

Abstract
TRPM8 plays a key role in COPD. The development of pulmonary hypertension (PH) in COPD adversely affects survival and exercise capacity. Thus, the aim of this study was to evaluate the possible association between single nucleotide polymorphisms (SNPs) in TRPM8 and COPD or PH in COPD among the Chinese Han population. A total of 513 COPD patients and 506 controls were enrolled in the study. Six tag SNPs (tSNPs) were genotyped. The relationship between COPD or PH in COPD and the six tSNPs was evaluated using the χ2 test and genetic model analysis. In the rs9789398 polymorphism, the T/C genotype was associated with an increased risk for COPD (P=0.005). Under the assumption of models of inheritance, there was an association between the rs9789398 polymorphism and COPD. In the rs9789675 polymorphism, the G/A genotype was associated with an increased risk for COPD (P=0.021). Furthermore, by the χ2 test, we found that the minor allele “A” of rs9789675 (odds ratio [OR] =0.63, 95% confidence interval [CI], 0.42–0.97, P=0.034) and the minor allele “C” of rs9789398 (OR =1.59, 95% CI, 1.03–2.44, P=0.034) were associated with a decreased risk of PH in COPD in allele models. In genetic models, the genotypes “GA” and “AA” of rs9789675 were associated with a decreased risk of PH in COPD. The genotypes “TC” and “CC” of rs9789398 were associated with a decreased risk of PH in COPD. Moreover, “CG” of rs1004478 was significantly associated with a decreased risk of PH in COPD. There was a significant association between the five SNPs (rs2362290, rs9789675, rs9789398, rs1003540, and rs104478) in the TRPM8 gene and the risk of PH in COPD. Our findings indicated that rs9789398 in the TRPM8 gene was significantly associated with the risk of COPD in the Chinese Han population. Moreover, rs9789675, rs9789398, and rs1004478 were significantly associated with the risk of PH in COPD. This study provides a novel insight into COPD and PH in the development of COPD.
Keywords: COPD, pulmonary hypertension, case–control study, single nucleotide polymorphism
Introduction
COPD is one of the leading causes of morbidity and mortality worldwide and is characterized by a partially reversible airflow limitation. Usually, when exposed to noxious particles or gases, COPD is progressive and associated with abnormal inflammatory responses of the lungs.1–4 Based on data from the World Health Organization, COPD may become the fourth leading cause of death by 2020,5–8 and in China, ∼9%–10% of adult patients and 8.2% of patients in the age group of 40 years would develop COPD.9,10
Recently, cigarette smoking is recognized as the major risk factor for COPD. However, only 10%–15% of smokers develop clinically significant COPD.11–14 The role of genetic factors in the development of COPD has been investigated by many studies such as family studies and hypothesis-free genome-wide association (GWA) studies,15,16 although the underlying COPD pathogenesis based on the molecular genetics is not well understood. Currently, many studies put forward that COPD is associated with both genetic and environmental factors. Based on the WHO classification, pulmonary hypertension (PH) is associated with lung diseases and/or hypoxemia.17 It has been reported that 5%–40% of COPD patients who underwent right heart catheterization (RHC) might develop an elevation of pulmonary arterial pressure.18 In addition, pulmonary arterial pressure is demonstrated as an important prognostic indicator for COPD.19
Transient receptor potential cation channel subfamily M member 8 (TRPM8) is activated by a temperature range of 8°C–22°C, which could evoke a sensation of coolness.20 Thus, respiratory autonomic responses might be introduced by breathing cold air, including airway constriction, cough, and mucosal secretion, which trigger an asthma attack or indeed exacerbate preexisting disease,21 such as COPD. Therefore, the aim of the current study was to investigate the potential relationship between single nucleotide polymorphisms (SNPs) in the TRPM8 gene and COPD or PH in COPD among the Chinese Han population.
Subjects and methods
Study participants
A total of 513 subjects diagnosed with COPD from the southern part of China were recruited (81 females, 432 males; median age, 68.02 years). COPD was diagnosed based on the criteria of the National Heart, Lung and Blood Institute (NHLBI)/WHO Global initiative for chronic Obstructive Lung Disease (GOLD).22 Patients with post-bronchodilator (BD) forced expiratory volume in 1 second (FEV1, 80% predicted) and FEV1/forced vital capacity (FVC, 0.7) were enrolled in the study. Patients were excluded from the study if they had other significant respiratory diseases, such as lung cancer, pulmonary tuberculosis, cystic fibrosis, and bronchial asthma.
PH was diagnosed by transthoracic Doppler echocardiography (TTE). The entry criteria for PH in COPD cases were tricuspid regurgitant (TR) velocity ≥2.8–2.9 m/sec and pulmonary artery systolic pressure (PASP) ≥40 mmHg. In our study, the patients diagnosed with COPD were divided into two groups according to the TTE: group 1, COPD with PH (24 females, 126 males; median age, 70.03 years), and group 2, COPD without PH (57 females, 363 males; median age, 67.19 years).
In addition, 506 healthy individuals were enrolled (92 females, 414 males; median age, 56.49 years) as a control group. All participants were from Han population of the southern part of China.
Spirometry and calculated method
COPD was defined as a post-BD FEV1/FVC ratio of 0.7 in patients aged ≥40 years, according to the GOLD guideline. Percentages of the predicted values were used to show the spirometry results. In addition, the results were calculated based on Morris’s predictive equations as follows (height in inches):
TTE and the calculated method
PH was defined by RHC showing precapillary PH with a mean pulmonary artery pressure of >25 mmHg and a normal pulmonary artery wedge pressure of <15 mmHg.23,24 RHC was recognized as a standard criteria for the diagnosis of PH, which could provide more accurate pulmonary vascular resistance and pulmonary arterial hypertension. When the pulmonary outflow was obstructed, TTE could provide an estimate of the right ventricular systolic pressure (RVSP), as the value was equivalent to the systolic pulmonary arterial pressure. Based on the velocity of TR jet, the RVSP and pulmonary arterial pressure can be calculated. The velocity of the TR jet can be used to calculate the RVSP using the simplified Bernoulli equation:
where v is the peak velocity of the TR jet in m/sec and RAP is the right atrial pressure that is estimated from inferior vena cava diameter and collapsibility with sniff.25 When pulmonic stenosis or right ventricular outflow obstruction was absent, PASP is equivalent to RVSP. The TR jet velocity of ≥2.8−2.9 m/sec and PASP of ≥40 mmHg were defined as abnormal.26
Demographic and clinical data
A standardized epidemiological questionnaire was designed to collect the demographic and personal data, and the information such as personal details (age, sex, ethnicity, and residential region), diet habit, smoking history, and family history of COPD was included.
Written informed consent was provided by all participants in the study. The study was approved by the ethics committee of the First Affiliated Hospital of Guangzhou Medical University. Approximately 5 mL of peripheral blood was taken from each subject.
Selection and genotyping of tag SNPs (tSNPs)
SNPs were selected from the regulatory region of TRPM8 gene which were from the coding regions within 3,000 bp. SNPs with minor allele frequency (MAF) >5% in the HapMap Chinese Han Beijing population were selected. tSNPs were selected based on the linkage disequilibrium (LD) analysis of 45 samples from controls. The methods for sequencing and SNP annotation were as follows: genomic DNA was extracted from whole blood using the QIAamp DNA Blood Mini Kit (QIAGEN Co. Ltd., Düsseldorf, Germany). DNA concentration was measured using a NanoDrop 2000 (Thermo Scientific, Fitchburg, WI, USA). tSNPs were genotyped with the SNPseq assay, an efficient multiple gene region enrichment/next-generation sequencing-based assay for SNP genotyping by Genesky Biotechnologies, Inc. (Shanghai, China). Briefly, segments of DNA containing tSNPs were amplified using the EasyTarget™ Amplification Kit (Genesky Biotechnologies, Inc.), which was developed using cycled primer extension and ligation-dependent amplification (CPELA). The CPELA method has been described as a fast and simple method for multiple gene region enrichment for massively parallel sequencing.27 Next-generation sequencing of the amplification products was carried out by HiSeq 2000 Sequencer (Illumina, Inc., San Diego, CA, USA) following the manufacturer’s standard sequencing protocols. Output sequence data were trimmed and then compared with fragment reference sequences (hg19) using the Blat program.28 Burrows- Wheeler Aligner (BWA, v0.7.5a) was used to map the reads,29 followed by Sequence Alignment/Map (SAM)-to-BAM conversion, sorting, and removal of duplicates using SAM tools (v0.1.19).30,31 Combined SNP calling was performed on the resulting BAM files using Genome Analysis Toolkit (GATK) and Varscan programs.32 The Annovar program was used for SNP annotation.33
Statistical analysis
Quantitative data were shown as median ± standard deviation. Student’s t-test was used to compare the differences of quantitative data, and χ2 test was applied for qualitative data comparison. Deviation from Hardy–Weinberg equilibrium (HWE) of genotypic distribution of each SNP in controls was analyzed using Fisher’s exact test. When P>0.05, we defined that the population were in genetic balance. The population would not be suitable for enrolling in the study as control if P<0.05. In addition, the Pearson’s χ2 and Fisher’s exact tests were used to calculate the allele frequencies of cases and controls, and MAF in controls was defined as baseline. After adjusting for age, sex, and smoking status, odds ratios (ORs) and 95% confidence interval (95% CI) were calculated using unconditional logistic regression analysis.34 The relationship between the selected SNPs and the risk of COPD was calculated using genotypic model analysis (codominant, dominant, recessive, overdominant, and log additive) by the website software program SNPStats.35 The analyses, including LD, haplotype construction, and genetic association at polymorphism loci, were performed using the Haploview software package (version 4.2) and SHEsis software platform22. In the LD analysis, pairwise distance among SNPs >500 kb was ignored. D′ value was used to evaluate the LD for each pair of SNPs, and variants with the red square indicated that the two related sites were in complete LD (D′=1). D′ value equaling to 0.8 indicated that the related tSNPs formed one block.36,37 Haplotypes were constructed using SNPs in the same LD block, and haplotype frequency of >0.05 was selected. Finally, one SNP in TRPM8 from each LD block was selected for the research.
Statistical analyses were performed using Microsoft Excel (Microsoft Corporation, Redmond, WA, USA) and SPSS 19.0 statistical package (IBM Corporation, Armonk, NY, USA). In the study, all the P-values were two-sided, and P<0.05 was defined as statistically significant.
Results
The characteristics of the enrolled patients are given in Table 1. A total of 531 COPD patients from the southern part of China were enrolled in the study, including 150 individuals with PH, and 506 healthy sex-matched volunteers were enrolled in the control group. No significant differences were found between the COPD and control groups in terms of sex (P=0.3128) or body mass index (BMI; P=0.575). However, COPD patients were significantly older than healthy controls (68.02±11.21 years vs 56.49±9.43 years, P<0.001). Patients with COPD had longer average smoking exposure (43.08 vs 39.45 pack-years, P<0.001) and significantly worse lung functions as compared with control subjects in terms of the predicted FEV1% (36.77% vs 95.77%, P<0.001) and FEV1/FVC ratio (0.43 vs 0.81, P<0.001).
Table 1.
Characteristics and grouping of the study population
| Characteristics | Cases | Controls | P-value | COPD with PH | COPD without PH | P-value |
|---|---|---|---|---|---|---|
| n | 513 | 506 | 150 | 363 | ||
| Age, years (mean±SD) | 68.02±11.21 | 56.49±9.43 | <0.001 | 70.03±12.37 | 67.19±10.60 | 0.190 |
| Male, n% | 432 (84.21) | 414 (81.82) | 0.318 | 126 (84.00) | 306 (84.30) | 1.000 |
| Pack-years | 43.08±21.71 | 39.45±19.58 | <0.001 | 41.93±21.65 | 40.47±23.51 | 0.467 |
| BMI, kg/m2 | 23.75±2.57 | 24.18±2.48 | 0.575 | 24.16±2.27 | 23.57±2.69 | 0.051 |
| FEV1 %, predicted | 36.77±16.94 | 95.77±12.98 | <0.001 | 35.67±16.10 | 37.23±18.64 | 0.138 |
| FEV1/FVC | 0.43±0.19 | 0.81±0.96 | <0.001 | 0.43±0.15 | 0.45±0.26 | 0.004 |
| GOLD classification | Level 3 | Level 3 | Level 3 | |||
| PASP, mmHg | 49.64±14.25 | 25.37±5.73 | <0.001 |
Note: P<0.05 indicates statistical significance.
Abbreviations: BMI, body mass index; GOLD, Global initiative for chronic Obstructive Lung Disease; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; PASP, pulmonary artery systolic pressure; PH, pulmonary hypertension.
Patients with COPD were divided into two groups based on the PASP value: COPD with PH and COPD without PH. For the group with PH in COPD, there were no significant differences in age, sex, BMI, and smoking status (P=0.190, 1.000, 0.051, and 0.467, respectively). Moreover, the two groups exhibited significantly different PASP values (49.64 vs 25.37, P<0.001) and FEV1/FVC (P=0.004).
Association of TRPM8 SNPs with risk of COPD
Table 2 summarizes the basic characteristics of the SNPs in the study population and shows that all the five SNPs from controls were in HWE (P>0.05). The differences in frequency distributions of alleles between cases and controls compared by Pearson’s χ2 test showed that the minor allele “C” of rs9789398 from TRPM8 was significantly associated with the risk of COPD in the study population (P=0.040, OR =1.33, 95% CI =1.01–1.74).
Table 2.
Basic characteristics of SNPs in the enrolled population
| SNPs | Chromosomes | Position | Gene | Allele | Risk of COPD
|
Risk of COPD with PH
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HWE-P | OR | 95% CI | Allele, P-value | HWE-P | OR | 95% CI | Allele, P-value | |||||
| rs2362290 | 2q37.1 | 233916724 | TRPM8 | AG | 0.061 | 1.03 | 0.85–1.24 | 0.781 | 0.291 | 0.8 | 0.60–1.07 | 0.130 |
| rs9789675 | 2q37.1 | 233916914 | TRPM8 | AG | 0.494 | 1.17 | 0.91–1.51 | 0.241 | 0.453 | 0.63 | 0.42–0.97 | 0.034* |
| rs9789398 | 2q37.1 | 233916979 | TRPM8 | CT | 0.922 | 1.33 | 1.01–1.74 | 0.040* | 0.609 | 1.59 | 1.03–2.44 | 0.034* |
| rs1003540 | 2q37.1 | 233917239 | TRPM8 | AG | 0.065 | 0.99 | 0.82–1.19 | 0.912 | 0.394 | 1.28 | 0.96–1.70 | 0.394 |
| rs1004478 | 2q37.1 | 233917250 | TRPM8 | CG | 0.06 | 0.96 | 0.80–1.16 | 0.683 | 0.182 | 1.21 | 0.91–1.62 | 0.194 |
Note:
P<0.05 indicates statistical significance.
Abbreviations: CI, confidence interval; HWE-P, P-value of Hardy–Weinberg equilibrium; OR, odds ratio; PH, pulmonary hypertension; SNP, single nucleotide polymorphism; TRPM8, transient receptor potential cation channel subfamily M member 8.
Comparing the possession of minor allele, it has been hypothesized that harboring the minor allele of each SNP was a risk factor. Table 3 shows the relationship between SNPs and the risk of COPD under various genetic models. Our analyses showed that crude analysis of rs9789398 was associated with an increased risk of COPD under the dominant model (P=0.024, OR =1.41, 95% CI =1.05–1.90), the overdominant model (P=0.021, OR =1.43, 95% CI =1.05–1.94), and log-additive model (P=0.037, OR =1.34, 95% CI =1.02–1.77). However, after adjusting for age, sex, and smoking status, no significant relationship was observed between rs9789398 and the risk of COPD under any different genotypic models.
Table 3.
Association between rs9789398 genotypes and the risk of COPD
| Model | Genotype | Control | Case | Without adjustment
|
With adjustment
|
||
|---|---|---|---|---|---|---|---|
| OR (95% CI) | P-valuea | OR (95% CI) | P-valueb | ||||
| Codominant | TT | 377 (79.2%) | 351 (73%) | 1.00 | 0.069 | 1.00 | 0.2 |
| TC | 93 (19.5%) | 124 (25.8%) | 1.43 (1.05–1.94) | 1.37 (0.96–1.95) | |||
| CC | 6 (1.3%) | 6 (1.2%) | 1.07 (0.34–3.36) | 1.29 (0.37–4.56) | |||
| Dominant | TT | 377 (79.2%) | 351 (73%) | 1.00 | 0.024* | 1.00 | 0.074 |
| TC–CC | 99 (20.8%) | 130 (27%) | 1.41 (1.05–1.90) | 1.37 (0.97–1.93) | |||
| Recessive | TT–TC | 470 (98.7%) | 475 (98.8%) | 1.00 | 0.99 | 1.00 | 0.77 |
| CC | 6 (1.3%) | 6 (1.2%) | 0.99 (0.32–3.09) | 1.20 (0.34–4.25) | |||
| Overdominant | TT–CC | 383 (80.5%) | 357 (74.2%) | 1.00 | 0.021* | 1.00 | 0.082 |
| TC | 93 (19.5%) | 124 (25.8%) | 1.43 (1.05–1.94) | 1.37 (0.96–1.94) | |||
| Log additive | 1.34 (1.02–1.77) | 0.037* | 1.32 (0.96–1.80) | 0.088 | |||
Notes:
P<0.05 indicates statistical significance.
P-values were calculated by two-sided χ2 tests or Fisher’s exact tests for each genotype distribution.
P-values were calculated by unconditional logistic regression adjusted for age, sex, and smoking status.
Abbreviations: CI, confidence interval; OR, odds ratio.
Association between COPD and smoking status was well documented. A smoking status stratification analysis was performed to eliminate potential confounding effects caused by differences in smoking history. As shown in Table 4, there was no significant association between SNPs and the risk of COPD in nonsmokers (n=247) or former smokers (n=454). However, SNP analysis among samples from current smokers (n=318) showed that the rs9789675 and rs9789398 polymorphisms were significantly associated with the risk of COPD after adjusting for age, sex, and pack-years. In the rs9789675 polymorphism, the GA genotype was associated with an increased risk of COPD (P=0.021). Under the assumption of models of inheritance, the rs9789675 polymorphism was significantly related to the risk of COPD (the dominant model: GA + AA vs GG: OR =2.00, 95% CI =1.08–3.72, P=0.026; the overdominant model: GA vs GG–AA: OR =1.94, 95% CI =1.02–3.67, P=0.04). Moreover, it was shown that the rs9789398 polymorphism was associated with the risk of COPD (the codominant model: TT vs TC vs CC: OR =2.48, 95% CI =1.25–4.93, P=0.022; the dominant model: TC–CC vs TT: OR =2.48, 95% CI =1.28–4.77, P=0.0057; the overdominant model: TC vs TT–CC: OR =2.43, 95% CI =1.23–4.80, P=0.0092). The TC genotype was associated with an increased risk of COPD (P=0.005).
Table 4.
Association between TRPM8 and COPD by smoking status: stratified analysis
| Reference SNP ID | Genotypes | Nonsmokers (n =247)
|
Former smokers (n =454)
|
Current smokers (n =318)
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| COPD; n=126 (%) |
Control; n=121 (%) |
P-valuea | COPD; n=214 (%) |
Control; n=240 (%) |
P-valueb | COPD; n=173 (%) |
Control; n=145 (%) |
P-valueb | ||
| rs2362290 | GG | 11 (8.9) | 9 (7.9) | 0.713 | 16 (7.6) | 24 (10.1) | 0.399 | 14 (8.1) | 9 (6) | 0.589 |
| GA | 61 (49.6) | 55 (48.2) | 0.759 | 106 (50.2) | 114 (48.1) | 0.865 | 69 (39.9) | 67 (44.7) | 0.474 | |
| AA | 51 (41.5) | 50 (43.9) | 89 (42.2) | 99 (41.8) | 90 (52) | 74 (49.3) | ||||
| rs9789675 | AA | 2 (1.6) | 1 (0.9) | 0.610 | 2 (1) | 3 (1.3) | 0.702 | 4 (2.4) | 2 (1.4) | 0.412 |
| GA | 40 (32.5) | 29 (27.4) | 0.376 | 42 (20.3) | 57 (24.6) | 0.276 | 45 (26.8) | 24 (16.2) | 0.021* | |
| GG | 81 (65.8) | 76 (71.4) | 163 (78.7) | 172 (74.1) | 119 (70.8) | 122 (82.4) | ||||
| rs9789398 | CC | 1 (0.8) | 1 (1) | 0.975 | 1 (0.5) | 3 (1.3) | 0.747 | 4 (2.5) | 2 (1.4) | 0.375 |
| TC | 40 (32.8) | 25 (23.8) | 0.138 | 41 (20.7) | 48 (21.3) | 0.840 | 43 (26.7) | 20 (13.7) | 0.005* | |
| TT | 81 (66.4) | 79 (75.2) | 156 (78.8) | 174 (77.3) | 114 (70.8) | 124 (84.9) | ||||
| rs1003540 | AA | 11 (9) | 8 (7.3) | 0.479 | 16 (7.6) | 23 (9.9) | 0.455 | 14 (8.2) | 9 (6) | 0.627 |
| AG | 62 (50.8) | 51 (46.4) | 0.393 | 105 (50) | 111 (47.8) | 0.838 | 65 (38.2) | 68 (45.3) | 0.257 | |
| GG | 49 (40.2) | 51 (46.4) | 89 (42.2) | 98 (42.2) | 91 (53.5) | 73 (48.7) | ||||
| rs1004478 | GG | 8 (6.6) | 13 (11.4) | 0.319 | 23 (10.9) | 17 (7.1) | 0.151 | 28 (16.5) | 18 (12) | 0.388 |
| CG | 60 (49.2) | 60 (52.6) | 0.124 | 97 (46.2) | 112 (46.9) | 0.775 | 73 (42.9) | 72 (48) | 0.603 | |
| CC | 54 (44.3) | 41 (36) | 90 (42.9) | 110 (46) | 69 (40.6) | 60 (40) | ||||
Notes:
P-values were calculated by logistic regression adjusted for age and sex in nonsmokers.
P-values were calculated by logistic regression adjusted for age, sex, and pack-years in former and current smokers.
P<0.05.
Abbreviations: SNP, single nucleotide polymorphism; TRPM8, transient receptor potential cation channel subfamily M member 8.
Based on haplotype analyses, rs2362290, rs9789675, rs9789398, rs1003540, and rs1004478 were in complete LD. However, no significant association between the selected haplotype and the risk of COPD was found.
Association of the TRPM8 SNPs with the risk of PH in COPD
Table 2 also shows the MAF of all SNPs genotyped in the COPD with PH group. HWE test revealed that the genotypic distribution of all SNPs conformed to HWE in the control group (P>0.05). Using the χ2 test, two SNPs (rs9789675 and rs9789398) were significantly associated with the risk of PH among COPD patients (OR =0.63, 95% CI, 0.42–0.97, P=0.034 and OR =1.59, 95% CI, 1.03–2.44, P=0.034, respectively).
As shown in Table 5, crude analysis revealed that the genotypes “GA” and “AA” of rs9789675 were associated with a decreased risk of PH in COPD under the dominant model (P=0.036, OR =0.62, 95% CI =0.39–0.98), the dominant model (P=0.013, OR =0.40, 95% CI =0.19–0.88), and the log-additive model (P=0.027, OR =0.62, 95% CI =0.23–0.97). “TC” and “CC” of rs9789398 were associated with a decreased risk of PH in COPD under the dominant model (P=0.027, OR =0.60, 95% CI =0.37–0.95), the overdominant model (P=0.04, OR =0.47, 95% CI =0.05–4.05), and log-additive model (P=0.026, OR =0.61, 95% CI =0.39–0.95). In addition, “CG” of rs1004478 was also significantly associated with a decreased risk of PH in COPD under the codominant model (P=0.048, OR =0.60, 95% CI =0.40–0.91), the dominant model (P=0.036, OR =0.66, 95% CI =0.45–0.97), and overdominant model (P=0.015, OR =0.61, 95% CI =0.41–0.91). After adjusting for sex, age, and smoking status, these three SNPs also showed a significant association with the risk of PH in COPD.
Table 5.
Association between genotypes (rs9789675, rs9789398, and rs1004478) and the risk of PH in COPD
| SNP | Model | Genotype | Control | Case | Without adjustment
|
With adjustment
|
||
|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P-valuea | OR (95% CI) | P-valueb | |||||
| rs9789675 | Codominant | GG | 248 (70.2%) | 115 (79.3%) | 1.00 | 0.084 | 1.00 | 0.110 |
| GA | 98 (27.8%) | 29 (20%) | 0.64 (0.40–1.02) | 0.64 (0.40–1.04) | ||||
| AA | 7 (2%) | 1 (0.7%) | 0.31 (0.04–2.53) | 0.35 (0.04–2.90) | ||||
| Dominant | GG | 248 (70.2%) | 115 (79.3%) | 1.00 | 0.036* | 1.00 | 0.045 | |
| GA–AA | 105 (29.8%) | 30 (20.7%) | 0.62 (0.39–0.98) | 0.6 (0.39–1.00) | ||||
| Recessive | GG–GA | 346 (98%) | 144 (99.3%) | 1.00 | 0.260 | 1.00 | 0.330 | |
| AA | 7 (2%) | 1 (0.7%) | 0.34 (0.04–2.82) | 0.39 (0.05–3.21) | ||||
| Overdominant | GG–AA | 255 (72.2%) | 116 (80%) | 1.00 | 0.066 | 1.00 | 0.076 | |
| GA | 98 (27.8%) | 29 (20%) | 0.65 (0.41–1.04) | 0.66 (0.41–1.05) | ||||
| Log additive | – | – | – | 0.62 (0.40–0.96) | 0.027* | 0.64 (0.41–0.99) | 0.037 | |
| rs9789398 | Codominant | TT | 237 (70.1%) | 114 (79.7%) | 1.00 | 0.082 | 1.00 | 0.100 |
| TC | 96 (28.4%) | 28 (19.6%) | 0.61 (0.38–0.98) | 0.61 (0.38–0.99) | ||||
| CC | 5 (1.5%) | 1 (0.7%) | 0.42 (0.05–3.60) | 0.47 (0.05–4.17) | ||||
| Dominant | TT | 237 (70.1%) | 114 (79.7%) | 1.00 | 0.027* | 1.00 | 0.034* | |
| TC–CC | 101 (29.9%) | 1 (1.7%) | 0.60 (0.37–0.95) | 0.60 (0.38–0.97) | ||||
| Recessive | TT–TC | 333 (98.5%) | 115 (80.4%) | 1.00 | 0.46 | 1.00 | 0.540 | |
| CC | 5 (1.5%) | 28 (19.6%) | 0.47 (0.05–4.05) | 0.53 (0.06–4.65) | ||||
| Overdominant | TT–CC | 242 (71.6%) | 1.00 | 0.04* | 1.00 | 0.045* | ||
| TC | 96 (28.4%) | 0.61 (0.38–0.99) | 0.62 (0.38–1.00) | |||||
| Log additive | – | – | – | 0.61 (0.39–0.95) | 0.026* | 0.62 (0.40–0.98) | 0.034* | |
| rs1004478 | Codominant | CC | 140 (39.4%) | 73 (49.7%) | 1.00 | 0.048* | 1.00 | 0.055 |
| CG | 175 (49.3%) | 55 (37.4%) | 0.60 (0.40–0.91) | 0.60 (0.40–0.92) | ||||
| GG | 40 (11.3%) | 19 (12.9%) | 0.91 (0.49–1.69) | 0.88 (0.47–1.64) | ||||
| Dominant | CC | 140 (39.4%) | 73 (49.7%) | 1.00 | 0.036* | 1.00 | 0.036* | |
| CG–GG | 215 (60.6%) | 74 (50.3%) | 0.66 (0.45–0.97) | 0.66 (0.44–0.92) | ||||
| Recessive | CC–CG | 312 (88.7%) | 128 (87.1%) | 1.00 | 0.600 | 1.00 | 0.690 | |
| GG | 40 (11.3%) | 19 (12.9%) | 1.17 (0.65–2.10) | 1.13 (0.62–2.03) | ||||
| Overdominant | CC–GG | 180 (50.7%) | 92 (62.6%) | 1.00 | 0.015* | 1.00 | 0.018* | |
| CG | 175 (49.3%) | 55 (37.4%) | 0.61 (0.41–0.91) | 0.62 (0.42–0.93) | ||||
| Log additive | – | – | – | 0.82 (0.61–1.10) | 0.190 | 0.82 (0.61–1.10) | 0.170 | |
Notes:
P<0.05 indicates statistical significance.
P-values were calculated by two-sided χ2 tests or Fisher’s exact tests for each genotype distribution.
P-values were calculated by unconditional logistic regression adjusted for age, sex, and smoking status.
Abbreviations: CI, confidence interval; OR, odds ratio; PH, pulmonary hypertension; SNP, single nucleotide polymorphism.
As shown in Figure 1, one haplotype block including five SNPs (rs2362290, rs9789675, rs9789398, rs1003540, and rs1004478) with D′=1 was detected in TRPM8 SNPs by haplotype analyses. Haplotypes with frequencies equaling to 1% were selected for the analysis. The five SNPs constructed four haplotypes (“AGTGG”, “GGTAC”, “AGTGC”, and “AACGC”). As shown in Table 6, there was a trend toward an increased risk of COPD and the haplotype “AGTGC” in the TRPM8 gene after adjusting for sex, age, and smoking status (OR =1.44, 95% CI =0.98–2.12, P=0.043).
Figure 1.
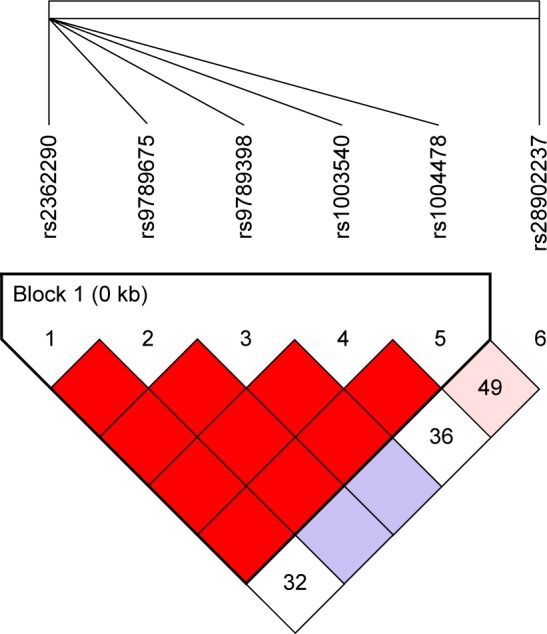
LD plots containing four SNPs from the TRPM8.
Notes: LD analysis of TRPM8 in the Chinese Han controls. LD is indicated using standard color schemes with red signifying very strong LD (LOD ≥2, D′=1) and pink signifying relatively weak LD (LOD ≥2, D′=1).
Abbreviations: LD, linkage disequilibrium; LOD, likelihood of odds; SNP, single nucleotide polymorphism; TRPM8, transient receptor potential cation channel subfamily M member 8.
Table 6.
Haplotype association with response (n=508, adjusted for smoking status + sex + age)
| rs2362290 | rs9789675 | rs9789398 | rs1003540 | rs1004478 | Frequency | OR (95% CI) | P-value | |
|---|---|---|---|---|---|---|---|---|
| 1 | A | G | T | G | G | 0.3463 | 1.00 | – |
| 2 | G | G | T | A | C | 0.3057 | 1.30 (0.92–1.84) | 0.14 |
| 3 | A | G | T | G | C | 0.201 | 1.44 (0.98–2.12) | 0.043* |
| 4 | A | A | C | G | C | 0.1298 | 0.71 (0.43–1.16) | 0.17 |
| Rare | * | * | * | * | * | 0.0173 | 1.40 (0.46–4.25) | 0.56 |
Note: Global haplotype association: P-value, 0.051.
P<0.05.
Abbreviations: CI, confidence interval; OR, odds ratio.
Discussion
Based on the data from 513 COPD patients and 506 controls from the Chinese Han population, the study demonstrated that rs9789398 in the TRPM8 gene was significantly associated with the risk of COPD in the Chinese Han population. Moreover, rs9789675, rs9789398, and rs1004478 in TRPM8 were significantly associated with the risk of PH in COPD.
Previous data demonstrated that TRP superfamily of existence of cation channels in a broad range of cell types played key roles in respiratory diseases such as COPD and asthma.38 Among this subfamily, TRPM2, TRPM4, and TRPM8 are emerging as possible targets in the treatment of respiratory diseases.39 In 2006, TRPM8 antagonists was used as potential therapeutic agents for the treatment of asthma, COPD, and allergic rhinitis.40 Subsequently, TRPM8 antagonists have been designed to treat several conditions such as COPD and PH. The possible treatment of respiratory disorders with TRPM8-specific ligands is currently being examined at an early exploratory stage. A Russian ethnic group showed that subjects with heterozygous genotype GC in SNP rs11562975 were characterized by increasing sensation of cold, hypometabolic response to local skin cooling, and non-temperature activation of TRPM8 ion channel by menthol, ie, decrease in total metabolism, pulmonary ventilation, and coefficient of oxygen extraction.40 Although no SNPs located in TRPM8 were reported, abundant genetic variation contributing to COPD susceptibility has been put forward. Using quantitative meta-analysis, Castaldi et al16 showed that four variants in GSTM1, TGFB1, TNF, and SOD3 would influence the development of COPD. The associations of HHIP on 4q31, FAM13A on 4q22, BICD1 on 12p11., and CHRNA3/5 on 15q25 with the risk of COPD were reported in the GWA study.15
COPD is widely recognized as a complex disease with genetic–environmental interactions, which has become one of the primary causes of morbidity.41–43 Traditionally, it is considered that cigarette smoking is an underlying risk factor for the development of COPD. However, data showed that only 10%–15% of chronic, heavy cigarette smokers would develop symptomatic airflow obstruction. In the current study, there was no significant association between studied SNPs and the risk of COPD among nonsmokers or former smokers. Notably, the analysis of the SNPs of the rs9789675 and the rs9789398 polymorphisms demonstrated that the two SNPs were significantly associated with the risk of COPD among current smokers, which might contribute to elucidate the pathogenesis of COPD and the high incidence of COPD among smokers.
Mild-to-moderate PH is a common complication of COPD. Previous evidence demonstrated that pulmonary vascular remodeling in COPD was the main reason for increased pulmonary artery pressure. Moreover, the remodeling was recognized as the result from the combined effects of hypoxia, inflammation, and loss of capillaries in severe emphysema.44 Notably, previous evidence demonstrated that TRPM8 might mediate secretion of bronchial epithelial cells introduced by cold temperature, including IL-1α, IL-1β, IL-4, IL-6, etc.45,46 Our data showed that three SNPs, including rs9789675, rs9789398, and rs1004478 located in TRPM8, were significantly associated with the risk of PH in COPD. Although no experimental evidence supports the association, further study will be taken into consideration focusing on the role of the SNPs in the risk of PH.
There are some limitations to be noted. First, genetic associations with COPD or PH in COPD were not able to elucidate causal mechanisms, and a larger sample size and a greater number of loci would be needed for additional evidence regarding the role of these genes in COPD and PH in COPD. Second, the effects of secondhand smoke or other smoke in the “non-smoking” individuals were not taken into account. Hence, our future research would incorporate the analysis of the relationship between COPD and the detailed smoking status, including smoking intensity, secondhand smoking, and air smog. Moreover, in the current study, the investigation was not performed in heterozygous patients due to the limited sample size. Further study enrolling larger sample size is needed to verify the strength of the study.
To the best of our knowledge, we are the first to identify one candidate locus (rs9789398) associated with an increased risk of COPD in the Chinese Han population. In addition, we are the first to report that three candidate loci rs9789398, rs9789675, and rs1004478 were associated with the risk of PH development in COPD. These findings might provide a novel insight for exploring the pathogenesis of COPD and new treatment strategies. In a further study, the functional changes introduced by mutated TRPM8 need to be explored, including the underlying mechanisms.
Acknowledgments
This study was funded by the Natural Science Foundation of Guangdong Province (no 1035101200300000). We are grateful to the COPD patients and control subjects for their participation in this study. We also thank the clinicians and hospital staff who contributed to the sample and data collection.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. doi: 10.1146/annurev.pathol.4.110807.092145. [DOI] [PubMed] [Google Scholar]
- 2.Lopez AD, Shibuya K, Rao C, et al. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J. 2006;27(2):397–412. doi: 10.1183/09031936.06.00025805. [DOI] [PubMed] [Google Scholar]
- 3.Trajkov D, Mirkovska-Stojkovikj J, Petlichkovski A, et al. Association of cytokine gene polymorphisms with chronic obstructive pulmonary disease in Macedonians. Iran J Allergy Asthma Immunol. 2009;8(1):31–42. [PubMed] [Google Scholar]
- 4.Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD) Lancet. 2004;364(9434):613–620. doi: 10.1016/S0140-6736(04)16855-4. [DOI] [PubMed] [Google Scholar]
- 5.Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS, GOLD Scientific Committee Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO global initiative for chronic obstructive lung disease (GOLD) workshop summary. Am J Respir Crit Care Med. 2001;163(5):1256–1276. doi: 10.1164/ajrccm.163.5.2101039. [DOI] [PubMed] [Google Scholar]
- 6.Mannino DM. COPD: epidemiology, prevalence, morbidity and mortality, and disease heterogeneity. Chest. 2002;121(5 Suppl):121S–126S. doi: 10.1378/chest.121.5_suppl.121s. [DOI] [PubMed] [Google Scholar]
- 7.Løkke A, Lange P, Vestbo J, Fabricius PG. Developing COPD – 25 years follow-up study of the general population. Ugeskr Laeger. 2006;168(50):4422–4424. [PubMed] [Google Scholar]
- 8.Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. doi: 10.1164/rccm.201204-0596PP. [DOI] [PubMed] [Google Scholar]
- 9.Halbert RJ, Natoli JL, Gano A, Badamgarav E, Buist AS, Mannino DM. Global burden of COPD: systematic review and meta-analysis. Eur Respir J. 2006;28(3):523–532. doi: 10.1183/09031936.06.00124605. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Li L, Xiao J, et al. Association of ADAM33 gene polymorphisms with COPD in a northeastern Chinese population. BMC Med Genet. 2009;10:132. doi: 10.1186/1471-2350-10-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rennard SI. Treatment of stable chronic obstructive pulmonary disease. Lancet. 2004;364(9436):791–802. doi: 10.1016/S0140-6736(04)16941-9. [DOI] [PubMed] [Google Scholar]
- 12.Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1(6077):1645–1648. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sethi JM, Rochester CL. Smoking and chronic obstructive pulmonary disease. Clin Chest Med. 2000;21(1):67–86. viii. doi: 10.1016/s0272-5231(05)70008-3. [DOI] [PubMed] [Google Scholar]
- 14.Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic obstructive pulmonary disease surveillance – United States, 1971–2000. Respir Care. 2002;47(10):1184–1199. [PubMed] [Google Scholar]
- 15.Nakamura H. Genetics of COPD. Allergol Int. 2011;60(3):253–258. doi: 10.2332/allergolint.11-RAI-0326. [DOI] [PubMed] [Google Scholar]
- 16.Castaldi PJ, Cho MH, Cohn M, et al. The COPD genetic association compendium: a comprehensive online database of COPD genetic associations. Hum Mol Genet. 2010;19(3):526–534. doi: 10.1093/hmg/ddp519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54(1 suppl):S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189–194. doi: 10.1164/rccm.200401-006OC. [DOI] [PubMed] [Google Scholar]
- 19.Kessler R, Faller M, Fourgaut G, Mennecier B, Weitzenblum E. Predictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;159(1):158–164. doi: 10.1164/ajrccm.159.1.9803117. [DOI] [PubMed] [Google Scholar]
- 20.Peier AM, Moqrich A, Hergarden AC, et al. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108(5):705–715. doi: 10.1016/s0092-8674(02)00652-9. [DOI] [PubMed] [Google Scholar]
- 21.Yoshihara S, Geppetti P, Hara M, et al. Cold air-induced bronchoconstriction is mediated by tachykinin and kinin release in guinea pigs. Eur J Pharmacol. 1996;296(3):291–296. doi: 10.1016/0014-2999(95)00719-9. [DOI] [PubMed] [Google Scholar]
- 22.Rabe KF, Hurd S, Anzueto A, et al. Global Initiative for Chronic Obstructive Lung Disease Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176(6):532–555. doi: 10.1164/rccm.200703-456SO. [DOI] [PubMed] [Google Scholar]
- 23.Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336(2):111–117. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- 24.Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. Turk Kardiyol Dern Ars. 2014;42(Suppl 1):45–54. [PubMed] [Google Scholar]
- 25.Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23(7):685–713. doi: 10.1016/j.echo.2010.05.010. quiz 786–788. [DOI] [PubMed] [Google Scholar]
- 26.Karas MG, Kizer JR. Echocardiographic assessment of the right ventricle and associated hemodynamics. Prog Cardiovasc Dis. 2012;55(2):144–160. doi: 10.1016/j.pcad.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Li C, Wu F, et al. Genomic variations of the mevalonate pathway in porokeratosis. Elife. 2015;4:e06322. doi: 10.7554/eLife.06322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kent WJ. BLAT – the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Handsaker B, Wysoker A, et al. 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bland JM, Altman DG. Statistics notes. The odds ratio. BMJ. 2000;320(7247):1468. doi: 10.1136/bmj.320.7247.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solé X, Guinó E, Valls J, Iniesta R, Moreno V. SNPStats: a web tool for the analysis of association studies. Bioinformatics. 2006;22(15):1928–1929. doi: 10.1093/bioinformatics/btl268. [DOI] [PubMed] [Google Scholar]
- 36.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 37.Teramoto S, Ishii T, Yamamoto H, Yamaguchi Y, Matsuse T. Xenobiotic enzymes and genetics of COPD. Chest. 2005;127(1):408–409. doi: 10.1378/chest.127.1.408. Author reply 409. [DOI] [PubMed] [Google Scholar]
- 38.Banner KH, Igney F, Poll C. TRP channels: emerging targets for respiratory disease. Pharmacol Ther. 2011;130(3):371–384. doi: 10.1016/j.pharmthera.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Gavva NR, Davis C, Lehto SG, Rao S, Wang W, Zhu DX. Transient receptor potential melastatin 8 (TRPM8) channels are involved in body temperature regulation. Mol Pain. 2012;8:36. doi: 10.1186/1744-8069-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozyreva TV, Tkachenko EIA, Potapova TA, Voevoda MI. Respiratory system response to cooling in subjects with single nucleotide polymorphism rs11562975 in gene of thermosensitive TRPM8 ion channel. Fiziol Cheloveka. 2014;40(2):94–98. [PubMed] [Google Scholar]
- 41.Burchfiel CM, Marcus EB, Curb JD, et al. Effects of smoking and smoking cessation on longitudinal decline in pulmonary function. Am J Respir Crit Care Med. 1995;151(6):1778–1785. doi: 10.1164/ajrccm.151.6.7767520. [DOI] [PubMed] [Google Scholar]
- 42.Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135(1):173–180. doi: 10.1378/chest.08-1419. [DOI] [PubMed] [Google Scholar]
- 43.Antó JM, Vermeire P, Vestbo J, Sunyer J. Epidemiology of chronic obstructive pulmonary disease. Eur Respir J. 2001;17(5):982–994. doi: 10.1183/09031936.01.17509820. [DOI] [PubMed] [Google Scholar]
- 44.Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J. 2008;32(5):1371–1385. doi: 10.1183/09031936.00015608. [DOI] [PubMed] [Google Scholar]
- 45.Sabnis AS, Reilly CA, Veranth JM, Yost GS. Increased transcription of cytokine genes in human lung epithelial cells through activation of a TRPM8 variant by cold temperatures. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L194–L200. doi: 10.1152/ajplung.00072.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sabnis AS, Shadid M, Yost GS, Reilly CA. Human lung epithelial cells express a functional cold-sensing TRPM8 variant. Am J Respir Cell Mol Biol. 2008;39(4):466–474. doi: 10.1165/rcmb.2007-0440OC. [DOI] [PMC free article] [PubMed] [Google Scholar]