

Abstract
Alloimmunity driving rejection in the context of solid organ transplantation can be grossly divided into mechanisms predominantly driven by either T cell-mediated rejection (TCMR) and antibody-mediated rejection (ABMR), though the co-existence of both types of rejections can be seen in a variable number of sampled grafts. Acute TCMR can generally be well controlled by the establishment of effective immunosuppression (1, 2). Acute ABMR is a low frequency finding in the current era of blood group and HLA donor/recipient matching and the avoidance of engraftment in the context of high-titer, preformed donor-specific antibodies. However, chronic ABMR remains a major complication resulting in the untimely loss of transplanted organs (3–10). The close relationship between donor-specific antibodies and ABMR has been revealed by the highly sensitive detection of human leukocyte antigen (HLA) antibodies (7, 11–15). Injury to transplanted organs by activation of humoral immune reaction in the context of HLA identical transplants and the absence of donor specific antibodies (17–24), strongly suggest the participation of non-HLA (nHLA) antibodies in ABMR (25). In this review, we discuss the genesis of ABMR in the context of HLA and nHLA antibodies and summarize strategies for ABMR management.
Keywords: HLA antibody, donor-specific HLA antibody, non-HLA antibody, antibody-mediated rejection, humoral immune system, in vitro B cell assay
Introduction
Organ transplantation improves the quality of life of patients with terminal dysfunction of organs, such as the kidney and pancreas, and is the most effective life support treatment for patients with heart, lung, and liver failure.
Although short-term prognoses for transplanted organs have improved significantly, long-term prognosis after 5–10 years remains insufficient, and reportedly reflects injury from chronic, indolent injury from sub-clinical antibody-mediated rejection (ABMR) (3–5, 15). Acute ABMR is a declining problem in organ transplantation as donor/recipient matching has improved (7, 16) and early acute ABMR is seen usually only in the context of ABO incompatible organ transplants (17, 18), and transplantation in highly sensitized patients with preformed donor-specific HLA antibodies (DSAs). Accordingly, preformed DSA are more likely to be produced before transplantation with histories of complications, such as pregnancy, previous transplant, blood transfusion, and prior organ transplantation (7, 19, 20). Hyper acute rejection, which can occur in the presence of preformed DSA, can be controlled using recently developed desensitization therapies (7).
Rejection due to de novo DSAs remains a major cause of transplanted organ loss, in the context of sub-clinical, chronic ABMR (21–24). Moreover, ABMR has also been reported in the absence of DSAs, leading to the discovery of specific non-HLA (nHLA) antigens that activate humoral immune responses in the graft. Potentially, nHLA antibody-mediated humoral immune responses develop acutely and chronically following transplantation and these antibodies may influence prognoses by participating in the onset and sequelae of rejection (16–18, 25–33). Although graft rejection has been reported among patients with nHLA antigens, one of challenges has been the discovery of the identity of these novel nHLA antigens and to correlate their presence and titers with ensuing mechanisms of transplant rejection.
Molecular Pathophysiology
During ABMR, antibodies for donor antigens are produced following activation of humoral immune responses, involving activated T cells and complement pathways.
As shown in Figure 1, naïve B cells differentiate into DSA-specific plasma cells (PCs) via germinal centers following exposure to antigens. This process involves initial uptake and surface presentation of donor antigens on antigen-presenting cells (APC) in response to an encounter of donor antigens, leading to activation of CD4+ effector T cells (34) and successive promotion of class-switching of naïve B cells and differentiation of memory B cells into PCs (35). Transmission of CD4+ effector T cell signals to B cells primarily involves association of major histocompatibility complex 1 (MHC-I) with T cell receptors (36). In addition, subordinate signaling pathways are activated by binding of CTLA4 (CD152), CD28, and CD40 ligand (CD40L) on T cell surfaces to the B7 (CD80/86) complex and CD40 on B-cell surfaces. Although CTLA-4 binds to B7, it reportedly downregulates T cell activity by binding to B7 with much greater affinity than CD28 (37–40). Intracellular CTLA-4 was closely related to the suppressor function of regulatory T cells (41–43) and reported the close relationship with autoimmune disease, including Graves’ disease, type 1 diabetes mellitus (DM) (44–48).
Figure 1.

The pathway of naïve B-cell differentiation into DSA-specific PCs. Naïve B cells differentiate into DSA-specific plasma cells (PCs) via germinal centers following exposure to antigens, herpes virus entry mediator; HVEM.
CD28 is expressed on CD4+ effector T cells and naive T cells (47), and promotes interleukin (IL)-2 production from B cells following binding to B7 complexes (48), leading to sustained naïve B cell differentiation into memory B cells (49). Conversely, CD40L mediates the class-switch of B cells in the germinal center by binding to CD40 expressing B cells (50) and support CD4+ effector T cells to help B cell differentiation (51, 52). Previous studies by Ettinger et al. (53) also showed that IL-21 induced PC phenotypes of human naïve and memory B cells following stimulation through B cell receptor (BCR) and CD40. Therefore, DSA-specific PCs developed and produced DSAs.
The Role of Costimulatory Pathway in the Clinical Field
CTLA-4Ig (immunoglobulin) binds to B7 and then suppresses the engagement of CD28. CTLA-4Ig can suppress the function of activated T cells through regulatory T cells, which may help suppress established chronic inflammatory disease (54).
In the field of transplantation, Belatacept, which links to the extracellular domain of CTLA-4, has been approved for the treatment of acute kidney rejection. In addition, an important problem in the field is to control antigen-specific memory B cells differentiation into PCs. The infusion of Belatacept might suppress DSAs development in a T-cell-independent manner because it has been reported that the infusion of CTLA-4Ig 1 week or more after transplantation could prevent DSAs development in a fully mismatched mouse cardiac transplant model but did not affect T-cell function (55). In addition, in the recent clinical BENEFIT trial of Belatacept induction by Vincenti et al. (56), there was a significant lower incidence of DSA development with Belatacept induction, when compared to the standard CNI arm, despite the higher incidence of acute rejection seen early with Belatacept induction.
The results might indicate that CTLA-4Ig could inhibit the growth and survival of DSA-specific memory B cells or PCs in a human model. About the other suppressive receptors related to CD28, PD-1 (programed death-1) has been reported to be expressed on the surface of T cells, and B and T lymphocyte attenuator (BTLA) has been reported to be expressed on the surface of both B and T cells, both of which have also been attracting attention as targets for treating autoimmune diseases and cancer (57).
In the field of autoimmune disease, the involvement of signaling through CD40–CD40L interaction in autoimmune diseases has been reported and dysregulation of CD40 may induce macrophage-mediated coronary artery disease (CAD); the blockade of CD40L may, thus, be an attractive therapeutic target to improve CAD (58). Recent studies have also implicated altered regulation of the CD40 axis and generation of pathogenic activating anti-CD40 antibody for the generation of podocyte injury in focal segmental glomerulosclerosis (FSGS) recurrence after kidney transplantation (59, 60). Further research is needed to better elucidate how the CD40 axis may help control other autoimmune diseases.
Human Leukocyte Antigen Antibodies
Histocompatibility analyses using cross-match, human leukocyte antigen (HLA) typing, and antibody tests are widely performed prior to transplantation in many laboratories, and are an accepted approach for limiting organ rejection. Recent developments in laboratory procedures, survey equipment, and technologies have led to highly sensitive detection of HLA antibodies.
Therefore, we could detect a very small amount of HLA antibodies and determine these antibody specificities; trace quantities of HLA antibodies recently provided useful prognostic information for ABMR and transplanted organ outcome and a judgment of transplant evaluation (61–65).
Major histocompatibility complex 1 class 1 (HLA-A, -B, and -C) and MHC class 2 (HLA-DR, -DP, and -DQ) have been identified as HLA antigens, and HLA antibodies can be detected in sera using FlowPRA® Class I & II Screening Tests (One Lamda) to identify Class I or/and Class II HLA antibodies. In further analyses, positive cases should be identified using HLA LAB Screen HLA Class I or/and Class II single antigen beads (One Lambda) with Luminex technology, which determines antibody profiles against HLA Class I or Class II and indicates the presence or absence of DSAs.
The Role of Preformed DSA in the Pathogenicity of Graft Injury
Donor-specific HLA antibodies that cause ABMR have been classified as those that are present before transplantation as well as those de novo that are produced after transplantation. Previous studies on kidney, heart, lung, and liver transplantation indicate that poor-prognosis is associated with the presence of DSAs before transplantation. We will next discuss the role of preformed DSAs in each organ transplant. With regard to kidney transplants, preformed DSAs have been recognized as one reason of hyper acute rejection. DSAs with high threshold MFI and DSAs with cross match-positive could predict ABMR onset after transplantation (7). With regard to pancreas transplants, we found a report describing that preformed DSAs did not affect graft prognosis (66) but DSAs could be detected from the sera with significantly higher probability than in recipients without a history of preformed DSAs after transplant. As a result, recipients sensitized by DSA before transplant had a history of DM more than 10 years after the transplant, so we should pay more attention to postoperative management, including blood sugar management (67).
With regard to liver, heart, and lung transplants, it is already known that preformed DSAs could affect graft outcome and patient mortality. In addition, preformed C1q binding DSAs have been reported to affect graft prognosis in liver and heart transplants and preformed DSAs with MFI ≥5000 and IgG3 DSAs could be risk factors for ABMR onset in liver transplant cases (19, 68, 69). Indeed, additional risk factors for ABMR in these patients include ABO incompatible and cross match-positive status, cases with a history of previous transplants, pregnancy, and blood transfusions (19, 70, 71). With regard to lung transplants, preformed DSAs have been reported to promote de novo DSA development early after transplant and patient survival (72–75). Therefore, these data on clinical correlations of DSA and rejection in different organ transplants suggests that improved screening and therapies, such as desensitization before transplant, may be of benefit across different types of solid organ transplants to limit subsequent postoperative complications (76).
Mechanisms of Onset of ABMR by Preformed DSA
Antibody-mediated rejection caused by preformed DSAs manifests as hyper acute rejection immediately after transplantation, leading to failure of the transplanted organ within several hours. In these cases, DSAs immediately bind to all capillary endothelium surfaces in the transplanted organ, and concomitant complement activation leads to the formation of fibrin clots and acceleration of blood coagulation. Subsequently, rapid peripheral circulation incompetence causes necrosis of vascular walls, intense bleeding of the transplant, and necrosis in neighboring tissues. Finally, inflammatory cells, such as neutrophils, infiltrate capillary endothelial surfaces, and further undermine the transplant (6, 77).
Management of ABMR by Preformed DSA
Improvements in desensitization therapy have enabled management of high risk recipients, such as those with cross match-positive phenotypes and high organ transplantation sensitivity; as a result, the prevalence of severe hyper acute rejection by preformed DSAs has decreased significantly (7). Accordingly, Ng et al. summarized desensitization protocols and complications using rituximab, bortezomib, eculizumab, and alemtuzumab, and reported promising graft survival in patients across various institutes (78). However, complications included anemia and thrombocytopenia, likely reflecting myelosuppression by these agents. In addition, various infections in some cases were detected, including cytomegalovirus (CMV), BK virus, and Epstein–Barr virus (EBV), indicating that desensitization therapy disposes patients to an increased risk of opportunistic viral infections. In addition, it was reported that induction with T-cell depleting agents (anti-thymocyte globulin) was closely associated with CMV, EBV, and BK polyomavirus (BKV) infections in comparison with IL-2a receptor antagonists (anti-CD25) (79). Therefore, the use of T-cell depleting agents should be avoided as an immunosuppressive reagent or induction. If possible, the use of IL-2a receptor antagonists or no induction should be considered (79, 80). Additionally, it was expected that these virus infection may contribute to the activation of immune responses in transplanted organs, and dose reductions of immunosuppressive agents may activate immune reactions to graft antigens.
To address this issue, prediction and early detection of viral infections is critical, and could be used to inform doses reductions of immunosuppressive agents. Concomitant administration of preventive and therapeutic antiviral agents is also critical in the management of these patients.
Desensitization Therapy
Prior to the introduction of rituximab, plasmapheresis and splenectomy were long recommended as desensitization therapies for patients with ABO incompatible kidney transplants. Subsequently, rituximab was shown to inhibit the onset of ABMR without splenectomy. Rituximab is a monoclonal antibody (mAb) against the protein CD20, which is expressed in immature and mature B cells. However, because CD20 is not expressed on PCs, rituximab may not inhibit the production of DSAs by PCs. In addition, recent studies show varying effects of rituximab on B cell phenotypes, with higher sensitivity of naïve B cells than memory B cells (81). Thus, although rituximab suppresses immune activation and may not provide protection from infection, memory B cells may remain viable.
In addition, posterior reversible encephalopathy syndrome (PRES) and acute respiratory distress syndrome (ARDS) was reportedly increased in patients treated with rituximab as severe adversity effect (82). These data warrant further clarification of the depletion mechanisms of rituximab in B cells.
Bortezomib is a proteasome inhibitor that was developed as a treatment for multiple myeloma, and the effectiveness of this agent against transplant rejection was reported in 2008. These studies showed downregulated immune responses to donor antigens, recovery of graft function, and long-term suppression of serum antibody levels. However, inhibition of the proteasome by bortezomib may be detrimental to healthy cells (83–88).
As an alternative, eculizumab is a recombinant humanized monoclonal IgG2/4 antibody that suppresses complement activation and inhibits production of C5, which is the final product of the complement pathway and activates inflammatory responses and ultimately results in apoptosis of infected cells (89). Accordingly, treatments with this agent led to severe infectious diseases, including meningitis (90).
Finally, alemtuzumab is a recombinant DNA-derived humanized IgG1 kappa mAb that is directed toward CD52 and is used is used to treat B-cell chronic lymphocytic leukemia (B-CLL) and multiple sclerosis patients, warranting consideration for the treatment of ABMR. As adverse effect, it has been associated with infusion-related events (91, 92).
Infection as a Trigger of Rejection
Cytomegalovirus Infection as a Trigger of Rejection
Cytomegalovirus is among the most common infections after solid-organ transplantation, and results in significant morbidity, graft loss, and adversity. Although numbers of CMV-seronegative (R−) cases have increased recently in healthy subjects, those with organ transplants from CMV-seropositive donors (D+) are at the highest risk of primary CMV disease, which can easily become serious causing the reactivation of latent virus transmitted in the allograft (93, 94). Additionally, a close relationship between CMV infection and allograft rejection has been reported in CMV D+/R− liver and kidney transplant patients (93, 95).
Laboratory Diagnosis of CMV
Nucleic Acid Testing
Nucleic acid testing (NAT) is widely used to detect and quantify CMV RNA and DNA.
Serology
Serological analyzes allow risk stratification of patients during the pre-transplant screening phase on the basis of tests for CMV IgG antibodies in both donors and recipients, and can indicate the presence of latent infection.
Antigenemia
The antigenemia assay detects the CMV pp65 antigen in infected leukocytes from peripheral blood, and has been used for rapid diagnosis of CMV infections in transplant recipients (96).
Treatment of CMV
In a previous study, valganciclovir was found to be more effective than oral ganciclovir at preventing CMV disease in solid organ transplant recipients (97), suggesting that extension of valganciclovir prophylaxis to 200 days may benefit high risk (D+/R−) kidney recipients. Following transplantation, CMV disease is predominantly treated using intravenous (IV) ganciclovir (5 mg/kg every 12 h) and oral valganciclovir (900 mg twice daily) (98).
BK Polyomavirus Infection as a Trigger of Rejection
More than 90% of healthy subjects become infected with BKV (99, 100), which is the major cause of polyomavirus-associated nephropathy (Py-VAN) and presents a 1–15% risk of allograft failure in kidney transplant patients (101–106). And it has been reported that BKV-activated antibody reactivity in recipients at the onset of immunosuppression (107). However, although number of BKV-seronegative (R−) cases has increased recently in healthy subjects, these patients are the most susceptible to BKV disease following transplantation from BKV seropositive donors (D+) (108, 109).
Laboratory Diagnosis
Screening for BKV replication should be performed at least every 3 months during the first 2 years after transplantation, and then annually until the fifth year.
Nucleic Acid Testing
Nucleic acid testing in polymerase chain reaction (PCR) is used to detect amplifications of BK DNA.
Urine Cytology
Urine cytology is sufficient to detect decoy cells, which are associated with BKV induced organ failure.
Treatment of BKV
First, reduction of immunosuppression should be considered (110, 111). In patients with sustained high-level plasma BKV loads despite dose reductions of immunosuppression agents, administration of antiviral agents (Cidofovir), and a replacement for mycophenolic acid (Leflunomide), intravenous immunoglobulin (IVIG), and anti-mycotic agents (Fluoroquinolones) should be considered.
Epstein–Barr Virus Infection as a Trigger of Rejection
Epstein–Barr virus contributes to the pathogenesis of post-transplant lymphoproliferative disease (PTLD) occurring cases early after transplantation in more than 90% of the cases, and small intestine transplantations are associated with higher risks than heart, lung, and liver transplantations (112, 113). The close relationship between EBV infection and ABMR has been reported in heart transplantation (114).
Laboratory Diagnosis
Nucleic Acid Testing
Epstein–Barr virus DNA monitoring for EBV D+/R− recipients should be recommended, with continued EBV load screening every 3–6 months until 2–3 years after transplantation. This monitoring is particularly important for EBV-seropositive recipients with intestinal transplants, and monitoring of EBV DNA every 2–4 weeks in the first 3 months should be performed, monthly until 6 months post-transplantation, and then every 3 months until the end of the first year.
Treatment of EBV
Antiviral prophylaxis for high risk patients (EBV D+/R−) is considered in some centers (99).
Treatment with acyclovir, ganciclovir, and IVIGs has shown some benefits in the prevention of PTLD among EBV-seronegative recipients who their donors are EBV-seropositive (113).
The Role of De Novo DSA in the Pathogenicity of Graft Injury
Recent reports show that DSAs play an important role in ABMR onset, and this has been shown by highly sensitive monitoring of HLA antibodies in the sera (11–13). However, DSAs may be absorbed into transplanted organs during the early phases of antibody production (115) (Figure 2). Accordingly, in the Guidelines of the Transplantation Society (TTS), post-kidney transplant DSA monitoring is not recommended for all patients beyond the first year (116). Hence, to avoid the influence of absorption, antibody production from PCs has been analyzed in vitro, because these antibodies may not be influenced by the absorption and provide us with further detailed illustrations that are available to clarify how these antibodies are produced in organ recipients. However, PCs are seldom found in blood and predominate in bone marrow and secondary lymphoid tissues, techniques for differentiating B cells into PCs, are required to investigate antibody production (117–119).
Figure 2.
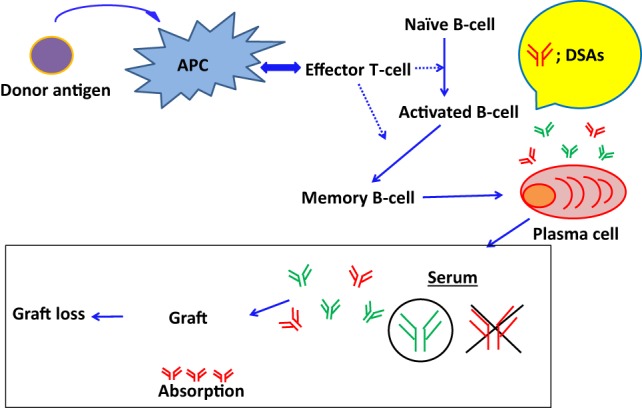
The absorption of DSAs into graft and the development of ABMR. DSAs may be absorbed into transplanted organs during the early phases of antibody production.
In Vitro B Cell Assays
Determination of HLA antibodies in supernatants of cultured B cells can better inform ABMR management than those in sera. Therefore, some researchers have suggested that if peripheral B cells could be differentiated into PCs in vitro, then the in vitro differentiation of peripheral B cells into PCs may facilitate the control of ABMR. However, unlike in vitro T cell assays that have long been used to control T cell-mediated rejection (TCMR), primary cultures of B cells are difficult to maintain and in vitro B cells assays have not long been established. Although memory B cells were reportedly differentiated into APC in vitro (120), these short comings require further improvements in the ease and convenience of B cell culture, and subsequent assay development that can be used to detect HLA antibodies in B cell culture supernatants to ultimately prevent transplant rejection.
Moreover, there are important points to detect HLA antibodies from the B cell culture supernatant. Peripheral B cells include naïve B cells and memory B cells (121, 122); and these B cells derived from PCs survive for varying durations and produce antibodies. However, HLA antibodies that cause ABMR are mainly produced by memory B cells (123–125), warranting establishment of in vitro B cell assays in which memory B cells are selectively differentiated into PCs in vitro and are used to produce antibodies. In addition, many reports have showed that long-lived PCs, which produce antibodies in the bone marrow for long periods, play an important role in ABMR (126–129). Therefore, to monitor the progression of antibody-mediated diseases, in vitro culture systems, in which B cells are differentiated to their terminal stage (long-lived PCs), are urgently required. On using a clinical specimen, the volume of B cells in peripheral blood is very low following immunosuppression or particularly when we could collect B cells from recipients who experienced desensitization therapy.
However, feeder cells can strongly activate human B cells to proliferate and differentiate in a cell–cell contact-dependent manner in these cases. Thus, in vitro B cell assays of HLA antibodies from the cultured supernatants may lead to drug sensitivity tests that are similar to those for T cells, may contribute to clinical applications of personalized immunosuppression, and the development of new immunosuppressant agents that control ABMR.
In Vitro Memory B Cell Assays
We found a report about immunosuppressive agent susceptibility for the differentiation of human B (CD19+) cells in vitro with a combination of IL-21, phosphorothioate CpG-oligodeoxynucleotide (CpG-ODN), histidine-tagged soluble recombinant human CD40 L and anti-polyhistidine mAb (130). IL-21 is produced by follicular helper T cells (131), which synergistically induce maximum Blimp-1 upregulation and optimal PC differentiation with CD40 L (132). TLR9 agonist CpG-ODN activates B cells proliferation and promotes PCs differentiation (133). This culture system induced IgG production but could not sustain the survival of PCs for a long period. It might indicate that the other cytokines play an important role in human B cells differentiation into mature PCs in vitro, because other groups have reported that APRIL and the B cell activating factor (BAFF) would support the survival of PBs and PCs recently (134–137). In addition, a previous report has shown that CD27+ memory B cells could be differentiated into long-lived PCs with supernatants from bone marrow stromal cell line M2-10B4 (138), which support the long-term culture of human bone marrow stem cells. The mechanisms by which M2-10B4 cells contribute to PCs survival has yet to be revealed, but it is suggested that CD27+ memory B cells demand well-balanced support from stromal cells (139–142). In addition, different environments or signal transmission might be required for the differentiation of CD27− naïve and CD27+ memory B cells into mature PCs. Therefore, we should improve the in vitro B cell assay to sustain CD27+ memory B cell-derived PC survival for a long-term selectively. For example, we should examine how any humoral factors, including growth factor or any cytokines from activated T cells could affect CD27+ memory B cell growth and survival in vitro, while referring to the reports that helper T-cells may mediate CD27+ memory B cell differentiation into PCs in vivo (143).
Risk Factors of ABMR from De Novo DSA
Not all DSAs participate in ABMR and transplanted organ prognosis (7), and although C1q binding DSAs are reported risk factors for ABMR onset, further studies of DSA characteristics are required to identify those with prognostic value. In addition, various other factors influence transplanted organ prognoses (ABMR onset, graft survival) and require further investigation. About the risk factors for graft loss, thrombotic microangiopathy (TMA), glomerulopathy, C4d deposition, and chronic injury change in histopathological diagnoses were reported.
As other factors besides histopathological findings, a history of subclinical ABMR and TCMR and a decline of graft function could be risk factors. This might indicate that a graft would fail with high probability when the humoral immune response toward a donor-specific antigen has proceeded to an irreversible stage.
C1q Binding DSA
C1q appear in the beginning of the classical complement pathway, and C1q binds directly to antigens and initiates classical complement pathway activation. Subsequent C1q-activated reactions include (i) antigen binding, (ii) binding to C-reactive protein, and (iii) binding to antigen–antibody complexes, and can lead to the activation of C3 convertase and the degradation of C3 to C3b and C3a (144, 145). Of these, C3b is the main effector of the complement pathway, while C3a activates inflammatory responses. Indeed, C1q may play important roles in the activation of inflammatory reactions against grafts. Accordingly, C1q binding to DSAs reportedly influences the frequency of ABMR onset and glomerulopathy in solid organ transplants, leading to increased chances of graft failure. Thus, binding of C1q to DSAs may be highly predictive of graft prognosis, warranting the development of interventions that decrease the presence of C1q binding DSAs. The C1qScreen™ (One Lamda) is a reliable tool for distinguishing complement-binding antibodies from non-complement-binding ones, and is widely applied using Luminex-based LABScan™ 100 flow fluorescence analyzers to determine relative amounts of C1q binding antibodies. The C1qScreen™ in combination with the Luminex-based LABScan™ can indicate the relative amount of C1q bound to DSAs and provides us with useful information from the sera (146). In addition, in C1q-positive cases despite being DSA-negative, graft survival is poor. It suggested that C1q could affect the transplanted organ prognosis by itself.
DSA Characteristics
In this study, we tabulated previously reported factors that participate in ABMR and graft loss (Table 1). Reports show that DSAs with higher mean fluorescent intensities (MFI) of ≥15,000 cause ABMR with higher probability than those with MFI of ≤5000, and higher level of DSAs may activate humoral immune reactions to donor antigens. In addition, many papers indicated that class II DSAs should be considered as a risk factor, particularly at the onset of ABMR. However, DSA specificities that activate humoral immune response to donor antigen may depend on the type of transplanted organ, and the further recognition about detailed association between DSAs and graft outcome is required in each solid organ transplantation.
Table 1.
Various other factors influence transplanted organ prognoses and require further investigation.
| Risk factors | Out come | |||
|---|---|---|---|---|
| Study size | Organ | ABMR | Graft loss | Reference |
| 226 | Kidney | Highly sensitized patients | ABMR-positive | (147) |
| DSA relative intensity scores greater than 17 | Thrombotic microangiopathy (TMA) positive | |||
| Presence of both class I and II DSAs at transplant | Induction with intravenous immunoglobulin and rituximab | |||
| 62 | C1q-positive | C1q-positive | (148) | |
| Both of DSA- and C1q-positive | ||||
| Transplant glomerulopathy | ||||
| Decline of eGFR | ||||
| 1016 | Complement-binding DSA DSA-positive | Complement-binding DSA | (149) | |
| DSA-positive | ||||
| 1307 | Subclinical ABMR | (150) | ||
| Subclinical TCMR | ||||
| 1365 | TCMR | TCMR diagnosed after the first year post-transplant | (151) | |
| Chronic histological injury | ||||
| Transplant glomerulopathy | ||||
| 67 (grafts) | Late aABMR | (152) | ||
| 885 | Capillary C4d-positive | (153) | ||
| 1054 | TCMR | Higher glomerulitis scores | (154) | |
| Higher C4d staining scores | ||||
| 1 | Plasma cell-rich rejection (PCRR) with ABMR | (155) | ||
| 237 | DSA-positive preformed DSA-positive | DSA-positive | (7) | |
| AMR | ||||
| DSA-positive/CXM-positive | ||||
| 234 | Microcirculation inflammation | (4) | ||
| 274 | C1q-fixing DSAs | (140) | ||
| 152 | Pancreas-kidney | De novo DSA-positive | (67) | |
| 439 | Pancreas | Elevated DSA | (156) | |
| Preformed DSA-positive | ||||
| 2631 | Liver | Preformed class II DSAs positive MFI ≧5000 | (19) | |
| 1270 | Preformed C1q-fixing class II DSA | IgG3 DSA-positive | (157) | |
| De novo IgG3 DSA | ||||
| 749 | De novo DSA development | (158) | ||
| 15 | Heart | SAB-C1q-positive DSA CDC-XM-positive | (9) | |
| 243 | De novo DSA-positive | (159) | ||
| Persistent DSA | (160) | |||
| 44 | Lung | DSA-positive | HLA-DQ DSA (>10,000) | (71) |
| 60 | ||||
| 546 | Early anti-HLA class II DSA | (72) | ||
| Pre-operative HLA antibodies | ||||
| Retransplantation | ||||
| Postoperative PGD | ||||
| 79 | Intestine | De novo DSA development early after transplant | (161) | |
| 291 | DSA-positive | DSA-positive | (162) | |
With regard to kidney transplants, DSAs with high threshold MFI and C1q binding DSAs have been reported to be closely related to TMA, glomerulopathy, microangiopathy, C4d deposition, extensive interstitial fibrosis, and tubular atrophy and these factors could affect graft prognosis in the long term (147–154). With regard to pancreatic transplants, elevated DSAs could affect graft prognosis (67, 156). With regard to liver transplants, IgG3 DSAs and C1qbinding DSAs were related to graft survival and class II DSAs were shown to be closely related to acute rejection early after a transplant (157). In addition, DSAs could affect graft outcome and reduce graft survival 1 year or more after a transplant by itself (158). With regard to cardiac transplants, C1q binding DSAs and cross match positivity could be risk factors for ABMR and de novo DSA development and persistent DSA were found to be closely related to graft loss (9, 159, 160). With regard to lung transplants, DSA has been related to ABMR, cellular rejection, and bronchiolitis obliterans and could significantly reduce postoperative survival 3 years later compared with that in DSA-negative recipients. In addition, de novo DSA (along with HLA-DR mismatch) development has been reported to reduce postoperative survival (71, 159). With regard to intestine transplants, de novo DSA development early after transplant could affect graft prognosis and might be effective for screening of acute rejection because DSA measurement has been shown to be closely related to histological findings (161, 162). The characteristics of DSA that could affect graft prognosis vary among the different types of organ transplant; we should, thus, understand these features well and make use of them for the postoperative management of transplant recipients.
Onset Mechanisms of ABMR from De Novo DSA
Antibody-mediated rejection caused by de novo DSAs typically appears several weeks to months after transplantation, but can develop at any time as far as a graft engrafts afterward.
Following the absorption of HLA antibodies onto capillary endothelial donor antigens (mainly HLA antigen), activation of pro-complement solidification and accumulation of inflammatory cytokines, macrophages, and neutrophils are caused successively. Therefore, it leads to microangiopathy and gradual annual declines in graft function (52, 163–165). Under these conditions, ABMR-mediated microangiopathy is chronic and sustained, although moderate inflammatory activities result in slow and irreversible disease progression.
Management Strategies of ABMR by De Novo DSA
In a previous section, we suggested that immunosuppressive therapy limits differentiation of naïve B cells to germinal center B cells by controlling CD4+ effector T cell stimulation. However, stronger immunosuppressive therapy is required to control B cell growth and survival after differentiation of naive B cells to memory B cells in germinal centers. As a result, chronic use of immunosuppressive agents after differentiation of naïve B cells into memory B cells corresponding to HLA antibodies may no longer affect B cells in germinal centers.
Thus, further attention should be paid to pancytopenia, anemia, and viral infection as well as to those concerning B cell differentiation, because more strong immunosuppressive therapy might be necessary to inhibit memory B cell growth and survival in comparison with naïve B cells. In particular, rituximab administration induces CD20+ memory B cell apoptosis (166), bortezomib therapy inhibits the production of DSAs production from PCs (167), and IVIG can be used to reduce circulating DSAs (168) (Figure 3). Hence, the development of new immunosuppressive agents that inhibit memory B cell growth and survival is warranted.
Figure 3.

The pathway of naïve B-cell differentiation into DSAs specific PCs and how immunosuppressive reagents suppress the development of ABMR.
In addition, the development of a diagnostic method for predicting the development of DSA specific memory B cells as soon as possible has been required.
Therefore, the control of ABMR is very difficult; the disease state progresses irreversibly and severely and is unresponsive to increasing immunosuppression following diagnosis using currently available methods. Although graft tissue biopsies are the most reliable diagnostic method for ABMR, it was hard to perform frequently because it is very invasive. Therefore, less invasive diagnostic approaches are urgently required to predict the development of DSA-specific memory B cells.
Histological studies of ABMR following solid organ transplantation show that the classical complement pathway is activated after adhesion of DSAs to capillary endothelia, and that C4d produced and deposited as the final product of this pathway is an important ABMR diagnostic factor before (169). In late years, the number of reported cases of C4d-negative ABMR has recently increased (15) and some DSA-related mechanisms that are independent of the classical complement pathway have been identified. In each organ transplantation, these observations necessitate revision of histologic diagnostic criteria for all organ transplant patients, and improved the understanding of ABMR (170–172).
Banff Score for Diagnosis of ABMR
Pathological diagnoses play important clinical roles, and diagnostic criteria have been revised for all organ transplants. In particular, Banff score are widely used as histologic methods for kidney transplantations, although diagnostic criteria were substantively revised in 2013; indeed, the roles of ABMR, DSAs, and C4d deposition in grafts received greater emphasis in the previous diagnostic criteria before the meeting in 2013. In the 2013 revised edition (Table 2), C4d-negative ABMR became the diagnostic focus especially, reflecting on the increased numbers of reported cases. Thus, we listed the important diagnostic criteria for ABMR in the revised edition in 2013, including confirmation of microangiopathy, evidence for DSA-capillary endothelial reactions, and detection of DSA in the serum. In particular, diagnosis of DSA-capillary endothelial reactions requires at least one of the following observations; (i) C4d deposition in peritubular capillaries (PTC), (ii) evidence of more than moderate microangiopathy and microvascular inflammation (MVI; g + ptc ≥ 2), and (iii) expression of endothelial activation (ENDAT) and injury transcripts.
Table 2.
Revised classification of antibody-mediated rejection.
| Acute/active ABMR | |
| 1 | Evidence of acute tissue injury, including one or more of the following |
| Microvascular inflammation (g > 0 and/or ptc > 0) | |
| Intimal or transmural arteritis (v > 0) | |
| Acute thrombotic microangiopathy, in the absence of any other cause | |
| Acute tubular injury, in the absence of any other cause | |
| 2 | Evidence of current/recent antibody interaction with vascular endothelium, including at least one of the following: |
| Linear C4d staining in ptc | |
| Moderate microvascular inflammation(g + ptc ≧ 2) | |
| Increased expression of gene transcripts indicative of endothelial injury | |
| 3 | Serologic evidence of DSAs |
| Chronic/active ABMR | |
| 1 | Evidence of chronic tissue injury, including one or more of the following |
| Transplant glomerulopathy(cg > 0) | |
| Severe ptc basement membrane multilayering (requires EM) | |
| Arterial intimal fibrosis of new onset, excluding other causes | |
| 2 | Evidence of current/recent antibody interaction with vascular endothelium, including at least one of the following |
| Linear C4d staining in ptc | |
| moderate microvascular inflammation(g + ptc ≧ 2) | |
| Increased expression of gene transcripts indicative of endothelial injury | |
| 3 | Serologic evidence of DSAs |
The bold font showed the most important factor to diagnose ABMR (Acute and Chronic).
DSAs, donor-specific HLA antibodies; EM, electron microscopy.
Furthermore, in the revised criteria, ABMR phenotypes have been classified as acute/active; chronic/active corresponding to the diagnostic criteria, which have been listed in detail.
Microvascular inflammation scores and C4d deposition in PTC are currently the most commonly used diagnostic criteria. However, according to this standard, ABMR diagnoses are recommended in the presence of strong MVI, even in specimens that are C4d-negative.
Thus, C4d deposition has not been necessary for ABMR diagnoses after the meeting in 2013.
By contrast, prior to 2013, ABMR was considered reflective of injury to capillary endothelia (165, 173), and C1q was the assumed trigger of the classical complement pathway following binding of DSAs to capillary endothelia. Although C4d is the final product (Figure 4), this classical pathway was not necessarily activated during ABMR in C4d-negative patients.
Figure 4.

The development of ABMR caused by DSAs. (A) Indirect injury via complement fixation or recruitment. C1q was the assumed trigger of the classical complement pathway following binding of DSAs to capillary endothelia. Although C4d is the final product. (B) Direct injury to the capillary endothelium. DSAs may directly promote vascular endothelial cell growth and proliferation, and inhibit apoptosis in capillary endothelia. (C) Recruitment of inflammatory cells with Fc receptors. DSAs have been shown to bind with Fcγ on the cell membrane surfaces of macrophages, natural killer cells, and neutrophils, and to induce inflammatory cytokine production and microangiopathy.
About the other pathways, DSAs may directly promote vascular endothelial cell (EC) growth and proliferation, and inhibit apoptosis in capillary endothelia (Figure 4); DSAs have been shown to bind with Fcγ on the cell membrane surfaces of macrophages, natural killer cells, and neutrophils, and to induce inflammatory cytokine production and microangiopathy (Figure 4).
Furthermore, in the revised criteria, ABMR phenotypes have been classified as acute/active, chronic/active corresponding to the diagnostic criteria, which have been listed in detail.
Among these, evidence of acute tissue injury as the diagnostic criteria of acute/active ABMR, and morphologic evidence of chronic tissue injury as the diagnostic criteria of chronic/active ABMR, is considered central. Therefore, effective management of ABMR entails varied treatments for differing levels of pathological progress, and these diagnostic criteria identify ABMR phenotypes with sufficient accuracy to inform treatments.
The Role of Non-HLA in the Pathogenicity of Graft Injury
Rejection by nHLAs was previously recognized as an unexpected hyper acute rejection of HLA identical transplants (174–177). Recently, it has been accepted that nHLA antibodies play an important role in acute and chronic rejection (178–184).
Moreover, in a report from 1997, antibodies against nHLA antigens were shown to activate humoral immune responses to graft antigens and cause graft injury (185). Subsequently, graft loss due to immunological factors occurred in 56% of cases, and 38% of factors were nHLA. Thus, because the probability of graft loss due to nHLA factors was shown to be greater than that due to HLA factors (186), in late years, more attention has been paid to nHLA factors in the transplantation field. For instance, the former have received increased attention in the transplantation field with the anti-MHC class I chain-related gene A (MICA) antibodies and nHLA antibodies to ECs in the presence of complement, as identified in numerous recent reports. Thus, it was expected that humoral response toward nHLA antigens is primarily activated to donor antigen on ECs.
However, while MICA and ECs was not expressed on lymphocyte membranes and was undetectable using cross-match studies (10), the HLA antibody tests LAB Screen Mixed Class I & II and LAB Screen MICA Single Antigen have been successfully used to detect anti-MICA antibodies in sera.
In addition, there are many reports on the other nHLAs that were associated with ABMR.
These reports showed that the type of nHLA antigens differed between patients with hyper acute rejection, acute rejection, and rejection due to chronic allograft injury (CAI), and it may predict graft success and that management plans could be informed by mapping nHLA antigens in recipient sera before transplantation, further indicating the utility of serum nHLA determinations in the diagnosis and management of ABMR.
Management Strategies of ABMR by Non-HLA
Previous studies have reported the detection of nHLA antibodies using ELISA and FACS. However, the clinical utility of these assays remains unclear, because they fail to distinguish antibodies from autoantibodies. Additionally, nHLA antibodies may be detectable in sera from patients with failed grafts but no immunological factors (26, 187). Thus, specific detection of nHLA antibodies that activate humoral immune responses to grafts requires more sensitive methods. For example, by using high-density protein array platforms (26), serum nHLA antibodies in transplant recipients may have up to 9000 different target proteins/antigens and these antibodies were screened immediately, indicating the importance of high throughput screening. In addition, nHLA genotyping of donor and recipient to estimate the risk of ABMR in recipients, such as HLA may be required; the specificities of these antibodies to nHLA should be identified in more details. For example, we should examine if nHLA has the capacity to bound complement or not and if these antibodies could activate humoral immune response toward the donor antigen by using donor-specific ECs.
Mechanism of participation of nHLA antibodies in ABMR and graft loss has not been investigated sufficiently. However, we could find reports that C4d deposition is related to ABMR causing from nHLA antibodies with high probability, indicating that this type of ABMR was caused by the activation of the complement classical pathway. Moreover, further studies are warranted to establish effective immunosuppressive therapies thereby clarifying the mechanism.
In addition, we summarized the association between the representative nHLA antibodies and graft prognosis (Table 3).
Table 3.
A list of selected nHLA antibodies and gene in transplantation.
| nHLA antibody (nHLA-ab) | Organ | Associated factors | Reference |
|---|---|---|---|
| Anti-protein kinase C zeta (PKCf) ab | Kidney | Graft loss | (188) |
| Steroid-resistant rejection and the hypertension | |||
| Mononuclear cell infiltrate of acute rejection | |||
| Anti-MHC I-related chain A (MICA) ab | Kidney | Poor graft survival with only MICA and significantly poor with both antibodies(MICA+/HLA+) | (189–197) |
| Kidney | Preformed MICA antibodies contributes to increasing frequency of graft loss | ||
| Kidney | Chronic rejection, poor graft survival | ||
| Kidney | Graft rejection, poor 1-year graft survival | ||
| Kidney | Poor graft survival | ||
| Heart | The incidence of transplant coronary artery disease | ||
| Heart | No negative effect on graft survival | ||
| Liver | Late graft rejection | ||
| Anti-angiotensin II type I receptor (AT1R) ab | Kidney | Refractory vascular rejection | (198–201) |
| Kidney | Cronic kidney disease | ||
| Kidney | Graft injury, graft loss | ||
| heart | Cellular and Ab-mediated rejection and early onset of microvasculopathy | ||
| Anti-endothelial antibodies (AECA) | Kidney | Cellular rejection | (177, 202–206) |
| Kidney | Hyperacute rejection | ||
| Kidney | Graft rejection | ||
| Kidney | Acute rejection | ||
| Kidney | Microvascular damage | ||
| Heart | Early acute rejection | ||
| Anti-endothelial-1 type A receptor (ETAR) ab | Kidney | Hyperacute rejection | (199, 207) |
| Kidney | Poor graft function early after transplant, hyperacute rejection | ||
| Kidney | Graft injury, graft loss | ||
| Heart | Cellular and Ab-mediated rejection and early onset of microvasculopathy | ||
| Anti-peroxisomal-trans-2-enoyl-coA-reductase (PECR) ab | Kidney | Transplant glomerulopathy | (79, 208) |
| Anti-PRKRIP1ab | Kidney | Cronic kidney disease | (32, 208) |
| Antivimentin ab | Heart | It did not correlate with early post-transplant rejection or graft survival | (32) |
| Non-HLA pigmy ab | Heart | Mortality | (209) |
| Antibodies against | Kidney | Acute ABMR | (210) |
| Endoglin | |||
| Epidermal growth factor (EGF)-like repeats | |||
| Discoidin I-like domains 3 | |||
| Intercellular adhesion molecule 4 | |||
| FMS-like tyrosine kinase-3 ligand | |||
| Antibodies against | Kidney | Chronic allograft injury (CAI) | (26) |
| MIG | |||
| ITAC | |||
| IFN-c | |||
| Glial-derived neurotrophic factor (GDNF) | |||
| Collagen type V, K-α1-tubulin | Lung | Graft disfunction, bronchiolitis obliterans syndrome | (207) |
| nHLA gene | |||
| FN-γ, IL-1B, IL-1RN, IL-2, IL-6, IL-7, IL-17, CCR9, ESR1, FAS Stem cell | GVHD ↑ | (206) | |
| IL-10, NOD2, toll-like receptors | GVHD ↑ or GVHD ↓ | ||
| VDR | GVHD ↑, mortality ↑ | ||
| CTLA4 | Acute GVHD ↑, survival ↑ | ||
| IL-7R, CXCL10 | Transplant-related mortality ↑ | ||
| IL-18 | Transplant-related mortality ↓ | ||
| Il-23R | Acute GVHD ↓ | ||
| HLA-E | Chronic GVHD ↓ | ||
| IL-1A | Chronic and acute GVHD ↑, transplant-related mortality ↑ | ||
| CCl-2 | Over roll survival ↓, transplant-related mortality ↑ | ||
| CXCL12 | Hematological recovery ↑ | ||
| TGFβ | Acute GVHD ↓, over roll survival ↓ | ||
| HMGB1 | Relapse ↑, relapse-related mortality ↑, transplant-related mortality ↑, over roll survival ↑, acute GVHD ↓, chronic GVHD ↑ | ||
| MICA | GVHD ↑ |
Conclusion
Absorption of DSAs has been regarded as the main cause of ABMR. However, numerous recent studies have characterized the involvement of nHLA antibodies, and have shown that DSA- and nHLA-mediated ABMR phenotypes likely require different management strategies.
Specifically, hyper acute rejections due to preformed DSAs may be avoided with improved desensitization therapy, while de novo DSA-mediated ABMR remains difficult to diagnose without invasive graft tissue biopsies prior to critical disease progression. Although the roles of nHLA antibodies have been identified in ABMR, ensuing that mechanisms remain insufficiently understood to inform improvements in management strategies. Moreover, because physiological nHLA antibodies are indistinguishable from those that are closely related to humoral immune reactions against graft antigens, highly sensitive methods that distinguish ABMR-relevant nHLAs are required for clinical diagnoses and management planning for organ transplant patients.
Author Contributions
YM and MS designed and wrote the paper. MS provided excellent advices.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Tara K. Sigdel for supporting them with literature search.
References
- 1.Cerottini JC, Brunner KT. Cell-mediated cytotoxicity, allograft rejection, and tumor immunity. Adv Immunol (1974) 18:67–132. 10.1016/S0065-2776(08)60308-9 [DOI] [PubMed] [Google Scholar]
- 2.Bunjes D, Hardt C, Röllinghoff M, Wagner H. Cyclosporin A mediates immunosuppression of primary cytotoxic T cell responses by impairing the release of interleukin 1 and interleukin 2. Eur J Immunol (1981) 11(8):657–61. 10.1002/eji.1830110812 [DOI] [PubMed] [Google Scholar]
- 3.Sellarés J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant (2012) 12(2):388–99. 10.1111/j.1600-6143.2011.03840.x [DOI] [PubMed] [Google Scholar]
- 4.Einecke G, Sis B, Reeve J, Mengel M, Campbell PM, Hidalgo LG, et al. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant (2009) 9(11):2520–31. 10.1111/j.1600-6143.2009.02799.x [DOI] [PubMed] [Google Scholar]
- 5.Piazza A, Poggi E, Borrelli L, Servetti S, Monaco PI, Buonomo O, et al. Impact of donor-specific antibodies on chronic rejection occurrence and graft loss in renal transplantation: posttransplant analysis using flow cytometric techniques. Transplantation (2001) 71(8):1106–12. 10.1097/00007890-200104270-00017 [DOI] [PubMed] [Google Scholar]
- 6.Patel R, Terasaki PI. Significance of the positive crossmatch test in kidney transplantation. N Engl J Med (1969) 280(14):735–9. 10.1056/NEJM196904032801401 [DOI] [PubMed] [Google Scholar]
- 7.Lefaucheur C, Suberbielle-Boissel C, Hill GS, Nochy D, Andrade J, Antoine C, et al. Clinical relevance of preformed HLA donor-specific antibodies in kidney transplantation. Am J Transplant (2008) 8(2):324. 10.1111/j.1600-6143.2007.02072.x [DOI] [PubMed] [Google Scholar]
- 8.Terasaki PI. A personal perspective: 100-year history of the humoral theory of transplantation. Transplantation (2012) 93(8):751. 10.1097/TP.0b013e3182483713 [DOI] [PubMed] [Google Scholar]
- 9.Zeevi A, Lunz J, Feingold B, Shullo M, Bermudez C, Teuteberg J, et al. Persistent strong anti-HLA antibody at high titer is complement binding and associated with increased risk of antibody-mediated rejection in heart transplant recipients. J Heart Lung Transplant (2012) 32(1):98. 10.1016/j.healun.2012.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Q, Cecka JM, Gjertson DW, Ge P, Rose ML, Patel JK, et al. HLA and MICA: targets of antibody-mediated rejection in heart transplantation. Transplantation (2011) 91(10):1153–8. 10.1097/TP.0b013e3182157d60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishida H, Hirai T, Kohei N, Yamaguchi Y, Tanabe K. Significance of qualitative and quantitative evaluations of anti-HLA antibodies in kidney transplantation. Transpl Int (2011) 24(2):150–7. 10.1111/j.1432-2277.2010.01166.x [DOI] [PubMed] [Google Scholar]
- 12.Terasaki PI, Cai J. Human leukocyte antigen antibodies and chronic rejection: from association to causation. Transplantation (2008) 86(3):377–83. 10.1097/TP.0b013e31817c4cb8 [DOI] [PubMed] [Google Scholar]
- 13.Lee PC, Terasaki PI, Takemoto SK, Lee PH, Hung CJ, Chen YL, et al. All chronic rejection failures of kidney transplants were preceded by the development of HLA antibodies. Transplantation (2002) 74(8):1192–4. 10.1097/00007890-200210270-00025 [DOI] [PubMed] [Google Scholar]
- 14.Nankivell BJ, Alexander SI. Rejection of the kidney allograft. N Engl J Med (2010) 363(15):1451. 10.1056/NEJMra0902927 [DOI] [PubMed] [Google Scholar]
- 15.Loupy A, Hill GS, Jordan SC. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nat Rev Nephrol (2012) 8:348–57. 10.1038/nrneph.2012.81 [DOI] [PubMed] [Google Scholar]
- 16.Montgomery RA. Therapeutics for antibody-mediated rejection: a slippery slope into confusion. Am J Transplant (2016) 16(5):1350–1. 10.1111/ajt.13721 [DOI] [PubMed] [Google Scholar]
- 17.Shin M, Kim SJ. ABO incompatible kidney transplantation-current status and uncertainties. J Transplant (2011) 2011:970421. 10.1155/2011/970421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toki D, Ishida H, Setoguchi K, Shimizu T, Omoto K, Shirakawa H, et al. Acute antibody-mediated rejection in living ABO-incompatible kidney transplantation: long-term impact and risk factors. Am J Transplant (2009) 9(3):567–77. 10.1111/j.1600-6143.2008.02538.x [DOI] [PubMed] [Google Scholar]
- 19.O’Leary JG, Kaneku H, Jennings LW, Bañuelos N, Susskind BM, Terasaki PI, et al. Preformed class II donor-specific antibodies are associated with an increased risk of early rejection after liver transplantation. Liver Transpl (2013) 19(9):973–80. 10.1002/lt.23687 [DOI] [PubMed] [Google Scholar]
- 20.Kaneku H, O’Leary JG, Banuelos N, Jennings LW, Susskind BM, Klintmalm GB, et al. De novo donor-specific HLA antibodies decrease patient and graft survival in liver transplant recipients. Am J Transplant (2013) 13(6):1541–8. 10.1111/ajt.12212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Süsal C, Wettstein D, Döhler B, Morath C, Ruhenstroth A, Scherer S, et al. Association of kidney graft loss with de novo produced donor-specific and non-donor-specific HLA antibodies detected by single antigen testing. Transplantation (2015) 99(9):1976–80. 10.1097/TP.0000000000000672 [DOI] [PubMed] [Google Scholar]
- 22.Hirai T, Furusawa M, Omoto K, Ishida H, Tanabe K. Analysis of predictive and preventive factors for de novo DSA in kidney transplant recipients. Transplantation (2014) 98(4):443–50. 10.1097/TP.0000000000000071 [DOI] [PubMed] [Google Scholar]
- 23.Brooks AM, Carter V, Liew A, Marshall H, Aldibbiat A, Sheerin NS, et al. De novo donor-specific HLA antibodies are associated with rapid loss of graft function following islet transplantation in type 1 diabetes. Am J Transplant (2015) 15(12):3239–46. 10.1111/ajt.13407 [DOI] [PubMed] [Google Scholar]
- 24.Banasik M, Boratyńska M, Kościelska-Kasprzak K, Mazanowska O, Krajewska M, Zabinska M, et al. The impact of de novo donor-specific anti-human leukocyte antigen antibodies on 5-year renal transplant outcome. Transplant Proc (2013) 45(4):1449–52. 10.1016/j.transproceed.2012.12.026 [DOI] [PubMed] [Google Scholar]
- 25.Li L, Chen A, Chaudhuri A, Kambham N, Sigdel T, Chen R, et al. Compartmental localization and clinical relevance of MICA antibodies after renal transplantation. Transplantation (2010) 89(3):312. 10.1097/TP.0b013e3181bbbe4c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butte AJ, Sigdel TK, Wadia PP, Miklos DB, Sarwal MM. Protein microarrays discover angiotensinogen and PRKRIP1 as novel targets for autoantibodies in chronic renal disease. Mol Cell Proteomics (2011) 10(3):M110.000497. 10.1074/mcp.M110.000497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Wadia P, Chen R, Kambham N, Naesens M, Sigdel TK, et al. Identifying compartment-specific non-HLA targets after renal transplantation by integrating transcriptome and “antibodyome” measures. Proc Natl Acad Sci U S A (2009) 106(11):4148. 10.1073/pnas.0900563106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sumitran-Holgersson S. Relevance of MICA and other non-HLA antibodies in clinical transplantation. Curr Opin Immunol (2008) 20(5):607. 10.1016/j.coi.2008.07.005 [DOI] [PubMed] [Google Scholar]
- 29.Opelz G. Non-HLA transplantation immunity revealed by lymphocytotoxic antibodies. Lancet (2005) 365(9470):1570. 10.1016/S0140-6736(05)66458-6 [DOI] [PubMed] [Google Scholar]
- 30.Sigdel TK, Li L, Tran TQ, Khatri P, Naesens M, Sansanwal P, et al. Non-HLA antibodies to immunogenic epitopes predict the evolution of chronic renal allograft injury. J Am Soc Nephrol (2012) 23(4):750. 10.1681/ASN.2011060596 [DOI] [PubMed] [Google Scholar]
- 31.Li L, Sigdel T, Vitalone M, Lee SH, Sarwal M. Differential immunogenicity and clinical relevance of kidney compartment specific antigens after renal transplantation. J Proteome Res (2010) 9(12):6715. 10.1021/pr1008674 [DOI] [PubMed] [Google Scholar]
- 32.Smith JD, Hamour IM, Burke MM, Mahesh B, Stanford RE, Haj-Yahia S, et al. A reevaluation of the role of IgM non-HLA antibodies in cardiac transplantation. Transplantation (2009) 87(6):864–71. 10.1097/TP.0b013e31819a65fa [DOI] [PubMed] [Google Scholar]
- 33.Le Bas-Bernardet S, Hourmant M, Coupel S, Bignon JD, Soulillou JP, Charreau B. Non-HLA-type endothelial cell reactive alloantibodies in pre-transplant sera of kidney recipients trigger apoptosis. Am J Transplant (2003) 3(2):167. 10.1034/j.1600-6143.2003.00021.x [DOI] [PubMed] [Google Scholar]
- 34.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. 10.1038/32588 [DOI] [PubMed] [Google Scholar]
- 35.Amoils S. Viral immunity: turning off class switching. Nat Rev Immunol (2000) 6:255. 10.1038/nri1834 [DOI] [Google Scholar]
- 36.den Haan JM, Bevan MJ. A novel helper role for CD4 T cells. Proc Natl Acad Sci U S A (2000) 97(24):12950–2. 10.1073/pnas.97.24.12950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med (1995) 182(2):459–65. 10.1084/jem.182.2.459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med (1996) 183(6):2541–50. 10.1084/jem.183.6.2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. 10.1016/1074-7613(94)90071-X [DOI] [PubMed] [Google Scholar]
- 40.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270(5238):985–8. 10.1126/science.270.5238.985 [DOI] [PubMed] [Google Scholar]
- 41.Tai X, Van Laethem F, Pobezinsky L. Basis of CTLA-4 function in regulatory and conventional CD4(+) T cells. Blood (2012) 119(22):5155–63. 10.1182/blood-2011-11-388918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci U S A (2010) 107(4):1524–8. 10.1073/pnas.0910341107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol (2009) 21(10):1105–11. 10.1093/intimm/dxp095 [DOI] [PubMed] [Google Scholar]
- 44.Ei Wafai RJ, Chmaisse HN, Makki RF, Fakhoury H. Association of HLA class II alleles and CTLA-4 polymorphism with type 1 diabetes. Saudi J Kidney Dis Transpl (2011) 22(2):273–81. [PubMed] [Google Scholar]
- 45.Mohamed Saleh H, Koeleman B, Szénási G, Rosivall L, Hamar P. Association of CTLA-4 polymorphisms with type 1 diabetes in the Egyptian population diabetes & metabolism. J Diabetes Metab (2013) 4:7. 10.4172/2155-6156.1000291 [DOI] [Google Scholar]
- 46.Du L, Yang J, Huang J, Ma Y, Wang H, Xiong T, et al. The associations between the polymorphisms in the CTLA-4 gene and the risk of Graves’ disease in the Chinese population. BMC Med Genet (2013) 14:46. 10.1186/1471-2350-14-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol (2003) 3:939–51. 10.1038/nri1248 [DOI] [PubMed] [Google Scholar]
- 48.Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med (1991) 173(3):721–30. 10.1084/jem.173.3.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rozanski CH, Arens R, Carlson LM. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. JEM (2011) 208(7):1435–46. 10.1084/jem.20110040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukocyte Biol (2000) 67:2–17. [DOI] [PubMed] [Google Scholar]
- 51.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature (1998) 393:478–80. 10.1038/30996 [DOI] [PubMed] [Google Scholar]
- 52.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature (1998) 393:480–3. 10.1038/31002 [DOI] [PubMed] [Google Scholar]
- 53.Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, et al. IL-21 induces differentiation of human naïve and memory B cells into antibody-secreting plasma cells. J Immunol (2005) 175:7867–79. 10.4049/jimmunol.175.12.7867 [DOI] [PubMed] [Google Scholar]
- 54.Deppong CM, Bricker TL, Rannals BD, Van Rooijen N, Hsieh CS, Green JM. CTLA4Ig inhibits effector T cells through regulatory T cells and TGF-β. J Immunol (2013) 191(6):3082–9. 10.4049/jimmunol.1300830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young JS, Chen J, Miller ML, Vu V, Tian C, Moon JJ, et al. Delayed CTLA4-Ig treatment reverses ongoing alloantibody responses and rescues allografts from acute rejection. Am J Transplant (2016) 16(8):2312–23. 10.1111/ajt.13761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and long-term outcomes in kidney transplantation. N Engl J Med (2016) 374(26):2600–1. 10.1056/NEJMoa1506027 [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Flies D. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13:227–42. 10.1038/nri3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jansen MF, Hollander MR, van Royen N, Horrevoets AJ, Lutgens E. CD40 in coronary artery disease: a matter of macrophages? Basic Res Cardiol (2016) 111(4):38. 10.1007/s00395-016-0554-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delville M, Sigdel TK, Wei C, Li J, Hsieh SC, Fornoni A, et al. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med (2014) 6(256):256ra136. 10.1126/scitranslmed.3008538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei C, Sigdel TK, Sarwal MM, Reiser J. Circulating CD40 autoantibody and suPAR synergy drives glomerular injury. Ann Transl Med (2015) 3(19):300. 10.3978/j.issn.2305-5839.2015.11.08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murphey CL, Forsthuber TG. Trends in HLA antibody screening and identification and their role in transplantation. Expert Rev Clin Immunol (2008) 4(3):391–9. 10.1586/1744666X.4.3.391 [DOI] [PubMed] [Google Scholar]
- 62.Cecka JM. The UNOS scientific renal transplant registry. Clin Transpl (1997):1–14. [PubMed] [Google Scholar]
- 63.Takemoto SK, Terasaki PI, Gjertson DW, Cecka JM. Twelve years’ experience with national sharing of HLA-matched cadaveric kidneys for transplantation. N Engl J Med (2000) 343(15):1078–84. 10.1056/NEJM200010123431504 [DOI] [PubMed] [Google Scholar]
- 64.Gjertson DW, Terasaki PI, Takemoto S, Mickey MR. National allocation of cadaveric kidneys by HLA matching. Projected effect on outcome and costs. N Engl J Med (1991) 324(15):1032–6. 10.1056/NEJM199104113241505 [DOI] [PubMed] [Google Scholar]
- 65.Terasaki PI, Cho Y, Takemoto S, Cecka M, Gjertson D. Twenty-year follow-up on the effect of HLA matching on kidney transplant survival and prediction of future twenty-year survival. Transplant Proc (1996) 28(3):1144–5. [PubMed] [Google Scholar]
- 66.Mittal S, Page SL, Friend PJ, Sharples EJ, Fuggle SV. De novo donor-specific HLA antibodies: biomarkers of pancreas transplant failure. Am J Transplant (2014) 14(7):1664–71. 10.1111/ajt.12750 [DOI] [PubMed] [Google Scholar]
- 67.Cantarovich D, De Amicis S, Akl A, Devys A, Vistoli F, Karam G, et al. Post transplant donor-specific anti-HLA antibodies negatively impact pancreas transplantation outcome. Am J Transplant (2011) 11(12):2737–46. 10.1111/j.1600-6143.2011.03729.x [DOI] [PubMed] [Google Scholar]
- 68.Murata K, Baldwin WM, III. Mechanisms of complement activation, C4d deposition, and their contribution to the pathogenesis of antibody-mediated rejection. Transplant Rev (2009) 23:139–50. 10.1016/j.trre.2009.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cerilli J, Brasile L, Galouzis T, Lempert N, Clarke J. The vascular endothelial cell antigen system. Transplantation (1985) 39(3):286–9. 10.1097/00007890-198503000-00016 [DOI] [PubMed] [Google Scholar]
- 70.O’Leary JG, Demetris AJ, Friedman LS, Gebel HM, Halloran PF, Kirk AD, et al. The role of donor-specific HLA alloantibodies in liver transplantation. Am J Transplant (2014) 14(4):779–87. 10.1111/ajt.12667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Leary JG, Klintmalm GB. Impact of donor-specific antibodies on results of liver transplantation. Curr Opin Organ Transplant (2013) 18(3):279–84. 10.1097/MOT.0b013e3283614a10 [DOI] [PubMed] [Google Scholar]
- 72.Hachem RR, Kamoun M, Budev M, Askar M, Vivek A, Levine DJ, et al. HLA antibodies after lung transplantation: early results of the HALT study. J Heart Lung Transplant (2013) 32(Suppl 4):76–7. 10.1016/j.healun.2013.01.191 [DOI] [Google Scholar]
- 73.Kim M, Townsend KR, Wood IG, Boukedes S, Guleria I, Gabardi S, et al. Impact of pretransplant anti-HLA antibodies on outcomes in lung transplant candidates. Am J Respir Crit Care Med (2014) 189(10):1234–9. 10.1164/rccm.201312-2160OC [DOI] [PubMed] [Google Scholar]
- 74.Smith JD, Banner NR, Hamour IM, Ozawa M, Goh A, Robinson D, et al. De novo donor HLA-specific antibodies after heart transplantation are an independent predictor of poor patient survival. Am J Transplant (2011) 11(2):312–9. 10.1111/j.1600-6143.2010.03383.x [DOI] [PubMed] [Google Scholar]
- 75.Ius F, Sommer W, Tudorache I, Kühn C, Avsar M, Siemeni T, et al. Early donor-specific antibodies in lung transplantation: risk factors and impact on survival. J Heart Lung Transplant (2014) 33(12):1255–63. 10.1016/j.healun.2014.06.015 [DOI] [PubMed] [Google Scholar]
- 76.Hachem RR, Reinsmoen NL. What is the definition of a clinically relevant donor HLA-specific antibody (DSA)? Am J Transplant (2015) 15(2):299–300. 10.1111/ajt.13079 [DOI] [PubMed] [Google Scholar]
- 77.Moreau A, Varey E, Anegon I, Cuturi MC. Effector mechanisms of rejection. Cold Spring Harb Perspect Med (2013) 3(11). 10.1101/cshperspect.a015461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ng YW, Singh M, Sarwal MM. Antibody-mediated rejection in pediatric kidney transplantation: pathophysiology, diagnosis, and management. Drugs (2015) 75:455–72. 10.1007/s40265-015-0369-y [DOI] [PubMed] [Google Scholar]
- 79.Issa NC, Fishman JA, Snydman DR. Infectious complications of antilymphocyte therapies in solid organ transplantation. Clin Infect Dis (2009) 48(6):772–86. 10.1086/597089 [DOI] [PubMed] [Google Scholar]
- 80.Epstein-Barr Virus and Posttransplant Lymphoproliferative Disorder in Solid Organ Transplantation. Special Issue: The American Society of Transplantation Infectious Diseases Guidelines 3rd Edition (2013) 13(S4):107–20. [DOI] [PubMed] [Google Scholar]
- 81.Maurer MA, Tuller F, Gredler V, Berger T, Lutterotti A, Lünemann JD, et al. Rituximab induces clonal expansion of IgG memory B-cells in patients with inflammatory central nervous system demyelination. J Neuroimmunol (2016) 290(15):49–53. 10.1016/j.jneuroim.2015.11.006 [DOI] [PubMed] [Google Scholar]
- 82.Wardrope KE, Manson L, Metcalfe W, Sullivan ED. Acute respiratory distress syndrome and posterior reversible encephalopathy syndrome following rituximab therapy. Case Rep Nephrol Dial (2016) 6:32–9. 10.1159/000444250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Everly MJ, Everly JJ, Susskind B, Brailey P, Arend LJ, Alloway RR, et al. Bortezomib provides effective therapy for antibody- and cell-mediated acute rejection. Transplantation (2008) 86(12):1754–61. 10.1097/TP.0b013e318190af83 [DOI] [PubMed] [Google Scholar]
- 84.Island ER, Gonzalez-Pinto IM, Tsai HL, Ruiz P, Tryphonopoulos P, Gonzalez ML, et al. Successful treatment with bortezomib of a refractory humoral rejection of the intestine after multivisceral transplantation. Clin Transpl (2009):465–9. [PubMed] [Google Scholar]
- 85.Eckman PM, Thorsgard M, Maurer D, Kim Y, Alloway RR, Woodle ES. Bortezomib for refractory antibody-mediated cardiac allograft rejection. Clin Transpl (2009):475–8. [PubMed] [Google Scholar]
- 86.Govil A, Walsh RC, Tevar A, Alloway R, Roy-Chaudhury P, Mogilishetty G, et al. Bortezomib-based treatment of antibody mediated rejection in pancreas allograft recipients. Clin Transpl (2009):443–53. [PubMed] [Google Scholar]
- 87.Raghavan R, Jeroudi A, Achkar K, Suki W, Gaber AO, Knight R, et al. Bortezomib in kidney transplant desensitization: a case report. Clin Transpl (2009):339–42. [PubMed] [Google Scholar]
- 88.Heidt S, Roelen DL, Vergunst M, Doxiadis II, Claas FH, Mulder A. Bortezomib affects the function of human B cells: possible implications for desensitization protocols. Clin Transpl (2009):387–92. [PubMed] [Google Scholar]
- 89.Wong EK, Kavanagh D. Anticomplement C5 therapy with eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Transl Res (2015) 165(2):306–20. 10.1016/j.trsl.2014.10.010 [DOI] [PubMed] [Google Scholar]
- 90.Bouts A, Monnens L, Davin JC, Struijk G, Spanjaard L. Insufficient protection by Neisseria meningitidis vaccination alone during eculizumab therapy. Pediatr Nephrol (2011) 26(10):1919–20. 10.1007/s00467-011-1929-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Caon C, Namey M, Meyer C, Mayer L, Oyuela P, Margolin DH, et al. Prevention and management of infusion-associated reactions in the comparison of alemtuzumab and rebif(®) efficacy in multiple sclerosis (CARE-MS) program. Int J MS Care (2015) 17(4):191–8. 10.7224/1537-2073.2014-030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Osterborg A, Karlsson C, Lundin J, Kimby E, Mellstedt H. Strategies in the management of alemtuzumab-related side effects. Semin Oncol (2006) 33(2 Suppl 5):S29–35. 10.1053/j.seminoncol.2006.01.027 [DOI] [PubMed] [Google Scholar]
- 93.Razonable RR. Strategies for managing cytomegalovirus in transplant recipients. Expert Opin Pharmacother (2010) 11:1983–97. 10.1517/14656566.2010.492395 [DOI] [PubMed] [Google Scholar]
- 94.Harvala H, Stewart C, Muller K, Burns S, Marson L, Mac-Gilchrist A, et al. High risk of cytomegalovirus infection following solid organ transplantation despite prophylactic therapy. J Med Virol (2013) 85:893–8. 10.1002/jmv.23539 [DOI] [PubMed] [Google Scholar]
- 95.Razonable RR, Rivero A, Rodriguez A, Wilson J, Daniels J, Jenkins G, et al. Allograft rejection predicts the occurrence of late-onset cytomegalovirus (CMV) disease among CMV-mismatched solid organ transplant patients receiving prophylaxis with oral ganciclovir. J Infect Dis (2001) 184(11):1461–4. 10.1086/324516 [DOI] [PubMed] [Google Scholar]
- 96.Razonable RR, Paya CV, Smith TF. Role of the laboratory in diagnosis and management of cytomegalovirus infection in hematopoietic stem cell and solid-organ transplant recipients. J Clin Microbiol (2002) 40(3):746–52. 10.1128/JCM.40.3.746-752.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paya C, Humar A, Dominguez E, Washburn K, Blumberg E, Alexander B, et al. Efficacy and safety of valganciclovir vs. oral ganciclovir for prevention of cytomegalovirus disease in solid organ transplant recipients. Am J Transplant (2004) 4:611–20. 10.1111/j.1600-6143.2004.00382.x [DOI] [PubMed] [Google Scholar]
- 98.Razonable RR, Humar A. AST infectious diseases community of practice. Cytomegalovirus in solid organ transplantation. Am J Transplant (2013) 13(Suppl 4):93–106. 10.1111/ajt.12103 [DOI] [PubMed] [Google Scholar]
- 99.Knowles WA, Pipkin P, Andrews N, Vyse A, Minor P, Brown DW, et al. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol (2003) 71:115–23. 10.1002/jmv.10450 [DOI] [PubMed] [Google Scholar]
- 100.Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, et al. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis (2009) 199:837–46. 10.1086/597126 [DOI] [PubMed] [Google Scholar]
- 101.Hirsch HH, Knowles W, Dickenmann M, Passweg J, Klimkait T, Mihatsch MJ, et al. Prospective study of polyomavirus type BK replication and nephropathy in renal-transplant recipients. N Engl J Med (2002) 347:488–96. 10.1056/NEJMoa020439 [DOI] [PubMed] [Google Scholar]
- 102.Nickeleit V, Klimkait T, Binet IF, Dalquen P, Del Zenero V, Thiel G, et al. Testing for polyomavirus type BK DNA in plasma to identify renal-allograft recipients with viral nephropathy. N Engl J Med (2000) 342:1309–15. 10.1056/NEJM200005043421802 [DOI] [PubMed] [Google Scholar]
- 103.Drachenberg CB, Hirsch HH, Papadimitriou JC, Gosert R, Wali RK, Munivenkatappa R, et al. Polyomavirus BK versus JC replication and nephropathy in renal transplant recipients: a prospective evaluation. Transplantation (2007) 84:323–30. 10.1097/01.tp.0000269706.59977.a5 [DOI] [PubMed] [Google Scholar]
- 104.Viscount HB, Eid AJ, Espy MJ, Griffin MD, Thomsen KM, Harmsen WS, et al. Polyomavirus polymerase chain reaction as a surrogate marker of polyomavirus-associated nephropathy. Transplantation (2007) 84:340–5. 10.1097/01.tp.0000275205.41078.51 [DOI] [PubMed] [Google Scholar]
- 105.Ramos E, Drachenberg CB, Papadimitriou JC, Hamze O, Fink JC, Klassen DK, et al. Clinical course of polyoma virus nephropathy in 67 renal transplant patients. J Am Soc Nephrol (2002) 13:2145–51. 10.1097/01.ASN.0000023435.07320.81 [DOI] [PubMed] [Google Scholar]
- 106.Wadei HM, Rule AD, Lewin M, Mahale AS, Khamash HA, Schwab TR, et al. Kidney transplant function and histological clearance of virus following diagnosis of polyomavirus-associated nephropathy (PVAN). Am J Transplant (2006) 6(1):1025–32. 10.1111/j.1600-6143.2006.01296.x [DOI] [PubMed] [Google Scholar]
- 107.Antonsson A, Pawlita M, Feltkamp MC, Bouwes Bavinck JN, Euvrard S, Harwood CA, et al. Longitudinal study of seroprevalence and serostability of the human polyomaviruses JCV and BKV in organ transplant recipients. J Med Virol (2013) 85(2):327–35. 10.1002/jmv.23472 [DOI] [PubMed] [Google Scholar]
- 108.Randhawa PS, Finkelstein S, Scantlebury V, Shapiro R, Vivas C, Jordan M, et al. Human polyoma virus-associated interstitial nephritis in the allograft kidney. Transplantation (1999) 67:103–9. 10.1097/00007890-199901150-00018 [DOI] [PubMed] [Google Scholar]
- 109.Binet I, Nickeleit V, Hirsch HH, Prince O, Dalquen P, Gudat F, et al. Polyomavirus disease under new immunosuppressive drugs: a cause of renal graft dysfunction and graft loss. Transplantation (1999) 67:918–22. 10.1097/00007890-199903270-00022 [DOI] [PubMed] [Google Scholar]
- 110.Schaub S, Hirsch HH, Dickenmann M, Steiger J, Mihatsch MJ, Hopfer H, et al. Reducing immunosuppression preserves allograft function in presumptive and definitive polyomavirusassociated nephropathy. Am J Transplant (2010) 10:2615–23. 10.1111/j.1600-6143.2010.03310.x [DOI] [PubMed] [Google Scholar]
- 111.Ginevri F, Azzi A, Hirsch HH, Basso S, Fontana I, Cioni M, et al. Prospective monitoring of polyomavirus BK replication and impact of pre-emptive intervention in pediatric kidney recipients. Am J Transplant (2007) 7:2727–35. 10.1111/j.1600-6143.2007.01984.x [DOI] [PubMed] [Google Scholar]
- 112.Walker RC, Marshall WF, Strickler JG, Wiesner RH, Velosa JA, Habermann TM, et al. Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin Infect Dis (1995) 20:1346–53. 10.1093/clinids/20.5.1346 [DOI] [PubMed] [Google Scholar]
- 113.Green M. Management of Epstein-Barr virus-induced posttransplant lymphoproliferative disease in recipients of solid organ transplantation. Am J Transplant (2001) 1:103–8. 10.1034/j.1600-6143.2001.001002103.x [DOI] [PubMed] [Google Scholar]
- 114.Fedrigo M, Abate D, Sgarabotto D, Feltrin G, Castellani C, Gambino A, et al. P761Antibody mediated rejection related with CMV and EBV infection in heart transplants recipients: a possible relation with infection and complement activation. Cardiovasc Res (2014) 103(Suppl 1):139. 10.1093/cvr/cvu098.180 [DOI] [Google Scholar]
- 115.Marrari M, Duquesnoy RJ. Detection of donor-specific HLA antibodies before and after removal of a rejected kidney transplant. Transpl Immunol (2010) 22(3–4):105–9. 10.1016/j.trim.2009.12.005 [DOI] [PubMed] [Google Scholar]
- 116.Morath C, Opelz G, Zeier M, Süsal C. Clinical relevance of HLA antibody monitoring after kidney transplantation. J Immunol Res (2014) 2014:845040. 10.1155/2014/845040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kunkel EJ, Butcher EC. Plasma-cell homing. Nat Rev Immunol (2003) 3(10):822–9. 10.1038/nri1203 [DOI] [PubMed] [Google Scholar]
- 118.Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med (2001) 194(1):45–56. 10.1084/jem.194.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science (1999) 286(5447):2098–102. 10.1126/science.286.5447.2098 [DOI] [PubMed] [Google Scholar]
- 120.Jourdan M, Caraux A, De Vos J, Fiol G, Larroque M, Cognot C, et al. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood (2009) 114:5173–81. 10.1182/blood-2009-07-235960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Agematsu K. Memory B cells and CD27. Histol Histopathol (2000) 15(2):573–6. [DOI] [PubMed] [Google Scholar]
- 122.Wu YC, Kipling D, Dunn-Walters DK. The relationship between CD27 negative and positive B cell populations in human peripheral blood. Front Immunol (2011) 2:81. 10.3389/fimmu.2011.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Aiba Y, Kometani K, Hamadate M, Moriyama S, Sakaue-Sawano A, Tomura M, et al. Preferential localization of IgG memory B cells adjacent to contracted germinal centers. Proc Natl Acad Sci U S A (2010) 107(27):12192–7. 10.1073/pnas.1005443107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Han M, Rogers JA, Lavingia B, Stastny P. Peripheral blood B cells producing donor-specific HLA antibodies in vitro. Hum Immunol (2009) 70(1):29–34. 10.1016/j.humimm.2008.10.013 [DOI] [PubMed] [Google Scholar]
- 125.Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity (2013) 39(1):136–47. 10.1016/j.immuni.2013.06.011 [DOI] [PubMed] [Google Scholar]
- 126.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived PCs. Immunity (1998) 8(3):363–72. 10.1016/S1074-7613(00)80541-5 [DOI] [PubMed] [Google Scholar]
- 127.Amanna IJ, Slifka MK. Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol Rev (2010) 236(1):125–38. 10.1111/j.1600-065X.2010.00912.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hiepe F, Dörner T, Hauser AE, Hoyer BF. Long-lived autoreactive PCs drive persistent autoimmune inflammation. Nat Rev Rheumatol (2011) 7(3):170–8. 10.1038/nrrheum.2011.1 [DOI] [PubMed] [Google Scholar]
- 129.Mei H, Radbruch A. Factors implicated in the generation and persistence of long-lived plasma cell-mediated autoimmunity. Autoimmun Rev (2011) 10(7):375–82. 10.1016/j.autrev.2010.12.007 [DOI] [PubMed] [Google Scholar]
- 130.Haneda M, Owaki M, Kuzuya T, Iwasaki K, Miwa Y, Kobayashi T. Comparative analysis of drug action on B-cell proliferation and differentiation for mycophenolic acid, everolimus, and prednisolone. Transplantation (2014) 97:405–12. 10.1097/01.TP.0000441826.70687.f6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, et al. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol (2007) 179(12):8180–90. 10.4049/jimmunol.179.12.8180 [DOI] [PubMed] [Google Scholar]
- 132.Ding BB, Bi E, Chen H, Yu JJ, Ye BH. IL-21 and CD40L synergistically promote plasma cell differentiation through upregulation of Blimp-1 in human B cells. J Immunol (2013) 190(4):1827–36. 10.4049/jimmunol.1201678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huggins J, Pellegrin T, Felgar RE, Wei C, Brown M, Zheng B, et al. CpG DNA activation and plasma-cell differentiation of CD27- naive human B cells. Blood (2007) 109(4):1611–9. 10.1182/blood-2006-03-008441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood (2008) 11(3):1004–12. [DOI] [PubMed] [Google Scholar]
- 135.O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199(1):91–8. 10.1084/jem.20031330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest (2003) 112:286–97. 10.1172/JCI18025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol (2006) 18(5):263–75. 10.1016/j.smim.2006.04.006 [DOI] [PubMed] [Google Scholar]
- 138.Cocco M, Stephenson S. In vitro generation of long-lived human PCs. J Immunol (2012) 189(12):5773–85. 10.4049/jimmunol.1103720 [DOI] [PubMed] [Google Scholar]
- 139.Chu VT, Beller A, Nguyen TTN. The long-term survival of PCs. Scand J Immunol (2011) 73(6):508–11. 10.1111/j.1365-3083.2011.02544.x [DOI] [PubMed] [Google Scholar]
- 140.Manz RA, Arce S, Cassese G, Hauser AE, Hiepe F, Radbruch A. Humoral immunity and long-lived PCs. Curr Opin Immunol (2002) 14(4):517–21. [DOI] [PubMed] [Google Scholar]
- 141.Belnoue E, Tougne C, Rochat AF, Lambert PH, Pinschewer DD, Siegrist CA. Homing and adhesion patterns determine the cellular composition of the bone marrow plasma cell niche. J Immunol (2012) 188(3):1283–91. 10.4049/jimmunol.1103169 [DOI] [PubMed] [Google Scholar]
- 142.Tangye SG. Staying alive: regulation of plasma cell survival. Trends Immunol (2011) 32(12):595–602. 10.1016/j.it.2011.09.001 [DOI] [PubMed] [Google Scholar]
- 143.McHeyzer-Williams LJ, McHeyzer-Williams MG. Antigen-specific memory B cell development. Annu Rev Immunol (2005) 23:487–513. 10.1146/annurev.immunol.23.021704.115732 [DOI] [PubMed] [Google Scholar]
- 144.Vlaicu SI, Tatomir A, Rus V, Mekala AP, Mircea PA, Niculescu F, et al. The role of complement activation in atherogenesis: the first 40 years. Immunol Res (2016) 64(1):1–13. 10.1007/s12026-015-8669-6 [DOI] [PubMed] [Google Scholar]
- 145.Frank MM. Complement deficiencies. Pediatr Clin North Am (2000) 47(6):1339–54. 10.1016/S0031-3955(05)70274-1 [DOI] [PubMed] [Google Scholar]
- 146.Lopez-Cepero M, Weston M, Leone JP, Wright L, Resto-Ruiz S. C1q assay: making sense of sensitivity. Hum Immunol (2013) 74:68. 10.1016/j.humimm.2013.08.103 [DOI] [Google Scholar]
- 147.Vo AA, Sinha A, Haas M, Choi J, Mirocha J, Kahwaji J, et al. Factors predicting risk for antibody-mediated rejection and graft loss in highly human leukocyte antigen sensitized patients transplanted after desensitization. Transplantation (2015) 99(7):1423–30. 10.1097/TP.0000000000000525 [DOI] [PubMed] [Google Scholar]
- 148.Fichtner A, Süsal C, Höcker B, Rieger S, Waldherr R, Westhoff JH, et al. Association of C1q-fixing DSA with late graft failure in pediatric renal transplant recipients. Pediatr Nephrol (2016) 31(7):1157–66. 10.1007/s00467-016-3322-8 [DOI] [PubMed] [Google Scholar]
- 149.Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med (2013) 369:1215–26. 10.1056/NEJMoa1302506 [DOI] [PubMed] [Google Scholar]
- 150.Loupy A, Vernerey D, Tinel C, Aubert O, Duong van Huyen JP, Rabant M, et al. Subclinical rejection phenotypes at 1 year post-transplant and outcome of kidney allografts. J Am Soc Nephrol (2015) 26(7):1721–31. 10.1681/ASN.2014040399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Naesens M, Kuypers DR, De Vusser K, Evenepoel P, Claes K, Bammens B, et al. The histology of kidney transplant failure: a long-term follow-up study. Transplantation (2014) 98(4):427–35. 10.1097/TP.0000000000000183 [DOI] [PubMed] [Google Scholar]
- 152.Dörje C, Midtvedt K, Holdaas H, Naper C, Strøm EH, Øyen O, et al. Early versus late acute antibody-mediated rejection in renal transplant recipients. Transplantation (2013) 96(1):79–84. 10.1097/TP.0b013e31829434d4 [DOI] [PubMed] [Google Scholar]
- 153.Kikić Ž, Kainz A, Kozakowski N, Oberbauer R, Regele H, Bond G, et al. Capillary C4d and kidney allograft outcome in relation to morphologic lesions suggestive of antibody-mediated rejection. Clin J Am Soc Nephrol (2015) 10(8):1435–43. 10.2215/CJN.09901014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chemouny JM, Suberbielle C, Rabant M, Zuber J, Alyanakian MA, Lebreton X, et al. De novo donor-specific human leukocyte antigen antibodies in nonsensitized kidney transplant recipients after T cell-mediated rejection. Transplantation (2015) 99(5):965–72. 10.1097/TP.0000000000000448 [DOI] [PubMed] [Google Scholar]
- 155.Hasegawa J, Honda K, Wakai S, Shirakawa H. Plasma cell-rich rejection after kidney transplantation and the role of donor-specific antibodies: a case report and review of the literature. Transplant Proc (2015) 47(8):2533–6. 10.1016/j.transproceed.2015.09.018 [DOI] [PubMed] [Google Scholar]
- 156.Redfield RR, Kaufman DB, Odorico JS. Diagnosis and treatment of pancreas rejection. Curr Transplant Rep (2015) 2(2):169–75. 10.1007/s40472-015-0061-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.O’Leary JG, Kaneku H, Bañuelos N, Jennings LW, Klintmalm GB, Terasaki PI. Impact of IgG3 subclass and C1q-fixing donor-specific HLA alloantibodies on rejection and survival in liver transplantation. Am J Transplant (2015) 15(4):1003–13. 10.1111/ajt.13153 [DOI] [PubMed] [Google Scholar]
- 158.Kaneku H, O’Leary JG, Banuelos N, Jennings LW, Susskind BM, Klintmalm GB, et al. De novo donor-specific HLA antibodies decrease patient and graft survival in liver transplant recipients. Am J Transplant (2013) 13(6):1541–8. 10.1111/ajt.12212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lobo LJ, Aris RM, Schmitz J, Neuringer IP. Donor-specific antibodies are associated with antibody-mediated rejection, acute cellular rejection, bronchiolitis obliterans syndrome, and cystic fibrosis after lung transplantation. J Heart Lung Transplant (2013) 32(1):70–7. 10.1016/j.healun.2012.10.007 [DOI] [PubMed] [Google Scholar]
- 160.Loupy A, Toquet C, Rouvier P, Beuscart T, Bories MC, Varnous S, et al. Late failing heart allografts: pathology of cardiac allograft vasculopathy and association with antibody-mediated rejection. Am J Transplant (2016) 16(1):111–20. 10.1111/ajt.13529 [DOI] [PubMed] [Google Scholar]
- 161.Kubal C, Mangus R, Saxena R, Lobashevsky A, Higgins N, Fridell J, et al. Prospective monitoring of donor-specific anti-HLA antibodies after intestine/multivisceral transplantation: significance of de novo antibodies. Transplantation (2015) 99(8):49–56. 10.1097/TP.0000000000000614 [DOI] [PubMed] [Google Scholar]
- 162.Tsai HL, Island ER, Chang JW, Gonzalez-Pinto I, Tryphonopoulos P, Nishida S, et al. Association between donor-specific antibodies and acute rejection and resolution in small bowel and multivisceral transplantation. Transplantation (2011) 92(6):709–15. 10.1097/TP.0b013e318229f752 [DOI] [PubMed] [Google Scholar]
- 163.Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant (2011) 11(11):2405–13. 10.1111/j.1600-6143.2011.03757.x [DOI] [PubMed] [Google Scholar]
- 164.Feucht HE, Felber E, Gokel MJ, Hillebrand G, Nattermann U, Brockmeyer C, et al. Vascular deposition of complement-split products in kidney allografts with cell-mediated rejection. Clin Exp Immunol (1991) 86(3):464–70. 10.1111/j.1365-2249.1991.tb02954.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat Rev Nephrol (2012) 8(11):670–8. 10.1038/nrneph.2012.212 [DOI] [PubMed] [Google Scholar]
- 166.Salama AD, Pusey CD. Drug insight: rituximab in renal disease and transplantation. Nat Clin Pract Nephrol (2006) 2:221–30. 10.1038/ncpneph0133 [DOI] [PubMed] [Google Scholar]
- 167.Perry DK, Burns JM, Pollinger HS, Amiot BP, Gloor JM, Gores GJ, et al. Proteasome inhibition causes apoptosis of normal human plasma cells preventing alloantibody production. Am J Transplant (2009) 9(1):201–9. 10.1111/j.1600-6143.2008.02461.x [DOI] [PubMed] [Google Scholar]
- 168.Jordan SC, Vo A, Tyan D, Toyota M. Desensitization therapy with intravenous gammaglobulin (IVIG): applications in solid organ transplantation. Trans Am Clin Climatol Assoc (2006) 117:199–211. [PMC free article] [PubMed] [Google Scholar]
- 169.Yabu JM, Higgins JP, Chen G, Sequeira F, Busque S, Tyan DB. C1q-fixing human leukocyte antigen antibodies are specific for predicting transplant glomerulopathy and late graft failure after kidney transplantation. Transplantation (2011) 91(3):342–7. 10.1097/TP.0b013e318203fd26 [DOI] [PubMed] [Google Scholar]
- 170.Haas M. Pathology of C4d-negative antibody-mediated rejection in renal allografts. Curr Opin Organ Transplant (2013) 18(3):319–26; Orandi BJ, Alachkar N, Kraus ES, Naqvi F, Lonze BE, Lees L, Presentation and outcomes of C4d-negative antibody-mediated rejection after kidney. Am J Transplant (2016) 16(1):213–20. 10.1097/MOT.0b013e32835d4daf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Haas M, Sis B, Racusen LC, Solez K, Glotz D, Colvin RB, et al. Banff meeting report writing committee. Banff 2013 meeting report: inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am J Transplant (2014) 14(2):272–83. 10.1111/ajt.12590 [DOI] [PubMed] [Google Scholar]
- 172.Racusen LC, Haas M. Antibody-mediated rejection in renal allografts: lessons from pathology. Clin J Am Soc Nephrol (2006) 1(3):415–20. 10.2215/CJN.01881105 [DOI] [PubMed] [Google Scholar]
- 173.Cohen D, Colvin RB, Daha MR, Drachenberg CB, Haas M, Nickeleit V, et al. Pros and cons for C4d as a biomarker. Kidney Int (2012) 81:628–39. 10.1038/ki.2011.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Paul LC, Baldwin WM, III, van Es LA. Vascular endothelial alloantigens in renal transplantation. Transplantation (1985) 40(2):117–23. 10.1097/00007890-198508000-00001 [DOI] [PubMed] [Google Scholar]
- 175.Jordan SC, Yap HK, Sakai RS, Alfonso P, Fitchman M. Hyperacute allograft rejection mediated by anti-vascular endothelial cell antibodies with a negative monocyte crossmatch. Transplantation (1988) 46(4):585–7. 10.1097/00007890-198810000-00024 [DOI] [PubMed] [Google Scholar]
- 176.Opelz G, Collaborative Transplant S. Non-HLA transplantation immunity revealed by lymphocytotoxic antibodies. Lancet (2005) 365(9470):1570–6. 10.1016/S0140-6736(05)66458-6 [DOI] [PubMed] [Google Scholar]
- 177.Sumitran-Karuppan S, Tyden G, Reinholt F, Berg U, Moller E. Hyperacute rejections of two consecutive renal allografts and early loss of the third transplant caused by non-HLA antibodies specific for endothelial cells. Transpl Immunol (1997) 5(4):321–7. 10.1016/S0966-3274(97)80016-0 [DOI] [PubMed] [Google Scholar]
- 178.Tait BD, Süsal C, Gebel HM, Nickerson PW, Zachary AA, Claas FH, et al. Consensus guidelines on the testing and clinical management issues associated with HLA and non-HLA antibodies in transplantation. Transplantation (2013) 95(1):19–47. 10.1097/TP.0b013e31827a19cc [DOI] [PubMed] [Google Scholar]
- 179.Terasaki PI. Humoral theory of transplantation. Am J Transplant (2003) 3(6):665–73. 10.1034/j.1600-6143.2003.00135.x [DOI] [PubMed] [Google Scholar]
- 180.Cai J, Terasaki PI. Humoral theory of transplantation: mechanism, prevention, and treatment. Hum Immunol (2005) 66(4):334–42. 10.1016/j.humimm.2005.01.021 [DOI] [PubMed] [Google Scholar]
- 181.Banasik M, Boratyńska M, Nowakowska B, Haloń A, Kościelska-Kasprzak K, Drulis-Fajdasz D, et al. Variability in donor-specific alloantibody production after transplantation. Transplant Proc (2007) 39(9):2715–7. 10.1016/j.transproceed.2007.08.054 [DOI] [PubMed] [Google Scholar]
- 182.Banasik M, Boratyńska M, Kościelska-Kasprzak K. The impact of de novo donor-specific anti-human leukocyte antigen antibodies on 5-year renal transplant outcome. Transplant Proc (2013) 45(4):1449–52. 10.1016/j.transproceed.2012.12.026 [DOI] [PubMed] [Google Scholar]
- 183.Crespo M, Torio A, Mas V, Redondo D, Pérez-Sáez MJ, Mir M, et al. Clinical relevance of pretransplant anti-HLA donor-specific antibodies: does C1q-fixation matter? Transpl Immunol (2013) 29(1–4):28–33. 10.1016/j.trim.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 184.Fidler SJ, Irish AB, Lim W, Ferrari P, Witt CS, Christiansen FT. Pre-transplant donor specific anti-HLA antibody is associated with antibody-mediated rejection, progressive graft dysfunction and patient death. Transpl Immunol (2013) 28(4):148–53. 10.1016/j.trim.2013.05.001 [DOI] [PubMed] [Google Scholar]
- 185.Hourmant M, Buzelin F, Dantal J, van Dixhoorn M, Le Forestier M, Coste M, et al. Late acute failure of well-HLA-matched renal allografts with capillary congestion and arteriolar thrombi. Transplantation (1995) 60(11):1252. 10.1097/00007890-199512000-00013 [DOI] [PubMed] [Google Scholar]
- 186.Terasaki PI. Deduction of the fraction of immunologic and non-immunologic failure in cadaver donor transplants. Clin Transpl (2003):449–52. [PubMed] [Google Scholar]
- 187.Ranganath P, Tripathi G, Sharma RK, Sankhwar SN, Agrawal S. Role of non-HLA genetic variants in end-stage renal disease. Tissue Antigens (2009) 74(2):147. 10.1111/j.1399-0039.2009.01276.x [DOI] [PubMed] [Google Scholar]
- 188.Sutherland SM, Li L, Sigdel TK, Wadia PP, Miklos DB, Butte AJ, et al. Protein microarrays identify antibodies to protein kinase Czeta that are associated with a greater risk of allograft loss in pediatric renal transplant recipients. Kidney Int (2009) 76(12):1277–83. 10.1038/ki.2009.384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Panigrahi A, Gupta N, Siddiqui JA, Margoob A, Bhowmik D, Guleria S, et al. Post transplant development of MICA and anti-HLA antibodies is associated with acute rejection episodes andrenal allograft loss. Hum Immunol (2007) 68(5):362–7. 10.1016/j.humimm.2007.01.006 [DOI] [PubMed] [Google Scholar]
- 190.Zou Y, Stastny P, Susal C, Dohler B, Opelz G. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med (2007) 357(13):1293–300. 10.1056/NEJMoa067160 [DOI] [PubMed] [Google Scholar]
- 191.Sumitran-Holgersson S, Wilczek HE, Holgersson J, Söderström K. Identification of the nonclassical HLA molecules, mica, as targets for humoral immunity associated with irreversible rejection of kidney allografts. Transplantation (2002) 74(2):268–77. 10.1097/00007890-200207270-00019 [DOI] [PubMed] [Google Scholar]
- 192.Mizutani K, Terasaki P, Rosen A, Esquenazi V, Miller J, Shih RN, et al. Serial ten-year follow-up of HLA and MICA antibody production prior to kidney graft failure. Am J Transplant (2005) 5(9):2265–72. 10.1111/j.1600-6143.2005.01016.x [DOI] [PubMed] [Google Scholar]
- 193.Zou Y, Stastny P, Süsal C, Döhler B, Opelz G. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med (2007) 357:1293–300. 10.1056/NEJMoa067160 [DOI] [PubMed] [Google Scholar]
- 194.Ciszek M, Foroncewicz B, Mucha K, Żochowska D, Ziarkiewicz-Wróblewska B, Krawczyk M, et al. Anti-HLA and anti-MICA antibodies in liver transplant recipients: effect on long-term graft survival. Clin Dev Immunol (2013) 2013:828201, 5. 10.1155/2013/828201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Smith JD, Brunner VM, Jigjidsuren S, Hamour IM, McCormack AM, Banner NR, et al. Lack of effect of MICA antibodies on graft survival following heart transplantation. Am J Transplant (2009) 9(8):1912–9. 10.1111/j.1600-6143.2009.02722.x [DOI] [PubMed] [Google Scholar]
- 196.Dragun D, Müller DN, Bräsen JH, Fritsche L, Nieminen-Kelhä M, Dechend R, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med (2005) 352(6):558. 10.1056/NEJMoa035717 [DOI] [PubMed] [Google Scholar]
- 197.Hiemann NE, Meyer R, Wellnhofer E, Schoenemann C, Heidecke H, Lachmann N, et al. Non-HLA antibodies targeting vascular receptors enhance alloimmune response and microvasculopathy after heart transplantation. Transplantation (2012) 94(9):919–24. 10.1097/TP.0b013e3182692ad2 [DOI] [PubMed] [Google Scholar]
- 198.Banasik M, Boratyńska M, Kościelska-Kasprzak K, Kaminska D, Zmonarski S, Mazanowska O, et al. Non-HLA antibodies: angiotensin II type 1 receptor (anti-AT1R) and endothelin-1 type A receptor (anti-ETAR) are associated with renal allograft injury and graft loss. Transplant Proc (2014) 46(8):2618–21. 10.1016/j.transproceed.2014.09.029 [DOI] [PubMed] [Google Scholar]
- 199.Grandtnerova B, Mackova N, Hovoricova B, Jahnova E. Hyperacute rejection of living related kidney grafts caused by endothelial cell-specific antibodies: case reports. Transplant Proc (2008) 40(7):2422. 10.1016/j.transproceed.2008.06.014 [DOI] [PubMed] [Google Scholar]
- 200.Breimer ME, Rydberg L, Jackson AM, Lucas DP, Zachary AA, Melancon JK, et al. Multicenter evaluation of a novel endothelial cell crossmatch test in kidney transplantation. Transplantation (2009) 87(4):549. 10.1097/TP.0b013e3181949d4e [DOI] [PubMed] [Google Scholar]
- 201.Jackson AM, Lucas DP, Melancon JK, Desai NM. Clinical relevance and IgG subclass determination of non-HLA antibodies identified using endothelial cell precursors isolated from donor blood. Transplantation (2011) 92(1):54. 10.1097/TP.0b013e31821b60e9 [DOI] [PubMed] [Google Scholar]
- 202.Jackson AM, Sigdel TK, Delville M, Hsieh SC, Dai H, Bagnasco S, et al. Endothelial cell antibodies associated with novel targets and increased rejection. J Am Soc Nephrol (2015) 26(5):1161–71. 10.1681/ASN.2013121277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shin YS, Yang CW, Ahn HJ, Park CW, Jin DC, Kim YS, et al. Clinical significance of anti-endothelial cell antibody in renal transplant recipients. Korean J Intern Med (2001) 16(1):24–9. 10.3904/kjim.2001.16.1.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lehle K, Kroher J, Kolat P, von Süßkind-Schwendi M, Schmid C, Haneya A, et al. Existence of circulating anti-endothelial cell antibodies after heart transplantation is associated with post-transplant acute allograft rejection. Heart Vessels (2016) 31(5):752–7. 10.1007/s00380-015-0666-0 [DOI] [PubMed] [Google Scholar]
- 205.Banasik M, Boratyńska M, Kościelska-Kasprzak K, Krajewska M, Mazanowska O, Kamińska D, et al. The impact of non-HLA antibodies directed against endothelin-1 type A receptors (ETAR) on early renal transplant outcomes. Transpl Immunol (2014) 30(1):24–9. 10.1016/j.trim.2013.10.007 [DOI] [PubMed] [Google Scholar]
- 206.Dinavahi R, George A, Tretin A, Akalin E, Ames S, Bromberg JS, et al. Antibodies reactive to non-HLA antigens in transplant glomerulopathy. J Am Soc Nephrol (2011) 22(6):1168–78. 10.1681/ASN.2010111183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cravedi P, Heeger PS. Immunologic monitoring in transplantation revisited. Curr Opin Organ Transplant (2012) 17(1):26–32. 10.1097/MOT.0b013e32834ee402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Young RK, Dale B, Russell SD, Zachary AA, Tedford RJ. Incidence and early outcomes associated with pre-transplant antivimentin antibodies in the cardiac transplantation population. Clin Transplant (2015) 29(8):685–8. 10.1111/ctr.12567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dickinson AM, Norden J. Non-HLA genomics: does it have a role in predicting haematopoietic stem cell transplantation outcome? Int J Immunogenet (2015) 42(4):229–38. 10.1111/iji.12202 [DOI] [PubMed] [Google Scholar]
- 210.Sigdel TK, Li L, Tran TQ, Khatri P, Naesens M, Sarwal P, et al. Non-HLA antibodies to immunogenic epitopes predict the evolution of chronic renal allograft injury. J Am Soc Nephrol (2012) 23(4):750. 10.1681/ASN.2011060596 [DOI] [PubMed] [Google Scholar]