

Abstract
Intravenous general anesthetics including propofol, etomidate, alphaxalone, and barbiturates, produce important actions by enhancing gamma-aminobutyric acid type A (GABA-A) receptor activation. Here we review scientific studies that have located and mapped IV anesthetic sites using photoaffinity labeling and substituted cysteine modification-protection. These anesthetics bind in transmembrane pockets between subunits of typical synaptic GABA-A receptors, and drugs that display stereoselectivity also show remarkably selective interactions with distinct interfacial sites. These results suggest strategies for developing new drugs that selectively modulate distinct GABA-A receptor subtypes.
Introduction
GABAARs are members of the pentameric ligand-gated ion channel (pLGIC) superfamily, the main inhibitory neurotransmitter receptors in the central nervous system, and major targets of many, but not all, general anesthetics (1,2). The GABAAR is assembled from five homologous subunits arranged pseudo-symmetrically around a central transmembrane chloride-conducting pore (Figure 1). Each subunit has an extracellular domain (ECD) with over 200 amino acids, a transmembrane domain with four membrane-spanning α-helices (M1 to M4), and a variable-size intracellular loop between the M3 and M4 helices (3). The physiological roles, pharmacological properties and distribution of GABAARs vary with the subunit composition. Most native GABAARs contain two α and two β subunits with the fifth subunit being another β, a γ, or a δ. Synaptic GABAARs mostly contain γ2-subunits, although these subunits are also found extrasynaptically. The δ-subunit is exclusively extrasynaptic (4,5). No high resolution crystal structures of heteromeric GABAARs are available. Homology models are based on crystal structures of related homomeric pLGICs from bacteria, nematodes, and human β3 GABAARs (6–8).
Figure 1. The locations of modulatory sites in a structural model of α1β3γ2L GABAARs.

The structure shown was obtained by homology modeling based on the GluCl chloride channel (3RHW.pdb) (7,26). The five subunits are arranged around a central ion–conducting pore and colored as follows: α1, yellow; β3, red, and γ2 green. The α-helices are represented as cylinders, β-sheets as flat planks and loops as strings. A. A side view of the α1β3γ2L GABAAR homology model shows the extracellular domain (ECD) and the transmembrane domain (TMD). The intracellular domain is not modeled because no structural information is available. B. The extracellular domain viewed from the synapse, showing residues involved in agonist and BDZ binding. By convention, subunit order is counted in an anticlockwise direction (arrow). Important amino acids associated with various sites are distinguished by the color of their carbon atoms: agonist site, dark green (α1 Phe-65; β3 Tyr-157 & −205, Phe-200); benzodiazepine site (BDZ), orange (α1 His-102 & −210); azietomidate site, cyan (β3 Met-286 & Val-290, α1 Met-236); main R–mTFD-MPAB site, goldenrod (γ2 Ser-301, β3 Met-227). Red and blue atoms are oxygen and nitrogen respectively.
GABAAR gating between resting (closed) and ion-conducting (open) states involves a widespread conformation change coupling GABA binding at two ECD agonist sites between subunits (both β+ – α− interfaces) and the transmembrane ion pore about 50 Å away (Figure 1). Volatile anesthetics, propofols, etomidates, barbiturates, steroids and alcohols all similarly enhance GABAAR–mediated currents, suggesting that GABAARs are major contributors to the anesthetic state (9–11). Convincing in vivo evidence that some high affinity general anesthetics act through GABAARs comes from studies of knock-in mice bearing a single amino acid mutation at position 265 in the GABAAR β3 subunit (β3N265M in the M2 helix) (12). This substitution reduces GABAAR sensitivity to propofol and etomidate in vitro (13) and β3N265M mice exhibit greatly reduced sensitivity to the anesthetic effects of etomidate, propofol, and pentobarbital (12,14), but not volatile or steroid anesthetics.
Anesthetics potentiate GABAAR gating in the presence of GABA by binding with much higher affinity to the open state than to the resting state, increasing the fraction of open channels and the duration of channel openings (15). In addition, high concentrations of anesthetics directly activate (agonize) GABAARs. Evidence suggests that both of these anesthetic actions are mediated by the same mechanism and the same allosteric sites (16). In essence, resting-state receptors have very low open probability and by binding with much higher affinity to the open state than to the resting state, high anesthetic concentrations induce a modest fraction of receptors to activate (allosteric agonism). When receptors are already partially activated by an orthosteric agonist such as GABA, much lower concentrations of anesthetics shift many more receptors toward opening (co-agonism). Quantitative allosteric co-agonist models can account for both modulation and agonism of synaptic GABAARs by etomidate and propofol (16,17). For anesthetics to exhibit different affinities for distinct functional receptor states, their binding sites must change shape during the state transition (18). This review summarizes progress toward our long-term goal to understand both where anesthetics interact with GABAARs and the conformational changes underlying their state-selective binding.
Overall Strategy for Mapping Functional General Anesthetic Sites
The overall strategy of our research program is 1) to identify GABAAR binding sites in an unbiased (hypothesis-free) manner using photolabeling with analogs of high-affinity anesthetics, and 2) to investigate the functional roles of specific amino acids at or near photolabeled sites using mutational, biochemical, and functional methods. These data are interpreted using both GABAAR structural homology models and the functional principles of allosterism briefly described above.
Photolabeling Strategy
Covalent photo-modification of target proteins overcomes weak affinity and transient binding site occupation by general anesthetics (19).
Our goal of identifying anesthetic binding sites in GABAARs has required a number of innovative strategic developments for each of the following steps:
Design, synthesize and pharmacologically characterize photo-reactive analogs of potent general anesthetics known to modulate GABAARs. Using multiple photolabel derivatives of the same parent drug reduces the likelihood of misinterpreting results.
Develop cell lines that express affinity-tagged GABAARs of physiologically significant subunit compositions, in quantities suitable for biochemical characterization, and Edman amino acid sequencing.
Develop methods to affinity-purify and functionally reconstitute expressed GABAARs while maintaining allosteric linkages to GABA and anesthetic sites.
Radiolabel (tritiate) the best photolabels, photolyze them while bound to GABAARs, purify radiolabeled peptides, and apply Edman sequencing to identify photo-adducted amino acids.
Assess the functional significance of photolabeled sites, using both photochemical and other techniques. Based on allosteric principles, GABA is expected to enhance anesthetic photo-incorporation. Parent drugs and those with overlapping sites are expected to competitively reduce rates of photolabel incorporation. Non-photochemical structure-function techniques include testing whether mutations at the photolabeled site alter sensitivity to parent drug, and SCAMP (see below).
Edman versus mass spectroscopy for photo-adduct location
Edman degradation is a biochemical method that sequentially cleaves one N-terminal amino acid at a time from a polypeptide. The free modified amino acid is then isolated and analyzed for the presence of radioactive photo-adducts or identification of its sidechain. We and others (related review by Woll et al. (20) in this issue of the journal) have also used mass spectroscopy (MS) to identify photo-adducted peptides and amino acids in molecular targets where anesthetics bind. MS precisely determines the mass:charge ratio of molecules moving through an electric field, and when coupled with methods for further fragmentation of peptides isolated in an ion trap (MS/MS), the amino acid sequence and the locations of photo-adducts can be determined.
MS requires far less protein than Edman degradation and target protein purification is also unnecessary. MS requires no radioactivity, although isotopic tracing (e.g. with deuterium) can improve adduct identification (21). For small soluble target proteins such as myokinase (MW ≈ 21 kDa) labeled at high efficiency, the precision of MS enables both identification of photo-incorporation sites, and the stoichiometry of sites (22). On the other hand, because MS depends on ionization of protein fragments, it is less successful in hydrophobic protein regions such as the transmembrane helices where anesthetics bind to GABAARs. Anesthetic adduction typically increases the hydrophobicity of peptides, exacerbating this problem. Another weakness of MS is that quantifying and comparing photo-adduction efficiency is very difficult (23). Methods used to fragment peptides during MS analysis can also degrade photo-adducts, complicating adduct identification.
In contrast to MS, a major advantage of combining tritiated photolabels with Edman sequencing is the ability to track and quantify radioactivity and normalize it to protein at every analytical step. Thus, photo-adduction levels (e.g. in cpm/pmol of protein) can be quantitatively compared in different receptor conformations (e.g. resting vs. GABA-bound). Competition with anesthetics can also be assessed under conditions reflecting initial photolabel incorporation rate. These advantages have led us to favor Edman sequencing for much of our photolabel research. Disadvantages of Edman sequencing include the additional synthetic chemistry required to produce stable high-level radiolabeling. Edman sequencing also needs large quantities of purified homogeneous GABAARs, achieved only recently with our stable inducible cell lines (24). Receptor yields from these cell lines are sufficient for identifying photo-adducted GABAAR amino acids in the resting and desensitized states (25,26), and for spectroscopic studies (27). Freeze-clamp photolabeling of GABAARs in the transient open state will require still more protein (28)
Choosing parent anesthetics
High affinity for targets is a key feature of successful anesthetic photolabels. Generally, drugs acting at concentrations above 50 µM show little target or site specificity, and their potency is predicted by hydrophobicity alone (29). For example, anesthetic potencies of methanol through 1-octanol are predicted by their octanol/water partition coefficients (30) (Figure 2). Clinical volatile anesthetics all act at free plasma concentrations above 100 µM, with potencies paralleling hydrophobicities, and can interact at many binding sites large enough to accommodate them (31,32). In contrast, for anesthetics with EC50s below 50 µM the correlation with hydrophobicity breaks down and significant stereoselectivity emerges (yellow zone, Figure 2). For further illustration, the volatile anesthetic bromoform (IC50 = 125 µM) occupies eleven sites in a crystal structure of a bacterial pLGIC, (33) (Figure 3A), whereas ketamine (IC50 = 58 µM) interacts stereoselectively with a single class of intrasubunit sites in another pLGIC (34) (Figure 3B). On this basis, we have focused on developing photolabels that act below 10 µM and, wherever possible, exhibit stereoselectivity, including derivatives of etomidate, propofol, barbiturates, and alphaxalone.
Figure 2. The importance of potency in governing selective binding to general anesthetic sites.

The correlation between EC50 for loss of righting reflexes (LoRR) in tadpoles and octanol/water partition coefficient holds well for anesthetics with EC50s above ~50 µM (Pearson correlation coefficient = −0.987; P < 0.0001). More potent agents display weak dependence on partition coefficient (Pearson correlation coefficient = −0.209; P = 0.49; yellow box). Some of the most successful photolabels lie in this group and are more potent than predicted by hydrophobicity (points with light blue fill). Data sources are given in (46).
Figure 3. Selectivity of general anesthetic binding to crystallized homomeric cation–conducting pLGICs is dependent on potency.

A. Bromoform (brown), a low potency drug, occupies eleven sites in three different classes on ELIC (33): (1) 5 homologous intrasubunit sites in the extracellular domain (ECD); (2) 5 homologous sites in the lipid-protein interface of the transmembrane domain, and (3) a single site in the channel lumen at the interface of all five subunits. B. Ketamine (carbons grey, nitrogen blue, chlorine green) occupies a single class of 5 intersubunit sites on GLIC (34), which potently and stereoselectively inhibits. Channel subunits are shown as ribbons and are colored arbitrarily. The anesthetics are shown space filled and colored conventionally.
Pharmacological characterization of anesthetic photolabels
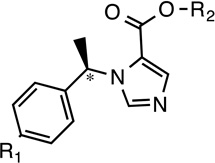
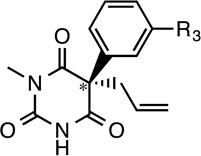
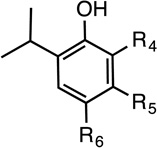
Table 1 shows the structures of photolabels based on etomidate, mephobarbital, and propofol that are described below.
Table 1.
Structures of Potent Anesthetics and Photolabel Derivatives
 |
 |
 |
||||||
|---|---|---|---|---|---|---|---|---|
| Name | R1 | R2 | Name | R3 | Name | R4 | R5 | R5 |
| R-Etomidate | H | R-Mephobarbital | H | Propofol |  |
H | H | |
| R-Azi- etomidate |
H | 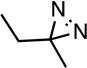 |
AziPm | H | 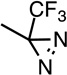 |
H | ||
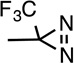
| R-TDBzl- etomidate |
H | 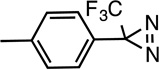 |
R–mTFD- Mephobarbital (R–mTFD- MPAB) |
 |
o-Propofol diazirine |
 |
H | H |
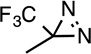
| R-pTFD- etomidate |
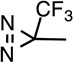 |
p-Azipentyl-Pro | 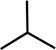 |
H | ||||
Chiral center
The first successful anesthetic photolabel in GABAARs, R-azietomidate, acts just like R-etomidate both in vivo and in vitro. Its potency for loss of righting reflexes (LoRR) in tadpoles (EC50 ≈ 2 µM) is identical to R-etomidate’s, and both S-enantiomers are about 10-fold less potent (Figure 2) (35). In GABAAR β3N265M mice, sleep times after intraperitoneal R-etomidate and R-azietomidate were equally attenuated relative to wild-type (36). Both drugs enhanced currents induced by low GABA concentrations in α1β2γ2L GABAARs with similar potency, efficacy and enantioselectivity (35).
A second–generation etomidate photolabel, R-TDBzl-etomidate, is an aromatic diazirine, reactive at a wider range of amino acid side chains than aliphatic diazirines. It is more potent than R-etomidate both in tadpoles (LoRR EC50 ≈ 700 nM) and in enhancing GABAAR currents (37).
R–mTFD-MPAB is a derivative of mephobarbital, but 25-fold more potent, with a tadpole LoRR EC50 of 3.7 µM, and its S-enantiomer is 10-fold less potent (38,39). R–mTFD-MPAB enhances GABAAR currents with an EC50 of 2.1 µM. It induces anesthesia in wild type and β3N265M mice with equal potency (personal communication from Robert Pearce, MD-PhD,Univ. Wisconsin at Madison), suggesting action via sites distinct from the etomidate sites.
Propofols are relatively small general anesthetics that nonetheless exhibit high potency and strong structure activity relationships (40). Three propofol photolabels have been described. AziPm (m-diaziryl-propofol) has potency similar to propofol in tadpoles (EC50 ≈ 3 µM), but modulates α1β2γ2L GABAAR with less efficacy than propofol (41). Ortho-propofol diazirine (o-PD) causes LoRR in rats with EC50 ≈ 14.7 mg/kg (vs. 4.7 mg/kg for propofol). It enhances α1β2γ2S receptor currents with efficacy comparable to propofol (21). o-PD is chemically unstable, making it unsuitable for approaches based on tritiation and Edman sequencing. It has been used for photolabeling with MS analysis (21). 4-Azipentyl-propofol contains an aliphatic diazirine at the para position. It produces tadpole LoRR with EC50 ≈ 3.2 µM and enhances α1β2γ2L receptor currents with efficacy similar to propofol (42).
Based on pharmacological interactions, sites where alphaxalone and related neurosteroids modulate GABAARs are likely different from other anesthetics (43). Photolabeling of GABAARs has succeeded with one steroid, 6-azi-pregnanolone (6-AziP). This compound enhances α1β2γ2L GABAAR currents with EC50 ≈ 3 µM (44).
Anesthetic Photolabeling Results in GABAARs
Potent anesthetic photolabels identify anesthetic sites in transmembrane subunit interfaces.
Etomidate binds at β+ – α− interfaces
Both [3H]azi-etomidate and [3H]TDBzl-etomidate photolabel α1β3 GABAARs at β3M286 in M3 and α1M236 in M1 (25,45). [3H]TDBzl-etomidate also labels β3V290, one helical turn intracellular from M286 in M3. Photoincorporation by these etomidate photolabels is inhibited by etomidate and propofol, but not mTFD-MPAB or alphaxalone (26). These residues all abut the inter-subunit spaces between β3-M3 helices (the “β3+” face) and α1-M1 helices (the “α1−” face; Figures 1 and 4). A photolabel moiety was added at the other end of etomidate, creating pTFD-etomidate (Table 1), but this compound did not modulate GABAARs (46), suggesting that the phenyl ring occupies a sterically restricted environment. In silico docking suggests the phenyl ring projects towards the M2 helices, coming close to β3N265, where mutations produce dramatic effects on etomidate sensitivity (SCAMP studies of this site are discussed below) (25,26,47).
Figure 4. Selectivity of general anesthetics for intersubunit sites in α1β3γ2L GABAARs.

The homology model and coloring are as in Figure 1, and the view of the transmembrane domain () is from the synaptic side with the extracellular domain removed. Subunit interfaces are labeled (note that “+” corresponds to M3 helices and “−” corresponds to M1 helices). Photolabels that selectively bind in each interface are identified in colored boxes, and white boxes identify the general anesthetics that compete with photolabels for occupancy of that interface.
R–mTFD-MPAB binds at γ+ – β− and α+ – β− interfaces
R–mTFD-MPAB does not photolabel the etomidate sites. Instead it photoincorporates at α1A291 and α1Y294 (both in M3), β3M227 (M1), and γ2S301 (M3) (26). These residues are homologs of those labeled by etomidate derivatives, but located in both γ2+ – β3− and α1+ – β3− interfaces of α1β3γ2L GABAARs (Figure 4). R-mTFD-MPAB photoincorporation is inhibited by mTFD-MPAB and propofol, but not by etomidate or alphaxalone. R–mTFD-MPAB modulates [3H]muscimol binding in α1β3γ2L and α1β3 GABAARs with EC50s of 2 and 30 µM respectively, suggesting that it has higher affinity for the γ2+ – β3− than for the interface α1+ – β3− interface (27).
Propofol binds at β+ – α−, γ+ – β−, and α+ – β− interfaces
Propofol binding sites have been explored with competition studies, described above, and two photolabel analogs. After o-PD photolabeling of β3 (homomeric) and α1β3 GABAARs, mass spectrometry detected a single adducted residue: β3H267 in M2 (21). However, propofol displacement of o-PD was not demonstrated. The location of β3H267 is at the top of M2 on the β− face (8). In contrast, propofol–displaceable photoincorporation of [3H]aziPm occurred at both in β3+ – α1− interfaces (β3M286, α1M236 and α1I239) and in α1+ – β3− interfaces (β3M227) in α1β3 GABAARs (47). These results suggest that AziPm binds to all four interfaces labeled by azietomidate and mTFD-MPAB. This inference is also supported by propofol displacement experiments (26) and quantitative analyses of α1β2γ2L GABAAR activity showing that etomidate acts via two equivalent sites, while propofol acts via more than two allosteric sites (16,17). Further research is needed to establish the relative affinities and efficacies of these sites, and their dependence on subunit composition.
6-AziP binds at β3+
Homomeric β3 GABAARs were photolabeled with 6-AziP (48). MS analysis identified β3F302 as the only photolabeled residue. This residue is located on β3-M3, three to four helical turns intracellular from β3M286 and β3V290, the residues photolabeled by TDBzl-etomidate. Displacement by other steroids such as alphaxalone was not demonstrated.
Do anesthetics bind at α+ – γ− interfaces?
No photolabeling has been detected in γ2-M1 using any of the anesthetic derivatives described above. This suggests, albeit inconclusively, that residues photolabeled on α1-M3 are from sites in the α+ – β− interfaces.
Anesthetic binding at β3+ – β3− interfaces
Azietomidate photolabels β3M227 in α1β3 GABAARs, but no incorporation is found in α1-M3 (25). Thus, this etomidate-displaceable labeling is in the β3+ – β3− interface, consistent with etomidate activation of β3 homomeric receptors (49). Interestingly, etomidate does not displace R-mTFD-MPAB photolabeling at β3M227 in α1β3 receptors, indicating that mTFD-MPAB binds very weakly to β3+ – β3− interfaces (26,47).
Substituted Cysteine Modification-Protection (SCAMP) Strategy
Anesthetic photolabeling has located a number of amino acid contacts in GABAAR inter-subunit pockets, but almost certainly has missed others. Thus, complementary techniques are needed to further probe anesthetic-receptor contacts near photolabel sites. One successful approach to extending the map of anesthetic contacts is SCAMP, illustrated in Figure 5. Briefly, a side-chain hypothesized to be in or near an anesthetic binding site is mutated to cysteine, providing a free sulfhydryl. These cysteine substituted receptors are then exposed to sulfhydryl-reactive chemical probes, and after washout of these reagents, electrophysiology is used to detect whether covalent bond formation produced an irreversible functional change. This is equivalent to real-time mutagenesis at the side-chain of interest. If a functional modification signal is observed, then the rates of modification in the absence and presence of anesthetic are compared. Drug occupancy will reduce the rate of covalent bond formation by steric competition if the substituted cysteine is in the binding pocket. This interpretation is strengthened if other classes of anesthetic fail to block modification. However, if all anesthetics inhibit modification, this could indicate a noncompetitive mechanism such as allosteric action.
Figure 5. Substituted Cysteine Modification and Protection (SCAMP).

Top: Transmembrane cross-sections of both open and closed receptors are diagrammed. Anesthetic site affinity or accessibility is enhanced by activating GABAA receptors with GABA. Bottom: A close-up view of one interfacial pocket is shown with engineered sulfhydryls, one of which is in the anesthetic site. Modification of both sulfhydryls by p-chloromercuribenzene sulfonate (pCMBS), and protection of one sulfhydryl by bound anesthetic are also depicted. Experiments are performed with one cysteine substitution at a time.
Several experimental criteria must be met when applying SCAMP to sites such as those for anesthetics that are coupled to channel gating (agonist sites). First, the anesthetic drug must still modulate or activate mutant receptors, signifying that normal drug-site interactions are retained. Second, the mix of receptor states in both control modification and anesthetic protection studies must be similar. Because anesthetics tend to activate receptors, this condition can often be met by including GABA in both control modification and anesthetic protection experiments. Third, protection is best studied using anesthetic concentrations that occupy a large fraction of sites, to more effectively block covalent modification. GABA also enhances anesthetic affinity and increases the fraction of drug-occupied binding sites. However, cysteine substitution may reduce GABA efficacy, requiring modified control conditions. Finally, GABA often increases the rate of modification at cysteine substituted positions in transmembrane helices, reducing the concentrations of probe chemicals needed in experiments.
Anesthetic SCAMP Results in GABAARs
The βM286 residue
SCAMP was used to investigate anesthetic sites in GABAARs several years before anesthetic photolabeling. Bali and Akabas (50) introduced cysteine at two β2 positions, β2N265 and β2M286, where other mutations were known to influence propofol sensitivity in α1β2γ2 receptors (Figure 6 shows these residues in β3, which is highly homologous to β2 in this region). Both residues are predicted to contribute to the “β+” transmembrane interface, although β2N265 may also be accessible from the “β−” interfacial pocket (51). To achieve comparable mixes of receptor states during control and protection experiments, Bali and Akabas used a strategy different from ours, likely resulting in under 50% anesthetic site occupancy. They found that propofol protected β2M286C from modification, but did not protect β2N265C. We later investigated the β2M286C mutation in a detailed study of etomidate protection, which revealed uniquely useful features of this mutation (52). The β2M286C mutation weakened etomidate modulation of GABA-elicited responses, and eliminated direct activation by high etomidate concentrations. Covalent bond formation between β2M286C and p-chloromercuribenzenesulfonate (pCMBS), a small water-soluble sulfhydryl-specific reagent, produced clear functional changes that were blocked in the presence of etomidate. The functional features of the βM286C mutation also enabled studies of etomidate-dependent protection in both the absence of GABA (almost entirely resting state receptors) and in the presence of GABA (activated and desensitized receptors). Protection studies revealed that etomidate concentrations occupying half of the receptor sites in the absence of GABA were about 10-fold higher than those in the presence of GABA, consistent with a quantitative functional model of etomidate co-agonism in α1β2M286Cγ2L GABAARs (16,52).
Figure 6. Combining photolabel and SCAMP results to map the β+ – α− interfacial anesthetic sites.

The homology model is based on the GluCl chloride channel (3RHW.pdb) (7,26). The five subunits are colored as follows: α1, yellow; β3, red, and γ2 green. The transmembrane helical backbones are depicted as ribbons. Residues in this site identified with either photolabeling or SCAMP are shown as space-filling atomic models and labeled. Contact residues are coded by coloring their carbons. Residues photolabeled by etomidate and propofol derivatives show cyan and pink carbons, respectively. Grey carbons indicate binding site residues identified by SCAMP with either etomidate or propofol. Red and blue atoms are oxygen and nitrogen respectively. Also shown in ball and stick mode are three residues in the γ+ – β− inter-subunit region that are discussed in the text: γ2S301, β3M227, and β3H267.
The α-M1 helix
Photolabeling at αM236 by azi-etomidate (45) provided the first hint that the α-M1 transmembrane helix abuts bound etomidate in β+–α− interfacial pockets. To further define the role of α-M1 in etomidate binding and modulation, we applied SCAMP to a series of 14 α1-M1 residues, from α1Q229 to α1Q242 spanning over three helical turns, in α1β2γ2L receptors (53). We found interesting functional phenotypes associated with these cysteine substitutions. For example, in α1M236Cβ2γ2L receptors GABA was a partial agonist, activating only about 25% of receptors, while etomidate alone activated more than 95% of receptors. Remarkably, these findings remained consistent with our allosteric co-agonist model. Most of the other α1-M1 cysteines we studied also displayed reduced receptor sensitivity to GABA, while retaining etomidate sensitivity. Thus, the entire α1-M1 region we studied was linked to channel gating.
For SCAMP studies of α1M236Cβ2γ2L, we established similar distributions of receptor states in both control modification and etomidate protection experiments by including alphaxalone (which does not bind at etomidate sites) with GABA in controls. This combination fully activated the mutant receptors, matching the effects of etomidate plus GABA. Etomidate protected α1M236C from covalent modification by pCMBS, confirming steric proximity. Etomidate protection was also observed at both α1L232C and α1T237C (Figure 6), neither of which was photolabeled by etomidate derivatives. Thus, etomidate apparently contacts one face of the outer α1-M1 helix over ~1.5 helical turns. Moreover, etomidate did not protect any cysteine substituted sidechains predicted to face the intra-subunit pocket formed by the four α1 transmembrane helices. This further supports the idea that sites where anesthetics act as allosteric agonists are located between adjacent subunits. Subsequent studies also demonstrated propofol protection at α1M236C (see below).
The βN265 residue
Despite the remarkable effects of mutations at βN265, Bali and Akabas (50) failed to demonstrate propofol protection and no anesthetic photolabels have adducted this locus. We examined etomidate interactions at βN265 using SCAMP (54), because βN265 mutations appear to affect etomidate sensitivity more than propofol. The β2N265C mutation eliminated modulation or activation by etomidate at concentrations (300 µM) more than100-fold higher than those affecting wild-type GABAARs. Similarly high concentrations of etomidate did not protect β2N265C from pCMBS modification. Because α1β2N265Cγ2L violates one requirement for interpretation of SCAMP protection data (sensitivity to the anesthetic), these negative results fail to reveal how the mutation alters etomidate-receptor interactions: it could obliterate etomidate binding; etomidate could bind without contacting β2N265, or the mutation could eliminate drug efficacy, by decoupling the site from channel gating.
To test for binding effects of βN265 mutations, we exploited α1M236C, already established as protected by etomidate (53). In a wild-type background, α1M236C was readily protected by etomidate (as low as 3 µM) and propofol, but not by alphaxalone. When the β2N265M mutation was added, etomidate concentrations below 300 µM did not protect α1M236C, indicating low anesthetic site occupancy in both the absence and presence of GABA. Similar results were found with propofol. These findings, although indirect, indicate that the β2N265 mutations strongly affect etomidate and propofol binding in the β+–α− interfacial pockets, supporting the hypothesis that β2N265 is a key contact point.
The βH267 residue
As noted above, photolabeling with o-PD identified β3H267 as a possible contact for propofol (21). We explored both the pharmacological effects of the β3H267C mutation, and the ability of four potent anesthetics to protect this sidechain from chemical modification (55). The α1β3H267Cγ2L receptors retain most of the gating features of wild-type: low spontaneous activation, normal GABA EC50, and high GABA efficacy. However, the β3H267C mutation selectively sensitizes receptors to direct activation by propofol and the barbiturate photolabel R-mTFD-MPAB. SCAMP experiments reveal that mTFD-MPAB protects β3H267C from pCMBS modification, while etomidate, alphaxalone, and propofol do not. Thus, while photolabeling shows that propofol and mTFD-MPAB sites overlap, β3H267 abuts the mTFD-MPAB binding site, but is far enough from bound propofol to allow pCMBS unimpeded access. As expected, β3H267 does not interact with either etomidate or alphaxalone, two anesthetics with sites that do not overlap with those of mTFD-MPAB.
This result also has implications for structural GABAAR homology models based on crystallized homomeric pLGICs. Homology models based on both β3 homomeric GABAARs (8) and on GLIC (6) have sidechains bridging the “β‒“ transmembrane interfaces, separating them into two non-contiguous pockets: one near the ion channel adjacent to βH267, and another near the lipid-protein interface that includes residues photolabeled by both mTFD-MPAB and azi-Pm (βM227, αA291, αY294, and γS301). SCAMP results indicate that mTFD-MPAB binds in a single contiguous pocket that abuts all of these residues, favoring homology models based on ivermectin-bound GluCl channels (7).
Summary and Conclusions
Anesthetic molecular mechanisms in GABAARs
The conclusion that etomidate, barbiturate and propofol binding sites on GABAARs are located in homologous pockets between adjacent subunits in the transmembrane domain is supported by both photolabeling and SCAMP studies. Mutations at photolabeled inter-subunit sites alter both receptor function and anesthetic sensitivity, supporting the pharmacological relevance of these sites (56–58). Molecular modeling studies also suggest that anesthetic binding to inter-subunit pockets in GABAARs correlates with drug potency (59). The identification of inter-subunit anesthetic sites is an important shift from previous hypotheses proposing anesthetic sites within the transmembrane four helix bundles of GABAAR subunits (i.e. intra-subunit sites) (60). Anesthetic sites on GABAARs are also distinguished from those on cationic channels of the pLGIC superfamily. In cationic channels, crystallography reveals general anesthetics binding in intra-subunit pockets within the four helix bundles of GLIC (61). In muscle type acetylcholine receptors, inhibitory sites are in the channel lumen, and four helix bundles (62–64).
Five homologous inter-subunit transmembrane sites are potentially formed by each GABAAR pentamer. In physiologically relevant α1β3γ2 heteropentamers, four classes of distinct interfacial sites are predicted (Figure 4), and anesthetic photolabeling has revealed remarkable selectivity for particular inter-subunit sites. In α1β3γ2 GABAARs, etomidate sites (β+ – α− interfaces) are favored over R-mTFD-MPAB sites by R- and S-etomidate (>100-fold) and propofol (1.5-fold), whereas the R–mTFD-MPAB sites (γ+ – β− and to a lesser extent α+ – β − interfaces) are favored by R-mTFD-MPAB (~60-fold), phenobarbital (13-fold), pentobarbital (8-fold) and thiopental (1.6-fold). However, the mTFD-MPAB sites cannot be regarded as universal barbiturate sites because brallobarbital favors the etomidate site (26). Preliminary SCAMP studies are consistent with these conclusions. The remaining α1+ – γ2− interface has not been unequivocally photolabeled, and SCAMP will be useful in testing whether any anesthetics bind near γ2-M1.
Subunit selective drug development
The subunit selectivity of various potent general anesthetics raises the possibility that new clinical drugs can be developed that act selectively on specific GABAAR subtypes. For example, etomidate acts selectively on receptors with β2 or β3 subunits, but not β1 (65). However, nearly all GABAARs contain β+ – α− interfaces, so etomidate derivatives act on a wide range of GABAA networks. In contrast, drugs like mTFD-MPAB may select for synaptic GABAARs, all of which contain γ2 subunits, rather than δ-subunit containing extrasynaptic receptors.
We do not yet know how the many physiological components underlying the general anesthetic state would respond to agents of these two classes, but the principle has already been illustrated for benzodiazepines and similar drugs that bind selectively at distinct ECD interfaces (66,67). These act in the αx+ – γ2 − and αx+ – βy− interfaces in the ECD (Figure 1). Studies in transgenic animals show that classical benzodiazepines act at α1+ – γ2 − interfaces to produce sedative and anticonvulsant effects, while α2+ – γ2 − interfaces mediate anxiolysis, and α5+ – γ2 − interfaces mediate learning and memory effects (68). Considering that the GABAA receptor family includes a large set of different subunit isoforms (α1–6; β1–3; γ1–3; δ; ε; π; ρ1–3; θ), that are distributed differentially throughout the central nervous system (4), it is clear that much remains to be discovered.
Structure activity relationships acquired during photolabel development have incidentally also proven useful in developing novel clinical anesthetics. Specifically, the development of different etomidate photolabels helped identify parts of the molecule that could be modified without loss of potency and efficacy (35,37,46). This information contributed to the design of short-acting etomidate derivatives for clinical use (69,70).
Future directions
Photolabeling and SCAMP are being applied to a number of important questions related to mechanisms of general anesthesia. One challenging goal is locating sites where neurosteroid anesthetics (e.g. alphaxalone) bind to heteromeric GABAARs. Another goal is to better understand state-dependent conformational changes and anesthetic binding, particularly in the transient open state. Sites where low affinity anesthetics act in heteromeric GABAARs are largely unmapped, but there is evidence that some bind in the inter-subunit pockets where potent IV drugs act (51,71). These approaches are also being applied to map inhibitory (convulsant) sites on GABAARs (72).
Focus and Scope.
This review focuses on progress in our research aimed at mapping binding sites for IV general anesthetics in heteromeric γ-aminobutyric acid type A receptors (GABAARs) using two complementary techniques: photolabeling and substituted cysteine modification-protection (SCAMP). We include sections on both research strategy and results for each approach. Results are discussed in the context of both models of GABAAR molecular structure and models of anesthetic-induced receptor modulation.
Acknowledgments
We wish to acknowledge research support from NIGMS (GM058448, GM089745). We also acknowledge the important contributions of colleagues in our research program: Shaukat Husain, D.Phil (Dept. of Anesthesia Critical Care & Pain Medicine, Massachusetts General Hospital), Karol Bruzik, PhD (Medicinal Chemistry, University Illinois Chicago) , Jonathan Cohen, PhD (Neurobiology, Harvard Medical School), David Chiara, MD-PhD (Neurobiology, Harvard Medical School) and Douglas Raines, MD (Dept. of Anesthesia Critical Care & Pain Medicine, Massachusetts General Hospital).
Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) (73).
Footnotes
Disclosures
Name: Stuart A. Forman, MD, PhD
Contribution: Prof. Forman contributed to the conception, writing, creation of figures, and editing of this manuscript.
Attestation: Stuart A. Forman approved the final manuscript.
Conflicts of Interest: Stuart A. Forman is a member of the ABP700 Scientific Advisory Board for The Medicines Company.
Name: Keith W. Miller, D.Phil
Contribution: Prof. Miller contributed to the conception, writing, creation of figures, and editing of this manuscript.
Attestation: Keith W. Miller approved the final manuscript.
Conflicts of Interest: None.
Contributor Information
Stuart A. Forman, Department of Anesthesia Critical Care & Pain Medicine, Massachusetts General Hospital, Boston, Massachusetts.
Keith W. Miller, Department of Anesthesia Critical Care & Pain Medicine, Massachusetts General Hospital, Boston, Massachusetts.
References
- 1.Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- 2.Sieghart W. Allosteric modulation of GABAA receptors via multiple drug-binding sites. Adv Pharmacol. 2015;72:53–96. doi: 10.1016/bs.apha.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci. 2010;31:161–174. doi: 10.1016/j.tips.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olsen RW, Sieghart W. GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141–148. doi: 10.1016/j.neuropharm.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 7.Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller PS, Aricescu AR. Crystal structure of a human GABAA receptor. Nature. 2014;512:270–275. doi: 10.1038/nature13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. [Review] Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 10.Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–510. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Olsen RW, Li G-D. GABAA receptors as molecular targets of general anesthetics: identification of binding sites provides clues to allosteric modulation. Can J Anaesth. 2011;58:206–215. doi: 10.1007/s12630-010-9429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003;17:250–252. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- 13.Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci USA. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeller A, Arras M, Jurd R, Rudolph U. Identification of a molecular target mediating the general anesthetic actions of pentobarbital. Mol Pharmacol. 2007;71:852–859. doi: 10.1124/mol.106.030049. [DOI] [PubMed] [Google Scholar]
- 15.Steinbach JH, Akk G. Modulation of GABA(A) receptor channel gating by pentobarbital. J Physiol. 2001;537:715–733. doi: 10.1111/j.1469-7793.2001.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rüsch D, Zhong H, Forman SA. Gating allosterism at a single class of etomidate sites on alpha1beta2gamma2L GABA-A receptors accounts for both direct activation and agonist modulation. J Biol Chem. 2004;279:20982–20992. doi: 10.1074/jbc.M400472200. [DOI] [PubMed] [Google Scholar]
- 17.Rüsch D, Neumann E, Wulf H, Forman SA. An Allosteric Coagonist Model for Propofol Effects on alpha1beta2gamma2L gamma-Aminobutyric Acid Type A Receptors. Anesthesiology. 2012;116:47–55. doi: 10.1097/ALN.0b013e31823d0c36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forman SA, Miller KW. Anesthetic sites and allosteric mechanisms of action on Cys-loop ligand-gated ion channels. Canadian journal of anaesthesia = Journal canadien d’anesthesie. 2011;58:191–205. doi: 10.1007/s12630-010-9419-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiser BP, Woll KA, Dailey WP, Eckenhoff RG. Mechanisms revealed through general anesthetic photolabeling. Curr Anesthesiol Rep. 2014;4:57–66. doi: 10.1007/s40140-013-0040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woll KA, Dailey WP, Brannigan G, Eckenhoff RG. Shedding Light on Anesthetic Mechanisms: Application of Photoaffinity Ligands. Anesth Analg. 2016 doi: 10.1213/ANE.0000000000001365. IN THIS ISSUE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yip GM, Chen ZW, Edge CJ, Smith EH, Dickinson R, Hohenester E, Townsend RR, Fuchs K, Sieghart W, Evers AS, Franks NP. A propofol binding site on mammalian GABAA receptors identified by photolabeling. Nat Chem Biol. 2013;9:715–720. doi: 10.1038/nchembio.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Addona GH, Husain SS, Stehle T, Miller KW. Geometric isomers of a photoactivable general anesthetic delineate a binding site on adenylate kinase. J Biol Chem. 2002;277:25685–25691. doi: 10.1074/jbc.M201303200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Miller KW. Dodecyl maltopyranoside enabled purification of active human GABA type A receptors for deep and direct proteomic sequencing. Mol Cell Proteomics. 2015;14:724–738. doi: 10.1074/mcp.M114.042556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dostalova Z, Liu A, Zhou X, Farmer SL, Krenzel ES, Arevalo E, Desai R, Feinberg-Zadek PL, Davies PA, Yamodo IH, Forman SA, Miller KW. High-level expression and purification of Cys-loop ligand-gated ion channels in a tetracycline-inducible stable mammalian cell line: GABA(A) and serotonin receptors. Protein Sci. 2010 doi: 10.1002/pro.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiara DC, Dostalova Z, Jayakar SS, Zhou X, Miller KW, Cohen JB. Mapping general anesthetic binding site(s) in human alpha1beta3 gamma-aminobutyric acid type A receptors with [(3)H]TDBzl-etomidate, a photoreactive etomidate analogue. Biochemistry. 2012;51:836–847. doi: 10.1021/bi201772m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, Cohen JB. Specificity of intersubunit general anesthetic binding sites in the transmembrane domain of the human alpha1beta3gamma2 GABAA receptor. J Biol Chem. 2013;288:19343–19357. doi: 10.1074/jbc.M113.479725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Jounaidi Y, Sarver LS, Zhou XJ, Cafiso DS, Miller KW. γ-Aminobutyric acid induced conformational change in GABAAR measured by pulsed electron paramagnetic resonance. Biophys J. 2014;106:547a. [Google Scholar]
- 28.Arevalo E, Chiara DC, Cohen JB, Miller KW. Photolabeling of the Open Channel State of the Nicotinic Acetylcholine Receptor. Biophys J. 2003;84:230a. [Google Scholar]
- 29.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alifimoff JK, Firestone LL, Miller KW. Anaesthetic potencies of primary alkanols: implications for the molecular dimensions of the anaesthetic site. Brit J Pharmacol. 1989;96:9–16. doi: 10.1111/j.1476-5381.1989.tb11777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byrem WC, Armstead SC, Kobayashi S, Eckenhoff RG, Eckmann DM. A guest molecule-host cavity fitting algorithm to mine PDB for small molecule targets. Biochim Biophys Acta. 2006;1764:1320–1324. doi: 10.1016/j.bbapap.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 32.Eckenhoff RG. Promiscuous ligands and attractive cavities: how do the inhaled anesthetics work? Molecular interventions. 2001;1:258–268. [PubMed] [Google Scholar]
- 33.Spurny R, Billen B, Howard RJ, Brams M, Debaveye S, Price KL, Weston DA, Strelkov SV, Tytgat J, Bertrand S, Bertrand D, Lummis SC, Ulens C. Multisite binding of a general anesthetic to the prokaryotic pentameric Erwinia chrysanthemi ligand-gated ion channel (ELIC) J Biol Chem. 2013;288:8355–8364. doi: 10.1074/jbc.M112.424507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan J, Chen Q, Willenbring D, Mowrey D, Kong XP, Cohen A, Divito CB, Xu Y, Tang P. Structure of the pentameric ligand-gated ion channel GLIC bound with anesthetic ketamine. Structure. 2012;20:1463–1469. doi: 10.1016/j.str.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Husain SS, Ziebell MR, Ruesch D, Hong F, Arevalo E, Kosterlitz JA, Olsen RW, Forman SA, Cohen JB, Miller KW. 2-(3-Methyl-3H–diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H–imidazole-5-carboxylate: A Derivative of the Stereoselective General Anesthetic Etomidate for Photolabeling Ligand-Gated Ion Channels. J Med Chem. 2003;46:1257–1265. doi: 10.1021/jm020465v. [DOI] [PubMed] [Google Scholar]
- 36.Liao M, Sonner JM, Husain SS, Miller KW, Jurd R, Rudolph U, Eger EI., 2nd R (+) etomidate and the photoactivable R (+) azietomidate have comparable anesthetic activity in wild-type mice and comparably decreased activity in mice with a N265M point mutation in the gamma-aminobutyric acid receptor beta3 subunit. Anesth Analg. 2005;101:131–135. doi: 10.1213/01.ANE.0000153011.64764.6F. [DOI] [PubMed] [Google Scholar]
- 37.Husain SS, Nirthanan S, Ruesch D, Solt K, Cheng Q, Li GD, Arevalo E, Olsen RW, Raines DE, Forman SA, Cohen JB, Miller KW. Synthesis of trifluoromethylaryl diazirine and benzophenone derivatives of etomidate that are potent general anesthetics and effective photolabels for probing sites on ligand-gated ion channels. J Med Chem. 2006;49:4818–4825. doi: 10.1021/jm051207b. [DOI] [PubMed] [Google Scholar]
- 38.Savechenkov PY, Zhang X, Chiara DC, Stewart DS, Ge R, Zhou X, Raines DE, Cohen JB, Forman SA, Miller KW, Bruzik KS. Allyl m-trifluoromethyldiazirine mephobarbital: an unusually potent enantioselective and photoreactive barbiturate general anesthetic. J Med Chem. 2012;55:6554–6565. doi: 10.1021/jm300631e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee-Son S, Waud BE, Waud DR. A comparison of the potencies of a series of barbiturates at the neuromuscular junction and on the central nervous system. J Pharmacol Exp Ther. 1975;195:251–256. [PubMed] [Google Scholar]
- 40.Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility. J Pharmacol Exp Ther. 2001;297:338–351. [PubMed] [Google Scholar]
- 41.Hall MA, Xi J, Lor C, Dai S, Pearce R, Dailey WP, Eckenhoff RG. m-Azipropofol (AziPm) a photoactive analogue of the intravenous general anesthetic propofol. J Med Chem. 2010;53:5667–5675. doi: 10.1021/jm1004072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart DS, Savechenkov PY, Dostalova Z, Chiara DC, Ge R, Raines DE, Cohen JB, Forman SA, Bruzik KS, Miller KW. p-(4-Azipentyl)propofol: A Potent Photoreactive General Anesthetic Derivative of Propofol. J Med Chem. 2011;54:8124–8135. doi: 10.1021/jm200943f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akk G, Covey DF, Evers AS, Steinbach JH, Zorumski CF, Mennerick S. Mechanisms of neurosteroid interactions with GABA(A) receptors Pharmacol Ther. 2007:35–57. doi: 10.1016/j.pharmthera.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Darbandi-Tonkabon R, Hastings WR, Zeng CM, Akk G, Manion BD, Bracamontes JR, Steinbach JH, Mennerick SJ, Covey DF, Evers AS. Photoaffinity labeling with a neuroactive steroid analogue 6-azi-pregnanolone labels voltage-dependent anion channel-1 in rat brain. J Biol Chem. 2003;278:13196–13206. doi: 10.1074/jbc.M213168200. [DOI] [PubMed] [Google Scholar]
- 45.Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Husain SS, Stewart D, Desai R, Hamouda AK, Li SG, Kelly E, Dostalova Z, Zhou X, Cotten JF, Raines DE, Olsen RW, Cohen JB, Forman SA, Miller KW. p-Trifluoromethyldiazirinyl-etomidate: a potent photoreactive general anesthetic derivative of etomidate that is selective for ligand-gated cationic ion channels. J Med Chem. 2010;53:6432–6444. doi: 10.1021/jm100498u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jayakar SS, Zhou X, Chiara DC, Dostalova Z, Savechenkov PY, Bruzik KS, Dailey WP, Miller KW, Eckenhoff RG, Cohen JB. Multiple propofol-binding sites in a gamma-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J Biol Chem. 2014;289:27456–27468. doi: 10.1074/jbc.M114.581728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen ZW, Manion B, Townsend RR, Reichert DE, Covey DF, Steinbach JH, Sieghart W, Fuchs K, Evers AS. Neurosteroid analog photolabeling of a site in the third transmembrane domain of the beta3 subunit of the GABA(A) receptor. Mol Pharmacol. 2012;82:408–419. doi: 10.1124/mol.112.078410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cestari IN, Min KT, Kulli JC, Yang J. Identification of an amino acid defining the distinct properties of murine beta1 and beta3 subunit-containing GABA(A) receptors. J Neurochem. 2000;74:827–838. doi: 10.1046/j.1471-4159.2000.740827.x. [DOI] [PubMed] [Google Scholar]
- 50.Bali M, Akabas MH. Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol. 2004;65:68–76. doi: 10.1124/mol.65.1.68. [DOI] [PubMed] [Google Scholar]
- 51.McCracken ML, Borghese CM, Trudell JR, Harris RA. A transmembrane amino acid in the GABAA receptor beta2 subunit critical for the actions of alcohols and anesthetics. J Pharmacol Exp Ther. 2010;335:600–606. doi: 10.1124/jpet.110.170472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stewart DS, Hotta M, Desai R, Forman SA. State-Dependent Etomidate Occupancy of Its Allosteric Agonist Sites Measured in a Cysteine-Substituted GABAA Receptor. Mol Pharmacol. 2013;83:1200–1208. doi: 10.1124/mol.112.084558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stewart DS, Hotta M, Li GD, Desai R, Chiara DC, Olsen RW, Forman SA. Cysteine Substitutions Define Etomidate Binding and Gating Linkages in the alpha-M1 Domain of gamma-Aminobutyric Acid Type A (GABAA) Receptors. J Biol Chem. 2013;288:30373–30386. doi: 10.1074/jbc.M113.494583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stewart DS, Pierce DW, Hotta M, Stern AT, Forman SA. Beta N265 in Gamma-Aminobutyric Acid Type A Receptors is Both a Binding and Efficacy Determinant for Etomidate and Propofol. PloS one. 2014 Oct 27;9(10):e111470. doi: 10.1371/journal.pone.0111470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stern AT, Forman SA. A Cysteine Substitution Probes beta3H267 Interactions with Propofol and Other Potent Anesthetics in alpha1beta3gamma2L gamma-Aminobutyric Acid Type A Receptors. Anesthesiology. 2016;124:89–100. doi: 10.1097/ALN.0000000000000934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eaton MM, Cao LQ, Chen ZW, Franks NP, Evers AS, Akk G. Mutational analysis of the putative high-affinity propofol binding site in human b3 homomeric GABAA receptors. Mol Pharmacol. 2015;88:736–745. doi: 10.1124/mol.115.100347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stewart DS, Desai R, Cheng Q, Liu A, Forman SA. Tryptophan mutations at azi-etomidate photo-incorporation sites on a1 or b2 subunits enhance GABAA receptor gating and reduce etomidate modulation. Mol Pharmacol. 2008;74:1687–1695. doi: 10.1124/mol.108.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bracamontes JR, Steinbach JH. Steroid interaction with a single potentiating site is sufficient to modulate GABA-A receptor function. Mol Pharmacol. 2009;75:973–981. doi: 10.1124/mol.108.053629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bertaccini EJ, Yoluk O, Lindahl ER, Trudell JR. Assessment of homology templates and an anesthetic binding site within the gamma-aminobutyric acid receptor. Anesthesiology. 2013;119:1087–1095. doi: 10.1097/ALN.0b013e31829e47e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer PJ. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- 62.Forman SA, Chiara DC, Miller KW. Anesthetics target interfacial transmembrane sites in nicotinic acetylcholine receptors. Neuropharmacology. 2015;96:169–177. doi: 10.1016/j.neuropharm.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bondarenko V, Mowrey DD, Tillman TS, Seyoum E, Xu Y, Tang P. NMR structures of the human alpha7 nAChR transmembrane domain and associated anesthetic binding sites. Biochimica et biophysica acta. 2014;1838:1389–1395. doi: 10.1016/j.bbamem.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jayakar SS, Dailey WP, Eckenhoff RG, Cohen JB. Identification of propofol binding sites in a nicotinic acetylcholine receptor with a photoreactive propofol analog. J Biol Chem. 2013;288:6178–6189. doi: 10.1074/jbc.M112.435909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hill-Venning C, Belelli D, Peters JA, Lambert JJ. Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br J Pharmacol. 1997;120:749–756. doi: 10.1038/sj.bjp.0700927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Varagic Z, Wimmer L, Schnurch M, Mihovilovic MD, Huang S, Rallapalli S, Cook JM, Mirheydari P, Ecker GF, Sieghart W, Ernst M. Identification of novel positive allosteric modulators and null modulators at the GABAA receptor alpha+beta- interface. Br J Pharmacol. 2013;169:371–383. doi: 10.1111/bph.12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varagic Z, Ramerstorfer J, Huang S, Rallapalli S, Sarto-Jackson I, Cook J, Sieghart W, Ernst M. Subtype selectivity of alpha+beta- site ligands of GABAA receptors: identification of the first highly specific positive modulators at alpha6beta2/3gamma2 receptors. Br J Pharmacol. 2013;169:384–399. doi: 10.1111/bph.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mohler H, Fritschy JM, Rudolph U. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 2002;300:2–8. doi: 10.1124/jpet.300.1.2. [DOI] [PubMed] [Google Scholar]
- 69.Cotten JF, Husain SS, Forman SA, Miller KW, Kelly EW, Nguyen HH, Raines DE. Methoxycarbonyl-etomidate: a novel rapidly metabolized and ultra-short-acting etomidate analogue that does not produce prolonged adrenocortical suppression. Anesthesiology. 2009;111:240–249. doi: 10.1097/ALN.0b013e3181ae63d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pejo E, Santer P, Jeffrey S, Gallin H, Husain SS, Raines DE. Analogues of Etomidate: Modifications around Etomidate’s Chiral Carbon and the Impact on In Vitro and In Vivo Pharmacology. Anesthesiology. 2014;121:290–301. doi: 10.1097/ALN.0000000000000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li GD, Chiara DC, Cohen JB, Olsen RW. Numerous classes of general anesthetics inhibit etomidate binding to gamma-aminobutyric acid type A (GABAA) receptors. J Biol Chem. 2010;285:8615–8620. doi: 10.1074/jbc.M109.074708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jayakar SS, Zhou X, Savechenkov PY, Chiara DC, Desai R, Bruzik KS, Miller KW, Cohen JB. Positive and Negative Allosteric Modulation of an alpha1beta3gamma2 gamma-Aminobutyric Acid Type A (GABAA) Receptor by Binding to a Site in the Transmembrane Domain at the gamma+-beta- Interface. J Biol Chem. 2015;290:23432–23446. doi: 10.1074/jbc.M115.672006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]