

Abstract
Colon cancer cells contain high levels of cystathionine-β-synthase (CBS). Its product, hydrogen sulfide (H2S), promotes the growth and proliferation of colorectal tumor cells. To improve the antitumor efficacy of the prototypical CBS inhibitor aminooxyacetic acid (AOAA), we have designed and synthesized YD0171, a methyl ester derivative of AOAA. The antiproliferative effect of YD0171 exceeded the antiproliferative potency of AOAA in HCT116 human colon cancer cells. The esterase inhibitor paraoxon prevented the cellular inhibition of CBS activity by YD0171. YD0171 suppressed mitochondrial respiration and glycolytic function and induced G0/G1 arrest, but did not induce tumor cell apoptosis or necrosis. Metabolomic analysis in HCT116 cells showed that YD0171 affects multiple pathways of cell metabolism. The efficacy of YD0171 as an inhibitor of tumor growth was also tested in nude mice bearing subcutaneous HCT116 cancer cell xenografts. Animals were treated via subcutaneous injection of vehicle or AOAA (0.1, 0.5 or 1 mg/kg/d) for 3 wks. Tumor growth was significantly reduced by 9 mg/kg/d AOAA, but not at the lower doses. YD0171 was more potent: tumor volume was significantly inhibited at 0.5 and 1 mg/kg/d. Thus, the in vivo efficacy of YD0171 is nine times higher than that of AOAA. YD0171 (1 mg/kg/d) attenuated tumor growth and metastasis formation in the intracecal HCT116 tumor model. YD0171 (3 mg/kg/d) also reduced tumor growth in patient-derived tumor xenograft bearing athymic mice. YD0171 (3 mg/kg/d) induced the regression of established HCT116 tumors in vivo. A 5-d safety study in mice demonstrated that YD0171 at 20 mg/kg/d (given in two divided doses) does not increase plasma markers of organ injury, nor does it induce histological alterations in the liver or kidney. YD0171 caused a slight elevation in plasma homocysteine levels. In conclusion, the prodrug approach improves the pharmacological profile of AOAA; YD0171 represents a prototype for CBS inhibitory anticancer prodrugs. By targeting colorectal cancer bioenergetics, an emerging important hallmark of cancer, the approach exemplified herein may offer direct translational opportunities.
INTRODUCTION
Colorectal cancer remains the third most prevalent cancer in the United States, with over 49,000 estimated deaths in 2016 (1). Over half of these patients are diagnosed with either regional nodal disease or distant organ metastasis, with a 5-year survival of 70% and 13%, respectively. In addition to surgical resection, these patients may be offered chemotherapeutic regimens, which may cause significant toxicity, and/or molecularly targeted therapies to epidermal growth factor receptor or vascular endothelial growth factor. Since not all patients respond to, or have cancers that are appropriate for treatment with standard and available therapies, novel targeted therapeutic options are needed.
We and others have shown that human colorectal cancers and cancer cells contain high levels of cystathionine-β-synthase (CBS); the product of this transsulfuration enzyme, hydrogen sulfide (H2S), promotes the growth and proliferation of colorectal cancer cells in vitro and in vivo (2–5). The prototypical CBS inhibitor aminooxyacetic acid (AOAA) suppresses the proliferation of colon cancer cells in vitro and reduces tumor growth in vivo (1,2). CBS and H2S have also been implicated in the pathogenesis of ovarian and breast cancer (6,7).
The potency of AOAA in recombinant CBS is markedly higher than the potency of the compound as an antiproliferative agent in the colon cancer cell line HCT116 in vitro (2,8). We hypothesized that the difference between enzyme potency and cell-based efficacy may be related to a limited cellular uptake of AOAA. Prodrug development represents a well-established pharmacological strategy to increase the cellular update of drugs or drug development candidates. Prodrugs are chemically modified versions of the pharmacologically active agent, which undergo transformation in vivo to release the active drug (9,10). The coupling serves to improve the pharmaceutical properties of the active principle (e.g., to improve cell uptake by increasing the lipophilicity of the compound).
Methyl or ethyl esters are some of the most common prodrugs in existence; addition of these groups substantially improves the physicochemical, biopharmaceutical and/or pharmacokinetic properties of the parent compound (9,10). Successful clinical examples of ester prodrugs include the antihypertensive drug enalapril, the antiviral drug oseltamivir and the antibiotics famciclovir and pivampicillin (9,10). Here we have designed and synthesized a methyl ester prodrug of AOAA (designated YD0171) and tested its efficacy, in comparison with the parent compound AOAA, in various in vitro and in vivo assays. YD0171 targets and inhibits cancer cell metabolism, a new hallmark of cancer (11). We show lower systemic toxicity with YD0171 administration compared with AOAA and demonstrate the translational therapeutic promise by inhibiting human colorectal cancer cell growth using patient-derived tumor xenografts.
MATERIALS AND METHODS
Synthesis and Chemical Characterization of YD0171
All chemicals were obtained from Sigma-Aldrich, unless stated otherwise. For YD0171 synthesis, all commercially available starting materials and solvents were reagent grade and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063–0.200 mm (70–230 mesh, flash). Analytical thin layer chromatography (TLC) was carried out employing silica gel 60 F254 plates (Merck). NMR spectra were recorded on a Bruker-300 (1H, 300 MHz; 13C, 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with tetramethylsilane (TMS) as an internal reference. Chemical shifts downfield from TMS were expressed in parts per million, and J values were given in Hertz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: nano ESI spray voltage was 1.8 kV, capillary temperature was 275°C and the resolution was 60,000; ionization was achieved by positive mode.
Synthesis of methyl 2-(aminooxy)acetate (YD0171) was achieved in a high yield of 97% through the reaction of AOAA hemihydrochloride with thionyl chloride in methanol at room temperature following the procedure of Woulfe and Miller (12). To a mixture of carboxymethoxylamine hemihydrochloride (2.10 g, 19.21 mmol) in MeOH (50 mL) SOCl2 (4.50 g, 38.42 mmol) was added at 0°C. The resulting mixture was stirred at room temperature for 12 h. The solvent was then removed under reduced pressure and the residue was dissolved in MeOH (50 mL). After the removal of the solvent, the crude product was crystallized and washed with ether (50 mL) to yield YD0171, which was obtained as a white solid (1.96 g, 97%). TLC: Rf = 0.3 (33% EtOAc/hexanes); 1H NMR (300 MHz, DMSO-d6): d 11.12 (s, 2H, br), 4.76 (s, 2H), 3.72 (s, 3H). 13C NMR (75 MHz, DMSO-d6): d 168.50, 70.64, 52.51. HRMS Calcd for C3H8NO3: [M + H]+ 106.0504; found 106.0498.
Water-octanol partition coefficient of AOAA and YD0171 was determined as described (13).
Measurement of H2S Production by Recombinant Human CBS
Full-length recombinant human CBS was produced as described (7). H2S production by CBS was measured in vitro using the 7-azido-4-methylcoumarin (AzMC) method (14) in a 96-well plate format. The volume of the activity buffer was set at 180 μL and contained 200 mmol/L Tris–HCl pH 8.0, 5 μmol/L pyridoxal 5′-phosphate (PLP), 10 mmol/L glutathione and 0.5 mg/mL BSA. AOAA or YD0171 was added to each well in 10 μL volume. CBS (5 μg/well) was added to all wells in a 10 μL volume. AzMc (10 μmol/L) and the CBS substrates L-cysteine and homocysteine (each at 2.5 mmol/L final concentration) were added in a 10 μL volume (i.e., total assay volume increased to 200 μL). The mixture was incubated at 37°C for 1 h, and fluorescence read at 450 nm (lex = 365 nm).
Cell Culture
The human colorectal carcinoma cell line, HCT116 was cultured in McCoy’s 5A medium supplemented with 10% FBS, 100 IU/mL penicillin and 100 mg/mL streptomycin as described (2,3). Cells were grown in a 37°C, 5% CO2 atmosphere.
Measurement of H2S Production and Methanol Production in HCT116 Cell Homogenates
HCT116 cells in culture were incubated with YD0171 or AOAA (10 μmol/L–1 mmol/L). Cells were then washed twice with phosphate-buffered saline (PBS) and homogenized in NP40 lysing buffer (1% NP40, 150 mmol/L NaCl, 50 mmol/L Tris–Cl, pH 8.0) on ice for 30 min. The protein concentration of the extract was ~10 mg/mL. Cell homogenates (20 μL) were incubated in 96-well plates with 5 μmol/L PLP and L-cysteine and homocysteine (each at 2.5 mmol/L), and H2S production was measured using the AzMC method as described above. In some experiments, cells were preincubated with the nonisoform-selective esterase inhibitor paraoxon (15) (1 mmol/L) for 30 min prior to the addition of YD0171.
Similar to other methyl ester prodrugs, the predicted cleavage and generation of AOAA from YD0171 would be expected to yield equimolar amounts of methanol. Cells were incubated with YD0171 (1 mmol/L), and methanol production was detected in cell homogenates with a colorimetric alcohol detection kit (Cell Biolabs).
Cell Proliferation Assays and Live Cell-Imaging Detection of H2S in HCT116 Cells In Vitro
For assessment of cell proliferation, the xCELLigence system (Roche) was used as described (2,3). Briefly, HCT116 cells were cultured until ~70% confluence. Cells were detached by trypsin–EDTA and resuspended in fresh culture media at a concentration of 30,000 cells/mL. Two hundred microliter of cell suspension was added to each well (6,000 cells/well) of an E-plate 96, a specially designed 96-well microtiter plate containing interdigitated microelectrodes to noninvasively monitor the cell proliferation by measuring the relative change in the electrical impedance of the cell monolayer, a unitless parameter named cell index. Cells were then treated with YD0171 or AOAA (10 μmol/L–1 mmol/L) and proliferation monitored for 48 h.
For detecting the effect of YD0171 on H2S production in live cells (3), 30,000 HCT116 cells were seeded in Lab-Tek II chamber coverglass system (Nalgen) and incubated at 37°C and 10% CO2 humidified incubator overnight. The cells were loaded with AzMC at 10 μmol/L final concentration for 30 min. YD0171 was added to the cells at 1 mmol/L and cells were further incubated for 1 h. Cells were washed three times with PBS, and dye’s specific fluorescence was visualized using Nikon eclipse 80i inverted microscope with Photometric CoolSNAP HQ2 camera and NIS-Elements BR 3.10 software.
Extracellular Flux Analysis and In Vitro Enzyme Activity Measurements
The XF24 Extracellular Flux Analyzer (Seahorse Bioscience) was used to measure bioenergetic function as described (2,16). Cells were treated with YD0171 (1 mmol/L) for 24 h, followed by extracellular flux analysis. Four key parameters of mitochondrial function (basal respiration, adenosine triphosphate (ATP) turnover, proton leak and maximal respiration) were assessed through the sequential use of 40 μmol/L oligomycin (ATP synthase inhibitor), 13.5 μmol/L FCCP (oxidative phosphorylation uncoupler) and 10 μmol/L rotenone + 1 μmol/L antimycin A (complex I and III inhibitors). The difference between the maximal and the basal respirations was considered the respiratory reserve capacity (the capacity of a cell to generate ATP via oxidative phosphorylation in response to increased demand for energy). A glycolytic stress test was used to estimate various parameters of cellular glycolysis (glycolysis, glycolytic capacity and glycolytic reserve), which was obtained with the sequential use of 25 mmol/L glucose, 5 μmol/L oligomycin (to block mitochondrial respiration and force the cells to rely on glycolysis for ATP production) and 100 mmol/L 2-deoxyglucose (2-DG, a glucose analog and inhibitor of glycolytic ATP production). Glycolytic reserve was calculated as the difference between the glycolytic capacity and the glycolysis; this parameter is indicative of the cellular ability to increase the glycolytic rate upon increased energy demand. Acidification of carbon dioxide, the end product of the tricarboxylic acid (TCA) cycle, which can be converted to bicarbonate, is considered a major contributor to nonglycolytic acidification.
The activity of glutamic oxaloacetic transaminase 1 (GOT1)/aspartate aminotransferase (AST), alanine aminotransaminase (ALT), succinyl-CoA-synthetase (SCS) and lactate dehydrogenase (LDH) was determined, respectively, by an AST Activity Assay Kit, an ALT Activity Assay Kit, a Succinyl-CoA Synthetase Activity Assay Kit with recombinant prokaryotic SCS (Megazyme), the LDH Activity Assay Kit with recombinant LDH (Abcam) and the α-ketoglutarate dehydrogenase activity using α-ketoglutarate dehydrogenase from porcine heart (all kits from Sigma-Aldrich). Recombinant human ornithine decarboxylase 1 activity (ODC, Origene) was determined via a modified spectrophotometric method as described (17). Enzymes were preincubated with various concentrations of AOAA or H2S (generated from NaHS) for 30 min, followed by the measurement of enzyme activity.
Cell Cycle Analysis
The Guava Cell Cycle Reagent (EMD-Millipore) was used for staining HCT-116 cells. Cell cycle analysis was conducted according to a previous protocol (18). Briefly, 48 h after nocodazole (G2/M block; 0.029 μg/mL) or YD0171 treatment, cells were trypsinized and centrifuged at 450g for 5 min and washed with PBS, fixed in 70% (vol/vol) ice-cold ethanol and finally stained with Guava Cell Cycle Reagent and subjected to flow cytometry using the Becton-Dickinson LSRFortessa Special Order Research Product (Becton Dickinson). DNA histograms were analyzed using ModFit LT software (Verity Software House).
Cell Death Assays
The detection of apoptosis/necrosis of HCT-116 treated with AOAA or YD0171 was performed 48 h after treatment by Annexin V-7AAD staining followed by flow cytometry using Annexin V-PE Apoptosis Detection Kit according to prior protocols (19,20) and following the manufacturer’s recommendation (Sigma-Aldrich). Briefly, control and treated cells were trypsinized, washed in ice-cold PBS and resuspended in 1 mL of binding buffer. Then, 1 × 105 cells (500 μL) were incubated with 5 μL annexin V PE and 5 μL 7AAD for 10 min at 25°C in the dark and analyzed immediately using a Guava EasyCyte Plus Flow Cytometer (Millipore). The early and late apoptotic cells, and as well as the necrotic cells, were estimated as the percentage of the total number of cells by CytoSoft 5.3 Software (Millipore) (21,22). Cells were incubated for 24 or 48 h with various concentrations (10 μmol/L–1 mmol/L) of YD0171 or AOAA, followed by the assessment of cell necrosis and apoptosis by flow cytometry. Necrotic cell death was also assessed by the LDH assay as described (23).
Quantification of Mitochondrial and Nuclear DNA Damage
Integrity (the level of the DNA damage) of the nuclear and the mitochondrial DNA was analyzed by semiquantitative, long-amplicon PCR assays (LA-PCR) using LongAmp Taq DNA Polymerase (New England BioLabs) (21,22). Total DNA was isolated using a DNase Blood and Tissue Kit (QIAGEN). Briefly, damage to nuclear DNA was estimated by quantification of the PCR amplification of the 10-kb nuclear-specific DNA fragment using PicoGreen fluorescent dye to detect amplified double-stranded DNA (Quant-iT PicoGreen; Life Technologies). Damage to the mitochondrial DNA was estimated by quantification of the PCR amplification of the 8.9-kb mitochondrial-specific DNA fragment using PicoGreen staining. Data were normalized by the secondary PCR amplification of 221-bp mitochondrial genome-specific fragment for correction of the multiple copies of the mitochondrial DNA.
Metabolomic Analysis
HCT116 cells in culture were treated with vehicle, or YD0171 or AOAA. An identical, high concentration of both compounds (1 mmol/L) was selected, at which concentration the antiproliferative effects of the two compounds are comparable. After 24 h, cells were washed with PBS, scraped and snap frozen in liquid nitrogen. Metabolomic analysis was performed by Metabolon Inc. as described (23).
Human Colon Cancer Cell Subcutaneous Tumor Xenograft Model
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of University of Texas Medical Branch (UTMB). Athymic male and female mice (8–10 wks) were injected subcutaneously in either the right or the left dorsum with 106 HCT116 cells as described (2). One week later, the mice were randomized and subcutaneous (SQ) injection of PBS, YD0171 (0.1, 0.5 or 1 mg/kg) or AOAA (1, 3 or 9 mg/kg/d) was performed 5 d/wk. Tumor dimensions were measured two or three times per week transcutaneously using a caliper, and tumor volume was calculated using the formula π/6 (HxWxL).
In a subset of parallel studies, HCT116 tumors were allowed to grow for 30 d; animals were then subjected to treatment with vehicle (PBS) or YD0171 (3 mg/kg/d) for 7 d, followed by harvesting of the tumor tissues (as well as liver tissues for normal control). CBS activity in homogenates was measured by the AzMC method as described above for cell homogenates.
To test whether YD0171 could induce regression of established tumors, we used a modified crossover experimental design. HCT116 tumors were initiated subcutaneously as described above. When a small palpable tumor was evident, the mice were randomized into two groups: vehicle- and YD0171-treated groups (3 mg/kg/d, s.q. 5 d/wk). At d 42, the YD0171 treatment group was switched to vehicle, and at d 50, the vehicle group was switched to YD0171. Tumor dimensions were measured transcutaneously with a caliper twice per week; tumor volumes were calculated using the formula π/6 (HxWxL).
Patient-Derived Colon Tumor Xenograft Model
To initiate patient-derived tumor xenografts (PDTXs), freshly resected human colorectal tumor tissue was collected under an IRB-approved protocol, rinsed in Dakin’s solution, minced into 2–3 mm3 pieces and implanted subcutaneously into two or three NOD/SCID/IL-2R–/– mice. Once the tumors from this initial cohort of mice (passage 0; P0) grew to ~100 mm3 in tumor volume, they were harvested, minced and implanted subcutaneously into a second cohort of athymic nude mice (passage 1; P1). Minced fragments of P1 PDTXs were then implanted into a final cohort of animal to generate P2 PDTXs for xenotrial experiments. Briefly, athymic male and female mice (8–10 wks) were implanted subcutaneously with a piece of P2 PDTXs measuring ~2–3 mm3 each as described (2). Ten days postimplant mice were randomized into two treatment groups and received either 3 mg/kg YD0171 dissolved in PBS (s.q. 5 d/wk) or PBS alone (vehicle) on the same schedule. Tumor size was measured transcutaneously twice per week.
Orthotopic Tumor Xenograft Model
Athymic male and female mice (8–10 wks) were anesthetized using isoflurane. A small laparotomy incision was made in the left lower quadrant of the abdomen, the cecum was exposed and 106 HCT116 cells (in 100 μL) were injected between the subserosal space of the bowel wall (24). The cecum was then returned to the abdomen, the abdominal muscle wall was closed using 6–0 vicryl suture and skin was closed using staples. Buprenorphine sustained release (1 mg/kg SQ) was administered for postprocedure pain control. On posttumor injection d 3, mice were randomized into SQ treatment with either PBS, YD0171 (1 mg/kg) or oxaliplatin (5 mg/kg) 5 d/wk. At the end of the treatment schedule (posttumor injection d 51), the cecum, liver, lung and lymph nodes were harvested, fixed with 10% formalin, paraffin embedded and sections (5 m thick) were cut either for hematoxylin/eosin (H&E) staining or immunohistochemistry with a human-specific mitochondrial antibody (Thermo Scientific, Cat# MA5-12014; 1:150 dilution). Tumor burden and metastasis were quantified using a weighted scoring system. Tumor deposits were classified as visible (visible tumors—brown stain—that could be visualized with the naked eye.), large (tumors with >100 cells counted using 100 × power), medium (tumors with 10–100 cells per 100 × field) and small (tumors with cells <10 per 100 × power). Each experiment was scored by four graders, blinded to the experimental groups and averaged.
Exploratory Safety Studies
C56BL/6 mice (20–25 g) were housed in a light-controlled room with a 12-h light–dark cycle and were allowed ad libitum access to food and water. Mice were randomly allocated into the following groups: (a) vehicle-treated group (n = 5); (b) YD0171-treated group (2 mg/kg, i.p.) (n = 5); (c) YD0171-treated group (6 mg/kg, i.p.) (n = 5); (d) YD0171-treated group (20 mg/kg, i.p.) (n = 5) and (e) YD0171- treated group (60 mg/kg, i.p.) (n = 5). The dose indicated represents the total daily dose, which was administered in two equal divided doses (in the morning and evening of each day). For instance, the highest active dosing group received 30 mg/kg, i.p. YD0171 in the morning and 30 mg/kg, i.p. YD0171 in the evening of each experimental day. The volume of saline administered to the vehicle-control animals was equal to the volume of YD0171 administered.
Treatment groups received vehicle or YD0171 in the morning as well as evening from the morning of the first day to until the evening of the fifth day. On the morning of the sixth day, animals were euthanized under deep anesthesia using isoflurane (5% inhalation). Blood samples (1 mL) were collected via cardiac puncture. Death was confirmed by opening of chest. Liver and kidney samples were collected immediately. Blood samples (150 μL) were analyzed by the Vetscan analyzer for biochemical parameters of organ injury within 1 h of collection as described (25). Plasma homocysteine levels were measured by a commercial Homocysteine Kit (Cell Biolabs) according to the manufacturer’s instructions. Liver and kidney tissues were fixed for 1 wk in buffered formaldehyde solution (10% in PBS) at room temperature, dehydrated by graded ethanol and embedded in Paraplast (Sherwood Medical). Tissue sections (thickness: 7 μm) were deparaffinized with xylene, stained with H&E and studied using light microscopy. Analysis of the histological slides was performed in a blinded fashion.
Statistical Analysis
All data are presented as mean ± SEM and were analyzed using GraphPad Prism software or SPSS. Statistical analyses included Student t test or one-way ANOVA followed by Bonferroni’s multiple comparisons.
All supplementary materials are available online at www.molmed.org.
RESULTS
Synthesis and In Vitro Characterization of an AOAA Prodrug, YD0171
A comprehensive attempt to modify the structure of AOAA to increase its inhibitory potency on recombinant CBS demonstrated that the structure of AOAA does not tolerate derivatization on either the amino or the linker at the α-carbon position. The α-carbon linker modification leads to a significant decrease of CBS inhibitory activity (Table 1, entries 2–7), while the free amino of AOAA has been confirmed an essential pharmacophore (26) required for interaction with and inhibition of CBS (Table 1, entries 8–9). Therefore, we have refocused our approach to synthesize prodrugs of AOAA through the carboxyl esterification with the intention of improving the compound’s lipophilicity and cell uptake.
Table 1.
Effect of covalent modifications of AOAA on recombinant CBS activity in vitro.
| Compound | Structure | CBS activity (% of control) at 100 μmol/L |
|---|---|---|
| AOAA | 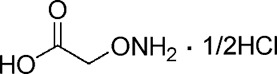 |
10 ± 3 |
| YD0135 | 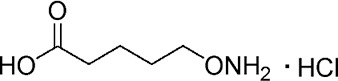 |
114 ± 5 |
| YD0151 | 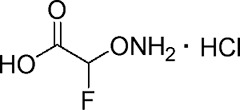 |
91 ± 2 |
| YD0154 | 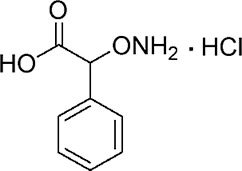 |
74 ± 5 |
| YD0163 | 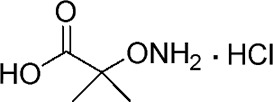 |
163 ± 12 |
| YD0164 | 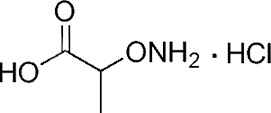 |
83 ± 9 |
| YD0177 | 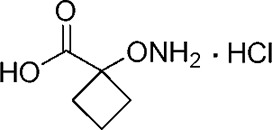 |
92 ± 3 |
| YD0196 | 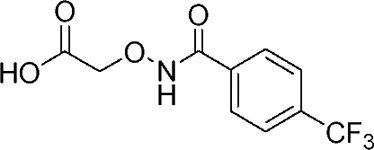 |
108 ± 11 |
| YD0197 | 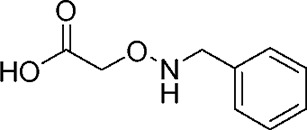 |
94 ± 3 |
Ester prodrugs are typically cleaved by intracellular esterases to yield the active principle. We hypothesized that this approach would generate intracellular AOAA and would improve cell-based potency. We synthesized a methyl ester derivative of AOAA (YD0171) by the route shown in Figure 1. The water/octanol coefficient of YD0171 was determined as 0.1210, whereas the water/octanol coefficient of AOAA was determined as 0.0019, indicating higher lipophilicity of the ester prodrug. YD0171 was a substantially weaker direct inhibitor of recombinant CBS when compared with the parent compound AOAA (Figure 2), supporting our prediction that the methyl ester group, similar to the covalent modifications in Table 1, sterically hinders the interactions between the AOAA moiety of YD0171 and CBS. However, when tested in a whole-cell homogenate of HCT116 colon cells, YD0171 inhibited H2S production, an effect that was blocked by pretreatment with the esterase inhibitor, paraoxon (1 mmol/L) (Figure 3). Esterase-mediated cleavage of a methyl ester yields methanol (9,10). Accordingly, incubation of YD0171 with HCT116 cell homogenates resulted in the transient production of methanol; methanol production was inhibited by paraoxon (Figure 4). The inhibitory effect of YD0171 on H2S production in HCT116 cells was confirmed using an AzMC-based live cell assay (Figure 5). These data demonstrate that YD0171 functions as an AOAA prodrug to inhibit cellular CBS activity in an esterase-dependent manner.
Figure 1.

Synthetic route of YD0171. AOAA, carboxymethoxylamine hemihydrochloride, or aminooxyacetic acid, or (aminooxy)acetate; YD0171, methyl 2-(aminooxy)acetate.
Figure 2.
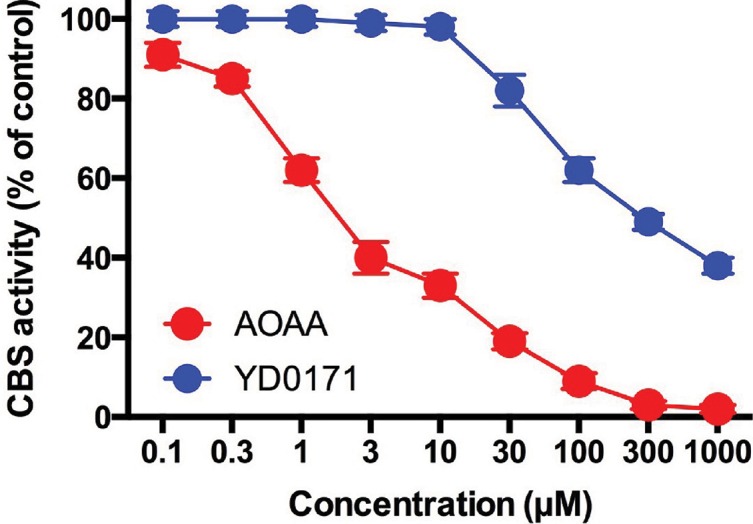
Effect of AOAA and YD0171 on the H2S-synthesizing activity of human recombinant full-length CBS enzyme. AOAA concentration dependently inhibited H2S production (detected by the AzMC method) with an IC50 of ~2 μmol/L, while YD0171 was a substantially weaker direct inhibitor of recombinant CBS activity (IC50: ~300 μmol/L). Data shown as mean ± SEM; n = 3.
Figure 3.
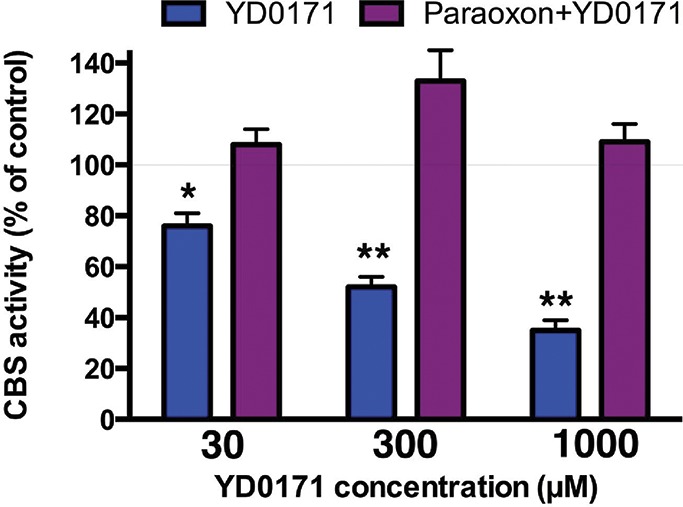
Inhibitory effect of YD0171 on the H2S-synthesizing activity of HCT116 cell homogenates: prevention of the inhibitory effect by the esterase inhibitor paraoxon. YD0171 concentration-dependently inhibited H2S production (detected by the AzMC method); the inhibitory effect was absent in cell homogenates pretreated with paraoxon (1 μmol/L). Data shown as mean ± SEM; n = 3.
Figure 4.
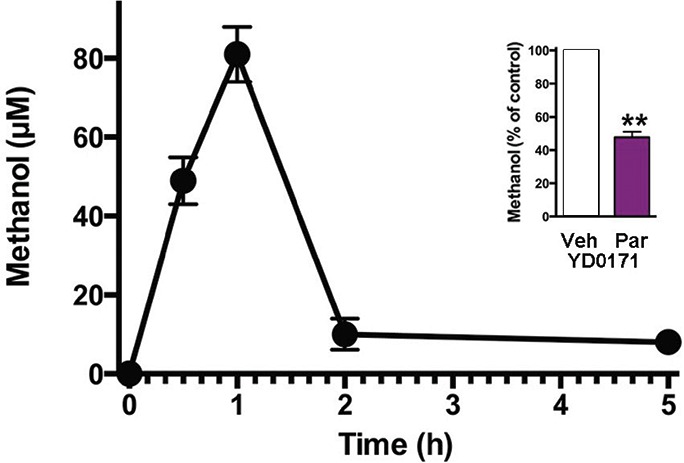
Methanol production in response to YD0171 in HCT116 cells. In response to YD0171 (1 mmol/L), the generation of methanol was noted in the HCT116 cells, with a peak at 1 h postexposure. Inset shows ethanol production in the presence of 1 mmol/L YD0171 at 2 h in the absence or presence of 1 mmol/L paraoxon pretreatment; **p < 0.01. Data shown as mean ± SEM; n = 3.
Figure 5.
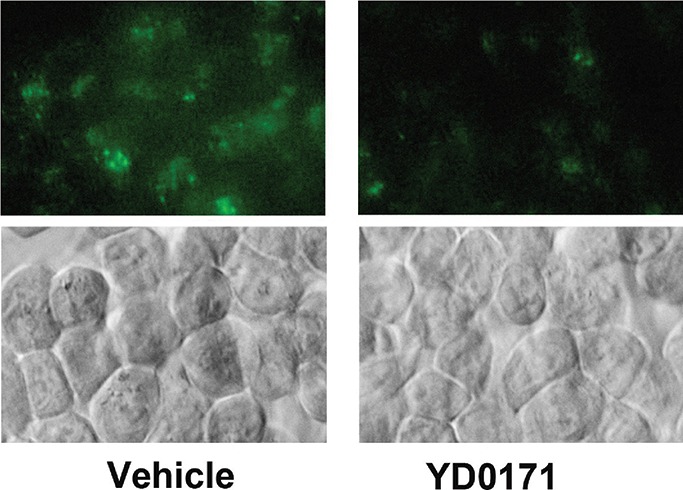
Inhibition of H2S production by YD0171 in HCT116 cells. Effect of YD0171 (1 mmol/L) on cellular H2S production is shown by the AzMC-based live cell assay. Top panel: green color shows AzMC fluorescence. Bottom panel: phase contrast images of the same visual field. Data shown are the representative of n = 3 independent determinations.
Effects of YD0171 and AOAA on HCT116 Cell Metabolome
The effect of YD0171 and AOAA on key metabolomic analytes is shown in Table 2; full metabolomic data are shown in Supplementary Table S1. YD0171 and AOAA treatment significantly affected the levels of multiple metabolites, with a high concordance between the effect of the two compounds (Figure 6, Table 3), supporting the conclusion that AOAA and YD0171 (after conversion into AOAA) affect similar molecular targets and pathways of cancer cells. As expected, both YD0171 and AOAA affected the pathways related to cysteine/homocysteine metabolism (Figure 7). Moreover, multiple alterations were detected in amino acid metabolism, glutamate metabolism, polyamine metabolism and glutathione homeostasis. The levels of numerous phospholipids were also affected as well as nicotinamide and thiamide metabolites. Lactate, imidazole lactate, phenyllactate and indolelactate levels were decreased as well as the levels of adenylosuccinate and 5-methyltetrahydrofolate. The largest quantitative increases were noted in spermine, mannitol/sorbitol, guanosine-5′-diphospate, nicotinamide riboside and cysteine sulfinic acid (Supplementary Table S1).
Table 2.
Metabolomic effects: modulation of selected cellular energetics-related metabolites in HCT116 cells by YD0171 and AOAA treatment (1 mmol/L, 24 h). Numbers represent fold changes in metabolite concentrations; red background indicates an increase, green background indicates a decrease in the concentration of the metabolite over vehicle-treated control.
| AOAA | YD0171 | |
|---|---|---|
| TCA cycle, oxidative phosphorylation | ||
| Citrate | 1.52 | 1.95 |
| Isocitrate | 1.66 | 3.02 |
| α-Ketoglutarate | 2.04 | 3.13 |
| Succinylcarnitine | 0.38 | 0.38 |
| Succinate | 0.42 | 0.56 |
| Fumarate | 0.12 | 0.19 |
| Malate | 0.12 | 0.27 |
| 2-Methylcitrate/homocitrate | 1.39 | 1.19 |
| Acetylphosphate | 1.49 | 1.53 |
| Phosphate | 1.13 | 1.07 |
| Glycolysis, gluconeogenesis, pyruvate metabolism | ||
| Glucose | 1.82 | 1.47 |
| Glucose 6-phosphate | 1.53 | 1.55 |
| Fructose-6-phosphate | 0.64 | 0.69 |
| Isobar: fructose 1,6-diphosphate, glucose 1,6-diphosphate, myo-inositol 1,4 or 1,3-diphosphate | 2.56 | 2.06 |
| 2,3-Diphosphoglycerate | 0.84 | 0.62 |
| Dihydroxyacetone phosphate | 1.87 | 0.86 |
| 3-Phosphoglycerate | 1.89 | 2.36 |
| Phosphoenolpyruvate | 3.07 | 3.31 |
| Pyruvate | 4.91 | 3.83 |
| Lactate | 0.53 | 0.62 |
| Glycerate | 1.07 | 0.85 |
| Pentose phosphate pathway | ||
| 6-Phosphogluconate | 1.55 | 1.66 |
| Ribose 1-phosphate | 0.38 | 0.31 |
| 5-Phosphoribosyl diphosphate | 3.42 | 1.22 |
| Sedoheptulose-7-phosphate | 1.77 | 2.08 |
| Creatine metabolism | ||
| Creatine | 1.13 | 1.19 |
| Creatinine | 0.81 | 0.88 |
| Creatine phosphate | 1.59 | 1.86 |
Figure 6.
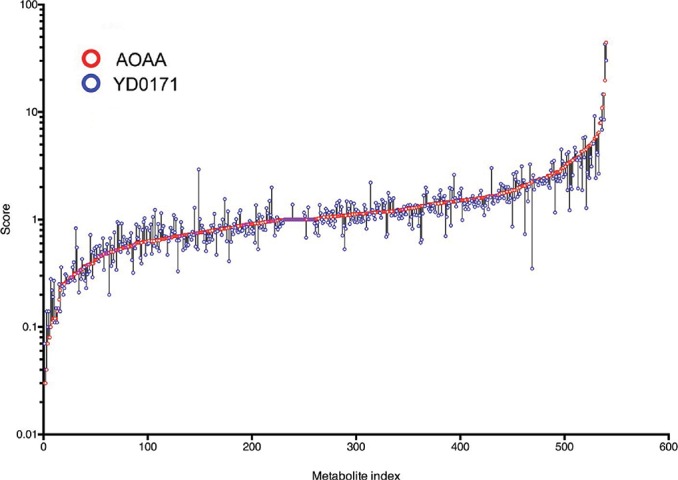
Effect of YD0171 on the metabolome of HCT116 cells. Cells were treated with YD0171 (1 mmol/L) or AOAA (1 mmol/L) for 24 h followed by metabolomic analysis. The horizontal scale shows analyte numbers. The vertical scale shows fold change; the value of 1 represents no change, a value of 0.1 represents a 90% decrease in the analyte value, a value of 10 represents a 10-fold increase in the analyte value in response to AOAA (red circles) or YD0171 (blue circles) treatment, was compared with vehicle control. Overlapping, circles or circles connected with short vertical black lines indicate that AOAA and YD0171 affected the analyte in a directionally similar way. Data are shown as mean values of n = 3.
Table 3.
Metabolomic effects of AOAA and YD0171: enrichment analysis. Clusters with >5 measured metabolites are presented. Concordance is calculated as the % of metabolites affected by AOAA that are also affected by YD0171 for clusters where more metabolites were affected by AOAA than YD0171 and concordance is calculated as the % of metabolites affected by YD0171 that are also affected by AOAA for clusters where more metabolites were affected by YD0171 than AOAA.
| Pathway cluster | Metabolites measured in cluster | Metabolites affected by AOAA (%) | Metabolites affected by YD0171 (%) | Concordance of the metabolites (%) |
|---|---|---|---|---|
| TCA cycle | 8 | 7 (88) | 7 (88) | 100 |
| Polyamine metabolism | 7 | 6 (86) | 4 (57) | 67 |
| Phospholipid metabolism | 43 | 33 (77) | 23 (53) | 70 |
| Plasmalogen | 10 | 7 (70) | 5 (50) | 71 |
| Glutamate metabolism | 10 | 6 (60) | 6 (60) | 100 |
| Nicotinate and nicotinamide metabolism | 10 | 6 (60) | 7 (70) | 86 |
| Alanine and aspartate metabolism | 6 | 4 (67) | 4 (67) | 100 |
| Fatty acid metabolism | 22 | 13 (59) | 9 (41) | 69 |
| Pyrimidine metabolism | 26 | 15 (58) | 17 (65) | 88 |
| Sphingolipid metabolism | 26 | 15 (58) | 7 (27) | 47 |
| Leucine, isoleucine and valine metabolism | 21 | 12 (57) | 7 (33) | 53 |
| Purine metabolism | 35 | 20 (57) | 18 (51) | 90 |
| Nucleotide sugar | 7 | 4 (57) | 3 (42) | 80 |
| Histidine metabolism | 9 | 5 (56) | 5 (56) | 100 |
| Vitamin B6 metabolism | 6 | 3 (50) | 3 (50) | 100 |
| γ-Glutamyl amino acid | 13 | 6 (46) | 5 (38) | 83 |
| Glycolysis, gluconeogenesis and pyruvate metabolism | 11 | 5 (45) | 3 (27) | 60 |
| Glutathione metabolism | 9 | 4 (44) | 4 (44) | 100 |
| Methionine, cysteine, SAM and taurine metabolism | 18 | 8 (44) | 4 (22) | 50 |
| Pentose metabolism | 7 | 3 (43) | 2 (29) | 67 |
| Lysine metabolism | 10 | 4 (40) | 2 (20) | 50 |
| PUFA metabolism | 13 | 5 (38) | 3 (23) | 60 |
| Urea cycle | 14 | 5 (38) | 7 (50) | 71 |
| Dipeptide | 18 | 6 (33) | 6 (33) | 83 |
| Phenylalanine and tyrosine | 10 | 3 (30) | 1 (10) | 33 |
| Glycine, serine and threonine | 7 | 2 (29) | 1 (14) | 50 |
| Aminosugar metabolism | 7 | 1 (14) | 1 (14) | 100 |
| Lysolipid | 26 | 3 (11) | 2 (7) | 67 |
Figure 7.

Effect of YD0171 on the transsulfuration pathway metabolites in HCT116 cells. Cells were treated with YD0171 (1 mmol/L) or AOAA (1 mmol/L) for 24 h, followed by metabolomic analysis. The left panel shows the relative position of the various measured metabolites in the transsulfuration pathway. *p < 0.05 and **p < 0.01 indicate significant effect of the test compounds compared with control. Data are shown as mean ± SEM values of n = 3.
The metabolomic signature of YD0171 included actions on targets of AOAA previously implicated in oncogenesis. GOT1, also known as aspartate transaminase or AST, is an essential enzyme in the malate/aspartate shuttle (27,28). GOT1 is inhibited by AOAA through a secondary action of the compound as a transaminase inhibitor (28). We confirmed that AOAA inhibited GOT1 in vitro; however, at the highest concentration tested, sodium hydrogen sulfide [NaHS (a H2S donor)] also decreased the activity of the enzyme (Figure 8A). The activity of another related transaminase, ALT, was also inhibited by AOAA, but not H2S (Figure 8B). SCS activity and α-ketoglutarate dehydrogenase activity were unaffected by either AOAA or H2S (Figure 8C, D). YD0171 induced an increase in pyruvate levels and simultaneous decrease in lactate levels in HCT116 cells (Table 1), suggesting an inhibition of lactate dehydrogenase A (LDH-A) activity. LDH-A is known to support the metabolism of various cancer cells including colorectal cancer (29). We have observed no direct inhibitory effect of AOAA on LDH activity in vitro but found that H2S directly stimulates LDH activity (Figure 8E). Similarly, ODC activity was directly increased by H2S incubation, although with an apparent inverse bell-shaped concentration-response curve, where the largest activation was noted at the lowest concentration of H2S tested; in addition, ODC activity was also attenuated by AOAA tested (Figure 8F). We conclude that the complex metabolomic signature of YD0171 is related to its conversion to AOAA, which then directly inhibits CBS activity (i.e., CBS-dependent H2S production) as well as other transaminases (e.g., GOT1, ALT).
Figure 8.

Direct effect of AOAA or H2S on selected enzyme activities in vitro. Effect of AOAA or H2S (produced as NaHS) on the enzymatic activity of (A) AST/GOT1, (B) ALT, (C) SCS (D) α-ketoglutarate dehydrogenase (E) LDH and (F) ODC. Data shown as mean ± SEM; n = 3–6; *p < 0.05 and **p < 0.01 indicate significant effects of AOAA or H2S compared with control.
Effects of YD0171 on Cellular Bioenergetics, Cell Cycle Regulation and Colon Cancer Cell Proliferation In Vitro
The decreases in succinate, fumarate and malate levels, simultaneously with increases in citrate, isocitrate and α-ketoglutarate predict significant alterations in mitochondrial function by the prodrug. Extracellular flux analysis confirmed that YD0171 inhibited cellular bioenergetics as evidenced by a suppression of mitochondrial electron transport and glycolytic activity (Figure 9). This was associated with a dose-dependent increase in the population of cells in G0/G1 phase of the cell cycle and a reduction in cells in S phase (Figure 10). However, YD0171, at concentrations where it inhibited HCT116 cell proliferation, did not induce significant cell death either by necrosis or by apoptosis (Figure 11). YD0171 induced an impairment of mitochondrial DNA integrity; at the highest concentration tested, YD0171 also impaired nuclear DNA integrity (Figure 12). In line with its effect on cellular bioenergetics, YD0171 inhibited the proliferation of HCT116 cells and it was more potent than AOAA in its in vitro antiproliferative potency; at 30 and 100 μmol/L, YD0171 already induced a significant inhibition of cell proliferation, while AOAA was without inhibitory effect; at 1 mmol/L the inhibitory dose-response curves converged (Figure 13).
Figure 9.

Inhibition of cellular bioenergetic parameters by YD0171 in HCT116 cells. Cells were treated with YD0171 (1 mmol/L) for 24 h, followed by extracellular flux analysis. (A) Left panel: mitochondrial function analysis. Four key parameters of mitochondrial function (basal respiration, ATP turnover, proton leak and maximal respiration) were assessed through the sequential use of 40 μmol/L oligomycin (ATP synthase inhibitor), 13.5 μmol/L FCCP (mitochondrial uncoupler) and 10 μmol/L rotenone + 1 μmol/L antimycin A (complex I and III inhibitors). The difference between the maximal and the basal respirations was considered the respiratory reserve capacity (the capacity of a cell to generate ATP via oxidative phosphorylation in response to increased demand for energy). Right panel: analysis of glycolytic function. A glycolytic stress test was used to estimate various parameters of cellular glycolysis (glycolysis, glycolytic capacity and glycolytic reserve), which was obtained with the sequential use of 25 mmol/L glucose, 5 μmol/L oligomycin (to block mitochondrial respiration and force the cells to rely on glycolysis for ATP production) and 100 mmol/L 2-deoxyglucose (2-DG, a glucose analog and inhibitor of glycolytic ATP production). Glycolytic reserve was calculated as the difference between the glycolytic capacity and glycolysis; this parameter is indicative of the cellular ability to increase the glycolytic rate upon increased energy demand. Acidification of carbon dioxide, the end product of the TCA cycle, which can be converted to bicarbonate, is considered a major contributor to nonglycolytic acidification. (B) Statistical analysis of the various bioenergetic parameters. Data shown as mean ± SEM; n = 6; *p < 0.05 and **p < 0.01 indicate significant inhibitory effects of YD0171 on the various bioenergetic parameters, compared with vehicle control.
Figure 10.

Effect of YD0171 on the cell cycle of HCT116 cells. Cells were treated with YD0171 (100, 300, 1 mmol/L), or the positive control nocodazole for 48 h, followed by flow cytometric cell cycle analysis. Typical profiles are shown in the gray panels; cell cycle distributions are shown by the bagel charts. Although nocodazole induced the expected G1 cell cycle arrest, YD0171 or AOAA did not induce cell cycle arrest but increased the G1 phase and reduced the G2 phase. Data shown are representative of n = 3 determinations.
Figure 11.

Effect of YD0171 on cell viability and cell death in HCT116 cells. Cells were treated with YD0171 (100, 300 or 1 mmol/L) for 48 h, followed by flow cytometry analysis of phospholipid redistribution by the annexin V/propidium iodide assay. Samples were analyzed for green fluorescence (FITC) and for red fluorescence (PI) by flow cytometry. Typical profiles are represented in the left panels. The bar graph on the right side shows the relative proportions of the live, necrotic, early apoptotic and late apoptotic cell populations; data are shown as mean ± SEM; n = 6.
Figure 12.

Effect of YD0171 on mitochondrial and nuclear DNA integrity in HCT116 cells. Cells were treated with YD0171 (100, 300 or 1 mmol/L) or AOAA (100, 300 or 1 mmol/L) for 48 h, followed by LA-PCR assays. *p < 0.05 and **p < 0.01 indicate significant reduction in DNA integrity compared with control. Data are shown as mean ± SEM; n = 6.
Figure 13.
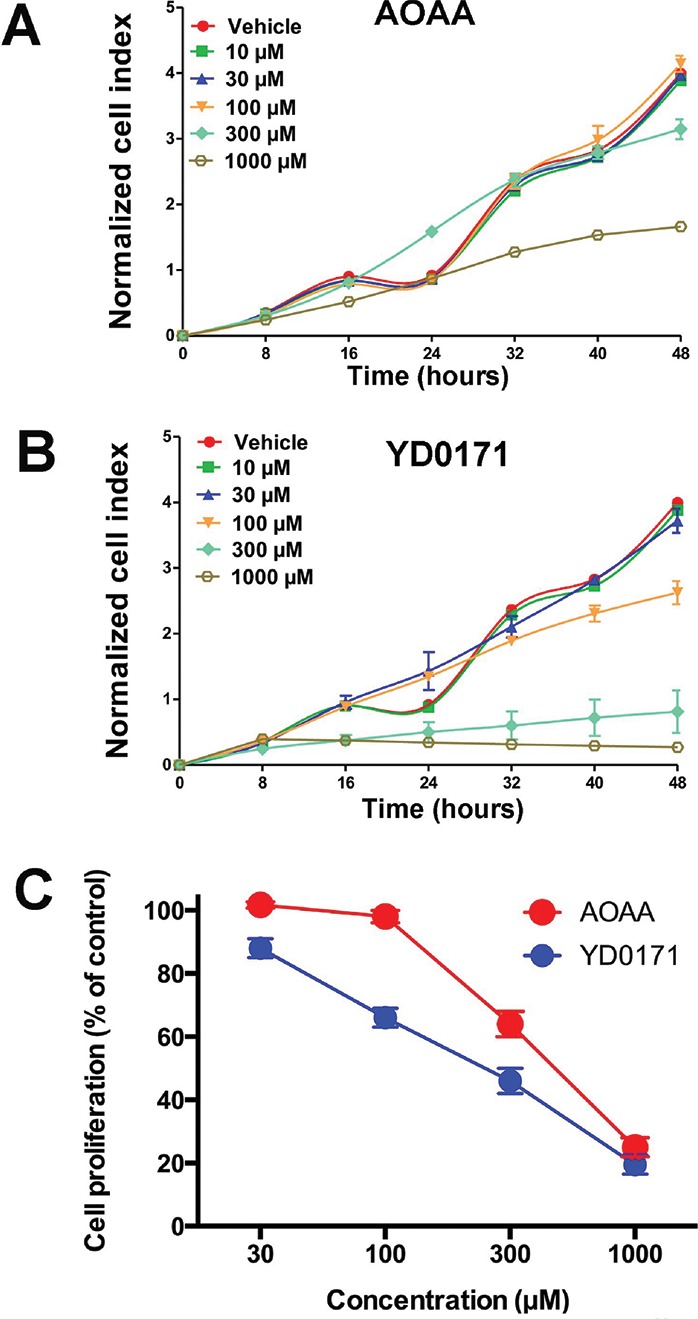
Inhibition of cell proliferation and H2S production by YD0171 in HCT116 cells. (A) Cells were treated with various concentrations of AOAA or YD0171 in 96-well xCELLigence plates, and proliferation was monitored for 48 h. Cell proliferation is shown over time for AOAA (top panel) and YD0171 (bottom panel) treated cells from one representative experimental run. Note that AOAA only inhibited cell proliferation at the highest concentration tested (1 mmol/L), while the inhibitory effect of YD0171 on cell proliferation was already apparent at 100–300 μmol/L. (B) Summary data (mean ± SEM) of n = 6 independent experiments testing the effect of AOAA and YD0171 in cultured HCT116 cells. Cell proliferation at 48 h is expressed as 100%. YD0171 inhibited the proliferation with an IC50 of 200 μmol/L, while the IC50 of AOAA was 500 μmol/L.
Evaluation of YD0171 in Preclinical Models of Colon Tumor Growth and Metastasis
To begin developing YD0171 toward a potential anticancer therapeutic agent, we evaluated its efficacy using the human colon cancer cell line HCT116 and tissue xenografts in immune-compromised (athymic nude) mice. The growth of HCT116 subcutaneous xenograft tumors was suppressed both by YD0171 and AOAA. However, YD0171 was more potent than AOAA, inducing an equivalent reduction in tumor volume at a 90% lower concentration (1 mg/kg/d YD0171 vs. 9 mg/kg/d AOAA) (Figure 14). Vehicle-treated animals and YD0171-treated animals (9 mg/kg/d) exhibited comparable mortality by the end of the experiments (8% and 14%, respectively), while AOAA (9 mg/kg/d) exhibited a higher, 53% mortality (p < 0.05). AOAA-treated animals also showed significant weight loss (11 ± 4%), while YD0171 animals maintained their weights similar to the vehicle-treated tumor-bearing controls, suggesting that YD0171 therapy is associated with less toxicity than AOAA.
Figure 14.
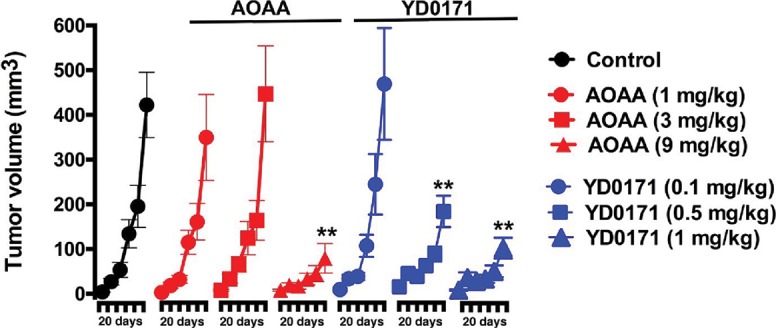
Efficacy of YD0171 in tumor-bearing mice. Comparison of the effect of AOAA or YD0171 on HCT116 tumor growth in nude mice. AOAA inhibited tumor growth at 9 mg/kg/d (but not at lower doses of 3 or 1 mg/kg/d), while YD0171 exerted an inhibitory effect at 0.5 and 1 mg/kg (but not at the lowest dose of 0.1 mg/kg). Data are shown as mean ± SEM of n = 10 animals per group; **p < 0.01 indicates a significant inhibitory effect.
The efficacy of YD0171 was further evaluated using PDTXs. Treatment with YD0171 (3 mg/kg/d), commencing at 7 d after tumor implantation, resulted in a marked suppression of tumor growth. Figure 15 shows the effect of YD0171 on the growth of three PDTX, obtained from three different patients. Intratumor CBS activity/H2S production was also measured ex vivo. YD0171 (3 mg/kg/d s.q. for 7 d) markedly reduced the H2S production in tumor homogenates, while H2S production in the liver (a tissue that expresses high levels of CBS) was not significantly affected (Figure 16).
Figure 15.
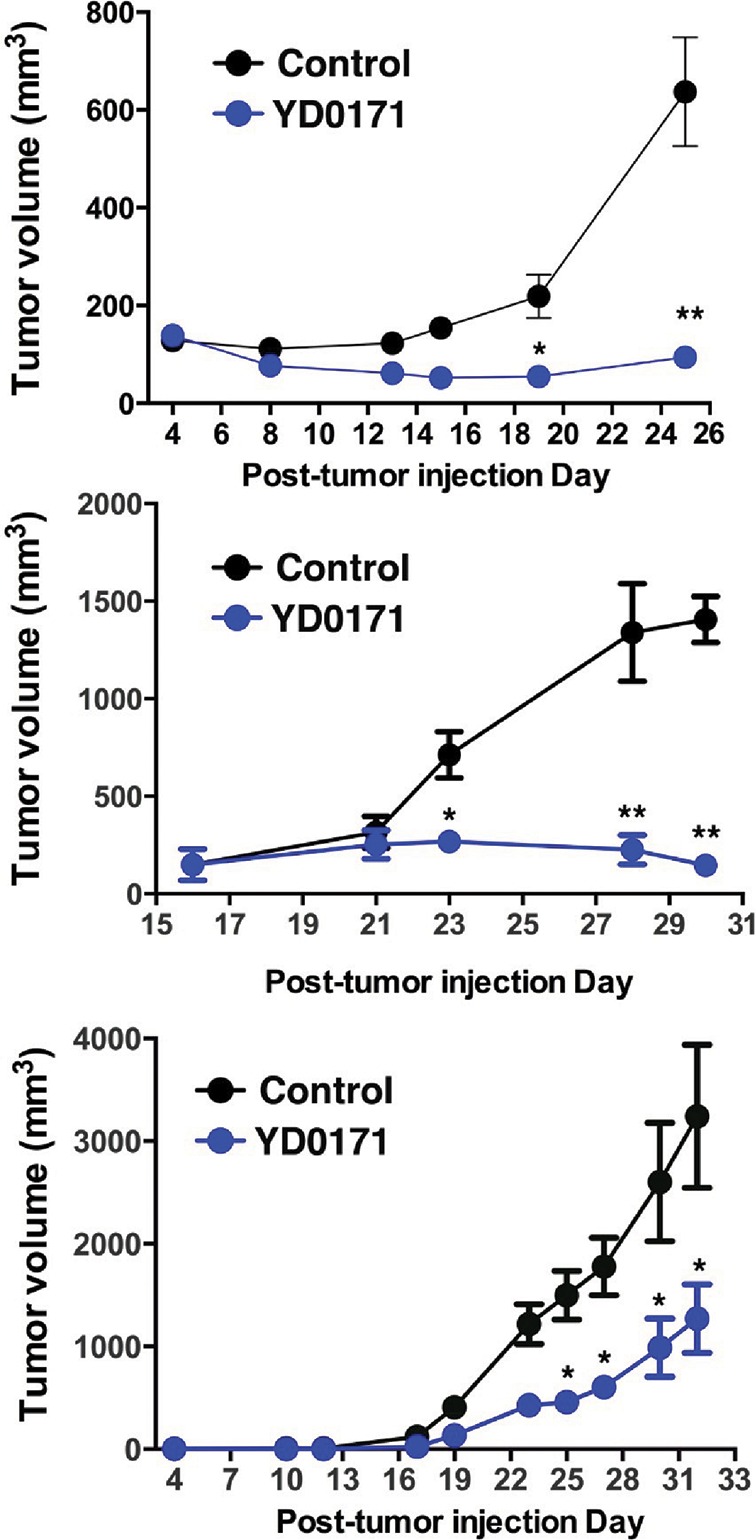
Effect of YD0171 on PDTX growth in nude mice. PDTX of three different colon cancer patients (top, middle and bottom panels) showing different growth rates. YD0171 (3 mg/kg/d) inhibited tumor growth in all experiments. *p < 0.05 and **p < 0.01 indicate a significant inhibitory effects. Data are shown as mean ± SEM values of n = 4–6 animals per group.
Figure 16.

YD0171 inhibits H2S production in HCT116 tumors of nude mice. Mice were subjected to implantation of PDTX of a colon cancer patient. When the tumor reached the size of 1,000 mm3, treatment with vehicle or YD0171 (3 mg/kg/d) started and continued for 7 d, at which point tumors were excised and liver tissues were collected. Tissues were homogenized and CBS-mediated H2S production was measured by the AzMC method. CBS activity was reduced by YD0171 (*p < 0.05) in the tumor tissue, but no significant inhibition was detected in the liver tissue. Data are shown as mean ± SEM values of n = 4 animals per group.
To test whether YD0171 could induce regression of established tumors, we used a modified crossover experimental design. YD0171 effectively suppressed tumor growth during the first 42 d of the experiment, whereas tumors in the vehicle treatment group grew significantly larger by 40 d (Figure 17). Once the YD0171 treatment was stopped and was switched to vehicle, tumors grew quickly, surpassing the size of the original vehicle group within 2 wks. In contrast, switching the original vehicle group to YD0171 resulted in a significant regression of the tumor volume within a week (Figure 17).
Figure 17.

YD0171 induces HCT116 tumor regression in nude mice. Mice were subjected to implantation of HCT116 tumors. When a small palpable tumor was evident at 7 d, the mice were randomized into a vehicle-treated group and a group treated with YD0171 (3 mg/kg/d). In the YD0171-treated animals, treatment discontinued on d 42. In vehicle-treated animals, on d 50, treatment with YD0171 commenced. Pretreatment with YD0171 at the onset of the study prevented the growth of the tumors; discontinuation of the YD0171 treatment allowed tumor growth to commence. Data are shown as mean ± SEM of n = 8 animals per group; *p < 0.05 and **p < 0.01 indicate a significant inhibitory effect of YD0171 compared with the corresponding vehicle control at the same time point.
To assess the effects of YD0171 on the metastatic spread of colon cancer cells, we used an orthotopic xenograft model that involves injecting HCT116 cells into the submucosal lamina propria of the cecum. The model recapitulates the metastatic dissemination of tumor cells from large bowel through the portal circulation to the liver. YD0171 (1 mg/kg/d) reduced the growth of the primary tumor in the cecum as well as reduced the number of liver metastases. The positive control, clinically used anticancer drug oxaliplatin, also reduced both primary tumor formation and metastasis formation in this model (Figure 18).
Figure 18.

YD0171 inhibits tumor growth and metastasis formation in nude mice subjected to intracecal HCT116 cell implantation. Mice were subjected to intracecal implantation of HCT116 cells. On posttumor injection d 3, mice were randomized into subcutaneous treatment with either vehicle, YD0171 (1 mg/kg) or oxaliplatin (5 mg/kg), each 5 d/wk. On posttumor injection d 51, the cecum, liver, lung and lymph nodes were harvested, and primary tumor size (cecum) and metastasis (liver, lung and lymph nodes) were quantified using a weighted score. Right side panels show representative photomicrographs and histological pictures of (A) the primary tumor and (B) the liver metastasis; human tumor tissue is highlighted by the brown staining. Data represent mean ± SEM values of n = 6–8 animals per group; *p < 0.05 and **p < 0.01 indicate a significant inhibitory effect of YD0171 compared with vehicle control.
Effect of YD0171 on Plasma Markers of Organ Damage and Plasma Homocysteine Levels
In an exploratory 5-d safety study in C56BL/6 mice, YD0171 (daily total dose: 2, 6 or 20 given in two divided doses i.p.) failed to increase any of the measured markers of organ injury. At the highest dose tested (60 mg/kg/d), a 23% increase in blood urea nitrogen and a 33% increase in total bilirubin was noted (Figure 19). The liver and the kidney samples of the YD0171-treated mice did not show major pathomorphological alterations. The liver and the kidney samples treated with either doses or vehicle present normal structure; no evidence of histological organ damage was noted (Figure 20). There was a 36% elevation in plasma homocysteine levels in mice treated with the highest dose of YD0171 (60 mg/kg/d): plasma homocysteine levels increased from 0.69 ± 0.14 to 0.94 ± 0.06 μg/mL (n = 5, p < 0.05).
Figure 19.
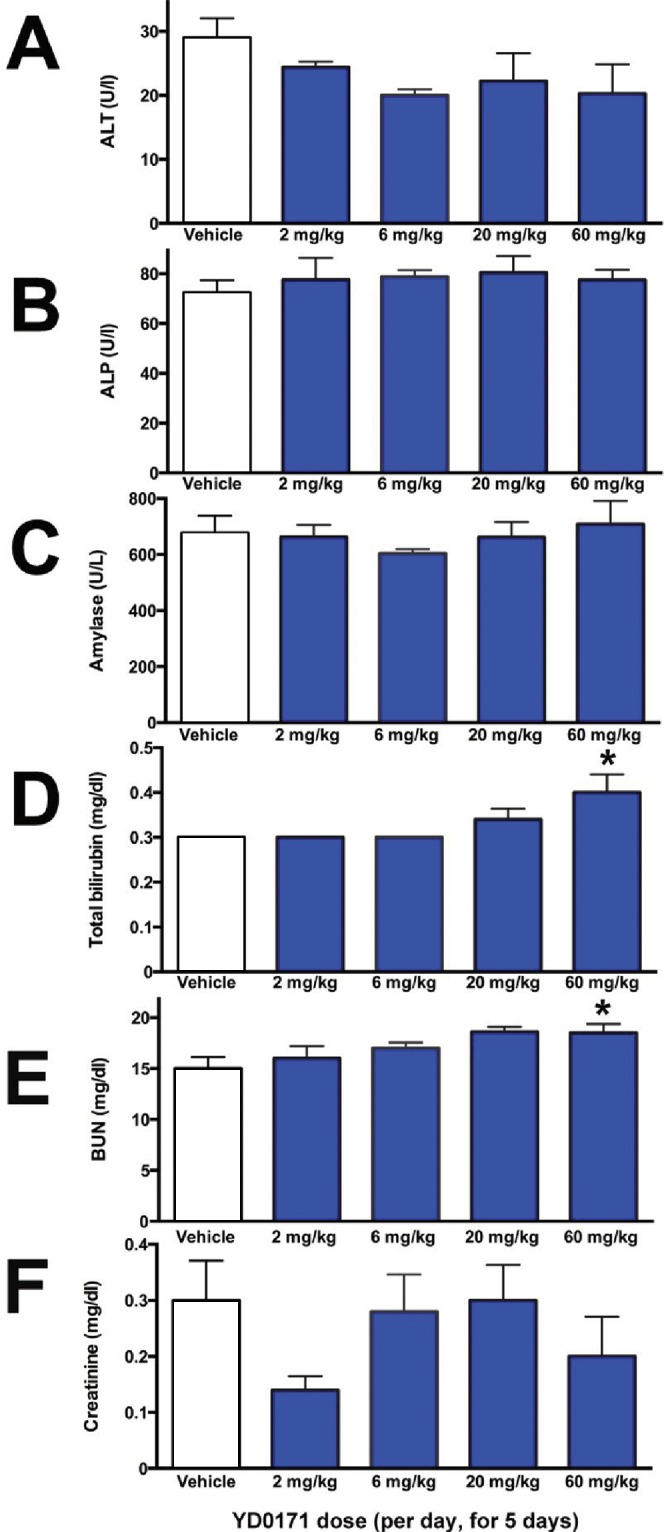
Effect of YD0171 treatment on plasma markers of organ damage in mice. Mice were subjected to intraperitoneal injection of YD0171 in two divided doses (total daily doses: 2, 6, 20 and 60 mg/kg/d) for five consequent days. On the morning of the sixth day, animals were killed and blood was collected and analyzed by the Vetscan system. Data are shown as mean ± SEM of n = 5 mice per group; *p < 0.05 indicates a significant effect of YD0171 compared with vehicle control.
Figure 20.

Liver and kidney histological pictures from mice treated with vehicle or YD0171. Mice were subjected to intraperitoneal injection of YD0171 in two divided doses (total daily doses: 2, 6, 20 and 60 mg/kg/d) for five consequent days. On the morning of the sixth day, animals were killed and tissues subjected to H&E staining and histological analysis. From each group, sections from three randomly selected animals are shown.
DISCUSSION
The main conclusions of the current report are the following: (a) The prodrug approach is an effective strategy to improve the antiproliferative potency of AOAA in vitro and in vivo and to reduce its toxicity in vivo. (b) The AOAA prodrug YD0171 suppresses the cellular bioenergetics and the growth of colon cancer cells without being directly cytotoxic. (c) YD0171 induces significant alterations in the metabolomic profile of colon cancer cells; its metabolomic signature is similar to the signature induced by its active metabolite (AOAA). (d) YD0171 induces a preferential inhibition of tumor CBS activity over parenchymal (hepatic) CBS activity. (e) YD0171 causes the regression of established colorectal cancer xenografts from patients in vivo. (f) A 5-d treatment of mice with YD0171 at doses >20–60 times higher than the effective antitumor dose does not cause organ injury but induces a slight degree of hyperhomocysteinemia.
Inhibition of tumor cell (colon, ovarian and breast) CBS activity is an emerging experimental therapeutic approach (4,5). CBS inhibition can be viewed as a “targeted therapy” because the tumor tissue expresses higher levels of CBS and produces higher amount of H2S than the normal margin of resected tissue (2). Nevertheless, CBS is also expressed in other tissues outside tumor cells (4). Therefore, systemic administration of a CBS inhibitor will also exert systemic effects, for example, inhibition of CBS in the liver (which would be expected to attenuate the physiological function of the liver to metabolize homocysteine). However, our results (Figure 13) show that the inhibition of CBS activity by YD0171 is more pronounced in the xenograft cancer tissue, compared with the liver tissue, suggesting cancer cell selectivity. The differential effect of cancer versus hepatic CBS inhibition by YD0171 may be either related to a differential turnover rate of hepatic versus intratumor CBS enzyme and/or due to the high efficiency of the hepatocytes to metabolize xenobiotics.
The prodrug approach is commonly used to increase the lipophilicity of various drugs, which often improves oral bioavailability and cell uptake, resulting in improved therapeutic efficacy (9,10). Controlled conversion of the prodrug to the active principle is also beneficial as it avoids high peak levels of the compound in the circulation, thereby reducing its toxicity (9,10). As mentioned in the Introduction, there are several methyl ester prodrugs in clinical use in diverse fields of medicine (9,10). The metabolism of a methyl ester prodrug yields low amounts of methanol as a by-product, which, in turn, are metabolized to formaldehyde and then to formic acid; our data, showing an early (1–2 h) increase in methanol formation after addition of YD0171 to HCT116 cells, followed by a decrease by 5 h, may be due to a simultaneous cellular production and elimination of methanol.
CBS inhibition suppresses colon cancer cell metabolic activity (2–6). Indeed, direct measurement of H2S production, as well the expected changes in transsulfuration pathway metabolites (demonstrated by metabolomic analysis), are consistent with a CBS inhibitory effect. AOAA also inhibits a second mammalian H2S-generating enzyme, cystathionine γ-lyase (8); this additional pharmacological action may also contribute to the observed changes in transsulfuration pathway intermediates. Moreover, AOAA, as an inhibitor of several other PLP-dependent enzymes (mainly, transaminases) (29), has pharmacological effects beyond the transsulfuration pathway, as confirmed by our metabolomic analysis. These effects include actions on targets of AOAA previously implicated in oncology—including GOT1, also known as aspartate transaminase or AST—an essential enzyme in the malate/aspartate shuttle (27,28). The complex metabolic signature obtained after YD0171 exposure of HCT116 cells are likely result from the inhibition of multiple PLP-dependent enzymes, including GOT1, as well as a related enzyme, ALT. GOT1 has been implicated in glutaminolysis, a metabolic pathway that partially selective to cancer cells, and which converts glutamine to glutamate, and then to α-ketoglutarate (26). However, YD0171 did not suppress the activity of this pathway, because both glutamate and α-ketoglutarate levels were increased. The YD0171-induced elevation of citrate, isocitrate and α- ketoglutarate levels, and concomitant decreases in succinate, fumarate and malate levels suggest a blockade of the Krebs cycle, most likely at the level of α-ketoglutarate dehydrogenase, even though we have been unable to detect any direct effect of either AOAA or H2S on the activity of this enzyme in vitro. Our direct measurements also show that SCS activity is unaffected by AOAA or H2S. (The blockade of the Krebs cycle by AOAA has previously been also noted in breast cancer cells and has been mainly attributed to the inhibition of AST activity [28].) The increases in glucose, glucose-6-phosphate, 3-phosphoglycerate, phosphoenolpyruvate and pyruvate observed after YD0171 exposure suggest that—in addition to inhibition of oxidative phosphorylation—glycolysis is also inhibited, an effect also confirmed by the extracellular flux analysis data. As glyceraldehyde 3-phosphate dehydrogenase (GAPDH), an essential enzyme in the glycolytic pathway, is known to be activated by H2S through posttranslational modification (sulfhydration) (30), we attribute the inhibition of glycolysis by YD0171, at least in part, to suppression of cellular H2S levels due to CBS inhibition by its intracellular metabolite, AOAA. As H2S was found to directly enhance LDH activity (Figure 8E), we attribute the YD0171-induced increase in pyruvate levels, and simultaneous decrease in lactate levels, to a cellular inhibition of LDH-A activity, secondary to the reduction of cellular H2S levels due to CBS inhibition. As H2S was found to directly enhance ODC activity, while AOAA was found to directly inhibit ODC activity (Figure 8F), we attribute the YD0171-induced changes in cellular polyamine levels to a combination of these actions. Based on the known role of LDH-A in cancer cells (31) and based on the known roles of polyamines in the cancer cell proliferation (32), we hypothesize that the above-discussed effects of AOAA (the active metabolite of YD0171) may, at least in part, contribute to the observed suppression of tumor cell proliferation. In the context of the above-described direct activating effects of H2S on various enzymes involved in cell metabolism (LDH, ODC), a recent study by Liang and colleagues (33) in cardiac myocytes should be emphasized, where several enzymes involved in glycolysis and the citric acid cycle were found to be activated by NaHS treatment (100 μmol/L), including LDH, as well as pyruvate kinase and—in contrast to our current findings—succinate dehydrogenase as well. By pharmacological inhibition of H2S biosynthesis, one would expect that these activating influences would be diminished, and this would be expected to contribute to the global inhibition of cell metabolism seen in our current study after treatment of the HCT116 cells with YD0171.
In addition to the modulation of the various bioenergetic pathways discussed above, YD0171 induced marked alterations in amino acid metabolism, polyamine metabolism (marked increases in spermine and spermidine and a marked decrease into putrescine); phospholipid/sphingolipid metabolism and vitamin B1/B6 metabolism; these alterations may also contribute to the functional alterations observed in the tumor cells after YD0171 exposure. Interestingly, although the transsulfuration pathway has been linked to glutathione biosynthesis, and in ovarian cancer cells CBS silencing and AOAA have been shown to suppress intracellular glutathione levels (resulting in an impairment of cellular antioxidant defense) (7), in the current experimental system, YD017 did not affect the cellular levels of oxidized or reduced glutathione.
YD0171 induced marked alterations in the metabolomic signature of cancer cells, and inhibited cellular metabolism and proliferation, as well an increase in the proportion of cells in G0/G1 phase of the cell cycle. Interestingly, a similar effect was noted in MCF7 breast cancer cells after incubation for 5 d in cocultures with activated macrophages in vitro: MCF7 cells with CBS silencing shifted a significant proportion of cells in the G0/G1 phase (7).
Despite of a variety of effects on the cellular metabolome, mitochondrial DNA integrity and cell cycle, YD0171 did induce overt HCT116 cell necrosis or apoptosis in vitro at concentrations, where it was markedly antiproliferative. YD0171 inhibited xenograft growth, and when applied in a delayed treatment protocol, produced the regression of established tumors. Moreover, when YD0171 was given early to mice with small xenograft tumors, after the discontinuation of YD0171 pretreatment, the cytostatic effect was removed and tumor growth commenced. These findings suggest that YD0171 acts as a targeted antiproliferative agent rather than a general chemotherapeutic/cytotoxic agent. The YD0171-dependent impairment of mitochondrial DNA integrity may be related to the previously established role of H2S in protecting against mitochondrial DNA damage (34).
Several lines of studies show that exogenously administered or endogenously produced H2S can induce pro-proliferative signaling pathways (including MAP kinases, Akt, ERK1 activation) (5–7,35–43). By inhibiting colon cancer cell H2S production, YD0171 may suppress these signaling processes, although this remains to be directly investigated in future studies. Likewise, the precise mechanistic link between the bioenergetic effects of YD0171 and its effects on various proliferative signaling pathways remains to be further elucidated.
CONCLUSION
In conclusion, the prodrug approach, as exemplified by YD0171, enhances the cell-based and in vivo potency of AOAA and may form the basis of future translational approaches based on CBS inhibition, or, more generally, on inhibition of tumor cell transaminase activity. We hypothesize that the primary mode of YD0171’s action—after conversion to AOAA—is a metabolic suppression of the cancer cell (affecting both oxygen-dependent pathways, i.e., the oxidative phosphorylation, and oxygen-independent pathways, i.e., glycolysis), which, in turn, leads to the suppression of macromolecule synthesis, cell growth and cell proliferation. This occurs through a combination of CBS inhibition (and consequent inhibition of cellular H2S levels, which, in turn, suppresses the activity of several other intracellular enzymes, e.g., GAPDH, LDH-A and ODC) as well as through actions on other PLP-dependent transaminases that are essential for tumor cell biology (e.g., GOT1). The anticancer approach introduced in the current report is in line with current concepts targeting key checkpoints in tumor cell bioenergetics (11,44) as well as with concepts aiming to convert cancer into a manageable “chronic disease” (45,46).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by R01CA175803 (CS, MRH), T32 DK007639-24 (MRH), a pilot project supported by UL1TR001439 from the National Center for Advancing Translational Sciences, National Institutes of Health and the Cancer Prevention Research Institute of Texas grant DP150074 (MRH, CS). The authors thank Dr. Steve Stirdivant (Metabolon Inc.) for helpful discussions during the interpretation of the metabolomic analysis data; Dr. Laixin Wang (Novabioassays LLC) for his contributions to the water/octanol partition study; Mr. Mark Griffin for the cell cycle analysis at the UTMB Flow Cytometry and Cell Sorting Core Facility and Dr. Andras Kiss (Semmelweis University, Department of Pathology, Budapest, Hungary) for the blinded evaluation of the histological slides from the safety study.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
CC, AP, JZ, MRH and CS are shareholders and/or officers in CBS Therapeutics Inc., an UTMB spin-off company involved in the development of CBS inhibitors.
Cite this article as: Chao C, et al. (2016) Cystathionine-β-Synthase inhibition for colon cancer: Enhancement of the efficacy of aminooxyacetic acid via the prodrug approach. Mol. Med. 22:361–79.
REFERENCES
- Rabeneck L, Horton S, Zauber AG, Earle C. Colorectal Cancer. In: Gelband H, Jha P, Sankaranarayanan R, Horton S, editors. Cancer: Disease Control Priorities, Third Edition. Volume 3. Washington, DC: The International Bank for Reconstruction and Development/The World Bank; 2015. Chapter 6. [PubMed] [Google Scholar]
- Szabo C, et al. Tumor-derived H2S, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation and angiogenesis in colon cancer; Proc Natl Acad Sci U S A; 2013. pp. 12474–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Módis K, et al. Effect of S-adenosyl-L- methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide. 2014;41:146–56. doi: 10.1016/j.niox.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich MR, Coletta C, Chao C, Szabo C. The therapeutic potential of cystathionine-β- synthase inhibition in cancer. Antioxid Redox Signal. 2015;22:424–48. doi: 10.1089/ars.2014.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat Rev Drug Discov. 2016;15:185–203. doi: 10.1038/nrd.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS One. 2013;8:e79167. doi: 10.1371/journal.pone.0079167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, et al. Role of cystathionine β-synthase in human breast cancer. Free Radic Biol Med. 2015;86:228–38. doi: 10.1016/j.freeradbiomed.2015.05.024. [DOI] [PubMed] [Google Scholar]
- Asimakopoulou A, et al. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE) Br J Pharmacol. 2013;169:922–32. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautio R, et al. Prodrugs: Design and clinical applications. Nat Rev Drug Discov. 2008;7:255–70. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- V. Stella, et al., editors. Prodrugs: Challenges and Rewards. New York: Springer; 2007. p. 1402. [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Woulfe SR, Miller MJ. Synthesis and biological activity of substituted [[3(S)-(acylamino)-2-oxo-1-azetidinyl]oxy]acetic acids. A new class of heteroatom-activated beta-lactam antibiotics. J Med Chem. 1985;28:1447–53. doi: 10.1021/jm00148a013. [DOI] [PubMed] [Google Scholar]
- Sangster J. Octanol-Water Partition Coefficients: Fundamentals and Physical Chemistry. Chichester: John Wiley &; Sons Ltd; 1997. p. 178. Vol. 2 of Wiley Series in Solution Chemistry. [Google Scholar]
- Thorson MK, Majtan T, Kraus JP, Barrios AM. Identification of cystathionine β-synthase inhibitors using a hydrogen sulfide selective probe. Angew Chem Int Ed Engl. 2013;52:4641–44. doi: 10.1002/anie.201300841. [DOI] [PubMed] [Google Scholar]
- Petroianu GA, Hasan MY, Arafat K, Nurulain SM, Schmitt A. Weak inhibitors protect cholinesterases from strong inhibitors (paraoxon): In vitro effect of tiapride. J Appl Toxicol. 2005;25:562–67. doi: 10.1002/jat.1097. [DOI] [PubMed] [Google Scholar]
- Coletta C, et al. Regulation of vascular tone, angiogenesis and cellular bioenergetics by the 3-mercaptopyruvate sulfurtransferase/H2S pathway: Functional impairment by hyperglycemia and restoration by DL-α-lipoic acid. Mol Med. 2015;21:1–14. doi: 10.2119/molmed.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luqman S, Masood N, Srivastava S, Dubey V, Luqman S. A modified spectrophotometric and methodical approach to find novel inhibitors of ornithine decarboxylase enzyme: A path through the maze. Protoc Exch. 2013 Published online; 22nd April. [Google Scholar]
- Gupta RS. Cross-resistance of nocodazole-resistant mutants of CHO cells toward other microtubule inhibitors: Similar mode of action of benzimidazole cabamate derivatives and NSC 181928 and TN-16. Mol Pharmacol. 1986;30:142–48. [PubMed] [Google Scholar]
- Wlodkowic D, Telford W, Skommer J, Darzynkiewicz Z. Apoptosis and beyond: Cytometry in studies of programmed cell death. Methods Cell Biol. 2011;103:55–98. doi: 10.1016/B978-0-12-385493-3.00004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virág L, et al. Peroxynitrite-induced thymocyte apoptosis: The role of caspases and poly (ADP-Ribose) synthetase (PARS) activation. Immunology. 1998;94:345–55. doi: 10.1046/j.1365-2567.1998.00534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oláh G, et al. Differentiation-associated downregulation of poly(ADP-ribose) polymerase-1 expression in myoblasts serves to increase their resistance to oxidative stress. PLoS One. 2014;10:e0134227. doi: 10.1371/journal.pone.0134227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesny B, Brunyanszki A, Olah G, Mitra S, Szabo C. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: Implications for the regulation of mitochondrial function. Nucleic Acids Res. 2014;42:13161–73. doi: 10.1093/nar/gku1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhri VK, et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol Cancer Res. 2013;11:579–92. doi: 10.1158/1541-7786.MCR-12-0437-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima N, Morikawa K, Fabra A, Bucana CD, Fidler IJ. Influence of organ environment on extracellular matrix degradative activity and metastasis of human colon carcinoma cells. J Natl Cancer Inst. 1990;82:1890–98. doi: 10.1093/jnci/82.24.1890. [DOI] [PubMed] [Google Scholar]
- Coletta C, et al. Endothelial dysfunction is a potential contributor to multiple organ failure and mortality in aged mice subjected to septic shock: Preclinical studies in a murine model of cecal ligation and puncture. Crit Care. 2014;18:511. doi: 10.1186/s13054-014-0511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler T, Churchich JE. Reactivity of the phosphopyridoxal groups of cystathionase. J Biol Chem. 1976;251:5267–71. [PubMed] [Google Scholar]
- Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–05. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg JM, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John RA, Charteris A. The reaction of amino-oxyacetate with pyridoxal phosphate-dependent enzymes. Biochem J. 1978;171:771–79. doi: 10.1042/bj1710771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augoff K, Hryniewicz-Jankowska A, Tabola R. Lactate dehydrogenase 5: An old friend and a new hope in the war on cancer. Cancer Lett. 2015;358:1–7. doi: 10.1016/j.canlet.2014.12.035. [DOI] [PubMed] [Google Scholar]
- Hixson LJ, et al. Ornithine decarboxylase and polyamines in colorectal neoplasia and mucosa. Cancer Epidemiol Biomarkers Prev. 1993;2:369–74. [PubMed] [Google Scholar]
- Liang M, Jin S, Wu DD, Wang MJ, Zhu YC. Hydrogen sulfide improves glucose metabolism and prevents hypertrophy in cardiomyocytes. Nitric Oxide. 2015;46:114–22. doi: 10.1016/j.niox.2014.12.007. [DOI] [PubMed] [Google Scholar]
- Szczesny B, et al. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide. 2014;41:120–30. doi: 10.1016/j.niox.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis; Proc Natl Acad Sci U S A; 2009. pp. 21972–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, et al. Hydrogen sulfide, a gaseous transmitter, stimulates proliferation of interstitial cells of Cajal via phosphorylation of AKT protein kinase. Tohoku J Exp Med. 2010;221:125–32. doi: 10.1620/tjem.221.125. [DOI] [PubMed] [Google Scholar]
- Coletta C, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium- dependent vasorelaxation; Proc Natl Acad Sci U S A; 2012. pp. 9161–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wei H, Kong G, Shim W, Zhang G. Hydrogen sulfide augments the proliferation and survival of human induced pluripotent stem cell-derived mesenchymal stromal cells through inhibition of BKCa. Cytotherapy. 2013;15:1395–1405. doi: 10.1016/j.jcyt.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Liu D, et al. Hydrogen sulfide promotes proliferation and neuronal differentiation of neural stem cells and protects hypoxia-induced decrease in hippocampal neurogenesis. Pharmacol Biochem Behav. 2014;116:55–63. doi: 10.1016/j.pbb.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Zhen Y, et al. Exogenous hydrogen sulfide promotes C6 glioma cell growth through activation of the p38 MAPK/ERK1/2-COX-2 pathways. Oncol Rep. 2015;34:2413–22. doi: 10.3892/or.2015.4248. [DOI] [PubMed] [Google Scholar]
- Coletta C, et al. Regulation of vascular tone, angiogenesis and cellular bioenergetics by the 3-mercaptopyruvate sulfurtransferase/H2S pathway: Functional impairment by hyperglycemia and restoration by DL-α-lipoic acid. Mol Med. 2015;21:1–14. doi: 10.2119/molmed.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonke E, et al. Inhibition of endogenous hydrogen sulfide production in clear-cell renal cell carcinoma cell lines and xenografts restricts their growth, survival and angiogenic potential. Nitric Oxide. 2015;49:26–39. doi: 10.1016/j.niox.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, et al. Hydrogen sulfide promotes cell proliferation of oral cancer through activation of the COX2/AKT/ERK1/2 axis. Oncol Rep. 2016;35:2825–32. doi: 10.3892/or.2016.4691. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S, Ng T. C2c: Turning cancer into chronic disease. Genome Med. 2014;6:38. doi: 10.1186/gm555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore ME, Larkin JMG. Challenges and opportunities for converting renal cell carcinoma into a chronic disease with targeted therapies. Br J Cancer. 2011;104:399–406. doi: 10.1038/sj.bjc.6606084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.