

Abstract
We describe a microfluidic device with an integrated nanochannel array to trap individual bacteria and monitor growth and reproduction of lineages over multiple generations. Our poly(dimethylsiloxane) device comprises a pneumatically actuated nanochannel array that includes 1280 channels with widths from 600 to 1000 nm to actively trap diverse bacteria. Integrated pumps and valves perform on-chip fluid and cell manipulations that provide dynamic control of cell loading and nutrient flow, permitting chemostatic growth for extended periods of time (typically 12 to 20 h). Nanochannels confine bacterial growth to a single dimension, facilitating high-resolution, time-lapse imaging and tracking of individual cells. We use the device to monitor the growth of single bacterial cells that undergo symmetric (Bacillus subtilis) and asymmetric (Caulobacter crescentus) division and reconstruct their lineages to correlate growth measurements through time and among related cells. Furthermore, we monitor the motility state of single B. subtilis cells across multiple generations by the expression of a fluorescent reporter protein and observe that the state of the epigenetic switch is correlated over five generations. Our device allows imaging of cellular lineages with high spatiotemporal resolution to facilitate the analysis of biological processes spanning multiple generations.
Graphical Abstract

Microfluidic systems are appealing platforms to study single-cell dynamics because these devices allow for individual cells to be confined, manipulated, and grown over multiple generations.1–4 These systems offer improved temporal and spatial resolution of single-cell measurements that can provide significant insights into population heterogeneity, as individual cells exhibit variation in gene expression, growth rate, and other phenotypes.5 Cellular lineages constructed from these observations provide valuable tools for epigenetic inheritance studies.6,7 Moreover, these lineages inform our understanding of cellular aging in both symmetrically8 and asymmetrically9 dividing bacteria.
A common approach to observing dynamic processes in bacterial lineages relies on collecting time-lapse images of growing microcolonies on agarose pads.10 Significant insights into biological processes overlooked by observation of the population averages have been gained from high-resolution, time-lapse images obtained by this approach.5,11,12 However, a limited number of divisions are observed with this technique owing to exponential growth of the immobilized cells, which causes colonies to quickly grow beyond the field of view or to overcrowd the agarose substrate and grow in multiple layers.10 Cells eventually deplete the nutrients in the agarose faster than diffusion can replenish them, forming concentration gradients across the colonies. Additionally, these closed systems preclude ready manipulation of the extracellular environment over the course of an experiment.
To overcome these limitations, micro- and nanofluidic systems that confine cells and supply fresh media over the course of the experiment have been developed. Devices that use chambers13,14 and membranes15,16 to trap cells have successfully imaged bacteria and yeast. These devices eliminate the depletion of nutrients, yet the cells are still confined in only one dimension and eventually grow beyond the camera field of view. A device that uses dead-end channels, called a mother machine,2 traps a mother cell indefinitely and can be used to easily create cellular lineages. However, such a device traps and maintains only the oldest bacterial cells. Bacterial aging occurs primarily over the first few generations as young cells progressively inherit the old cell pole.17 Beyond this point, aging phenotypes effectively plateau, presumably due to a limited capacity for aging cells to accumulate damage. Many existing microfluidic platforms, therefore, offer limited utility in aging studies.18
Recent work uses nanochannels that are positioned orthogonal to microfluidic channels to trap and culture cells.3,4 The dimensions of the nanochannels constrain cell growth to an open-ended, one-dimensional line, and the microfluidic channels supply fresh media to the cells on both sides of the nanochannels. Because cells can exit both ends of the nanochannel, the trapped population is constantly replenished with fresh cells of diverse reproductive ages. In one example,3 nanochannels are formed in agarose to trap the cells. Agarose is an attractive substrate for the nanochannels, as the interactions between agarose and cells are reasonably well understood. However, we have found that cells often grow into the soft agarose (data not shown), and the porosity of the agarose makes the rapid exchange of media impossible. Similar nanochannels have been formed in poly(dimethylsiloxane) (PDMS).4 However, cells were inserted into the nanochannels by hydrostatic pressure, and the nanochannels had short lengths of ~20 μm which limit the number of generations observed in the channel.
We have developed an automated PDMS-based microfluidic device with an integrated nanochannel array. Our device uses a pneumatically actuated nanochannel array to trap cells and integrated pumps and valves to perform fluid and cell manipulations.19–21 Our device immobilizes cells for high-resolution, time-lapse fluorescence imaging and tracking, has dynamic control of cell loading and nutrient flow, and can rapidly exchange media. To perform required fluid and cell manipulations, we have integrated pneumatically actuated valves and peristaltic pumps. The individually addressable valves separate the nanochannel array from input and output reservoirs. This device supports growth of bacterial species of different sizes (e.g., Caulobacter crescentus and Bacillus subtilis) over extended periods of time (12 to 20 h). We use the device to analyze two biological phenomena: (1) life history parameters of symmetrically and asymmetrically dividing cells and (2) epigenetic inheritance of a bistable motility switch.
EXPERIMENTAL SECTION
Materials
We purchased glass slides (50 mm × 75 mm) from Corning, Inc.; poly(dimethylsiloxane) (PDMS, Sylgard 184) from Dow Corning, Inc.; No. 1.5 Gold Seal coverslips (48 mm × 60 mm) from VWR International, LLC; SU-8 2010 photoresist and SU-8 Developer from MicroChem Corp.; and titanium diisopropoxide bis(2,4-pentanedionate) from Gelest, Inc. All other chemicals were purchased from Sigma-Aldrich Co.
Cell Strains and Preparation
Peptone yeast extract (PYE) was prepared as described previously.22 Lysogeny broth (LB) medium was prepared as described by Miller,23 and the pH was adjusted to 7.0. Caulobacter crescentus strain YB6474 bears several mutations that facilitate growth and imaging in nanochannels: a frameshift mutation in the hfsA locus prevents synthesis of the adhesive holdfast;24 deletion of the creS locus renders the cell body straight, rather than curved;25 deletion of the f lgE locus eliminates flagellar motility;26 and insertion of a Tn7 transposon bearing the gene for the DsRedExpress protein results in constitutive cytoplasmic fluorescence.27 Several mutations similarly facilitate nanochannel analysis of Bacillus subtilis DK1437: deletion of the hag gene cluster prevents motility;28 interruption of the epsH locus reduces exopolysaccharide-mediated adhesion;29 an isopropyl-beta-D-thiogalactopyranoside (IPTG)-inducible mCherry allele permits imaging of the cytoplasm; and a fusion of the gf p gene to the flagellar hag promoter reports transcriptional activity at the hag locus.30 We initially tested the growth of wild-type B. subtilis and C. crescentus in our devices and observed that cells adhered to the PDMS and glass. Because of this adhesion, cells accumulated in the microchannels and eventually blocked media flow through the device. Consequently, we chose to use nonadherent strains to facilitate long-term growth and observation on the devices. There are no known reports that the aforementioned mutations in adherence or other phenotypes impact cell growth rate or other life history parameters measured here.
Device Fabrication
The microfluidic device comprises two layers of PDMS (fluid and control) and a glass coverslip. The fluid layer (Figure 1a) contains the micro- and nanochannels, is sandwiched between the control layer and coverslip, and is deformable to permit valving, pumping, and cell trapping. The fluid-layer master was fabricated by a combination of electron-beam (e-beam) lithography and standard photolithography in the negative-tone resist SU-8. Glass substrates were cleaned with methanol, dried with nitrogen, and spin-coated with 2% titanium diisopropoxide (bis-2,4-pentanedionate) in isopropyl alcohol to promote adhesion of the SU-8 2010 resist. To increase the robustness of the master, a 20-μm thick layer of SU-8 was spin-coated onto the substrate at 1000 rpm for 45 s. The SU-8 film was then prebaked at 95 °C for 4 min and exposed to 200 mJ/cm2 of UV light from an exposure system (205S, Optical Associates, Inc.). After exposure, the substrate was postbaked at 95 °C for 5 min. To create the nanochannels, the SU-8 2010 resist was diluted to 50% (w/v) in cyclopentanone and spin-coated at 1500 rpm onto the glass substrates for 30 s to form a 1.0-μm thick layer. The SU-8 was prebaked at 95 °C for 1 min, and a scanning electron microscope (SEM; Quanta 600F, FEI Company) equipped with Nanometer Pattern Generation System (JC Nabity Lithography Systems) was used to write the nanochannel patterns into the SU-8 layer. An e-beam dose of 2 μC/cm2 at a 30 kV accelerating potential exposed the nanochannel pattern, which consisted of 1280 channels written in a 4 × 4 grid, where each section of the grid had 80 nanochannels. After e-beam exposure, the substrate was postbaked for 2 min at 95 °C and developed in SU-8 Developer for 1 min.
Figure 1.

(a) Schematic of the fluid layer of the microfluidic device with an integrated nanochannel array for monitoring growth and development of single cells over multiple generations. Open circles represent reservoirs, and closed circles represent valves. Three valves in series are used as peristaltic pumps and are labeled pumps 1 and 2. A large open region in the control layer above the nanochannel array (~4 mm × 3 mm) actuates the cell trapping region. (b) Scanning electron microscope (SEM) image of a section of the SU-8 fluid-layer master that shows 80 nanochannels with widths from 600 to 1000 nm positioned orthogonal to the microchannels, which deliver nutrients to the nanochannels. This set of 80 nanochannels is one of 16 sets of channels arranged in a 4 × 4 grid. (c) SEM image of a subset of nanochannels replicated in poly(dimethylsiloxane) (PDMS). Schematic of pneumatic actuation with (d) vacuum applied to raise the nanochannel array and load cells and (e) pressure applied to lower the nanochannel array and trap cells for growth and analysis. The control layer, fluid layer, and coverslip constitute the three layers of the device.
Microchannels for the fluid- and control-layer masters were fabricated by photolithography. To form microchannels on top of the nanochannel array in the fluid layer, an additional 20-μm of SU-8 2010 resist was spin-coated over the nanochannels and prebaked at 95 °C for 4 min. After the prebake, the SU-8 was exposed to 250 mJ/cm2 of UV light through the fluid-layer photomask (International Phototool Company, LLC) to create microchannels perpendicular to the nanochannels (Figure 1b). For the control-layer master, a 40-μm thick layer of SU-8 2010 resist was spin-coated onto a substrate without nanochannels, prebaked at 95 °C for 5 min, and exposed to 350 mJ/cm2 of UV light through the control-layer photomask. The masters were then postbaked for 6 min at 95 °C, developed for 2 min in SU-8 Developer, rinsed with isopropyl alcohol, and dried with nitrogen. To ensure clean and repeated release of PDMS from the SU-8 surface, the masters were coated with (tridecafluoro-1,1,2,2-tetrahydrooctyl) trichlorosilane by vapor deposition in a desiccator.
PDMS replicas of the control layer were prepared by creating a boundary around the master with tape and dispensing 9 mL of uncured PDMS (10:1 ratio) onto the master. The PDMS was degassed under vacuum for 30 min and then cured at 65 °C for 2 h. Pneumatic access ports were punched through the control layer with a 2 mm biopsy punch. To create the PDMS replicas of the fluid layer (Figure 1c), 3 mL of uncured PDMS was spread onto the master and degassed under vacuum for 30 min. The master was spun at 1000 rpm for 30 s to form a ~100-μm thick layer of PDMS, which was partially cured at 65 °C for 15 min. The control layer was aligned with the partially cured fluid layer under a stereoscope (S6E, Leica, Inc.), and the two substrates were brought together and cured at 65 °C for 4 h to ensure a strong bond between the control and fluid layer. The bonded control and fluid layers were carefully peeled off the master, and fluid input/outlet reservoirs were punched through the PDMS with a 2 mm biopsy punch.
Prior to bonding the PDMS replica to the cover glass, we used the SEM to determine the channel widths of the SU-8 nanochannel master and PDMS replica. Because SU-8 and PDMS charge under high vacuum, the images presented in Figure 1b,c were obtained under low vacuum at an accelerating potential of 5 kV and a beam current of 10 pA. An atomic force microscope (AFM; MFP-3D, Asylum Research) was used to measure the channel heights of the SU-8 nanochannel master. Due to the narrow widths of the nanochannels in the PDMS, we were not able to measure their depths directly with the AFM. The nanochannels were 1.06 ± 0.07 μm deep and 620 ± 10, 720 ± 10, 830 ± 10, 900 ± 10, and 1010 ± 10 nm wide. Below, we use nominal values of 600, 700, 800, 900, and 1000 nm for the widths.
A coverslip (50 mm × 45 mm) was cleaned in a solution of ammonium hydroxide, hydrogen peroxide, and water (2:1:2) at 70 °C for 20 min, rinsed with ultrapure water, and dried with nitrogen. The bonded control and fluid layers were rinsed with methanol and dried. All valves were covered with a plastic mask prior to plasma cleaning to prevent irreversible adhesion that would render the valves nonfunctional. The bonded control and fluid layers and coverslip were plasma treated (PDC-32G, Harrick), the plastic mask was removed, and the substrates were brought into direct contact with each other to form an irreversible bond between the PDMS and the glass. To prevent an irreversible bond between the glass and the nanochannel array, vacuum was applied to the control layer to raise the nanochannel array during bonding. The bonded device was coupled to an AssayMark Controller (Microfluidic Innovations, LLC) through a stainless steel manifold and was operated as previously described.19
On-Device Cell Culture
Cell loading was performed on-chip in manual operation mode, whereas growth and tracking was performed over hours with an automated program. Prior to the introduction of cells, the channels in the fluid layer were treated with 1% bovine serum albumin (BSA), which acts as a passivation layer; the cells were also incubated with media that contained 0.1% BSA. Addition of 1 mM IPTG in experiments with B. subtilis induced expression of mCherry protein for visualizing the cell body by epifluorescence microscopy. A saturated culture of cells (~15 μL) was added to the cell reservoir and pumped into the nanochannel region by pump 1 (Figure 1a). During cell loading, vacuum was applied to the control layer above the nanochannel array to allow cells to flow underneath the nanochannels (Figure 1d). Subsequently, positive pressure applied to the control layer above the nanochannel array trapped inoculated cells in the nanochannels (Figure 1e). We typically used vacuum of 100 mbar to raise the nanochannel array and pressure of 100 mbar to lower the nanochannel array and trap the cells. Positive pressures >250 mbar appeared to partially collapse the nanochannels and negatively impact cell growth. However, we were not able to achieve precise control of the channel depth by pressure actuation. In cases where the density of trapped cells was either too low or too high, we simply adjusted the cell inoculum and actuated the nanochannel array again in order to achieve the desired density.
Once the cells were trapped, media from the media reservoir was pumped through the nanochannel region by pump 1 to flush away excess cells and provide fresh media to the trapped cells. After all excess cells were flushed away, the control program was switched to a continuous cycle that alternately flushed the nanochannel region with fresh media for 2 min (0.9 μL/min) and then paused for 3 min during image acquisition. For peristaltic pumping, the three valves in series were actuated simultaneously, and four-cycle operation opened (○) and closed (●) the first (V1), second (V2), and third (V3) valves in the following sequences: V1 = ○-○-●-●; V2 = ●-○-○-●; V3 = ●-●-○-○. The pump operated at 1.04 Hz with a delay between each set of valve actuations of 240 ms.
Time-Lapse Image Acquisition
Phase contrast and epifluorescence microscopy of the trapped cells was performed with a Nikon Ti-E fluorescence microscope equipped with a Plan Apo 60x/1.40 NA Oil Ph3 DM objective. All images were captured with an Andor iXon3 DU-885 EMCCD camera operated with Nikon Elements software (Nikon, Inc.). Images were acquired at 12 locations for C. crescentus and 16 locations for B. subtilis across the nanochannel array at 5 min intervals over 12 h for C. crescentus and 20 h for B. subtilis. The nanochannel array was maintained at 30 °C with an objective warmer.
Image Analysis
We developed a MATLAB (The Math-Works, Inc.) program to track individual cells and reconstruct their lineages. The task of identifying and tracking individual cells over many generations is quite challenging; however, we took advantage of cell confinement in the nanochannel by reducing the collected 2-D images to 1-D line profiles, greatly decreasing the computational complexity. Adjacent frames were compared to help identify individual cells, and cells between frames were linked based on cell length and fluorescence intensity to construct a lineage. After cell lineages were reconstructed, the program performed various measurements on individual cells and lineages based on cell area and fluorescence intensity. The program determined rates of cell division and growth, quantitated fluorescent proteins, and correlated these measurements through time, with channel position, and among related cells.
RESULTS AND DISCUSSION
Device Design
We fabricated a nanochannel array that contains a broad range of channel widths to trap cells with various dimensions. We have successfully trapped bacteria of diverse sizes, ranging from 500 nm wide Caulobacter crescentus to 850 nm wide Bacillus subtilis (presented here), as well as the intermediately sized Escherichia coli (600 nm) and Agro-bacterium tumefaciens (800 nm; data not shown). To minimize physical stress to the confined cells, the nanochannels must be similar in width to the bacterial cells of interest. The nanochannels must also be deep enough to permit diffusion of fresh media into the nanochannels yet shallow enough to confine the cells in the z-dimension and prevent cells from standing on-end. The SU-8 masters have raised features that are 1 μm tall and 600, 700, 800, 900, and 1000 nm wide to accommodate bacteria of diverse size. To ensure that a sufficient number of cells are trapped for high-throughput data collection, the nanochannel array comprises 16 80-channel sections of the five channel widths for 1280 total nanochannels.
The distance between microchannels determines the length of the nanochannels. Longer channels permit cells to be trapped for more generations before they are pushed out of the nanochannels by the growth of neighboring cells. We have observed robust growth in channels up to 140 μm long (i.e., the distance between adjacent microchannels), which can trap 6 to 8 generations simultaneously without the loss of confinement or excessive stress to the cells. Our results suggest that rod-shaped bacteria fit best in the nanochannels; consequently, we either chose bacteria that were already rod-shaped (B. subiltis) or ensured the bacteria had a rod shape (ΔcreS mutant of C. crescentus). Moreover, bacterial size varies with growth stage and environmental conditions.31 Because the channel dimensions of our devices are fixed, we sought to minimize variation in cell size during our experiments by inoculating with bacteria grown in the same culture medium used on-chip.
Symmetric Bacterial Growth
To determine whether our devices provided a chemostatic environment for long-term study of cells over multiple generations, we trapped the bacterium B. subtilis and observed its growth for 20 h from multiple positions in the nanochannel array. B. subtilis cells were initially trapped in all channel widths in the array. However, in the narrower 600–800 nm wide channels, B. subtilis cells (~850 nm wide in prepared media) pushed under the PDMS and were no longer contained by the nanochannels after a few generations of growth or had their growth significantly perturbed (e.g., filamenting) by the channel walls (data not shown). Consequently, we report only B. subtilis growth data from channels 900 and 1000 nm wide. Individual bacterial cells were tracked throughout the experiment to determine division times, growth rates, and position within the nanochannel. Figure 2a shows a kymograph (i.e., a single nanochannel over the first 600 min of the experiment) of growing B. subtilis cells. Movie S1 in the Supporting Information shows robust growth and division of cells in a region of the nanochannel array. Cells periodically become trapped between nanochannels, but as shown in the movie, these cells do not negatively impact other cells growing in neighboring nanochannels. Over the duration of the experiment, individual cells are tracked (Figure 2c), their growth and division rates are determined, and lineages spanning several generations are reconstructed (Figure 2d).
Figure 2.

Bacillis subtilis growth in nanochannels. (a) Kymograph of a single channel of B. subtilis cells expressing constitutively induced, cytoplasmic mCherry over the first 600 min of growth. (b) Kymograph of the same cells in panel (a) that are expressing gfp fused to a promoter (Phag) for the motility gene hag. (c) Subset of cells from the highlighted area in panel (a). (d) Cell lineages of the cells in panel (c) that were constructed by a custom MATLAB program to track individual cells throughout the experiment and analyze division time, growth rate, and cell position within the nanochannel.
With the device design employed in Figure 1a, media was pumped through the microchannels and delivered into the orthogonal nanochannel array by diffusive transport. One benefit of this design is the trapped cells were not subjected to fluid flow during the experiment, because the nanochannels did not have a pressure gradient along them. However, to determine whether nutrient depletion occurred in the nanochannel array, we measured rates of division (Figure 3a,b) and growth (Figure S1a,b) of the trapped bacteria as a function of time and position within the nanochannels. We found that rates of division and growth varied early in the experiment before the channels had completely filled with cells (~200 min; data not shown). However, subsequent growth in full channels yielded uniform rates of division and growth over time and length of the nanochannel, which suggests that the cells require some time to adapt to the device. Consequently, our analysis began 200 min after the start of the experiment.
Figure 3.
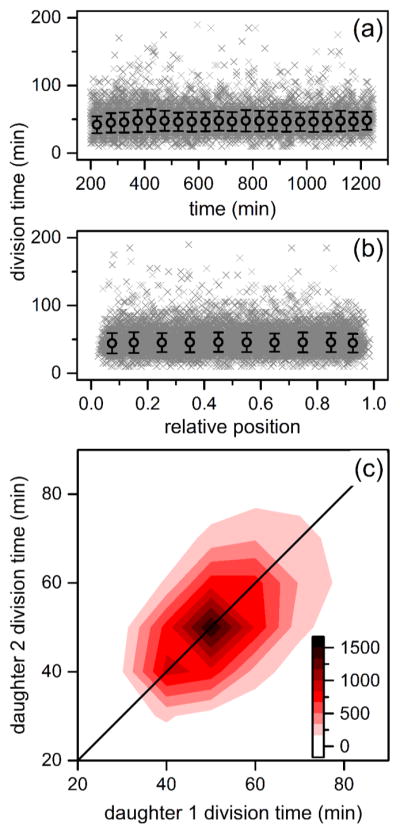
Variation of division times of B. subtilis cells in the nanochannels with (a) time and (b) relative position within the channel. Time is from the start of the experiment, and relative positon is the normalized pixel location of the cell. The gray crosses represent 18921 individual divisions, and the black circles are averages binned in 50 min intervals and 0.1 channel positions. Error bars are ± σ. (c) Heat map of division times of daughter cell pairs derived from the same mother cell where division events increase from white to black. Upon division of a mother cell, each resulting daughter was tracked until its subsequent division to determine daughter division time.
Linear fits of the individual rates of division and growth as a function of time and relative position from the center of the channel were performed to confirm cellular growth remains unchanged in the device. Linear fits returned slopes and standard errors of 2.78 × 10−3 ± 3.55 × 10−4 min of division time per min of the experiment and 0.841 ± 0.828 min of division time per relative cell position from the middle of the channel, and growth rates had slopes of −7.16 × 10−6 ± 6.13 ×10−7 μm2 of cell area per min of the experiment and −5.23 ×10−4 ± 1.43 × 10−3 μm2 of cell area per relative cell position from the middle of the channel. The slopes of the linear fits are very close to 0, which indicates that growth is constant and that a nutrient gradient does not form between the center of the nanochannel and the microchannel. Most importantly, a chemostatic environment is maintained within the device. Furthermore, whereas the cell population exhibits marked variation in reproductive rate, daughter pairs derived from the same division show correlated division times (r = 0.559, Figure 3c). The observed correlation suggests epigenetic inheritance of factors driving growth in sibling pairs.
Asymmetric Bacterial Growth
To demonstrate the versatility of our device, we trapped and observed growth of the asymmetrically dividing bacterium C. crescentus. The life cycle of C. crescentus (Figure 4) starts with a nonreproductive, motile swarmer cell. Subsequent differentiation yields a sessile, reproductively mature stalked cell that produces new swarmer cells through growth and division.32 Before the cell can enter the division cycle, the swarmer cell must go through a complex development process in which the cell extrudes a stalk tipped with an adhesive holdfast, ejects its flagellum, and disassembles its pili. At this point, the cell is considered a stalked cell and commences DNA replication.
Figure 4.

Caulobacter crescentus cell cycle. Newborn motile swarmer cells (SW) have pili and a flagellum at the same pole and are unable to undergo DNA replication. After a motile period of 20–30 min, swarmer cells differentiate into sessile stalked cells (ST) by shedding their flagellum, retracting their pili, and synthesizing an adhesive holdfast and stalk from the same pole. Stalked cells initiate DNA replication, elongate, and synthesize a flagellum at the opposite pole of the stalk, forming a predivisional cell (PD). The predivisional cell subsequently divides into two daughter cells: one swarmer cell and one stalked cell. Repeated division of the stalked cell thus yields one new swarmer cell for each round of reproduction.
The width of C. crescentus (~500 nm under our growth conditions) is substantially smaller than the width of B. subtilis (~850 nm). C. crescentus grew in all five nanochannel widths, but in the widest nanochannels, the cells quickly began to grow alongside each other (data now shown). Therefore, we analyzed only data generated from the 600 and 700 nm wide nanochannels, which gave robust growth of Caulobacter cells constrained along their widths in a single line. Observing Caulobacter growth for 12 h and performing the same linear regression analysis as with B. subtilis growth, we found that stalked cell division and growth rates were constant over time and nanochannel position (Figure 5a,b and Figure S2a,b), which further demonstrates our ability to maintain a chemostatic environment within the device. Linear fits returned slopes and standard errors of −3.72 × 10−3 ± 1.25 × 10−3 min of division time per min of the experiment and −0.993 ± 7.11 min of division time per relative cell position from the middle of the channel, and growth rates had slopes of −4.14 × 10−6 ± 2.72 × 10−6 μm2 of cell area per min of the experiment and 4.84 ×10−4 ± 1.55 × 10−3 μm2 of cell area per relative cell position from the middle of the channel.
Figure 5.
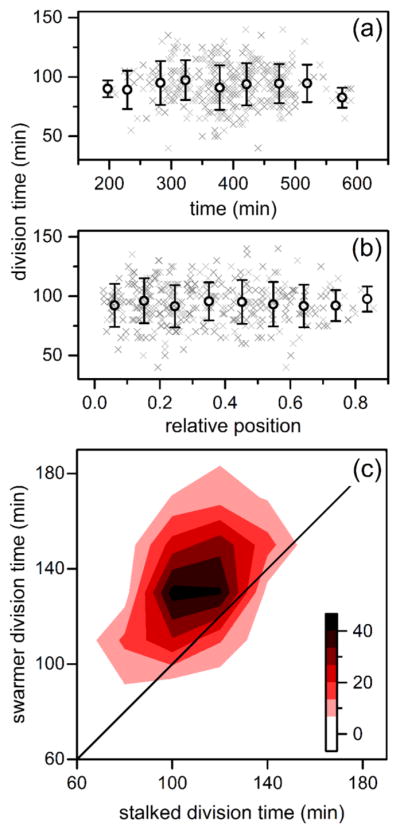
Variation of division times of C. crescentus stalk cells in the nanochannels with (a) time and (b) relative position within the channel. The gray crosses represent 362 individual divisions, and the black circles are averages binned in 50 min intervals and 0.1 channel positions. Error bars are ± σ. (c) Heat map of division times of paired stalked cells and swarmer cells where division events increase from white to black. Upon division of a stalked cell, the resulting stalked and swarmer cell were tracked until subsequent division of each cell to determine its respective division time.
To capture asymmetric division of C. crescentus cells, we needed to track at least three divisions to determine stalked and swarmer cell identity without specific morphological or cell identity markers. The first division event in a lineage identified new and old cell poles, while not yet determining cell stage. Upon a second division event, both old poles had reached the stalked stage, establishing stalked and swarmer cell identity in subsequent divisions. Tracked nanochannel lineages successfully captured the Caulobacter developmental delay, as swarmer cells differentiate prior to reproduction and, thus, take longer to divide (Figure 5c). Additionally, we observed a correlation within stalked-swarmer pairs in Figure 5c, whereby slowly dividing stalked cells tend to derive from lineages producing slowly dividing swarmer counterparts. This epigenetic division time effect agrees with previously published data obtained with flow cells.7
Epigenetic Inheritance
Our device provides a continuous nutrient environment that yields chemostatic bacterial growth. Accordingly, we are able to measure individual bacterial growth for multiple generations. Moreover, we sought to demonstrate our ability to quantitate protein expression and its epigenetic inheritance within distinct cell lineages of a clonal population. To observe this epigenetic inheritance, we used a B. subtilis strain that contained a fluorescent reporter to monitor motility state over multiple generations.
Exponentially growing B. subtilis exists as a mixed population of two discrete cell types: individual motile cells and long chains of sessile cells.30 This population heterogeneity arises from the transcription of the genes for motility and autolysin production for cell separation. Expression of these genes is governed by RNA polymerase and the alternative sigma factor σD such that cells with high σD activity are motile and cells with low activity are nonmotile.28,30,33 Switching between these states is thought to serve as a bet-hedging strategy, where motile cells pursue new sources of nutrients and nonmotile cells form colonies in which the population currently exists.34,35
We observed the motility state of individual B. subtilis cells by monitoring expression of Phag-gfp, a fluorescent gene reporter for transcriptional activity of the flagellin gene (hag), which is colored green in Figure 2b. Movie S2 in the Supporting Information depicts the same region in the nanochannel array as Movie S1 but shows the switching between motile and nonmotile states. Motile cells are ON (green) for flagellin expression and synthesize a flagellum in the wild-type genetic background, whereas nonmotile cells are OFF (dark) and do not undergo flagellum synthesis.30 A previous study looked at the dynamics of B. subtilis cells switching between each developmental state and found that cells do not exhibit control over the amount of time spent in the motile state, whereas cells exit the sessile state at a defined rate governed by a network of regulatory proteins.36 That study used short, dead-end channels similar to a mother machine2 to observe the cell fates of only the oldest cell at the closed end of each channel.
Here, we wanted to track lineages that span a range of ages and form new progeny cells and compare them to those cells that have aged over several generations. We were also interested in determining whether the state of this epigenetic switch for motility was inherited across multiple generations. We, therefore, compared the fluorescence intensities of corresponding lineage pairs. To establish these lineage pairs, an initial progenitor cell was identified, and its two progeny were arbitrarily designated lineage “a” or lineage “b” (Figure 6a). To determine whether the state of the epigenetic switch was inherited across multiple generations, we first identified progenitor cells that were ON and OFF. We established cells as being ON or OFF by classifying the brightest 20% of cells expressing Phag-gfp as ON and the dimmest 20% of cells as OFF. From these cells, the expression of the Phag-gfp construct was tracked across 5 generations and over 8800 cell divisions. Fluorescence intensities from all cells derived from the progenitor cell were averaged for each side of the lineage pair one frame prior to division for each lineage. A linear correlation (Pearson’s r) of the average fluorescence intensities for lineage “a” and lineage “b” was performed for each generation. We found that cells starting in both the ON and OFF states exhibited marked but gradually decreasing epigenetic inheritance spanning several generations. However, initially OFF lineages exhibited stronger inheritance, as indicated by higher correlation coefficients at each generation (Figure 6b). This higher correlation of the OFF state and lower correlation of the ON state agrees with an earlier report of B. subtilis switching dynamics in which the OFF state persists for a defined interval, whereas the ON state exhibits no such temporal regulation.36 This correlation in gene expression across multiple generations suggests epigenetic inheritance of the motility state. Furthermore, these results establish the ability of our microfluidic device and automated tracking analysis to accurately reconstruct cell lineages and to measure critical factors to study epigenetic inheritance.
Figure 6.

Expression of the Phag-gfp construct shown in Figure 2b tracked over 8800 cell divisions across 5 generations. Lineages were initiated from the brightest 20% and dimmest 20% of cells from 22,000 total cell divisions. (a) Lineage pairs were identified from the two daughter cells from a single division event and were arbitrarily chosen to be lineage “a” (red) or lineage “b” (blue). Subsequent growth and divisions were tracked throughout the experiment, and fluorescence intensities were determined one frame prior to division for each lineage (dashed boxes). (b) Variation of correlation coeffcients with generation for lineages that were initially identified as expressing the Phag-gfp construct (ON) and those lineages that were not (OFF).
CONCLUSION
Here, we have shown that our microfluidic device with an integrated nanochannel array allows for a continuous nutrient environment that produces chemostatic bacterial communities in both B. subtilis and C. crescentus. Our device offers dynamic control over the trapping of cells through the use of a pneumatically actuated nanochannel array and integrated pumps and valves that complete on-chip fluid and cell manipulations. Moreover, we demonstrated our ability to accurately reconstruct cell lineages and measure fundamental life history parameters, such as division time and growth rate, with our automated analysis program. Lineage reconstruction permits the observation of epigenetic effects, as demonstrated by correlations in division times for C. crescentus and in the motility state for B. subtilis. We foresee that our microfluidic device will serve as a general platform to study aging and other epigenetic phenomena in diverse bacteria.
Supplementary Material
Acknowledgments
This work was supported in part by NIH R01 GM113172 to S.C.J. and Y.V.B. and NIH R01 GM51986 to Y.V.B. The authors thank Prof. Daniel Kearns at Indiana University for kindly engineering and providing B. subtilis DK1437. The authors also thank the Indiana University Nanoscale Characterization Facility for the use of its instruments.
Footnotes
The authors declare the following competing financial interest(s): S.C.J. has an interest in Microfluidic Innovations, LLC, which markets the AssayMark Controller.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.6b00889.
Plots of cell growth rates for B. subtilis and C. crescentus (PDF)
Time-lapse video of B. subtilis cells growing and dividing in a section of the nanochannel array (Movie S1) (AVI) Time-lapse video of B. subtilis cells switching between motile and nonmotile states (Movie S2) (AVI)
References
- 1.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 2.Wang P, Robert L, Pelletier J, Dang WL, Taddei F, Wright A, Jun S. Curr Biol. 2010;20:1099–1103. doi: 10.1016/j.cub.2010.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moffitt JR, Lee JB, Cluzel P. Lab Chip. 2012;12:1487–1494. doi: 10.1039/c2lc00009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long Z, Nugent E, Javer A, Cicuta P, Sclavi B, Cosentino Lagomarsino M, Dorfman KD. Lab Chip. 2013;13:947–954. doi: 10.1039/c2lc41196b. [DOI] [PubMed] [Google Scholar]
- 5.Locke JCW, Elowitz MB. Nat Rev Microbiol. 2009;7:383–392. doi: 10.1038/nrmicro2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufmann BB, Yang Q, Mettetal JT, van Oudenaarden A. PLoS Biol. 2007;5:e239. doi: 10.1371/journal.pbio.0050239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegal-Gaskins D, Crosson S. Biophys J. 2008;95:2063–2072. doi: 10.1529/biophysj.108.128785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stewart EJ, Madden R, Paul G, Taddei F. PLoS Biol. 2005;3:e45. doi: 10.1371/journal.pbio.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ackermann M, Stearns SC, Jenal U. Science. 2003;300:1920–1920. doi: 10.1126/science.1083532. [DOI] [PubMed] [Google Scholar]
- 10.Young JW, Locke JCW, Altinok A, Rosenfeld N, Bacarian T, Swain PS, Mjolsness E, Elowitz MB. Nat Protoc. 2011;7:80–88. doi: 10.1038/nprot.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrell JE, Jr, Machleder EM. Science. 1998;280:895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 12.Skotheim JM, Di Talia S, Siggia ED, Cross FR. Nature. 2008;454:291–U12. doi: 10.1038/nature07118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groisman A, Lobo C, Cho HJ, Campbell JK, Dufour YS, Stevens AM, Levchenko A. Nat Methods. 2005;2:685–689. doi: 10.1038/nmeth784. [DOI] [PubMed] [Google Scholar]
- 14.Grunberger A, Paczia N, Probst C, Schendzielorz G, Eggeling L, Noack S, Wiechert W, Kohlheyer D. Lab Chip. 2012;12:2060–2068. doi: 10.1039/c2lc40156h. [DOI] [PubMed] [Google Scholar]
- 15.Wong I, Atsumi S, Huang WC, Wu TY, Hanai T, Lam ML, Tang P, Yang JA, Liao JC, Ho CM. Lab Chip. 2010;10:2710–2719. doi: 10.1039/c004719h. [DOI] [PubMed] [Google Scholar]
- 16.Charvin G, Cross FR, Siggia ED. PLoS One. 2008;3:e1468. doi: 10.1371/journal.pone.0001468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kysela DT, Brown PJ, Huang KC, Brun YV. Annu Rev Microbiol. 2013;67:417–35. doi: 10.1146/annurev-micro-092412-155622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rang CU, Peng AY, Chao L. Curr Biol. 2011;21:1813–1816. doi: 10.1016/j.cub.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 19.Madren SM, Hoffman MD, Brown PJB, Kysela DT, Brun YV, Jacobson SC. Anal Chem. 2012;84:8571–8578. doi: 10.1021/ac301565g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amin A, Thakur R, Madren S, Chuang HS, Thottethodi M, Vijaykumar TN, Wereley S, Jacobson S. Microfluid Nanofluid. 2013;15:647–659. doi: 10.1007/s10404-013-1180-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shang Y, Zeng Y, Zeng Y. Sci Rep. 2016;6:20297. doi: 10.1038/srep20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ely B. In: Methods in Enzymology. Jeffrey HM, editor. Vol. 204. Academic Press; 1991. pp. 372–384. [DOI] [PubMed] [Google Scholar]
- 23.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1972. p. 466. [Google Scholar]
- 24.Marks ME, Castro-Rojas CM, Teiling C, Du L, Kapatral V, Walunas TL, Crosson S. J Bacteriol. 2010;192:3678–3688. doi: 10.1128/JB.00255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gitai Z, Dye N, Shapiro L. Proc Natl Acad Sci U S A. 2004;101:8643–8648. doi: 10.1073/pnas.0402638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullin DA, Ohta N, Mullin AH, Newton A. Mol Genet Genomics. 2001;265:445–454. doi: 10.1007/s004380000432. [DOI] [PubMed] [Google Scholar]
- 27.Lambertsen L, Sternberg C, Molin S. Environ Microbiol. 2004;6:726–732. doi: 10.1111/j.1462-2920.2004.00605.x. [DOI] [PubMed] [Google Scholar]
- 28.Mirel DB, Chamberlin MJ. J Bacteriol. 1989;171:3095–3101. doi: 10.1128/jb.171.6.3095-3101.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. Proc Natl Acad Sci U S A. 2001;98:11621–11626. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kearns DB, Losick R. Genes Dev. 2005;19:3083–3094. doi: 10.1101/gad.1373905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chien AC, Hill NS, Levin PA. Curr Biol. 2012;22:R340–R349. doi: 10.1016/j.cub.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curtis PD, Brun YV. Microbiology and Molecular Biology Reviews. 2010;74:13–41. doi: 10.1128/MMBR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dubnau D, Losick R. Mol Microbiol. 2006;61:564–572. doi: 10.1111/j.1365-2958.2006.05249.x. [DOI] [PubMed] [Google Scholar]
- 34.Acar M, Mettetal JT, van Oudenaarden A. Nat Genet. 2008;40:471–475. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- 35.Veening JW, Stewart EJ, Berngruber TW, Taddei F, Kuipers OP, Hamoen LW. Proc Natl Acad Sci U S A. 2008;105:4393–4398. doi: 10.1073/pnas.0700463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norman TM, Lord ND, Paulsson J, Losick R. Nature. 2013;503:481–486. doi: 10.1038/nature12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.