

Abstract
Glomerular disease is characterized by morphologic changes in podocyte cells accompanied by inflammation and fibrosis. Thymosin β4 regulates cell morphology, inflammation, and fibrosis in several organs and administration of exogenous thymosin β4 improves animal models of unilateral ureteral obstruction and diabetic nephropathy. However, the role of endogenous thymosin β4 in the kidney is unknown. We demonstrate that thymosin β4 is expressed prominently in podocytes of developing and adult mouse glomeruli. Global loss of thymosin β4 did not affect healthy glomeruli, but accelerated the severity of immune-mediated nephrotoxic nephritis with worse renal function, periglomerular inflammation, and fibrosis. Lack of thymosin β4 in nephrotoxic nephritis led to the redistribution of podocytes from the glomerular tuft toward the Bowman capsule suggesting a role for thymosin β4 in the migration of these cells. Thymosin β4 knockdown in cultured podocytes also increased migration in a wound-healing assay, accompanied by F-actin rearrangement and increased RhoA activity. We propose that endogenous thymosin β4 is a modifier of glomerular injury, likely having a protective role acting as a brake to slow disease progression.
Keywords: cytoskeleton, fibrosis, glomerulus, inflammation, podocyte
End-stage renal failure is a devastating condition requiring lifelong dialysis and transplantation and a risk factor for all-cause mortality and cardiovascular disease.1 Many cases are due to disruption of the glomerular filtration barrier, consisting of epithelial podocytes, the endothelium, the mesangium, and the glomerular basement membrane.2 Podocytes have a unique shape maintained by a complex cytoskeleton,3 with branched foot process extensions that abut each other at slit diaphragms. During glomerular injury, the podocyte architecture is perturbed, resulting in defective filtration and proteinuria,2, 3, 4 often with inflammatory components characterized by leukocyte infiltration followed by glomerulosclerosis and tubulointerstitial fibrosis.5
Thymosin β4 (Tmsb4x) is a naturally occurring peptide. It is the major G-actin sequestering protein in mammalian cells,6 with critical roles in maintaining the cell cytoskeleton. In animal models, exogenous Tmsb4x has beneficial effects in diverse pathologies including myocardial infarction,7 stroke,8 dry eye,9 and inflammatory lung disease,10 and there are clinical trials assessing Tmsb4x treatment in wound healing and cardioprotection.11 The utility of Tmsb4x in these pathologies has been attributed to modulation of several cellular functions including cell motility,12 differentiation,13 survival,14 angiogenesis,15 inflammation,16 and fibrosis.10
Recent studies investigated exogenous Tmsb4x as a treatment for kidney disease.17 Tmsb4x reduced renal tubulointerstitial fibrosis after unilateral ureteral obstruction in mice, potentially through decreasing plasminogen activator inhibitor-1 expression and dampening transforming growth factor-β1 signaling.18 In KK Cg-Ay/J mice, a model of type 2 diabetes mellitus, daily Tmsb4x treatment for 3 months reduced albuminuria and attenuated renal pathology.19 Furthermore, N-acetyl-seryl-aspartyl-lysyl-proline, the N-terminal tetrapeptide generated by Tmsb4x cleavage,20 has beneficial effects on fibrosis and inflammation in unilateral ureteral obstruction, remnant kidneys, diabetic nephropathy, and glomerulonephritis.18, 21, 22, 23
Tmsb4x transcripts are detectable by in situ hybridization in developing and adult glomeruli,24 with strong expression in podocytes.24, 25 Furthermore, in rat remnant kidneys, proteomic analysis of laser-capture dissected glomeruli demonstrated significantly increased Tmsb4x in sclerotic versus normal glomeruli.26 Despite such evidence of expression and beneficial renal effects, the functional importance of endogenous Tmsb4x in the kidney during health and disease is completely unknown.
In this study, we confirmed that Tmsb4x is highly expressed in the kidney glomerulus, predominantly in podocytes. Using global Tmsb4x knockout mice,27 we demonstrated that endogenous Tmsb4x was dispensable in healthy glomeruli. Furthermore, in an experimental model of glomerular damage, lack of Tmsb4x worsened disease progression by (i) enhancing podocyte migration, facilitating their redistribution from the glomerular tuft to the Bowman capsule, and (ii) increasing periglomerular inflammation and interstitial fibrosis. Thus, we provide the first evidence that endogenous Tmsb4x is critical in the progression of glomerular disease.
Results
Tmsb4x is expressed in mouse glomerular podocytes
Tmsb4x mRNA levels were assessed in the spleen, liver, heart, and whole kidney of healthy adult mice. The highest transcript levels were found in the spleen with ∼10 times less Tmsb4x in the kidney (Figure 1a). Using Dynabead perfusion (Thermo Fisher Scientific, Waltham, MA),28 we isolated glomeruli and found that Tmsb4x levels were significantly enriched in glomeruli compared with the rest of the kidney (Figure 1a). In situ hybridization detected Tmsb4x expression in immature glomeruli of embryonic day 16.5 developing kidneys, predominantly in podocytes (Figure 1b and c). The protein expression of Tmsb4x was also assessed by immunohistochemistry, and we found strong localization in glomerular podocytes at embryonic day 18 (Figure 1d). This expression pattern was maintained in 1-week-old postnatal (Figure 1e) and 8-week-old adult kidneys (Figure 1f). Tmsb4x podocyte expression was further confirmed by colocalization of Tmsb4x with nephrin (Nphs1), a slit diaphragm component29 (Figure 1g–i), and nestin, an intermediate filament protein expressed in mature podocytes30 (Supplementary Figure S1A–C). In contrast, Tmsb4x did not colocalize with the pan-endothelial marker Cd31 (Supplementary Figure S1D–F).
Figure 1.

Tmsb4x expression in the mouse glomerulus. (a) Quantification of thymosin β4 (Tmsb4x) mRNA levels in adult mouse spleen, liver, heart, and kidney by quantitative reverse transcriptase polymerase chain reaction. Tmsb4x expression was also quantified in glomeruli-depleted [glom (-)] and glomeruli-enriched [glom (+)] kidney homogenates. The bars represent the mean of 3 samples ± SEM. (b,c) In situ hybridization for Tmsb4x on embryonic day (E) 16.5 mouse kidney sections. Cells positive for Tmsb4x are indicated by arrows. Immunohistochemistry for Tmsb4x in the mouse glomerulus at E18 (d), 1 week (e), and 8 weeks (f) of age. Cells positive for Tmsb4x are indicated by arrows. Representative images of Tmsb4x (g) and nephrin (Nphs1) (h) staining in the mouse adult wild-type glomerulus visualized by fluorescent microscopy. (i) Merged images showing Nphs1 (red) and Tmsb4x (green) staining; areas of colocalization are indicated by arrows. Bar = 20 μm. **P ≤ 0.01.
Lack of endogenous Tmsb4x has no effect on healthy glomeruli
We examined mice with a global loss of Tmsb4x27 to assess the importance of endogenous Tmsb4x in healthy glomeruli. As Tmsb4x is mapped to the X chromosome,31 we crossed hemizygous null male mice (Tmsb4x−/y) with heterozygous Tmsb4x+/− adult female mice (Figure 2a). We found no lethal developmental abnormalities with the offspring conforming to Mendelian ratios (Supplementary Table S1). Tmsb4x−/y mice had albumin excretion (Figure 2b) and blood urea nitrogen (Figure 2c) levels similar to those of male wild-type Tmsb4x+/y mice at the ages of 1, 3, and 6 months. There was no difference in body weight at 1 (Tmsb4x+/y = 23.8 ± 0.7 g; Tmsb4x−/y = 23.4 ± 0.4 g), 3 (Tmsb4x+/y = 30.0 ± 0.5 g; Tmsb4x−/y = 29.2 ± 0.7 g), or 6 (Tmsb4x+/y = 35.8 ± 1.0 g; Tmsb4x−/y = 35.3 ± 1.6 g) months of age. Using semiquantitative analysis of light microscopy images, we found no differences in glomerular morphology between 6-month-old Tmsb4x+/y and Tmsb4x−/y mice (Figure 2d–f). This was confirmed by transmission electron microscopy with normal foot process architecture, laminar structure of the basement membrane, and the presence of endothelial fenestrae in both Tmsb4x+/y and Tmsb4x−/y mice (Figure 2g and h). We demonstrated the loss of Tmsb4x protein in glomeruli of Tmsb4x−/y mice compared with Tmsb4x+/y (Figure 2i and j), along with undetectable Tmsb4x mRNA levels in whole kidneys (Figure 2k). We examined whether there was any compensation for the lack of Tmsb4x by other β-thymosins, but found no changes in the renal mRNA levels of Tmsb10, Tmsb15a, Tmsb15b, and Tmsb15l (Figure 2l–o). We also assessed mRNA levels of genes involved in actin polymerization and found no differences in profilin (Pfn) 1 and 2 and destrin (Dstn) between Tmsb4x−/y and Tmsb4x+/y mice (Supplementary Figure S2A–C). In contrast, cofilin 1 (Cfl1) mRNA levels were significantly increased in Tmsb4x−/y kidneys by ∼30% compared with Tmsb4x+/y mice (Supplementary Figure S2D). We specifically examined Cfl1 mRNA levels in podocytes and found no change after knock down of endogenous Tmsb4x by small, interfering RNA (siRNA) (Supplementary Figure S2E). Furthermore, there was no difference in Cfl1 mRNA levels in primary podocytes isolated from Tmsb4x−/y and Tmsb4x+/y mice (Supplementary Figure S2F). Finally, the mRNA levels of genes important for podocyte function (Nphs1, Nphs2, Synpo, Cd2ap, and Wt1) were unchanged in Tmsb4x−/y and Tmsb4x+/y kidneys (Supplementary Figure S3A–E).
Figure 2.

Renal function and glomerular morphology in Tmsb4x+/yand Tmsb4x−/ymice. (a) Breeding scheme: heterozygous female mice (Tmsb4x+/−) were bred with hemizygous null male mice (Tmsb4x−/y). Male wild-type (Tmsb4x+/y) and Tmsb4x−/y mice were compared in all subsequent experiments. (b) Twenty-four hour albumin excretion in urine of Tmsb4x+/y (n = 14–15) and Tmsb4x−/y (n = 9–10) mice collected at 1, 3, and 6 months of age. Data were log-transformed before analysis. (c) Blood urea nitrogen (BUN) concentration in Tmsb4x+/y (n = 9–15) and Tmsb4x−/y mice (n = 7–9) at 1, 3, and 6 months of age. Data were log-transformed before analysis. Representative images of periodic acid–Schiff staining of paraffin-embedded sections from Tmsb4x+/y (d) and Tmsb4x−/y(e) kidneys. Bar = 20 μm. The glomerular score (f) was quantified as explained in the Methods section (Tmsb4x+/y: n = 4; Tmsb4x−/y: n = 5). Representative images of the glomerular architecture of Tmsb4x+/y (g) and Tmsb4x−/y (h) kidneys assessed by transmission electron microscopy. An average of 5 glomeruli were examined per animal (Tmsb4x+/y, n = 4; Tmsb4x−/y, n = 3). Representative images of immunohistochemistry for thymosin β4 (Tmsb4x) on paraffin-embedded sections from Tmsb4x+/y (i) and Tmsb4x−/y (j) kidneys from 6-month-old mice. Tmsb4x-positive cells are indicated by arrows. Note: nonspecific staining in tubules in Tmsb4x+/y mice and Tmsb4x−/y mice. Quantification of Tmsb4x (k), Tmsb10 (l), Tmsb15a (m), Tmsb15b (n), and Tmsb15l (o) mRNA levels in whole-kidney homogenates of Tmsb4x+/y (n = 8) and Tmsb4x−/y (n = 4) mice by quantitative reverse transcriptase polymerase chain reaction. Data are presented as mean ± SEM. ***P ≤ 0.001. Bar = 20 μm. EC, endothelial cell; GBM, glomerular basement membrane; HPRT, hypoxanthine-guanine phosphoribosyltransferase; Pod, podocyte; Tmsb4x, thymosin β4; Tmsb10, thymosin β10; Tmsb15a, thymosin β15a; Tmsb15b, thymosin β15b; Tmsb15b1, thymosin β15b-like.
Lack of endogenous Tmsb4x worsens renal function and glomerular injury in nephrotoxic serum nephritis
Our results suggest that the lack of Tmsb4x does not affect the function and morphology of healthy glomeruli. Therefore, we investigated whether Tmsb4x has a role in glomerular disease. We utilized the nephrotoxic serum (NTS) nephritis model, which replicates some of the pathologic features of human crescentic glomerulonephritis.32 NTS nephritis involves the injury of intrinsic glomerular cells, including podocytes, as well as leukocyte infiltration, glomerulosclerosis, and tubulointerstitial fibrosis,33 processes in which Tmsb4x has been implicated.10, 16 We predicted that the lack of global Tmsb4x may exacerbate NTS nephritis severity and examined this in 3-month-old Tmsb4x−/y and Tmsb4x+/y mice (Figure 3a). Albuminuria and the albumin/creatinine ratio were significantly increased in Tmsb4x+/y mice 21 days after NTS administration compared with the levels before immunization (P < 0.001 in both cases) (Figure 3b and c). Strikingly, both albuminuria and the albumin/creatinine ratio were further enhanced by ∼5- and 7-fold, respectively, when NTS was injected in Tmsb4x−/y mice compared with Tmsb4x+/y mice (P < 0.01 in both cases) (Figure 3b and c). Administration of NTS to Tmsb4x−/y mice also significantly increased plasma creatinine (P < 0.01) (Figure 3d), impaired creatinine clearance (P < 0.01) (Figure 3e), and raised the blood urea nitrogen level (P < 0.05) (Figure 3f) compared with Tmsb4x+/y mice with NTS nephritis. We found no difference in Tmsb4x levels in whole kidneys obtained from Tmsb4x+/y mice without disease or administered NTS (Supplementary Figure S4A). Furthermore, in kidney biopsy specimens obtained from patients with either rapidly progressive glomerulonephritis or lupus nephritis, there was no change in glomerular or tubulointerstitial TMSB4X mRNA levels compared with living donor control kidneys (Supplementary Figure S4B and C).
Figure 3.

Renal function in Tmsb4x+/yand Tmsb4x−/ymice after the induction of nephrotoxic nephritis. (a) Outline of experimental strategy. Twenty-four–hour albumin excretion in urine (b), urinary albumin to urinary creatinine ratio (c), plasma creatinine concentration (d), creatinine clearance (e), and blood urea nitrogen (BUN) concentration (f) of Tmsb4x+/y and Tmsb4x−/y mice. Samples were collected before immunization with Freund’s adjuvant (Tmsb4x+/y, control group) and 21 days after administration of nephrotoxic serum (NTS) (Tmsb4x+/y + NTS and Tmsb4x−/y + NTS); n = 12 in each group. Data were log-transformed before analysis and presented as mean ± SEM. (g) Glomerular score was quantified as described in the Methods section in Tmsb4x+/y (n = 5), Tmsb4x+/y + NTS (n = 9), and Tmsb4x−/y + NTS (n = 6) mice. Data are presented as mean ± SEM. Representative images of periodic acid–Schiff staining in glomeruli from control mice (Tmsb4x+/y; h) and mice administered NTS; Tmsb4x+/y + NTS (i) and Tmsb4x−/y + NTS (j) are shown. Bar = 20 μm. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. s.c., subcutaneous; Tmsb4x, thymosin β4.
Seven days after NTS administration, we observed mild glomerular injury in Tmsb4x−/y and Tmsb4x+/y mice, with some glomeruli containing hyaline deposits, increased mesangial matrix, and occasional adhesion of the glomerular tuft to the Bowman capsule (Supplementary Figure S5A–C). After 21 days, there was a range of abnormalities in Tmsb4x−/y and Tmsb4x+/y mice injected with NTS including collapse of capillary loops, segmental or global glomerulosclerosis, adhesion of the glomerular tuft to the Bowman capsule, and glomerular epithelial hyperplasia lesions, a feature of early crescent formation in this model.34 Semiquantitative histologic scoring (Supplementary Figure S6A–E) by 2 blinded observers revealed that Tmsb4x+/y mice injected with NTS had a significantly increased mean glomerular score compared with Tmsb4x+/y mice without disease (P < 0.001) (Figure 3g–j). Glomeruli of Tmsb4x−/y administered NTS had an even higher glomerular mean score that was significantly greater than that in Tmsb4x+/y mice with NTS nephritis (P < 0.05). This was associated with an increased proportion of sclerotic glomeruli and an increased incidence of epithelial hyperplasia lesions in Tmsb4x−/y compared with Tmsb4x+/y mice administered NTS (Supplementary Figure S6F).
We examined whether the difference in disease severity between Tmsb4x−/y and Tmsb4x+/y mice administered NTS could be due to a decrease in binding of the antiglomerular antibody but found no difference in the amount of sheep IgG deposited within the glomerulus (Supplementary Figure S7A–C). We assessed whether the lack of Tmsb4x changes the humoral immune response to sheep IgG. NTS injection led to significantly increased production of circulating murine IgG1 and IgG2a, but not IgG2b or IgG3, against sheep IgG compared with Tmsb4x+/y mice without disease. However, there was no difference in the plasma titers of any of the IgG subclasses between Tmsb4x−/y and Tmsb4x+/y mice administered NTS (Supplementary Figure S7D–G).
Changes in podocyte distribution in Tmsb4x−/y glomeruli after NTS nephritis
After NTS nephritis, we found that Tmsb4x still colocalized with Nphs1 (Figure 4a–c) and subsequently examined the effect that a lack of Tmsb4x had on podocytes in this model. First, we quantified Wilms Tumor 1 postive (WT1+) podocyte numbers,35 inside and outside of the glomerular tuft (Figure 4d–f). The number of glomerular tuft WT1+ cells was unchanged in Tmsb4x+/y mice after NTS injection compared with mice without disease. However, NTS administration to Tmsb4x−/y mice significantly reduced the number of WT1+ glomerular tuft cells compared with Tmsb4x+/y mice with NTS nephritis (P < 0.001) (Figure 4g). This finding persisted after normalizing the WT1+ cell number to the glomerular tuft area (P < 0.05) (Figure 4h). In contrast, there was an increased number of WT1+ cells outside the glomerular tuft in both Tmsb4x+/y and Tmsb4x−/y mice after NTS administration; this was more prominent and significantly different in the Tmsb4x−/y animals (P < 0.05 compared with Tmsb4x+/y without disease) (Figure 4i). The total number of podocytes in the whole glomerulus did not differ between any of the groups (Figure 4j), suggesting that lack of Tmsb4x leads to podocyte redistribution from the glomerular tuft toward the Bowman capsule rather than affecting podocyte cell death. To support this, we assessed podocyte apoptosis using terminal deoxynucleotidyltransferase–mediated dUTP nick end-labeling (TUNEL)36 in combination with WT1 staining (Supplementary Figure S8A–C). Administration of NTS increased the number of glomerular apoptotic cells compared with healthy mice, and this effect was significant in Tmsb4+/y mice with glomerular disease (P < 0.05) (Supplementary Figure S8D). However, the number of TUNEL+/WT1+ cells was not significantly different between any of the groups (Supplementary Figure S8E).
Figure 4.

Podocyte assessment in Tmsb4x+/yandTmsb4x−/ymice after induction of nephrotoxic nephritis. Representative image of nephrin (Nphs1; podocytes) (a) and Tmsb4x (b) staining in the adult wild-type mouse glomerulus 21 days after injection of nephrotoxic serum (NTS). Cells positive for both Nphs1 (green) and Tmsb4x (red) are indicated by arrows in the merged image (c). Representative images of glomeruli from Tmsb4x+/y (d), Tmsb4x+/y + NTS (e), and Tmsb4x−/y + NTS (f) mice stained for WT1. The glomerular tuft areas are indicate the dotted lines. (g–j) Graphs showing the number of WT1+ cells in the glomerular tuft (g), the number of WT1+ cells in the glomerular tuft normalized to the glomerular area (h), the number of WT1+ cells in the area of the glomerulus surrounding the glomerular tuft (i), and the number of WT1+ cells in the whole glomerulus (j). Cells were counted as 50 glomeruli per sample, except in (h), where cells were counted and normalized to the glomerular area as 15 glomeruli per sample. Data are presented as mean ± SEM. Tmsb4x+/y (n = 5), Tmsb4+/y + NTS (n = 9), and Tmsb4x−/y + NTS (n = 6) mice. Bar = 20 μm. Tmsb4x, thymosin β4.
Lack of Tmsb4x induces migration and modulates the cytoskeleton of podocytes in vitro
We postulated that the redistribution of podocytes in nephrotoxic nephritis may be due to changes in cell migration driven by lack of Tmsb4x. Therefore, we transfected cultured differentiated mouse podocytes37 with siRNA against Tmsb4x (Figure 5a); this resulted in >90% knockdown in Tmsb4x levels (Figure 5b) (P < 0.001). Knockdown of endogenous Tmsb4x did not affect podocyte viability (Figure 5c) but increased the number of cells that migrated into the wound area in a wound-healing assay (P < 0.05) (Figure 5d and e). Because the cytoskeleton is essential for cell movement,38 we visualized podocyte actin by phalloidin staining and classified the filament organization as either cytoplasmic stress fibers (Figure 5f) or cortical actin (Figure 5g). Knockdown of endogenous Tmsb4x significantly increased the percentage of cells with stress actin fiber organization (P < 0.001) (Figure 5h). Finally, we assessed the effects of Tmsb4x knockdown on the activation of RhoA and Cdc42, which regulate actin dynamics and cell migration.39 There was increased RhoA activity in podocytes transfected with Tmsb4x siRNA compared with control siRNA (P < 0.05) (Figure 5i), whereas Cdc42 activity was unaffected (Figure 5j).
Figure 5.

Effects of downregulating endogenous Tmsb4x expression in podocytes in vitro. (a) Podocytes grown in vitro under permissive conditions were differentiated for 14 days before transfecting them with control siRNA or siRNA targeting Tmsb4x. (b) Quantification of Tmsb4x mRNA levels in podocytes 48 hours after transfection. (c) Cell viability after knockdown of endogenous Tmsb4x was assessed by MTT assay. (d) Podocyte migration after knockdown of endogenous Tmsb4x was assessed by a wound-healing assay, and the number of cells that migrated into the wound area was counted. (e) Representative images of podocytes transfected with control or Tmsb4x siRNA 0, 6, and 24 hours after wound formation. Representative images showing a podocyte with cytoplasmic stress fiber F-actin distribution (f) or cortical F-actin distribution (g). The percentage of cells with predominantly cytoplasmic stress fibers or cortical actin formation was quantified 48 hours after transfection (h). Quantification of active RhoA (i) and active Cdc42 (j) 48 hours after transfection. All experiments were repeated 3 to 4 times, and the data are presented as mean ± SEM. *P ≤ 0.05, ***P ≤ 0.001. IFN-γ, interferon-γ; Lip, lipofectamine; OD, optical density; siRNA, small, interfering RNA; TB, Tmsb4x, thymosin β4.
Macrophage accumulation and increased fibrosis in Tmsb4x−/y glomeruli after NTS nephritis
Tmsb4x is expressed in macrophages18, 40 and reduces inflammation in several disease settings.10, 16, 18, 41 As immune cell infiltration plays a critical role in the initiation and progression of crescentic glomerulonephritis,42, 43, 44 we decided to examine this in the NTS nephritis model. We found expression of Tmsb4x in F4/80+ macrophages surrounding the glomeruli and occasionally within the glomerular tuft (Figure 6a–c) and went on to examine the effect of Tmsb4x loss on glomerular inflammation in our experimental model.
Figure 6.

Assessment of inflammation in nephrotoxic nephritis. Representative images of F4/80 (macrophages) (a) and thymosin β4 (Tmsb4x) staining (b). Cells positive for both F4/80 (green) and Tmsb4x (red) are shown in the merged image (c) and are indicated by arrows. Images were taken by fluorescent microscopy. Bar = 20 μm. Representative images showing Cd3 (T-cell marker) staining in the glomerular tuft (arrows) and in the periglomerular area (arrowheads) of Tmsb4x+/y controls (d) and Tmsb4x+/y (e) and Tmsb4x−/y (f) mice 21 days after administration of nephrotoxic serum (NTS). The number of Cd3+ cells in the glomerular tuft (g) and in the periglomerular area (h) was counted as 50 glomeruli per sample, and the average number was calculated (Tmsb4x+/y, n = 5; Tmsb4x+/y + NTS, n = 9; Tmsb4x−/y + NTS, n = 6). Representative images showing F4/80 (activated macrophage marker) staining in the glomerular tuft and the periglomerular area of Tmsb4x+/y control (i), Tmsb4x+/y + NTS (j), and Tmsb4x−/y + NTS (k) mice. The number of F4/80+ cells in the glomerular tuft (l) and in the periglomerular area (m) was counted as 50 glomeruli per sample, and the average number was calculated (Tmsb4x+/y, n = 5; Tmsb4x+/y + NTS, n = 8; Tmsb4x−/y + NTS, n = 6). Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Bar = 20 μm. Tmsb4x, thymosin β4.
We measured the number of Cd3+ (T cells) and F4/80+ cells in Tmsb4x−/y and Tmsb4x+/y glomeruli. Twenty-one days after NTS injection, there was a significant increase in Cd3+ cells in the glomerular tuft of Tmsb4x−/y mice compared with Tmsb4x+/y mice without disease (P < 0.05) but no significant difference when comparing Tmsb4x−/y and Tmsb4x+/y mice with nephrotoxic nephritis (Figure 6d–g). There was also no difference in the number of periglomerular Cd3+ cells between experimental groups (Figure 6h). Seven days after NTS administration, the number of F4/80+ glomerular tuft cells was similar in all experimental groups, but were significantly increased in the periglomerular area of both NTS-injected Tmsb4x+/y and Tmsb4x−/y mice compared with healthy mice (P < 0.01) (Supplementary Figure S9A and B). The accumulation of F4/80+ cells persisted in Tmsb4x−/y mice 21 days after NTS administration, with increased numbers in both the glomerular tuft and periglomerular area compared with Tmsb4x+/y mice with or without disease (P < 0.01) (Figure 6i–m). mRNA levels of the pan-macrophage marker Cd68 were also significantly higher 21 days after NTS injection in whole-kidney homogenates obtained from Tmsb4x−/y compared with Tmsb4x+/y mice (Supplementary Figure S10A). Cd68 is expressed by all macrophages, but these comprise a diverse group that includes a broad spectrum of cellular phenotypes, often characterized as pro-inflammatory (M1 type) and tissue repair (M2 type) macrophages. We quantified the mRNA levels of M1 (Mcp1, Cd86) and M2 markers (Cd206, Arg1) and found all of these genes were significantly upregulated in Tmsb4x−/y mice compared with Tmsb4x+/y mice after NTS administration (Supplementary Figure S10B–E). This suggests that there is a global increase in macrophages in Tmsb4x−/y kidneys after NTS rather than a shift toward an M1 or M2 phenotype.
Finally, the increased accumulation of macrophages in the periglomerular area in Tmsb4x−/y mice with NTS was associated with increased periglomerular fibrosis, as shown by increased staining for both collagen IV (Figure 7a–c) and α-smooth muscle actin (Figure 7e–g) in sections from Tmsb4x−/y compared with Tmsb4x+/y mice injected with NTS along with increased whole-kidney mRNA levels of Col4a1 (P < 0.05) (Figure 7d) and Acta2 (P < 0.05) (Figure 7h).
Figure 7.

Assessment of fibrosis in nephrotoxic nephritis. Representative images showing collagen IV staining in glomeruli of Tmsb4x+/y (a), Tmsb4x+/y + nephrotoxic serum (NTS) (b), and Tmsb4x−/y + NTS (c) mice. (d) Quantification of collagen IV (Col4a1) mRNA levels in whole-kidney homogenates of Tmsb4x+/y (n = 8), Tmsb4x+/y + NTS (n = 9), and Tmsb4x−/y + NTS (n = 6) mice. Representative images showing α-smooth muscle actin (α-SMA) staining in glomeruli of Tmsb4x+/y (e) Tmsb4x+/y + NTS (f) and Tmsb4x−/y + NTS (g) mice. (h) Quantification of α-SMA (Acta2) mRNA levels in whole-kidney homogenates of Tmsb4x+/y (n = 8), Tmsb4x+/y + NTS (n = 8), and Tmsb4x−/y + NTS (n = 5) mice. Data are presented as mean ± SEM. *P ≤ 0.05, **P ≤ 0.01. Bar = 20 μm. Tmsb4x, thymosin β4.
Discussion
In this study, we found endogenous Tmsb4x was not required to maintain glomerular structure and function in healthy adult mice. However, in an experimental model of NTS nephritis, glomerular disease was exacerbated in mice lacking Tmsb4x accompanied by changes in the distribution of podocytes within the glomerulus, increased periglomerular macrophage accumulation, and enhanced fibrosis. These findings provide the first evidence that endogenous Tmsb4x modifies glomerular injury, likely having a protective role acting as a brake to slow disease progression.
We showed that Tmsb4x is expressed in developing and adult mouse glomeruli, predominantly localized to podocytes. Previous studies also found Tmsb4x in glomerular podocytes,25 but others reported the complete absence of Tmsb4x in the glomeruli of human fetal and adult kidneys45 and rat kidneys.26 These discrepancies may be due to differences in the antibodies and fixation methods used.46 Importantly, we obtained similar results for both Tmsb4x mRNA and protein and confirmed the specificity of antibody staining using tissue from Tmsb4x−/y mice as an additional negative control.
Given that Tmsb4x plays a role in actin binding,6 we initially hypothesized that the lack of endogenous Tmsb4x might disrupt the highly branched architecture of glomerular podocytes and impair renal function.3 However, the lack of Tmsb4x did not affect glomerular morphology or podocyte architecture of normal healthy mice in vivo. We found upregulation of Cfn1, which severs actin filaments, in whole Tmsb4x−/y kidneys. This could partly compensate for the lack of Tmsb4x and maintain actin dynamics47; however, Cfn1 was not specifically altered in podocytes lacking Tmsb4x, thus making this unlikely.
A significant finding of our study was that severity of glomerular disease induced by NTS was greater in Tmsb4x−/y mice compared with wild-type littermates. We postulate that endogenous Tmsb4x has a protective role in the setting of NTS, a prediction supported by a study showing that exogenous administration of Ac-SDKP ameliorated rat glomerulonephritis.22 However, we found that whole-mouse kidney Tmsb4x mRNA levels were unchanged with NTS nephritis, and this was mirrored when we assessed TMSB4X mRNA levels in glomerular and tubulointerstitial extracts from human kidneys affected by rapidly progressive glomerulonephritis or lupus nephritis. In contrast, a previous study in rat remnant kidneys in which nephron loss results in focal segmental glomerulosclerosis showed that Tmsb4x protein levels were significantly increased in sclerotic versus normal glomeruli.26 The discrepancy between these findings may be due to the different renal injury models and time points examined.
There are likely to be multiple mechanisms by which the lack of endogenous Tmsb4x results in increased glomerular injury in our experimental model. During nephrotoxic nephritis, podocytes switch from a terminally differentiated cell to a migratory cell that forms bridges between the glomerular tuft and the Bowman capsule34 and populates glomerular crescents.30, 48 The lack of Tmsb4x increased the number of glomeruli with adhesion of the glomerular tuft to the Bowman capsule and glomerular epithelial hyperplasia lesions, a feature of early crescent formation in this model.34 We also found that there was a redistribution of podocytes from the glomerular tuft, where they contribute to filtration barrier integrity toward the Bowman capsule. Our in vitro data demonstrate that downregulation of endogenous Tmsb4x in podocytes increases migration, and we predict that this may promote their redistribution in the nephrotoxic nephritis model. The increased podocyte migration was associated with increased actin stress fibers and activation of RhoA, which has been linked to podocyte stress fiber formation.49, 50 Moreover, podocyte-specific overexpression of RhoA induces proteinuria,50, 51 whereas RhoA inhibition improves renal injury in mouse models of nephrectomy52 and nephrotoxic nephritis,53 demonstrating the functional importance of this pathway in glomerular function. Other studies have shown that RhoA activation inhibits podocyte migration,54 but these experiments used a constitutively active form of RhoA permanently in the guanosine triphosphate-bound state. This would result in a high degree of RhoA activity, which has been associated with inhibition of migration.55 In contrast, Tmsb4x knockdown led to a 2-fold upregulation of RhoA activity in podocytes. It has been postulated that this lesser degree of RhoA activation promotes contractile stress fiber formation, facilitating cell detachment in migrating cells39 and enhancing lamellipodia formation driving cell motility.55 It was previously reported that activation of other Rho GTPases, Cdc42 and Rac1, may increase podocyte migration.56 However, we found that downregulation of endogenous Tmsb4x did not affect Cdc42 activation in podocytes. Rac1 activity was not assessed in this study, and it would be interesting to explore its involvement in the future.
Tmsb4x is also expressed in macrophages,18, 40 including in our nephrotoxic nephritis model, but its precise function has yet to be determined. It could be postulated that loss of the macrophage Tmsb4x may regulate the actin cytoskeleton, which has been implicated in both phenotypic polarization57 and migration.58 In our study, the loss of Tmsb4x did not alter macrophage polarization or the number of activated macrophages found in the glomerular area in the early stages of nephrotoxic nephritis. However, the number of activated macrophages in the periglomerular area at the late stage of the disease was increased in Tmsb4x−/y mice, suggesting a deficiency in the resolution of inflammation resulting in persistent macrophage accumulation. Macrophage accumulation may result from an absence of the Tmsb4x-derivative thymosin β4 sulfoxide, which has been shown to disperse inflammatory macrophages at the injury site in zebrafish and mouse models of heart injury.59 We also found that periglomerular fibrosis was enhanced in Tmsb4x−/y mice after NTS administration compared with wild-type mice. This may represent a secondary effect of enhanced inflammation and glomerular damage. However, previous studies showed that Tmsb4x can alter both plasminogen activator inhibitor-1 and transforming growth factor-β1,18, 27 both of which are drivers of fibrosis and play important roles in the progression of nephrotoxic nephritis.60, 61
In summary, we have provided the first evidence that the lack of endogenous Tmsb4x does not affect healthy glomeruli but exacerbates renal function impairment, periglomerular inflammation, and fibrosis in the context of nephrotoxic nephritis. These findings suggest that modulating Tmsb4x could be a potential therapeutic target in immune-mediated glomerular disease.
Methods
Experimental animals and procedures
C57Bl/6 hemizygous null male mice (Tmsb4x−/y) were bred with heterozygous Tmsb4x+/− adult female mice to generate male wild-type (Tmsb4x+/y) and null mice.27 To induce glomerular disease, Tmsb4+/y and Tmsb4x−/y mice were preimmunized by subcutaneous injection of sheep IgG (250 μg) in complete Freund’s adjuvant, followed by i.v. administration of sheep NTS (250 μl) 5 days later.62 All procedures were approved by the UK Home Office.
Renal function
Urine was collected from mice by housing them individually in metabolic cages. Blood samples were collected from the lateral saphenous vein. Albumin concentrations were measured by enzyme-linked immunosorbent assay28, 63 (Bethyl Laboratories, Montgomery, TX). Urinary and plasma creatinine concentrations were measured using isotope dilution electrospray mass spectrometry.64 Creatinine clearance (μl/min per gram of body weight) was derived from the formula urinary creatinine × urine volume × 1440 min−1 × plasma creatinine−1 × body weight (g)−1.63 Blood urea nitrogen was assessed using a commercially available assay kit, validated in mice (BioAssay Systems, Hayward, CA).65
Histologic analysis and immunohistochemistry
Kidneys were fixed in 4% paraformaldehyde and embedded in paraffin, and 5-μm sections were cut and stained with periodic acid–Schiff reagent. Fifty glomeruli per sample were scored by 2 blinded observers using the following system: 0, normal glomerular structure; 1, increased mesangial matrix deposition and hypercellularity with some loss of capillary loops; 2, increased matrix deposition and focal areas of sclerosis; 3, >50% of glomerulus sclerotic with very few capillary loops; 4, >75% of glomerulus sclerotic and the presence of glomerular epithelial hyperplasia lesions (Supplementary Figure S5). An average score was obtained for each kidney.
Immunohistochemistry or immunofluorescence was performed66 using antibodies against Tmsb4x (A9520, Immundiagnostik, Bensheim, Germany), collagen IV (ab19808, Abcam, Cambridge, UK), Cd3 (ab16669, Abcam), α-smooth muscle actin (M0851, Dako, Ely, UK), F4/80 (MCA497R, AbD Serotec, Oxford, UK), Nphs1 (GP-N2, Progen, Heidelberg, Germany), nestin (NB100-1604, Novus Biologicals, Littleton, CO), WT1 (AP15857PU-S, Acris Antibodies, Herford, Germany), Cd31 (MA3105, Thermo Fisher Scientific, Waltham, MA), and sheep IgG (A11016, Thermo Fisher Scientific). CD3+ and F4/80+ cells were counted in 50 glomeruli per sample. To assess glomerular sheep IgG deposition, mean fluorescence intensity was measured using ImageJ67 (30 glomeruli/sample). The number of WT1+ cells found within or outside (in glomerular crescents or lining the Bowman capsule) the glomerular tuft was counted in 50 consecutive glomeruli per sample. To account for any changes in the glomerular tuft area, the number of WT1+ cells in the glomerular tuft was normalized to the glomerular area (measured using ImageJ67) in 15 glomeruli per sample. Apoptosis was identified using TUNEL (Roche, Burgess Hill, UK). The number of TUNEL+ and WT1+/TUNEL+ cells was counted in 50 glomeruli per sample.
Measurement of murine IgG subclasses specific for sheep IgG
The titers of murine IgG subclasses specific for sheep IgG were measured by enzyme-linked immunosorbent assay in plasma, as described,68 using alkaline phosphatase subclass-specific antibodies for IgG1, IgG2b, and IgG3 (SouthernBiotech, Birmingham, AL) and IgG2a (Bethyl Laboratories).
In situ hybridization
In situ hybridization on paraffin sections was performed as described,27 using a digoxigenin-labeled antisense riboprobe specific for the 3′UTR of Tmsb4x, alongside a sense control.
Electron microscopy
For transmission electron microscopy, kidney cortex specimens (1 mm3) were postfixed in osmium tetroxide, dehydrated in acetone, and embedded in epoxy resin. Ultrathin sections were stained with uranyl acetate and lead citrate and examined.
Quantitative real-time polymerase chain reaction
RNA was extracted from mouse whole-kidney or glomerular extracts (isolated by Dynabeads [Thermo Fisher Scientific]28); 500 ng was used to prepare cDNA (iScript kit, Bio-Rad, Hemel Hempstead, UK), and quantitative real-time polymerase chain reaction was performed as described28 with Hprt as a housekeeping gene. All measurements were performed in duplicate. Renal biopsy specimens from patients with rapidly progressive glomerulonephritis (n = 12), lupus nephritis (n = 12), and living donor controls (n = 7) were collected within the framework of the European Renal cDNA Bank–Kroener–Fresenius Biopsy Bank69 after informed consent and local ethics committee approval. Unfixed tissue was transferred to RNase inhibitor and manually microdissected into glomerular and tubulointerstitial compartments. Total RNA was isolated and quantitative real-time polymerase chain reaction performed as reported,69 with 18S rRNA (Applied Biosystems, Foster City, CA) as the reference gene. Primer details are available on request.
Cell culture
Mouse podocytes37 were cultured as described70 and allowed to differentiate for 14 days. Cells were transfected with 10 nM siRNA specific for Tmsb4x or with a nontargeting control (both from Santa Cruz Biotechnology, Dallas, TX) using the transfection reagent Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Cell viability was assessed 24, 48, and 72 hours post-transfection using the methyltetrazolium assay. To assess migration, podocytes were plated to confluence, and a scratch was created using a pipette tip. Images (4 fields per condition) were taken 0, 6, and 24 hours later and the number of cells that migrated into the wound area counted. To visualize F-actin filaments 48 hours post-transfection, podocytes were fixed in 4% paraformaldehyde and 4% sucrose and stained with AlexaFluor-594 phalloidin (Thermo Fisher Scientific). The arrangement of actin filaments (either cytoplasmic stress fibers or cortical actin) was assessed in 30 cells per condition. RhoA and Cdc42 activity was quantified in podocyte lysates by G-LISA Small G-protein Activation Assays (Cytoskeleton, Denver, CO).
Glomeruli from Tmsb4x+/y and Tmsb4x−/y mice were isolated by Dynabeads (Thermo Fisher Scientific)28 and cultured in Matrigel-coated plates (Corning, Tewksbury, MA) in Dulbecco’s modified Eagle’s medium:F12 with 10% fetal calf serum, 1% Insulin-Transferrin-Selenium, and 100 μg/ml penicillin (Thermo Fisher Scientific). On day 7, when podocytes had grown out of the glomeruli, they were detached using trypsin–ethylenediamine tetraacetic acid and separated from glomeruli using 40-μm cell strainers (Corning). Primary podocytes obtained with this method were >90% pure, as judged by cell morphology and staining using podocyte (nephrin, nestin) markers.
Statistical methods
All samples were assessed blinded to treatment. Data are presented as mean ± SEM and were analyzed using GraphPad Prism (GraphPad Software, La Jolla, CA). When differences between 2 groups were evaluated, data were analyzed using a t test. When ≥3 groups were assessed, 1-way analysis of variance with Bonferroni’s multiple comparison post hoc tests were used. Data affected by 2 variables were analyzed using 2-way analysis of variance with Bonferroni’s multiple comparison post hoc tests. Statistical significance was accepted at P ≤ 0.05.
Disclosure
All the authors declared no competing interests.
Acknowledgments
We thank UCL Biological Services, Hortensja Brzoska, and Sabrina Pacheco for their assistance with animal experiments and Neil Dalton (King’s College London) for creatinine measurements. This work was supported by a Kidney Research UK (KRUK) Senior Non-Clinical Fellowship (SF1/2008 to DAL), a KRUK Postdoctoral Fellowship (PDF8/2015 to EV), a Medical Research Council New Investigator Award (MR/J003638/1 to DAL) (MR/J003638/1), and by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. CDC and MTL are supported by the Else Kröner Fresenius Foundation. We thank all participating centers of the European Renal cDNA Bank–Kroener–Fresenius Biopsy Bank (ERCB-KFB) and their patients for their cooperation. Active members at the time of the study are listed in Martini et al.71
Footnotes
Figure S1. Representative images of nestin (A) and Tmsb4x (B) staining in the mouse adult wild-type glomerulus visualized by fluorescent microscopy. (C) Merged images showing Tmsb4x (red) and nestin (green) staining; areas of colocalization are indicated by arrows. Representative images of Tmsb4x (D) and Cd31 (E) staining in the mouse adult wild-type glomerulus visualized by fluorescent microscopy. (F) Merged images showing Tmsb4x (green) and Cd31 (red) staining. Bar = 20 μm.
Figure S2. Quantification of profilin 1(Pfn1) (A), profilin 2 (Pfn2) (B), destrin (Dstn) (C), and cofilin 1 (Cfl1) (D) mRNA in whole-kidney homogenates of Tmsb4x+/y (n = 8) and Tmsb4x−/y (n = 4) mice by quantitative reverse transcriptase polymerase chain reaction. Quantification of cofilin 1 (Cfl1) mRNA in podocytes 48 hours after transfection with control siRNA or siRNA targeting Tmsb4x (n = 4) (E) and in podocytes isolated from Tmsb4x+/y or Tmsb4x−/y mice (n = 6) (F). Data are presented as mean ± SEM. *P < 0.05.
Figure S3. Quantification of nephrin (Nphs1; A), podocin (Nphs2; B), synaptopodin (Synpo; C), cd2 associated protein (Cd2ap) (D) and Wilms tumor 1 (Wt1) (E) mRNA in whole-kidney homogenates of Tmsb4x+/y (n = 8) and Tmsb4x−/y (n = 4) mice by quantitative reverse transcriptase polymerase chain reaction. Data are presented as mean ± SEM.
Figure S4. (A) Quantification of Tmsb4x mRNA levels in whole-kidney homogenates of Tmsb4x+/y (n = 8) and Tmsb4x+/y + NTS (n = 9) mice by quantitative reverse transcriptase polymerase chain reaction. (B and C) Quantification of TMSB4X mRNA levels in homogenates from human glomeruli (B) or tubulointerstitium (C). LD, living donor; RPGN, rapidly progressive glomerulonephritis; SLE, lupus nephritis. Data are presented as mean ± SEM.
Figure S5. Representative images of the histology observed 7 days after NTS administration showing a normal glomerulus (A), a glomerulus with hyaline deposits indicated by arrowheads (B), and a glomerulus with a bridge formed between the glomerular tuft and the Bowman capsule indicated by an arrow (C). Bar = 20 μm.
Figure S6. Representative images of the different scoring categories for glomerular injury showing a normal glomerulus, score 0 (A); a glomerulus with increased mesangial matrix deposition and some loss of capillary loops, score 1 (B); a glomerulus with increased mesangial matrix deposition and focal areas of sclerosis, score 2 (C); a glomerulus with >50% sclerotic area and very few capillary loops, score 3 (D); and a sclerotic glomerulus with an epithelial hyperplastic lesion (*) and a bridge formed between the glomerular tuft and the Bowman capsule indicated by an arrow, score 4 (E). Bar = 20 μm. (F) Proportions of glomeruli in the different scoring categories of glomerular injury in Tmsb4x+/y (n = 5), Tmsb4x+/y + NTS (n = 9), and Tmsb4x−/y + NTS (n = 6) mice. Bars represent the mean ± SEM.
Figure S7. Representative images showing sheep IgG immunoreactivity in glomeruli of Tmsb4x+/y (A) and Tmsb4x−/y (B) mice injected with nephrotoxic serum (NTS). (C) Quantification of mean fluorescence in 30 glomeruli per sample (Tmsb4+/y + NTS, n = 9; Tmsb4x−/y + NTS, n = 6). Plasma titers of murine IgG subclasses (D, IgG1; E, IgG2a; F, IgG2b; G, IgG3) against sheep IgG in control, not immunized Tmsb4x+/y mice (n = 5) and in Tmsb4x+/y (n = 9) and Tmsb4x−/y (n = 6) mice 21 days after NTS injection. Data are presented as mean ± SEM. Bar = 20 μm, *P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Figure S8. Representative image showing staining for TUNEL (A) and WT-1 (B) in the glomerulus. A TUNEL-positive podocyte is identified by an arrowhead (C). The number of TUNEL-positive cells (D) and TUNEL-positive WT-1–positive cells per glomerulus was counted in 50 glomeruli per sample, and the average number was calculated (Tmsb4x+/y, n = 4; Tmsb4x+/y, + NTS, n = 9; Tmsb4x−/y + NTS, n = 6). Data are presented as mean ± SEM. Bar = 20 μm. *P ≤ 0.05.
Figure S9. The number of F4/80+ cells in the glomerular tuft (A) and in the periglomerular area (B) 7 days after administration of nephrotoxic serum (NTS) was counted in 50 glomeruli per sample, and the average number was calculated (Tmsb4x+/y, n = 5; Tmsb4x+/y + NTS+, n = 6; Tmsb4x−/y + NTS, n = 8). Data are presented as mean ± SEM. **P ≤ 0.01.
Figure S10. Quantification of Cd68 (A), Mcp1 (B), Cd86 (C), Cd206 (D), and Arg1 (E) mRNA in whole-kidney homogenates of Tmsb4+/y + NTS (n = 9) and Tmsb4x−/y + NTS (n = 6) mice. NTS = nephrotoxic serum.
Table S1. Proportions of Tmsb4x+/−, Tmsb4x−/−, Tmsb4x+/y, and Tmsb4x−/y mice born after crossing adult male Tmsb4x−/y mice with Tmsb4x+/− adult female mice.
Supplementary material is linked to the online version of the paper at www.kidney-international.org.
Supplementary Material
Figure S1.

Representative images of nestin (A) and Tmsb4x (B) staining in the mouse adult wild-type glomerulus visualized by fluorescent microscopy. (C) Merged images showing Tmsb4x (red) and nestin (green) staining; areas of colocalization are indicated by arrows. Representative images of Tmsb4x (D) and Cd31 (E) staining in the mouse adult wild-type glomerulus visualized by fluorescent microscopy. (F) Merged images showing Tmsb4x (green) and Cd31 (red) staining. Bar = 20 μm.
Figure S2.

Quantification of profilin 1(Pfn1) (A), profilin 2 (Pfn2) (B), destrin (Dstn) (C), and cofilin 1 (Cfl1) (D) mRNA in whole-kidney homogenates of Tmsb4x+/y (n = 8) and Tmsb4x−/y (n = 4) mice by quantitative reverse transcriptase polymerase chain reaction. Quantification of cofilin 1 (Cfl1) mRNA in podocytes 48 hours after transfection with control siRNA or siRNA targeting Tmsb4x (n = 4) (E) and in podocytes isolated from Tmsb4x+/y or Tmsb4x−/y mice (n = 6) (F). Data are presented as mean ± SEM. *P < 0.05.
Figure S3.
Quantification of nephrin (Nphs1;A), podocin (Nphs2; B), synaptopodin (Synpo; C), cd2 associated protein (Cd2ap) (D) and Wilms tumor 1 (Wt1) (E) mRNA in whole-kidney homogenates of Tmsb4x+/y (n = 8) and Tmsb4x−/y (n = 4) mice by quantitative reverse transcriptase polymerase chain reaction. Data are presented as mean ± SEM.
Figure S4.

(A) Quantification of Tmsb4x mRNA levels in whole-kidney homogenates of Tmsb4x+/y (n = 8) and Tmsb4x+/y + NTS (n = 9) mice by quantitative reverse transcriptase polymerase chain reaction. (B and C) Quantification of TMSB4X mRNA levels in homogenates from human glomeruli (B) or tubulointerstitium (C). LD, living donor; RPGN, rapidly progressive glomerulonephritis; SLE, lupus nephritis. Data are presented as mean ± SEM.
Figure S5.

Representative images of the histology observed 7 days after NTS administration showing a normal glomerulus (A), a glomerulus with hyaline deposits indicated by arrowheads (B), and a glomerulus with a bridge formed between the glomerular tuft and the Bowman capsule indicated by an arrow (C). Bar = 20 μm.
Figure S6.
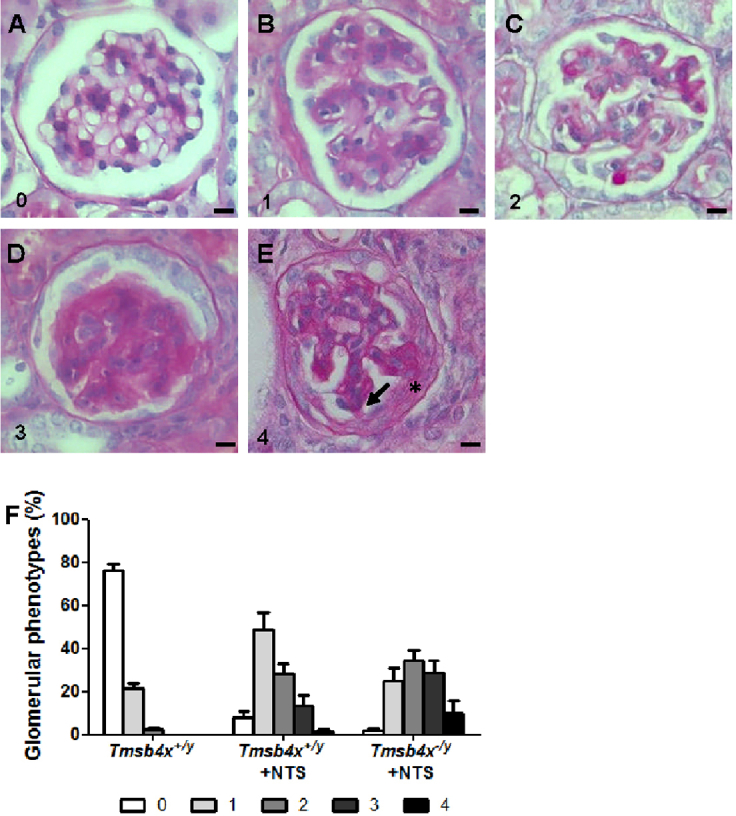
Representative images of the different scoring categories for glomerular injury showing a normal glomerulus, score 0 (A); a glomerulus with increased mesangial matrix deposition and some loss of capillary loops, score 1 (B); a glomerulus with increased mesangial matrix deposition and focal areas of sclerosis, score 2 (C); a glomerulus with >50% sclerotic area and very few capillary loops, score 3 (D); and a sclerotic glomerulus with an epithelial hyperplastic lesion (*) and a bridge formed between the glomerular tuft and the Bowman capsule indicated by an arrow, score 4 (E). Bar = 20 μm. (F) Proportions of glomeruli in the different scoring categories of glomerular injury in Tmsb4x+/y (n = 5), Tmsb4x+/y + NTS (n = 9), and Tmsb4x−/y + NTS (n = 6) mice. Bars represent the mean ± SEM.
Figure S7.

Representative images showing sheep IgG immunoreactivity in glomeruli of Tmsb4x+/y (A) and Tmsb4x−/y (B) mice injected with nephrotoxic serum (NTS). (C) Quantification of mean fluorescence in 30 glomeruli per sample (Tmsb4+/y + NTS, n = 9; Tmsb4x−/y + NTS, n = 6). Plasma titers of murine IgG subclasses (D, IgG1; E, IgG2a; F, IgG2b; G, IgG3) against sheep IgG in control, not immunized Tmsb4x+/y mice (n = 5) and in Tmsb4x+/y (n = 9) and Tmsb4x−/y (n = 6) mice 21 days after NTS injection. Data are presented as mean ± SEM. Bar = 20 μm, *P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
Figure S8.

Representative image showing staining for TUNEL (A) and WT-1 (B) in the glomerulus. A TUNEL-positive podocyte is identified by an arrowhead (C). The number of TUNEL-positive cells (D) and TUNEL-positive WT-1–positive cells per glomerulus was counted in 50 glomeruli per sample, and the average number was calculated (Tmsb4x+/y, n = 4; Tmsb4x+/y, + NTS, n = 9; Tmsb4x−/y + NTS, n = 6). Data are presented as mean ± SEM. Bar = 20 μm. *P ≤ 0.05.
Figure S9.

The number of F4/80+ cells in the glomerular tuft (A) and in the periglomerular area (B) 7 days after administration of nephrotoxic serum (NTS) was counted in 50 glomeruli per sample, and the average number was calculated (Tmsb4x+/y, n = 5; Tmsb4x+/y + NTS+, n = 6; Tmsb4x−/y + NTS, n = 8). Data are presented as mean ± SEM. **P ≤ 0.01.
Figure S10.

Quantification of Cd68 (A), Mcp1 (B), Cd86 (C), Cd206 (D), and Arg1 (E) mRNA in whole-kidney homogenates of Tmsb4+/y + NTS (n = 9) and Tmsb4x−/y + NTS (n = 6) mice. NTS = nephrotoxic serum.
Proportions of Tmsb4x+/−, Tmsb4x−/−, Tmsb4x+/y, and Tmsb4x−/y mice born after crossing adult male Tmsb4x−/y mice with Tmsb4x+/− adult female mice.
References
- 1.Eckardt K.U., Coresh J., Devuyst O. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet. 2013;382:158–169. doi: 10.1016/S0140-6736(13)60439-0. [DOI] [PubMed] [Google Scholar]
- 2.Greka A., Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welsh G.I., Saleem M.A. The podocyte cytoskeleton–key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2012;8:14–21. doi: 10.1038/nrneph.2011.151. [DOI] [PubMed] [Google Scholar]
- 4.Brinkkoetter P.T., Ising C., Benzing T. The role of the podocyte in albumin filtration. Nat Rev Nephrol. 2013;9:328–336. doi: 10.1038/nrneph.2013.78. [DOI] [PubMed] [Google Scholar]
- 5.Kurts C., Panzer U., Anders H.J. The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol. 2013;13:738–753. doi: 10.1038/nri3523. [DOI] [PubMed] [Google Scholar]
- 6.Sanders M.C., Goldstein A.L., Wang Y.L. Thymosin beta 4 (Fx peptide) is a potent regulator of actin polymerization in living cells. Proc Natl Acad Sci U S A. 1992;89:4678–4682. doi: 10.1073/pnas.89.10.4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smart N., Bollini S., Dube K.N. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011;474:640–644. doi: 10.1038/nature10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morris D.C., Cui Y., Cheung W.L. A dose-response study of thymosin beta4 for the treatment of acute stroke. J Neurol Sci. 2014;345:61–67. doi: 10.1016/j.jns.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sosne G., Qiu P., Ousler G.W., 3rd Thymosin beta4: a potential novel dry eye therapy. Ann N Y Acad Sci. 2012;1270:45–50. doi: 10.1111/j.1749-6632.2012.06682.x. [DOI] [PubMed] [Google Scholar]
- 10.Conte E., Genovese T., Gili E. Thymosin beta4 protects C57BL/6 mice from bleomycin-induced damage in the lung. Eur J Clin Invest. 2013;43:309–315. doi: 10.1111/eci.12048. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein A.L., Hannappel E., Sosne G. Thymosin beta4: a multi-functional regenerative peptide. Basic properties and clinical applications. Expert Opin Biol Ther. 2012;12:37–51. doi: 10.1517/14712598.2012.634793. [DOI] [PubMed] [Google Scholar]
- 12.Fan Y., Gong Y., Ghosh P.K. Spatial coordination of actin polymerization and ILK-Akt2 activity during endothelial cell migration. Dev Cell. 2009;16:661–674. doi: 10.1016/j.devcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santra M., Zhang Z.G., Yang J. Thymosin beta4 up-regulation of microRNA-146a promotes oligodendrocyte differentiation and suppression of the Toll-like proinflammatory pathway. J Biol Chem. 2014;289:19508–19518. doi: 10.1074/jbc.M113.529966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bock-Marquette I., Saxena A., White M.D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature. 2004;432:466–472. doi: 10.1038/nature03000. [DOI] [PubMed] [Google Scholar]
- 15.Smart N., Risebro C.A., Melville A.A. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature. 2007;445:177–182. doi: 10.1038/nature05383. [DOI] [PubMed] [Google Scholar]
- 16.Sosne G., Qiu P., Christopherson P.L. Thymosin beta 4 suppression of corneal NFkappaB: a potential anti-inflammatory pathway. Exp Eye Res. 2007;84:663–669. doi: 10.1016/j.exer.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasilopoulou E., Winyard P.J., Riley P.R. The role of thymosin-beta4 in kidney disease. Expert Opin Biol Ther. 2015;15:S187–S190. doi: 10.1517/14712598.2015.1009891. [DOI] [PubMed] [Google Scholar]
- 18.Zuo Y., Chun B., Potthoff S.A. Thymosin beta4 and its degradation product, Ac-SDKP, are novel reparative factors in renal fibrosis. Kidney Int. 2013;84:1166–1175. doi: 10.1038/ki.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu J., Su L.P., Zhou Y. Thymosin beta4 attenuates early diabetic nephropathy in a mouse model of type 2 diabetes mellitus. Am J Ther. 2015;22:141–146. doi: 10.1097/MJT.0b013e3182785ecc. [DOI] [PubMed] [Google Scholar]
- 20.Cavasin M.A., Rhaleb N.E., Yang X.P. Prolyl oligopeptidase is involved in release of the antifibrotic peptide Ac-SDKP. Hypertension. 2004;43:1140–1145. doi: 10.1161/01.HYP.0000126172.01673.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao T.D., Yang X.P., D'Ambrosio M. N-acetyl-seryl-aspartyl-lysyl-proline attenuates renal injury and dysfunction in hypertensive rats with reduced renal mass: council for high blood pressure research. Hypertension. 2010;55:459–467. doi: 10.1161/HYPERTENSIONAHA.109.144568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omata M., Taniguchi H., Koya D. N-acetyl-seryl-aspartyl-lysyl-proline ameliorates the progression of renal dysfunction and fibrosis in WKY rats with established anti-glomerular basement membrane nephritis. J Am Soc Nephrol. 2006;17:674–685. doi: 10.1681/ASN.2005040385. [DOI] [PubMed] [Google Scholar]
- 23.Rhaleb N.E., Pokharel S., Sharma U. Renal protective effects of N-acetyl-Ser-Asp-Lys-Pro in deoxycorticosterone acetate-salt hypertensive mice. J Hypertension. 2011;29:330–338. doi: 10.1097/HJH.0b013e32834103ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guinobert I., Viltard M., Piquemal D. Identification of differentially expressed genes between fetal and adult mouse kidney: candidate gene in kidney development. Nephron Physiol. 2006;102:81–91. doi: 10.1159/000090054. [DOI] [PubMed] [Google Scholar]
- 25.Brunskill E.W., Georgas K., Rumballe B. Defining the molecular character of the developing and adult kidney podocyte. PLoS One. 2011;6:e24640. doi: 10.1371/journal.pone.0024640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu B.J., Shyr Y., Liang X. Proteomic patterns and prediction of glomerulosclerosis and its mechanisms. J Am Soc Nephrol. 2005;16:2967–2975. doi: 10.1681/ASN.2005030262. [DOI] [PubMed] [Google Scholar]
- 27.Rossdeutsch A., Smart N., Dube K.N. Essential role for thymosin beta4 in regulating vascular smooth muscle cell development and vessel wall stability. Circ Res. 2012;111:e89–e102. doi: 10.1161/CIRCRESAHA.111.259846. [DOI] [PubMed] [Google Scholar]
- 28.Long D.A., Kolatsi-Joannou M., Price K.L. Albuminuria is associated with too few glomeruli and too much testosterone. Kidney Int. 2013;83:1118–1129. doi: 10.1038/ki.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X., Chuang P.Y., D'Agati V.D. Nephrin preserves podocyte viability and glomerular structure and function in adult kidneys. J Am Soc Nephrol. 2015;26:2361–2377. doi: 10.1681/ASN.2014040405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thorner P.S., Ho M., Eremina V. Podocytes contribute to the formation of glomerular crescents. J Am Soc Nephrol. 2008;19:495–502. doi: 10.1681/ASN.2006101115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X., Zimmerman A., Copeland N.G. The mouse thymosin beta 4 gene: structure, promoter identification, and chromosome localization. Genomics. 1996;32:388–394. doi: 10.1006/geno.1996.0133. [DOI] [PubMed] [Google Scholar]
- 32.Pippin J.W., Brinkkoetter P.T., Cormack-Aboud F.C. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009;296:F213–F229. doi: 10.1152/ajprenal.90421.2008. [DOI] [PubMed] [Google Scholar]
- 33.Khan S.B., Cook H.T., Bhangal G. Antibody blockade of TNF-alpha reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int. 2005;67:1812–1820. doi: 10.1111/j.1523-1755.2005.00279.x. [DOI] [PubMed] [Google Scholar]
- 34.Le Hir M., Keller C., Eschmann V. Podocyte bridges between the tuft and Bowman's capsule: an early event in experimental crescentic glomerulonephritis. J Am Soc Nephrol. 2001;12:2060–2071. doi: 10.1681/ASN.V12102060. [DOI] [PubMed] [Google Scholar]
- 35.Guo J.K., Menke A.L., Gubler M.C. WT1 is a key regulator of podocyte function: reduced expression levels cause crescentic glomerulonephritis and mesangial sclerosis. Human Mol Genet. 2002;11:651–659. doi: 10.1093/hmg/11.6.651. [DOI] [PubMed] [Google Scholar]
- 36.Davis B., Dei Cas A., Long D.A. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol. 2007;18:2320–2329. doi: 10.1681/ASN.2006101093. [DOI] [PubMed] [Google Scholar]
- 37.Mundel P., Reiser J., Zuniga Mejia Borja A. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 38.Pollard T.D., Cooper J.A. Actin, a central player in cell shape and movement. Science. 2009;326:1208–1212. doi: 10.1126/science.1175862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raftopoulou M., Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 40.Hannappel E., Xu G.J., Morgan J. Thymosin beta 4: a ubiquitous peptide in rat and mouse tissues. Proc Natl Acad Sci U S A. 1982;79:2172–2175. doi: 10.1073/pnas.79.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng H., Xu J., Yang X.P. Thymosin-beta4 prevents cardiac rupture and improves cardiac function in mice with myocardial infarction. Am J Physiol Heart Circ Physiol. 2014;307:H741–H751. doi: 10.1152/ajpheart.00129.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duffield J.S., Tipping P.G., Kipari T. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang X.R., Tipping P.G., Apostolopoulos J. Mechanisms of T cell-induced glomerular injury in anti-glomerular basement membrane (GBM) glomerulonephritis in rats. Clin Exp Immunol. 1997;109:134–142. doi: 10.1046/j.1365-2249.1997.4091307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tipping P.G., Huang X.R., Qi M. Crescentic glomerulonephritis in CD4- and CD8-deficient mice. Requirement for CD4 but not CD8 cells. Am J Pathol. 1998;152:1541–1548. [PMC free article] [PubMed] [Google Scholar]
- 45.Nemolato S., Cabras T., Fanari M.U. Immunoreactivity of thymosin beta 4 in human foetal and adult genitourinary tract. Eur J Histochem. 2010;54:e43. doi: 10.4081/ejh.2010.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knop J., App C., Hannappel E. Antibodies in research of thymosin beta4: investigation of cross-reactivity and influence of fixatives. Ann N Y Acad Sci. 2012;1270:105–111. doi: 10.1111/j.1749-6632.2012.06659.x. [DOI] [PubMed] [Google Scholar]
- 47.Bravo-Cordero J.J., Magalhaes M.A., Eddy R.J. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol. 2013;14:405–415. doi: 10.1038/nrm3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moeller M.J., Soofi A., Hartmann I. Podocytes populate cellular crescents in a murine model of inflammatory glomerulonephritis. J Am Soc Nephrol. 2004;15:61–67. doi: 10.1097/01.asn.0000102468.37809.c6. [DOI] [PubMed] [Google Scholar]
- 49.Asanuma K., Yanagida-Asanuma E., Faul C. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 50.Wang L., Ellis M.J., Gomez J.A. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012;81:1075–1085. doi: 10.1038/ki.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu L., Jiang R., Aoudjit L. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22:1621–1630. doi: 10.1681/ASN.2010111146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Babelova A., Jansen F., Sander K. Activation of Rac-1 and RhoA contributes to podocyte injury in chronic kidney disease. PLoS One. 2013;8:e80328. doi: 10.1371/journal.pone.0080328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hidaka T., Suzuki Y., Yamashita M. Amelioration of crescentic glomerulonephritis by RhoA kinase inhibitor, Fasudil, through podocyte protection and prevention of leukocyte migration. Am J Pathol. 2008;172:603–614. doi: 10.2353/ajpath.2008.070196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian D., Jacobo S.M., Billing D. Antagonistic regulation of actin dynamics and cell motility by TRPC5 and TRPC6 channels. Sci Signal. 2010;3:ra77. doi: 10.1126/scisignal.2001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ridley A.J. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103–112. doi: 10.1016/j.ceb.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gee H.Y., Saisawat P., Ashraf S. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest. 2013;123:3243–3253. doi: 10.1172/JCI69134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McWhorter F.Y., Wang T., Nguyen P. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A. 2013;110:17253–17258. doi: 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Man S.M., Ekpenyong A., Tourlomousis P. Actin polymerization as a key innate immune effector mechanism to control Salmonella infection. Proc Natl Acad Sci U S A. 2014;111:17588–17593. doi: 10.1073/pnas.1419925111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evans M.A., Smart N., Dube K.N. Thymosin beta4-sulfoxide attenuates inflammatory cell infiltration and promotes cardiac wound healing. Nat Commun. 2013;4:2081. doi: 10.1038/ncomms3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feng L., Tang W.W., Loskutoff D.J. Dysfunction of glomerular fibrinolysis in experimental antiglomerular basement membrane antibody glomerulonephritis. J Am Soc Nephrol. 1993;3:1753–1764. doi: 10.1681/ASN.V3111753. [DOI] [PubMed] [Google Scholar]
- 61.Huang X.R., Chung A.C., Zhou L. Latent TGF-beta1 protects against crescentic glomerulonephritis. J Am Soc Nephrol. 2008;19:233–242. doi: 10.1681/ASN.2007040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brown H.J., Lock H.R., Sacks S.H. TLR2 stimulation of intrinsic renal cells in the induction of immune-mediated glomerulonephritis. J Immunol. 2006;177:1925–1931. doi: 10.4049/jimmunol.177.3.1925. [DOI] [PubMed] [Google Scholar]
- 63.Dessapt-Baradez C., Woolf A.S., White K.E. Targeted glomerular angiopoietin-1 therapy for early diabetic kidney disease. J Am Soc Nephrol. 2014;25:33–42. doi: 10.1681/ASN.2012121218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greenberg N., Roberts W.L., Bachmann L.M. Specificity characteristics of 7 commercial creatinine measurement procedures by enzymatic and Jaffe method principles. Clin Chem. 2012;58:391–401. doi: 10.1373/clinchem.2011.172288. [DOI] [PubMed] [Google Scholar]
- 65.Kolatsi-Joannou M., Price K.L., Winyard P.J. Modified citrus pectin reduces galectin-3 expression and disease severity in experimental acute kidney injury. PLoS One. 2011;6:e18683. doi: 10.1371/journal.pone.0018683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang J.L., Woolf A.S., Kolatsi-Joannou M. Vascular endothelial growth factor C for polycystic kidney diseases. J Am Soc Nephrol. 2016;27:69–77. doi: 10.1681/ASN.2014090856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown H.J., Sacks S.H., Robson M.G. Toll-like receptor 2 agonists exacerbate accelerated nephrotoxic nephritis. J Am Soc Nephrol. 2006;17:1931–1939. doi: 10.1681/ASN.2005111167. [DOI] [PubMed] [Google Scholar]
- 69.Cohen C.D., Frach K., Schlondorff D. Quantitative gene expression analysis in renal biopsies: a novel protocol for a high-throughput multicenter application. Kidney Int. 2002;61:133–140. doi: 10.1046/j.1523-1755.2002.00113.x. [DOI] [PubMed] [Google Scholar]
- 70.Yates L.L., Papakrivopoulou J., Long D.A. The planar cell polarity gene Vangl2 is required for mammalian kidney-branching morphogenesis and glomerular maturation. Hum Mol Genet. 2010;19:4663–4676. doi: 10.1093/hmg/ddq397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Martini S., Nair V., Keller B.J. Integrative biology identifies shared transcriptional networks in CKD. J Am Soc Nephrol. 2014;25:2559–2572. doi: 10.1681/ASN.2013080906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proportions of Tmsb4x+/−, Tmsb4x−/−, Tmsb4x+/y, and Tmsb4x−/y mice born after crossing adult male Tmsb4x−/y mice with Tmsb4x+/− adult female mice.