosen for further investigation as it has been previously implicated in the tumorigenesis of a number of solid malignancies (19,20); however, its role in EAC is currently unknown. MiR-187 was significantly decreased in pretreatment biopsies from EAC patients who had a subsequent nonresponse to neoadjuvant CRT, highlighting miR-187 as a potential predictive biomarker of treatment response (Figure 1A).
Chromothripsis is a phenomenon that describes catastrophic chromosomal events (25), and a recent study demonstrated that chromothripsis is a frequent occurrence in EAC (26). Loss of heterozygosity on chromosome 18 has previously been demonstrated in EAC (27,28), suggesting the involvement of genes encoded in this chromosomal region in the disease pathogenesis. MiR-187 is encoded at 18q12.2, suggesting that loss of copy number in this region may be a mechanism underlying decreased expression of miR-187 in tumors from nonresponders. Supporting this, CytoScan HD microarray analysis demonstrated copy number loss in the region encoding miR-187 in EAC in vitro, highlighting chromosomal deletions as a mechanism of altered miR-187 expression in EAC cells (Figure 2). This further highlights genomic alterations as a potentially important mechanism of altered miRNA expression in EAC tumors and suggests for the first time that loss of miR-187 at 18q12.2 may be a mechanism for resistance to CRT in EAC.
In vitro experiments demonstrated a functional role for miR-187 in modulating the response of EAC cells to a clinically relevant dose of X-ray radiation (Figure 3). Importantly, this radiosensitizing effect was demonstrated in both a model of acquired radioresistance and inherent radioresistance, further supporting miR-187 as a critical modulator of the radioresponse in EAC. Furthermore, miR-187 was demonstrated to significantly sensitize EAC cells to the chemotherapeutic cisplatin, suggesting a common role for miR-187 in the cellular response to radiation and chemotherapy (Figure 3D). Taken together, this demonstrates for the first time a functional role for miR-187 in modulating sensitivity to radiation and cisplatin in EAC cells, which suggests downregulation of miR-187 as a mechanism of CRT resistance in vivo in EAC tumors.
This study is the first to demonstrate a role for miR-187 as a radio- and cisplatin- sensitizer. Kim and colleagues previously demonstrated that miR-187 is decreased in taxol-resistant ovarian cell lines (29), supporting the data presented here. However, in breast cancer cells, miR-187 was demonstrated to have no effect on sensitivity to tamoxifen or fulvestrant (30). These conflicting data may be due to the tumor- and/or tissue-specific function of miR-187 or may result from the differing modes of action of these cytotoxic agents. DNA damage is a common mechanism of action of both X-ray radiation and cisplatin, suggesting that miR-187 may be involved in the regulation of pathways related to DNA damage and repair in EAC. Supporting this, DGE analysis demonstrated that several genes involved in DNA damage response (nuclear protein 1 [NUPR1], SP100 nuclear antigen [SP100] and interferon γ-inducible protein 16 [IFI16]) were downregulated following miR-187 overexpression (Supplementary Table S2). In vivo data suggest that loss of miR-187 may be important for progression of EAC. This is supported by several studies highlighting a tumor suppressor role for miR-187. In ovarian cancer, decreased miR-187 is associated with poorer survival and was demonstrated to regulate cancer progression via its negative regulation of disabled homolog-2, which plays an important role in epithelial–mesenchymal transition (31). This is also supported in clear cell renal cell carcinoma, where decreased miR-187 expression is associated with poorer survival (20).
Elucidating the targets and interactions of miR-187 is crucial to further uncover its role in the tumor response to neoadjuvant CRT. Numerous computational algorithms for miRNA target prediction have been developed; however, these usually generate hundreds of potential targets that contain many false positives (32). In this study, we used a nonbiased, transcriptome-wide sequencing approach to identify mRNA targets of miR-187. Interestingly, PTEN and TNF were demonstrated to be upregulated following miR-187 overexpression (Figure 4A). TNF is a cytokine that regulates multiple cellular processes including proliferation and apoptosis. PTEN acts as a tumor suppressor and regulates the PI3K/AKT pathway, which has been identified as a radiation response pathway (33). The upregulation of PTEN enhances radiosensitivity via the downregulation of the PI3K/AKT pathway (33). Gene expression profiling identified 162 upregulated genes with miR-187 overexpression, supporting increasing evidence that miRNA can also stimulate gene expression (34,35) (Supplementary Table S2). However, it is highly likely that the upregulation of some of these genes is an indirect consequence of miR-187-mediated target regulation of upstream signaling pathways. Complement component 3 (C3), C-X-C motif chemokine 10 (CXCL10), complement factor B (CFB), tetratricopeptide repeat and ankyrin repeat containing 1 (TRANK1), apolipoprotein L 2 (APOL2), 2′-5′-oligoadenylate synthetase 1 (OAS1), interleukin 28A (IL28A) and interferon β1 (IFNβ1) were validated to be downregulated following overexpression of miR-187, supporting miR-187-mediated negative regulation of these genes (Figure 4B). The expression of CXCL10 has been reported previously as a prognostic marker of patient response to radiation (36,38). In patients with squamous cell carcinoma of the tongue, high CXCL10 expression was associated with poor response to radiotherapy (36). Conversely, high CXCL10 expression was associated with good response to neoadjuvant CRT in rectal cancer patients and overexpression of CXCL10 in HeLa cells enhanced cellular sensitivity to radiation through cell cycle redistribution (37,38). APOL2 is a member of the apolipoprotein L family, the function of this lipid-binding protein is not known (39). APOL1, 3 and 6 were also downregulated with miR-187 overexpression. APOL1 and APOL6 proteins have been identified as regulators of autophagy (40,41). OAS1 encodes a protein of the 2′-5′ OAS family, which is involved in the innate immune response, the OAS2 and OASL genes were also downregulated with miR-187 overexpression. The OAS genes are induced by interferons and have been identified previously as part of an IFN-related DNA damage resistance signature (42). Interestingly, radiation resistance has been linked to an overexpression of IFN-related genes and a vast number of genes identified in the gene expression analysis presented here are IFN-related genes (Supplementary Table S2) (42).
C3 is a central protein of the complement activation cascade (43), which is an essential component of the innate immune system and acts as an important bridge between innate and adaptive immunity (44). The altered expression of C3 in vitro and in vivo further supports its negative regulation by miR-187. Mir-187 is not predicted to target C3, suggesting that miR-187-mediated downregulation of C3 may be indirect. Although the altered expression of C3 in EAC tumors requires further investigation in a larger independent patient cohort, the increased expression of C3 in pretreatment tumors from nonresponders suggests a role for C3 in the tumor response to neoadjuvant CRT (Figure 5). These data support the previous work from our unit, which demonstrated that pretreatment sera levels of C3a, a cleavage product of C3, are significantly increased in esophageal cancer patients who subsequently had a nonresponse to neoadjuvant CRT (22). This may suggest that abrogation of miR-187-mediated negative regulation of the complement system in EAC tumors results in increased circulating levels of C3a in nonresponders, highlighting C3a as a minimally invasive predictive biomarker of response to neoadjuvant CRT in EAC. Inflammation is now considered a hallmark of cancer, impacting all stages of tumorigenesis (45). Chronic inflammation is also implicated in the resistance of tumors to therapy (46); however, the role of the complement system in resistance to anticancer therapy is largely unknown. Our data are supported by one recent study, which demonstrated a role for the complement system in radioresistance in mouse models of lymphoma and breast cancer (47). However, this study is the first to highlight a potential role for C3 in the resistance of EAC to CRT.
CONCLUSION
In this study, we performed global miRNA profiling of EAC patient tumors to identify miRNAs that may be involved in tumor resistance to neoadjuvant CRT. We identified miR-187 as a potential novel biomarker of treatment response and a potential therapeutic target that could enhance patient sensitivity to radiotherapy and chemotherapy. Importantly, this study demonstrates for the first time a functional role for miR-187 in modulating sensitivity to X-ray radiation and cisplatin in EAC. Gene expression analysis identified 303 differentially expressed genes, including genes involved in the complement cascade, as a result of miR-187 overexpression in EAC cells. Furthermore, C3 was increased in pretreatment EAC tissue biopsies from nonresponder patients, supporting miR-187-mediated regulation of C3 in vivo. Although further work is required to fully elucidate the role of miR-187 and C3 in the resistance of EAC to CRT, this study highlights for the first time a potential role for miR-187 and C3 as predictive markers of response to neoadjuvant CRT in EAC.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the Health Research Board, Ireland (HRA_POR/2011/63 to SGM), and the Cancer and Polio Research Fund, United Kingdom. BASB is supported by a University of Hull PhD scholarship.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Lynam-Lennon N, et al. (2016) Low MiR-187 expression promotes resistance to chemoradiation therapy in vitro and correlates with treatment failure in patients with esophageal adenocarcinoma. Mol. Med. 22:388–97.
REFERENCES
- Arnold M, Soerjomataram I, Ferlay J, Forman D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut. 2015;64:381–7. doi: 10.1136/gutjnl-2014-308124. [DOI] [PubMed] [Google Scholar]
- Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–12. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- Thrift AP, Whiteman DC. The incidence of esophageal adenocarcinoma continues to rise: Analysis of period and birth cohort effects on recent trends. Ann Oncol. 2012;23:3155–62. doi: 10.1093/annonc/mds181. [DOI] [PubMed] [Google Scholar]
- Murphy TJ, Ravi N, Reynolds JV. Treatment options for esophageal cancer. Expert Opin Pharmacother. 2008;9:3197–210. doi: 10.1517/14656560802560237. [DOI] [PubMed] [Google Scholar]
- Reynolds JV, et al. Long-term outcomes following neoadjuvant chemoradiotherapy for esophageal cancer. Ann Surg. 2007;245:707–16. doi: 10.1097/01.sla.0000254367.15810.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hagen P, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366:2074–84. doi: 10.1056/NEJMoa1112088. [DOI] [PubMed] [Google Scholar]
- Geh JI, Crellin AM, Glynne-Jones R. Preoperative (neoadjuvant) chemoradiotherapy in oesophageal cancer. Br J Surg. 2001;88:338–56. doi: 10.1046/j.1365-2168.2001.01670.x. [DOI] [PubMed] [Google Scholar]
- Lynam-Lennon N, Maher SG, Reynolds JV. The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc. 2009;84:55–71. doi: 10.1111/j.1469-185X.2008.00061.x. [DOI] [PubMed] [Google Scholar]
- Orellana EA, Kasinski AL. MicroRNAs in cancer: A historical perspective on the path from discovery to therapy. Cancers (Basel) 2015;7:1388–405. doi: 10.3390/cancers7030842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Sakai NS, Samia-Aly E, Barbera M, Fitzgerald RC. A review of the current understanding and clinical utility of miRNAs in esophageal cancer. Semin Cancer Biol. 2013;23:512–21. doi: 10.1016/j.semcancer.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Skinner HD, et al. A validated miRNA profile predicts response to therapy in esophageal adenocarcinoma. Cancer. 2014;120:3635–41. doi: 10.1002/cncr.28911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynam-Lennon N, et al. MicroRNA-31 modulates tumour sensitivity to radiation in oesophageal adenocarcinoma. J Mol Med (Berl) 2012;90:1449–58. doi: 10.1007/s00109-012-0924-x. [DOI] [PubMed] [Google Scholar]
- Bibby BA, Reynolds JV, Maher SG. MicroRNA-330-5p as a putative modulator of neoadjuvant chemoradiotherapy sensitivity in oesophageal adenocarcinoma. PLoS One. 2015;10:e0134180. doi: 10.1371/journal.pone.0134180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher SG, et al. Gene expression analysis of diagnostic biopsies predicts pathological response to neoadjuvant chemoradiotherapy of esophageal cancer. Ann Surg. 2009;250:729–37. doi: 10.1097/SLA.0b013e3181bce7e1. [DOI] [PubMed] [Google Scholar]
- Mandard AM, et al. Pathologic assessment of tumor regression after preoperative chemoradiotherapy of esophageal carcinoma. Clinicopathologic correlations. Cancer. 1994;73:2680–6. doi: 10.1002/1097-0142(19940601)73:11<2680::aid-cncr2820731105>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Lynam-Lennon N, et al. Alterations in DNA repair efficiency are involved in the radioresistance of esophageal adenocarcinoma. Radiat Res. 2010;174:703–11. doi: 10.1667/RR2295.1. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Casanova-Salas I, et al. Identification of miR-187 and miR-182 as biomarkers of early diagnosis and prognosis in patients with prostate cancer treated with radical prostatectomy. J Urol. 2014;192:252–9. doi: 10.1016/j.juro.2014.01.107. [DOI] [PubMed] [Google Scholar]
- Zhao J, et al. MicroRNA-187, down- regulated in clear cell renal cell carcinoma and associated with lower survival, inhibits cell growth and migration though targeting B7-H3. Biochem Biophys Res Commun. 2013;438:439–44. doi: 10.1016/j.bbrc.2013.07.095. [DOI] [PubMed] [Google Scholar]
- Calin GA, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers; Proc Natl Acad Sci U S A; 2004. pp. 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher SG, et al. Serum proteomic profiling reveals that pretreatment complement protein levels are predictive of esophageal cancer patient response to neoadjuvant chemoradiation. Ann Surg. 2011;254:809–16. doi: 10.1097/SLA.0b013e31823699f2. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. Lin28 mediates radiation resistance of breast cancer cells via regulation of caspase, H2A.X and Let-7 signaling. PLoS One. 2013;8:e67373. doi: 10.1371/journal.pone.0067373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, et al. DICER-dependent biogenesis of let-7 miRNAs affects human cell response to DNA damage via targeting p21/p27. Nucleic Acids Res. 2015;43:1626–36. doi: 10.1093/nar/gku1368. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang CZ, Leibowitz ML, Pellman D. Chromothripsis and beyond: Rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013;27:2513–30. doi: 10.1101/gad.229559.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nones K, et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat Commun. 2014;5:5224. doi: 10.1038/ncomms6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett MT, Galipeau PC, Sanchez CA, Emond MJ, Reid BJ. Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene. 1996;12:1873–8. [PubMed] [Google Scholar]
- Barrett MT, Schutte M, Kern SE, Reid BJ. Allelic loss and mutational analysis of the DPC4 gene in esophageal adenocarcinoma. Cancer Res. 1996;56:4351–3. [PubMed] [Google Scholar]
- Kim YW, et al. Differential microRNA expression signatures and cell type-specific association with Taxol resistance in ovarian cancer cells. Drug Des Devel Ther. 2014;8:293–314. doi: 10.2147/DDDT.S51969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulrane L, et al. miR-187 is an independent prognostic factor in breast cancer and confers increased invasive potential in vitro. Clin Cancer Res. 2012;18:6702–13. doi: 10.1158/1078-0432.CCR-12-1420. [DOI] [PubMed] [Google Scholar]
- Chao A, et al. Regulation of ovarian cancer progression by microRNA-187 through targeting Disabled homolog-2. Oncogene. 2012;31:764–75. doi: 10.1038/onc.2011.269. [DOI] [PubMed] [Google Scholar]
- Maziere P, Enright AJ. Prediction of microRNA targets. Drug Discov Today. 2007;12:452–8. doi: 10.1016/j.drudis.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Zhao L, Bode AM, Cao Y, Dong Z. Regulatory mechanisms and clinical perspectives of miRNA in tumor radiosensitivity. Carcinogenesis. 2012;33:2220–7. doi: 10.1093/carcin/bgs235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, et al. A KLF4-miRNA-206 autoregulatory feedback loop can promote or inhibit protein translation depending upon cell context. Mol Cell Biol. 2011;31:2513–27. doi: 10.1128/MCB.01189-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan S. Posttranscriptional upregulation by microRNAs. Wiley Interdiscip Rev RNA. 2012;3:311–30. doi: 10.1002/wrna.121. [DOI] [PubMed] [Google Scholar]
- Rentoft M, et al. Expression of CXCL10 is associated with response to radiotherapy and overall survival in squamous cell carcinoma of the tongue. Tumour Biol. 2014;35:4191–8. doi: 10.1007/s13277-013-1549-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, et al. CXCL10 mRNA expression predicts response to neoadjuvant chemoradiotherapy in rectal cancer patients. Tumour Biol. 2014;35:9683–91. doi: 10.1007/s13277-014-2234-0. [DOI] [PubMed] [Google Scholar]
- Yang LL, Wang BQ, Chen LL, Luo HQ, Wu JB. CXCL10 enhances radiotherapy effects in HeLa cells through cell cycle redistribution. Oncol Lett. 2012;3:383–6. doi: 10.3892/ol.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo-Moreno J, et al. Apolipoprotein L2 contains a BH3-like domain but it does not behave as a BH3-only protein. Cell Death Dis. 2014;5:e1275. doi: 10.1038/cddis.2014.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaorigetu S, Yang Z, Toma I, McCaffrey TA, Hu CA. Apolipoprotein L6, induced in atherosclerotic lesions, promotes apoptosis and blocks Beclin 1-dependent autophagy in atherosclerotic cells. J Biol Chem. 2011;286:27389–98. doi: 10.1074/jbc.M110.210245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. 2008;4:1079–82. doi: 10.4161/auto.7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichselbaum RR, et al. An interferon- related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer; Proc Natl Acad Sci U S A; 2008. pp. 18490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pio R, Ajona D, Lambris JD. Complement inhibition in cancer therapy. Semin Immunol. 2013;25:54–64. doi: 10.1016/j.smim.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis. 2009;30:1073–81. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- Multhoff G, Radons J. Radiation, inflammation, and immune responses in cancer. Front Oncol. 2012;2:58. doi: 10.3389/fonc.2012.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvington M, et al. Complement- dependent modulation of antitumor immunity following radiation therapy. Cell Rep. 2014;8:818–30. doi: 10.1016/j.celrep.2014.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.