

Abstract
Introduction:
Well-differentiated systemic mastocytosis (WDSM) is a rare, recently recognized provisional subvariant of systemic mastocytosis (SM). We report a case of WDSM that showed excellent clinical and cutaneous response to imatinib in the absence of known molecular genetic abnormalities.
Clinical Findings/Diagnoses:
We present a 24-year-old woman with childhood onset of skin manifestations that progressed to mediator-related systemic events, and a gastrointestinal tract mastocytoma. A subsequent bone marrow examination showed WDSM. Treatment with imatinib resulted in complete resolution of cutaneous lesions and systemic symptoms, which relapsed with the discontinuation of the drug. Targeted next-generation sequencing-based mutation analysis did not demonstrate any mutations in the coding regions of KIT or other genes commonly associated with myeloid neoplasms.
Conclusions:
The diagnosis of WDSM is challenging in the absence of spindle-shaped mast cells, CD2 or CD25 expression, and KIT D816 mutation. This case illustrated the need for recognizing this unique variant of SM for diagnostic and therapeutic implications.
Keywords: imatinib, KIT D816, systemic mastocytosis
1. Introduction
Well-differentiated systemic mastocytosis (WDSM) is a rare, recently described provisional subvariant of systemic mastocytosis (SM),[1–4] with distinct morphologic, immunophenotypic, and molecular features.[4,5] WDSM accounts for approximately 5% to 7% of SM, shows a female predominance, often childhood onset and tendency for familial predisposition. Based on the proposed Spanish Network on Mastocytosis (REMA) algorithm for the diagnosis and classification of mastocytosis, diagnosis of WDSM could be made according to the following criteria: skin involvement, compact bone marrow (BM) mast cell aggregates (>15 mast cells), mast cells with round-shape and larger CD25 negative mast cells, elevated serum tryptase level (>20 ng/mL), despite lacking KIT D816V mutation.[5]
However, patients with WDSM usually present with a low mast cell burden that progresses over time, and have a prolonged disease course.[4] Due to the low disease burden, especially early in the disease course, some cases with low serum tryptase levels (minor criteria) may not meet the 2008 World Health Organization (WHO) diagnostic criteria for SM, since the other 3 minor criteria (spindle-morphology, aberrant CD2/CD25 expression, KIT D816 mutation) are missing in this subvariant SM.[6] However, recognition of this entity is of significance, not only for diagnosis but also for treatment. WDSM has been reported to show excellent responses to imatinib therapy.[2,3,7–9] Of note, the responders often showed KIT mutations not involving the D816V loop. Here, we report a case of WDSM and highlight the clinical, pathologic, and immunophenotypic findings of this entity, and demonstrate excellent clinical and cutaneous responses to imatinib even in the absence of KIT mutation by next-generation sequencing (NGS)-based testing.
2. Case presentation
A 24-year-old woman presented with a 7-year history of intermittent nausea, vomiting, abdominal pain, joint pains, and progressive weight loss. The patient was diagnosed with a gastric mastocytoma (5 cm) on endoscopic biopsy 7 months before this presentation and had a partial gastrectomy afterward. The diagnosis of mastocytoma was based on the presence of sheets of mature, nonatypical mast cells involving the submucosa; no increased number of mitoses was seen. The patient had a long-standing history of maculopapular skin rash over the chest and back since the age of 4. On admission, physical examination revealed diffuse faint maculopapular rash, mainly on upper chest and back. A complete blood count was within normal limits. Serum tryptase level was elevated at 47.1 (normal <11.5 ng/mL). A BM biopsy showed loose aggregates and interstitial infiltrate of round mast cells with abundant cytoplasmic granules, representing about 20% of the marrow cellularity. The mast cells accounted for about 5% of the cells in the aspirate smears. Flow cytometry immunophenotypic (FCI) analysis on the BM showed an aberrant mast cell population representing 0.2% of total analyzed cells, positive for CD117, and CD63 (partial); but negative for CD2, CD25, and CD69. Mutation-specific quantitative real-time PCR for D816V mutation of the KIT gene was negative. Nested PCR analysis for FIP1L1–PDGFRA fusion transcript was negative. Conventional cytogenetic analysis showed no clonal chromosomal abnormality. A diagnosis of SM was rendered using the 2008 WHO classification, based on the presence of 1 major criterion (multifocal mast cell aggregates) and 1 minor criterion (elevated serum tryptase levels).
The patient was treated with imatinib mesylate 400 mg daily, and the symptoms resolved over the course of 8 months. Unfortunately, the patient had poor compliance and started to miss doses after 8 months of continuous therapy, and developed intermittent constitutional symptoms over time. The patient completely stopped imatinib on her own about 2 years after treatment initiation. She started to experience fatigue, nausea and vomiting, and weight loss. The patient returned to our institution with symptoms, 7 months after stopping therapy. A complete blood count revealed minimal leukopenia without anemia. Repeat serum tryptase levels were elevated, ranging between 31.7 and 36.1 ng/mL. A repeat BM examination was performed. BM aspirates showed numerous intact round and well-granulated mast cells, representing about 4% to 5% of the cellularity. This time, many mast cells showed erythrophagocytosis (Fig. 1 A and B). BM biopsy showed extensive paratrabecular and interstitial aggregates of round mast cells with abundant cytoplasm and coarse granules without spindle or atypical morphology, involving about 50% of the cellularity (Fig. 1C). The mast cells were positive for tryptase and CD30 by immunohistochemistry (Fig. 1D). Reticulin stain showed increased fibrosis and trichrome stain showed focal collagen deposition. FCI studies on BM showed a discrete mast cell population representing 0.5% of total analyzed cells that were positive for CD22 (dim), CD123 (dim), CD117 (bright), HLA-DR (dim), and CD30 (66%, dim); and negative for CD2, CD19, CD25, and CD34 (Fig. 2). No aberrant mast cells were detected in the peripheral blood by FCI. Fluorescence in situ hybridization for BCR–ABL1 fusion was negative. Conventional cytogenetic studies showed no clonal chromosomal abnormality. Microarray-based comparative genomic hybridization did not show any submicroscopic abnormalities. NGS-based analysis for the detection of somatic mutations in the coding regions of a total of 28 genes related with myeloid neoplasms (ABL1, ASXL1, BRAF, DNMT3A, FGFR, EZH2, FLT3, GATA1, GATA2, HRAS, IDH1, IDH2, IKZF2, JAK2, KIT, KRAS, MDM2, MLL, MPL, MYD88, NOTCH1, NPM1, NRAS, PTPN11, RUNX1, TET2, TP53, and WT1) was performed on the DNA extracted from BM aspirate in our CLIA-certified molecular diagnostic laboratory. No mutation was detected including KIT (exons 1–21). Similar to the previous sample, no FIP1L1–PDGFRA gene fusion transcript was detected by qualitative nested RT-PCR analysis. Patient was restarted on imatinib 400 mg daily.
Figure 1.
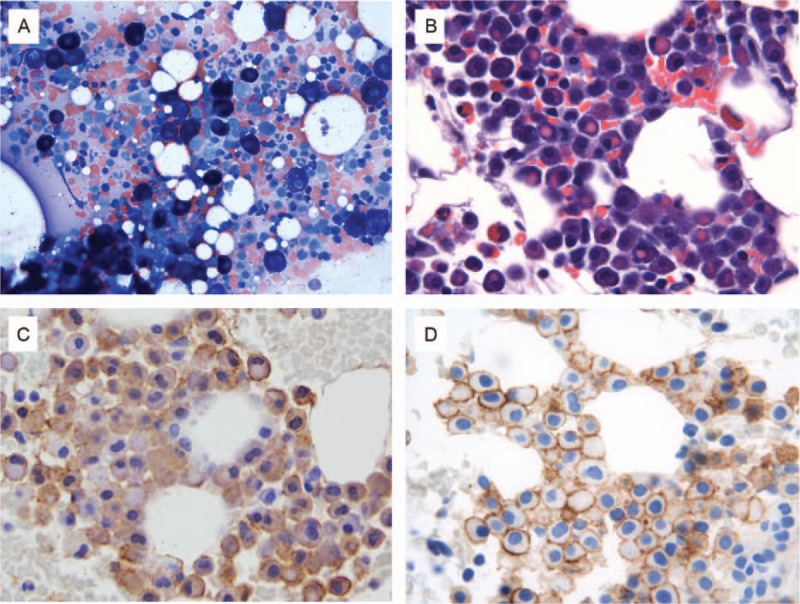
Histologic and cytomorphologic bone marrow features. (A) Aspirate smear, Wright Giemsa stain 500× magnification; (B) clot section: erythrophagocytosis, hematoxylin, and eosin stain 500× magnification; (C) clot section, mast cell tryptase immunohistochemistry, 500× magnification; (D) clot section, CD30 immunohistochemistry, 500× magnification.
Figure 2.
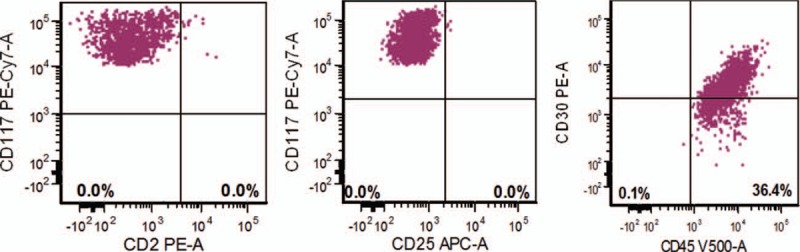
Flow cytometry immunophenotype shows that mast cells are positive for CD30 (66%) and CD117, and negative for CD2 and CD25. All plots are set based on controls.
3. Discussion
Here, we present a case of WDSM that manifested initially at childhood, with skin lesions that progressed to mediator-related systemic events including musculoskeletal complaints and constitutional symptoms, and a gastric mastocytoma diagnosed at adult age. Although childhood-onset mastocytosis is usually a skin-limited disease that commonly spontaneously regresses before puberty, a small subset of patients show progression to aggressive disease.[10–12] In this patient, the diagnosis of systemic involvement by mastocytosis, WDSM subvariant was made at 24 years of age, based on the BM examination, however, BM involvement before this cannot be excluded. The findings met 1 major and 1 minor diagnostic criteria of SM, reportedly elevated serum tryptase levels over 20 at the time of diagnosis and relapse.
The relapsed BM had a higher degree of BM mast cell infiltration by morphology and flow cytometry (0.5%) than when performed at diagnosis (0.2% by flow). However, the degree of the serum tryptase level elevation, measured at multiple time points over a period of 18 days before restarting therapy, was lower than the diagnostic sample. This illustrates that the serum tryptase level reflects the degree of mast cell activation, and not merely quantitative mast cell burden. Hence, correlation between BM mast cell burden and serum tryptase levels may not be absolute. One of the reasons is sampling error due to patchy MC infiltration in the marrow. Secondly, serum tryptase levels correspond to mast cell activation of the total body mast cells in various organs. In this patient, the extent of skin and BM involvement at 2 different time points could have been different. Further, the degree of mast cell activation, and thereby serum tryptase levels can be dependent on time, external stimuli, etc.[13] In this case, unlike serum tryptase testing at the time of relapse, serum tryptase level was performed only at 1 time point. Another interesting finding at relapse was overt erythrophagocytosis observed in mast cells while the patient was neither anemic nor being transfused. Erythrophagocytosis in mast cells has been reported in both neoplastic and normal mast cells from human and animal species, but the significance is unknown.[14–16]
The mast cell immunophenotype of the current case is similar to previous studies that have shown the neoplastic mast cells in WDSM to show variable expression of CD22, CD30, CD63, CD117, CD123, and very dim HLA-DR, while they show negativity for CD2, CD19, CD25, and CD34.[4,17,18] In this case, CD30 was expressed in ∼50% of cells by BM immunohistochemistry and 66% by FCI. CD30 is rarely expressed in normal mast cells,[18] but frequently expressed in SM.[18–21] CD30 has been found highly expressed in WDSM both by FCI and IHC.[4,18] This is important since WDSM is often negative for CD2 and CD25, and aberrant CD30 expression may be a surrogate marker of mast cell clonality for this variant SM. In addition, CD30 may serve as a therapeutic target marker.[22,23] CD30 is a transmembrane glycoprotein receptor found on certain hematopoietic cells and activated T-cells. It has been implicated both in cell death and proliferation. As we learn from this current case and previous reports, CD30 is expressed in the cytoplasm and on the cell surface of the neoplastic mast cells and is therapeutically targetable. Brentuximab vedotin (BV) is a CD30-directed antibody–drug conjugate (ADC), that selectively induces cell cycle arrest and apoptotic death of the CD30-positive tumor cells. In vitro studies in SM have confirmed the effectiveness of this drug in targeting neoplastic mast cells with CD30 expression. This is currently being evaluated in a phase II trial in patients with CD30-positive advanced SM (clinicaltrials.gov identifier NCT01807598).[22,23] However, the utility of this drug has not yet been demonstrated in WDSM patients, and could be of value and worth exploring.
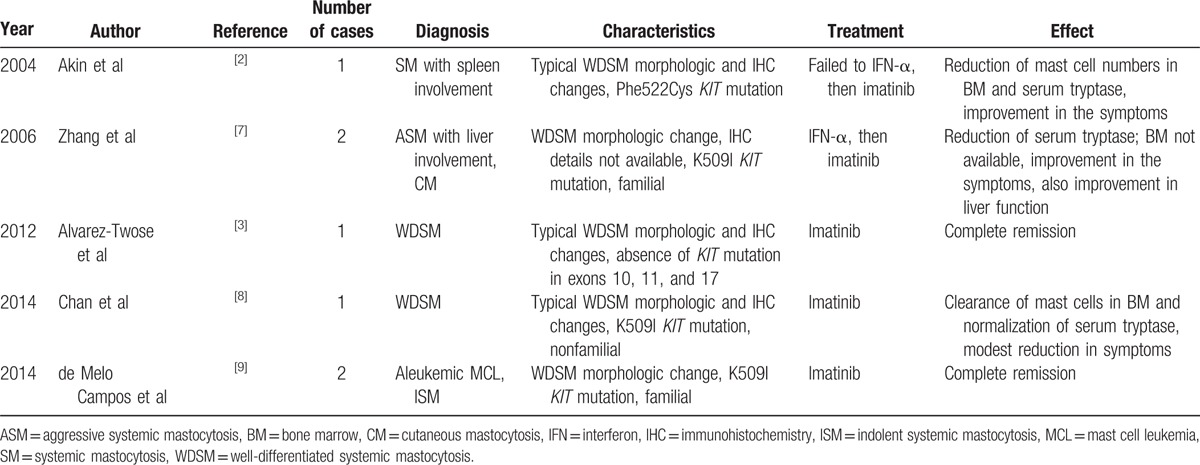
Imatinib is the only SM treatment currently approved by the Food and Drug Administration (FDA).[2,3,8,9] It is an effective target agent that inhibits activated tyrosine kinase, and has been shown to be effective in SM with KIT mutations occurring outside exon 17, but not with mutations in the KIT D816V loop. KITD816V mutation causes conformational changes in the enzymatic domain of the protein, thereby preventing binding of imatinib.[24,25] From the literature review, we identified 5 reported cases of WDSM with excellent response to imatinib: 4 of 5 cases had KIT non-D816 mutation involving Phe522Cys and K509I; 1 case showed absence of KIT mutation in exons 10, 11, and 17. Our patient showed excellent clinical and cutaneous response to imatinib with complete resolution of cutaneous lesions and mast cell-mediated release symptoms (summarized in Table 1). Following the discontinuation of the drug, the patient developed progression of symptoms, and BM confirmed relapse. However, no mutation was detected upon sequencing the entire coding region of KIT. Furthermore, our patient had a normal karyotype, and did not show any mutations in the other myeloid-related genes included in our NGS panel. Recently, mutations in TET2, SRSF2, ASXL1, CBL, and RUNX1 have been reported in SM patients who also had KIT D816V mutation, and are associated with a worse overall survival.[26] The observed clinical response to imatinib in the absence of a mutation causing kinase activation is intriguing. The mechanism behind the response of imatinib may be related with a nonspecific inhibition of tyrosine kinase due to yet undefined mechanisms.
Table 1.
Use of imatinib in the treatment on WDSM patients: review of the literature.
4. Conclusion
The diagnosis of WDSM is challenging in the absence of spindle-shaped mast cells, CD2 or CD25 expression, and KIT D816 mutation. In such cases, aberrant CD30 expression can help in the diagnosis and may serve as a potential therapeutic target. The excellent clinical and cutaneous responses to imatinib treatment as observed in this case of WDSM suggest the need for diagnosis of this variant of SM and empirical treatment with tyrosine kinase inhibitors, despite no identifiable molecular genetic alterations.
Footnotes
Abbreviations: ADC = antibody–drug conjugate, BM = bone marrow, BV = Brentuximab vedotin, FCI = flow cytometry immunophenotypic, FDA = the Food and Drug Administration, NGS = next-generation sequencing, REMA = Spanish Network on Mastocytosis, SM = systemic mastocytosis, WDSM = well-differentiated systemic mastocytosis, WHO = World Health Organization.
Written informed consent was obtained from the patient for publication of this Case Report and all accompanying images.
Author's contributions: LH, SAW, and RK-S conceived the study, participated in the design, analyzed the data, and drafted the manuscript. EJ provided clinical information and was responsible for patient management. All authors contributed vital strategies, participated in discussions, and provided scientific input. All authors read and approved the final manuscript.
The authors have no conflicts of interest to disclose.
References
- 1.Akin C. Well-differentiated systemic mastocytosis∗1A new disease variant with mature mast cell phenotype and lack of codon 816 c-Kit mutations. J Allergy Clin Immunol 2004; 113:S327. [Google Scholar]
- 2.Akin C, Fumo G, Yavuz AS, et al. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood 2004; 103:3222–3225. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez-Twose I, Gonzalez P, Morgado JM, et al. Complete response after imatinib mesylate therapy in a patient with well-differentiated systemic mastocytosis. J Clin Oncol 2012; 30:e126–e129. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez-Twose I, Jara-Acevedo M, Morgado JM, et al. Clinical, immunophenotypic, and molecular characteristics of well-differentiated systemic mastocytosis. J Allergy Clin Immunol 2016; 137:168.e1–178.e1. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez-Munoz L, Alvarez-Twose I, Garcia-Montero AC, et al. Evaluation of the WHO criteria for the classification of patients with mastocytosis. Mod Pathol 2011; 24:1157–1168. [DOI] [PubMed] [Google Scholar]
- 6.Horny HP, Metcalfe DD, Bennett JM. Swerdlow SH, Campo E, Harris NL, et al. Mastocytosis. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Lyon: International Agency for Research and Cancer (IARC); 2008. 54–63. [Google Scholar]
- 7.Zhang LY, Smith ML, Schultheis B, et al. A novel K509I mutation of KIT identified in familial mastocytosis—in vitro and in vivo responsiveness to imatinib therapy. Leuk Res 2006; 30:373–378. [DOI] [PubMed] [Google Scholar]
- 8.Chan EC, Bai Y, Kirshenbaum AS, et al. Mastocytosis associated with a rare germline KIT K509I mutation displays a well-differentiated mast cell phenotype. J Allergy Clin Immunol 2014; 134:178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Melo Campos P, Machado-Neto JA, Scopim-Ribeiro R, et al. Familial systemic mastocytosis with germline KIT K509I mutation is sensitive to treatment with imatinib, dasatinib and PKC412. Leuk Res 2014; 38:1245–1251. [DOI] [PubMed] [Google Scholar]
- 10.Hannaford R, Rogers M. Presentation of cutaneous mastocytosis in 173 children. Australas J Dermatol 2001; 42:15–21. [DOI] [PubMed] [Google Scholar]
- 11.Georgin-Lavialle S, Lhermitte L, Dubreuil P, et al. Mast cell leukemia. Blood 2013; 121:1285–1295. [DOI] [PubMed] [Google Scholar]
- 12.Ryan RJ, Akin C, Castells M, et al. Mast cell sarcoma: a rare and potentially under-recognized diagnostic entity with specific therapeutic implications. Mod Pathol 2013; 26:533–543. [DOI] [PubMed] [Google Scholar]
- 13.Quintas-Cardama A, Sever M, Cortes J, et al. Bone marrow mast cell burden and serum tryptase level as markers of response in patients with systemic mastocytosis. Leuk Lymphoma 2013; 54:1959–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mezger J, Permanetter W, Gerhartz H, et al. Philadelphia chromosome-negative acute hematopoietic malignancy: ultrastructural, cytochemical and immunocytochemical evidence of mast cell and basophil differentiation. Leuk Res 1990; 14:169–175. [DOI] [PubMed] [Google Scholar]
- 15.Khan KN, Sagartz JE, Koenig G, et al. Systemic mastocytosis in a goat. Vet Pathol 1995; 32:719–721. [DOI] [PubMed] [Google Scholar]
- 16.Lin TY, Hamberg A, Pentecost R, et al. Mast cell tumors in a llama (Lama glama). J Vet Diagn Invest 2010; 22:808–811. [DOI] [PubMed] [Google Scholar]
- 17.Teodosio C, Garcia-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol 2010; 125:719–726.726.e1–726.e4. [DOI] [PubMed] [Google Scholar]
- 18.Morgado JM, Perbellini O, Johnson RC, et al. CD30 expression by bone marrow mast cells from different diagnostic variants of systemic mastocytosis. Histopathology 2013; 63:780–787. [DOI] [PubMed] [Google Scholar]
- 19.Sotlar K, Cerny-Reiterer S, Petat-Dutter K, et al. Aberrant expression of CD30 in neoplastic mast cells in high-grade mastocytosis. Mod Pathol 2011; 24:585–595. [DOI] [PubMed] [Google Scholar]
- 20.Valent P, Sotlar K, Horny HP. Aberrant expression of CD30 in aggressive systemic mastocytosis and mast cell leukemia: a differential diagnosis to consider in aggressive hematopoietic CD30-positive neoplasms. Leuk Lymphoma 2011; 52:740–744. [DOI] [PubMed] [Google Scholar]
- 21.van Anrooij B, Kluin PM, Oude Elberink JN, et al. CD30 in systemic mastocytosis. Immunol Allergy Clin North Am 2014; 34:341–355. [DOI] [PubMed] [Google Scholar]
- 22.Gotlib J. Tyrosine kinase inhibitors and therapeutic antibodies in advanced eosinophilic disorders and systemic mastocytosis. Curr Hematol Malig Rep 2015; 10:351–361. [DOI] [PubMed] [Google Scholar]
- 23.Blatt K, Cerny-Reiterer S, Schwaab J, et al. Identification of the Ki-1 antigen (CD30) as a novel therapeutic target in systemic mastocytosis. Blood 2015; 126:2832–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arock M, Akin C, Hermine O, et al. Current treatment options in patients with mastocytosis: status in 2015 and future perspectives. Eur J Haematol 2015; 94:474–490. [DOI] [PubMed] [Google Scholar]
- 25.Ma Y, Zeng S, Metcalfe DD, et al. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood 2002; 99:1741–1744. [DOI] [PubMed] [Google Scholar]
- 26.Schwaab J, Schnittger S, Sotlar K, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013; 122:2460–2466. [DOI] [PubMed] [Google Scholar]