

Abstract
Background
Clostridium bolteae and Clostridium clostridioforme, previously included in the complex C. clostridioforme in the group Clostridium XIVa, remain difficult to distinguish by phenotypic methods. These bacteria, prevailing in the human intestinal microbiota, are opportunistic pathogens with various drug susceptibility patterns. In order to better characterize the two species and to obtain information on their antibiotic resistance genes, we analyzed the genomes of six strains of C. bolteae and six strains of C. clostridioforme, isolated from human infection.
Results
The genome length of C. bolteae varied from 6159 to 6398 kb, and 5719 to 6059 CDSs were detected. The genomes of C. clostridioforme were smaller, between 5467 and 5927 kb, and contained 5231 to 5916 CDSs. The two species display different metabolic pathways. The genomes of C. bolteae contained lactose operons involving PTS system and complex regulation, which contribute to phenotypic differentiation from C. clostridioforme. The Acetyl-CoA pathway, similar to that of Faecalibacterium prausnitzii, a major butyrate producer in the human gut, was only found in C. clostridioforme. The two species have also developed diverse flagella mobility systems contributing to gut colonization. Their genomes harboured many CDSs involved in resistance to beta-lactams, glycopeptides, macrolides, chloramphenicol, lincosamides, rifampin, linezolid, bacitracin, aminoglycosides and tetracyclines. Overall antimicrobial resistance genes were similar within a species, but strain-specific resistance genes were found. We discovered a new group of genes coding for rifampin resistance in C. bolteae. C. bolteae 90B3 was resistant to phenicols and linezolide in producing a 23S rRNA methyltransferase. C. clostridioforme 90A8 contained the VanB-type Tn1549 operon conferring vancomycin resistance. We also detected numerous genes encoding proteins related to efflux pump systems.
Conclusion
Genomic comparison of C. bolteae and C. clostridiofrome revealed functional differences in butyrate pathways and in flagellar systems, which play a critical role within human microbiota. Most of the resistance genes detected in both species were previously characterized in other bacterial species. A few of them were related to antibiotics inactive against Clostridium spp. Some were part of mobile genetic elements suggesting that these commensals of the human microbiota act as reservoir of antimicrobial resistances.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3152-x) contains supplementary material, which is available to authorized users.
Keywords: Clostridium bolteae, Clostridium clostridioforme, Comparative genomics, Antimicrobial resistance, Butyrate pathway
Background
Clostridium bolteae and Clostridium clostridioforme are members of the normal intestinal microbiota of humans, which can cause intra-abdominal infections when the natural intestinal barrier is altered. C. bolteae is present in stools of most children, but counts are significantly higher in autistic children than in controls [1]. Chronic diarrheal episodes associated with some forms of autism could be attributed to an overabundance of C. bolteae and release of end products of metabolism, such as butyrate, propionate and acetate, that alter the motility and contraction rate of the gastrointestinal tract [2].
The two opportunistic pathogens have been isolated in intra-abdominal infections, bacteremia and in abscesses and various drug susceptibility patterns have been reported [3]. Resistance to penicillin G is common but only a few C. clostridioforme produce a beta-lactamase, as detected by the nitrocefin test [4]. Most of the strains are susceptible to ampicillin-sulbactam, piperacillin-tazobactam, imipenem, and metronidazole. Resistance to clindamycin and moxifloxacin was previously reported in the complex C. clostridioforme [5]. In general, C. bolteae appears more resistant than C. clostridioforme, with higher MICs of penicillin G, ampicillin-sulbactam, ticarcilllin, piperacillin and piperacillin-tazobactam and more strains producing beta-lactamases [6]. Resistance to aminopenicillins, lincosamides and quinolones has been reported but not documented at genetic level.
Comparative analysis of 16S rRNA gene sequences of clostridial strains place C. clostridioforme in the subcluster XIVa of clostridia, including several non-spore-forming cocci and mesophilic spore-forming rod shaped bacteria [7]. C. bolteae was previously reported as a member of the C. clostridioforme complex including C. clostridioforme (formally), Clostridium aldenense, Clostridium citroniae and Clostridium hathewayi [5, 6]. A divergence of 3 % in 16S rRNA separates C. bolteae from C. clostridioforme, but few phenotypic characters distinguish the two species, such as lactose fermentation, which is a key phenotypic test [8] and little is known about their genetics.
Commensal anaerobes of gastrointestinal tract have been proposed as reservoir for various antibiotic resistance determinants [9]. However, the sequencing effort for Clostridium spp., mainly concerns C. difficile a cause of post-antibiotic diarrhoea. Only two genomes of C. bolteae (strains BAA613 & WAL-14578) and three of C. clostridioforme (strains 2149FAA.1, WAL-7855 and CM201.1) are available in public databases.
The aim of this study was to analyse the genomes of six clinical isolates of C. bolteae and six clinical isolates of C. clostridioforme in order to identify genetic specificities between these closely related species. We focused our analysis on (CDSs coding for) functions, which could have a physiological effect within microbiota, and provided the catalogue of antimicrobial resistance genes of the two opportunistic pathogens.
Results and discussion
General features of genomes reveal intra and interspecies variations
A total of 1 to 21 contigs were generated from assembly of reads from Illumina (134 to 185-fold coverage) for the six strains of C. bolteae (Table 1). A total of 10 to 48 contigs were generated (82 to 264-fold coverage) for the six strains of C. clostridioforme. Total genome size varied between species and strains. The size of C. bolteae ranged from 6159 kb for strain 90A7 to 6480 kb for strain 90B3 with 5833 and 6059 DNA-coding sequences (CDSs), respectively, and four 16S rRNA genes. The genome size of C. clostridioforme was smaller, from 5467 kb for strain 90A3 to 5970 kb for strain 90A6 with 5231 to 5916 CDSs, respectively, and four 16S rRNA genes. The phylogenetic tree based on the 16S rRNA sequences showed that the C. bolteae and C. clostridioforme studied were closely related to C. hathewayi, C. aldenense, C. citroniae, C. saccharolyticum and C. symbiosum, members of the Clostridium cluster XIVa of Firmicutes, as previously reported [6, 7] (data not shown).
Table 1.
Sequencing statistics and genome information
| Strain name | Year of isolation | Origin | CIP | Average sequencing coverage (times) | No. of contigs | Genome size (kb) | Predicted no. of genes (Broad Institute) | Predicted no. of proteins (Broad Institute) | No. of strain-specific CDSs (singletons & paralogs) | No. of strain-specific CDSs (unique) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C. bolteae | 90A5 | 2009 | perineal abcess | 110254 | 176 | 12 | 6394 | 5833 | 5716 | 84 | 73 |
| C. bolteae | 90A7 | 2009 | intraabdominal fluid | 110239 | 185 | 8 | 6159 | 5719 | 5592 | 743 | 735 |
| C. bolteae | 90A9 | 2009 | douglas abcess | 110242 | 134 | 1 | 6367 | 5833 | 5731 | 163 | 161 |
| C. bolteae | 90B3 | 2009 | pelvis abcess | 110247 | 176 | 4 | 6480 | 6059 | 5949 | 239 | 188 |
| C. bolteae | 90B7 | 2010 | pleural abcess | 110255 | 165 | 19 | 6398 | 5876 | 5761 | 120 | 105 |
| C. bolteae | 90B8 | 2010 | ureteral abcess | 110256 | 76 | 21 | 6449 | 6023 | 5901 | 905 | 846 |
| C. clostridioforme | 90A1 | 2009 | intraabdominal fluid | 110245 | 145 | 16 | 5769 | 5669 | 5546 | 130 | 72 |
| C. clostridioforme | 90A3 | 2009 | sigmoid abcess | 110246 | 135 | 11 | 5467 | 5231 | 5096 | 31 | 21 |
| C. clostridioforme | 90A4 | 2009 | bone biopsy | 110238 | 264 | 48 | 5820 | 5726 | 5547 | 97 | 72 |
| C. clostridioforme | 90A6 | 2009 | intraabdominal fluid | 110240 | 167 | 22 | 5970 | 5851 | 5711 | 307 | 273 |
| C. clostridioforme | 90A8 | 2009 | intraabdominal fluid | 110249 | 82 | 47 | 5927 | 5916 | 5734 | 1190 | 1006 |
| C. clostridioforme | 90B1 | 2009 | rectal abcess | 110244 | 174 | 10 | 5533 | 5308 | 5184 | 48 | 36 |
Genomes of C. bolteae and C. clostridioforme are large genomes, where genetic redundancy is prevalent (data not shown). The redundant genes were involved in a variety of metabolic pathways, including carbon metabolism, transport, iron metabolism and amino acid biosynthesis. The differences in the number of CDSs between genomes reflected variation in genetic redundancy more than gain or loss of particular functions. In addition, genomes integrated mobile elements i.e. transposons, Insertion Sequences, plasmids or phages (integrase, capsid protein,…) indicative of lateral gene transfers. Some of them carried antimicrobial resistance genes (see below).
To examine the pangenome of the two species, we compared the 97,210 CDSs obtained from the 12 newly sequenced genomes with those of five other genomes (C. bolteae BAA613, C. bolteae WAL-14578, C. clostridioforme CM201.1, C. clostridioforme 2149FAA.1, and C. clostridioforme WAL-7855). All CDSs were clustered using the BlastClust algorithm at high stringency, above a 90 % sequence identity cut-off and 90 % length overlap. A total of 10,530 clusters were found. Only 2294 (21.78 %) clusters were shared by the two species.
In using only genomes newly sequenced, we estimated the (species) core genome and (strain-specific) genes of the six C. bolteae, and the six C. clostridioforme (Table 1). A total of 3714 genes formed the core genome of C. bolteae. The number of strain-specific genes in this species varied from 73 to 846. In C. clostridioforme, 3660 genes defined the core genome. A total of 2409 clusters were shared by the two species; 1305 genes were specific to C. bolteae and 1251 to C. clostridioforme.
C. bolteae 90A7 and 90B8 had the largest number of unique genes for this species (735 and 846, respectively). C. clostridioforme 90A8, with 1006 (17 %) unique genes had the largest number of strain specific genes in this study. These strains integrated a high number of mobile elements. Some unique CDSs were annotated as transporters or regulators. Few of them were involved in defence mechanisms (antimicrobial resistance genes..) or metabolic pathways. Most of them, often surrounded by CDSs from phages or transposons, were of unknown functions (data not shown).
Functional differences between species in the core genomes
The challenge of our study was to provide reliable information from draft genomes. Therefore, we focused our analysis on the core genomes. The classification of the CDSs according to the Clusters of Orthologous Groups (COGs) system allowed to give an overview of the functions displayed by the two species. The core genomes of C. bolteae and C. clostridioforme were enriched (over 7 % of total COG matched counts) in COG categories K, E, G and R relative to Transcription (309 and 290 CDSs), Amino acid transport and metabolism (335 and 276 CDSs), Carbohydrate transport and metabolism (431 and 425 CDSs) and General function prediction (366 and 331CDSs) (Table 2).
Table 2.
Functional profile ( COG categories ) of C. bolteae and C. clostridioforme
| Core genome of C. bolteae | Core genome of C. clostridioforme | Shared between the two species | Specific of C. bolteae | Specific of C. clostridioforme | |
|---|---|---|---|---|---|
| Total Number of Orthologs groups | 3714 | 3660 | 2409 | 1305 | 1251 |
| Ortholog groups in COGs | 3060 | 2861 | 2151 | 909 | 710 |
| No Hits on COGs | 660 | 805 | 264 | 396 | 541 |
| INFORMATION STORAGE AND PROCESSING | |||||
| Translation, ribosomal structure and biogenesis (J) | 151 | 148 | 137 | 14 | 11 |
| Transcription (K) | 309 | 290 | 191 | 118 | 99 |
| Replication, recombination and repair (L) | 111 | 135 | 85 | 26 | 50 |
| CELLULAR PROCESSES AND SIGNALING | |||||
| Cell cycle control, cell division, chromosome partitioning (D) | 43 | 43 | 36 | 7 | 7 |
| Defense mechanisms (V) | 84 | 72 | 52 | 32 | 20 |
| Signal transduction mechanisms (T) | 180 | 139 | 101 | 79 | 38 |
| Cell wall/membrane/envelope biogenesis (M) | 122 | 137 | 97 | 25 | 40 |
| Cell motility (Cell motility and secretion) (N) | 34 | 18 | 0 | 34 | 18 |
| Intracellular trafficking, secretion, and vesicular transport (U) | 12 | 17 | 9 | 3 | 8 |
| Posttranslational modification, protein turnover, chaperones (O) | 84 | 83 | 72 | 12 | 11 |
| METABOLISM | |||||
| Energy production and conversion (C) | 194 | 155 | 120 | 74 | 35 |
| Carbohydrate transport and metabolism (G) | 431 | 425 | 296 | 135 | 129 |
| Amino acid transport and metabolism (E) | 335 | 276 | 238 | 97 | 38 |
| Nucleotide transport and metabolism (F) | 115 | 94 | 85 | 30 | 9 |
| Coenzyme transport and metabolism (H) | 99 | 89 | 72 | 27 | 17 |
| Lipid transport and metabolism (I) | 50 | 60 | 42 | 8 | 18 |
| Inorganic ion transport and metabolism (P) | 122 | 129 | 94 | 28 | 35 |
| Secondary metabolites biosynthesis, transport and catabolism (Q) | 12 | 0 | 0 | 12 | 0 |
| POORLY CHARACTERIZED | |||||
| General function prediction only (R) | 366 | 331 | 256 | 110 | 75 |
| Function unknown (S) | 187 | 178 | 150 | 37 | 28 |
While C. bolteae and C. clostridioforme are phenotypically related, the pattern of functions obtained through change in COG annotation differed between the two species (Table 2). 30 additional CDSs of the Nucleotide transport and metabolism (F), 97 CDSs of the Amino acid transport and metabolism (E) and 79 CDSs coding for Signal transduction mechanisms (T) categories were specific for C. bolteae. 40 CDSs coding for the Cell wall/membrane/envelope biogenesis (M), 50 CDSs for Replication, recombination and repair (L) and 18 CDSs for the Lipid transport and metabolism (I) categories were specific for C. clostridioforme. Differences between metabolic pathways in C. clostridioforme and C. bolteae seem to be large enough to support delineation of the species.
Among carbohydrate pathways, C. bolteae and C. clostridioforme harboured different systems for the assimilation of lactose, which differ in their phosphorylation states, intermediate metabolites, and bioenergetics (Additional file 1: Table S1). Genes coding for a β-galactosidase, which hydrolyzes lactose yielding glucose and galactose, were found in both species. An alternative lactose catabolic pathway, the lactose/cellobiose dependent phosphotransferase system (lac/cell-PTS) was found in almost all genomes of C. bolteae. The lac/cell-PTS operon, previously described in C. acetobutylicum [10], consists of genes for the 6-phospho-β-galactosidase, phosphoglycerate mutase, and lichenan operon transcriptional antiterminator and of two copies of genes for lactose/cellobiose family IIC, IIB and IIA components. By such a system, lactose is phosphorylated at the C-6 carbon and the internalized lactose 6-phosphate is degraded in galactose 6-phosphate and glucose by the 6-phospho-β-galactosidase. In addition, the gene for the 6-phospho-β-galactosidase and the genes for the lactose/cellobiose family components were lacking in C. bolteae 90A7. It is likely that this system, inducible by cellobiose or lactose and regulated by several repressors (described in other Gram positive bacteria [11, 12]) accounts for the lactose-negative phenotype in C. bolteae [6]. By using our annotation system, we detected a galactose operon repressor (GalR) among lacI-family regulators, in C. clostridioforme (all, except 90A8), but not in C. bolteae. Laboratory experiments are needed to determine how the transcription factors from the two species mediate preferences in the utilization of certain carbohydrates over others.
Other distinctive features between the two species were CDSs coding for secondary metabolites biosynthesis and transport and catabolism which were only found in C. bolteae (Table 2).
Interestingly, the number of genes of the cell motility and secretion category (N) (34 and 18 CDSs) was different between the two species. Among them, we found CDSs encoding flagella motility recognized as essential virulence factors for most motile pathogens. Overall, twenty-four genes (46 clusters + 3 orphans) represented the flagellar operon in the genomes of C. bolteae. Among them, genes for flagellin (fliC) and flagellar cap (fliD), one of the multiple cell-surface adhesins of the bacteria, revealed cluster specificity and microevolution. Genes coding for fliD were represented by one cluster and two additional genes in C. bolteae 90A7 and 90B8. FliC sequences from C. bolteae 90A9, 90B3 and 90B8 formed one cluster, those from 90A5 and 90B7 clustered in another group, and sequences from C. bolteae 90A7 remained orphans (unique genes) after clustering (Fig. 1). They were closely related to flagellin sequences of C. citroniae and C. hathewayi, other Clostridium spp. of the group XIVa, isolated occasionally from human infections. In addition, C. bolteae 90A9 and 90B3 shared a second operon of only 19 genes in syntheny, including a flagellin gene (flaA) closely related to those of C. clostridioforme (63 % identity). Based on conserved residues L87, Q88, R89 and Q96 critical for TLR5 signalling and flagellin polymerisation, these proteins were predicted to have pro-inflammatory properties [13]. In C. clostridioforme, twenty genes (57 other clusters) organised in a single operon encoded the flagellar apparatus. FlaA sequences from C. clostridioforme belonged to a phylogenetic group closely related to flagellin sequences from Eubacterium cellulosovens isolated from the rumen. Overall, flagellin genes and loci organization related to flagella were different between species (Additional file 2: Table S2 and Additional file 3: Table S3), suggesting that motility, chemotaxis, and occurrence of potential interactions with the colonic mucosa are species specific [14].
Fig. 1.
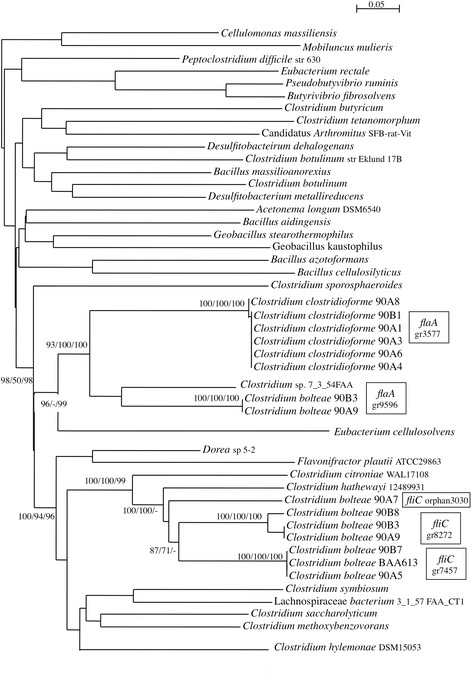
Distance-based phylogenetic tree of flagellin genes. Values at nodes corresponded to bootstrap percents obtained from phylogenetic trees based on multiple sequence alignments by ClustalW, Tcoffee or Promals, respectively. Gene names and, orphan or cluster numbers were indicated
Species differences in pathways for butyrate synthesis
Comparison of whole genome sequences revealed that pathways for butyrate synthesis, which play a key role in colonic health in humans, were present in C. bolteae and C. clostridioforme.
The two species were butyrate producers through different and complementary ways (Fig. 2, Additional file 4: Table S4). All C. clostridioforme, except 90A8, carried a locus coding for the Acetyl-CoA pathway (from Acetyl-CoA to butyryl-CoA), including genes for the beta-hydroxylbutyrylCoA dehydrogenase (hbd), thiolase (thl), crotonase (cro), butyryl-CoA dehydrogenase (bcd) and two electron transfer proteins (ETF alpha, ETF beta) (Additional file 4: Table S4). Only, C. bolteae 90A8 and C. clostridioforme 2149FAA.1 contained another putative bcd (74.9 % identity) in their genomes (data not shown). The locus composition and arrangement were similar to that in Faecalibacterium prausnitzii, a major butyrate producer of the human large intestine [15]. The Acetyl-CoA pathway was not found in C. bolteae. Both species shared genes for the two hydroxy-glutaryl-CoA dehydrogenase (HgCoAd) and the glutaconyl-CoA decarboxylase (Gcd) from the Glutarate pathway that can lead to crotonyl CoA and to butyryl-CoA via bcd genes [16].
Fig. 2.
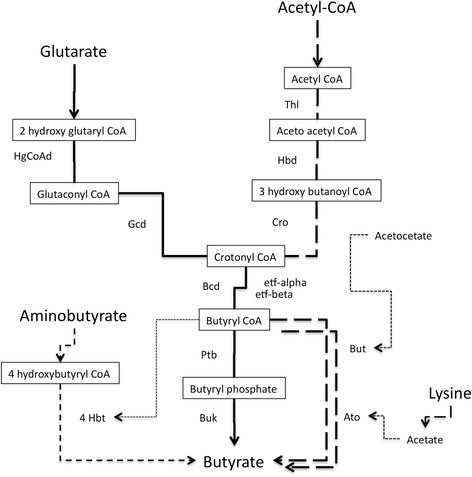
Different pathways for butyrate synthesis potentially present in genomes of C. clostridioforme and C. bolteae. In solid lines : found in C. bolteae and C. clostridioforme; In bold dashed lines : only found in C. clostridioforme ; In fine dashed lines : only found in C. bolteae 90A5 and 90B7 (for more details, see Additional file 4: Table S4). Genes (protein names) are displayed. Ato, Acetyl-CoA acetyltransferase ; Bcd, butyryl-CoA deshydrogenase ; Buk, butyrate kinase; But, Butyrate-acetoacetate CoA-transferase ; Cro, crotonase ; Etf, electron transfer protein ; Gcd, glutaconyl-CoA decarboxylase ; Hbd, Acetoacetyl-CoA reductase ; 4Hbt, 4-hydroxybutyrate CoA transferase ; HgCoAd, 3-hydroxybutyryl-CoA dehydrogenase ; Ptb, phosphate butyryltranferase; Thl, thiolase
The final conversion from butyryl-CoA to butyrate can be performed by the butyrate kinase (buk) and the phosphotransbutyrylase (ptb) present in both species (Additional file 4: Table S4). The group of buk sequences from C. clostridioforme branched in the vicinity of the buk sequence from C. citroniae on the phylogenetic tree (Additional file 5: Figure S1). Buk sequences from C. bolteae formed distinct monophyletic groups and sequences distributed among the phylogenetic trees, suggesting polymorphism and/or functional variations of the enzyme in this species. Other genes for transferases from the lysine pathway (ato—alpha and beta subunit ; but—acetate CoA transferase), detected near the butyrate locus in six genomes of C. clostridioforme, can be involved as final enzymes. Genes from the 4-aminobutyrate pathway (4hbt) can be another alternative for the terminal step in C. bolteae 90A5, 90B7, WAL14578, and BAA613 [16].
The butyryl-CoA:acetate CoA-transferase (but) of the acetyl-CoA pathway, the final step in butyrate production predominant in Clostridium XIVa, was neither found in the common core genomes nor in the genomes of the C. bolteae studied [17]. In the human gut, previous studies on colonic isolates of healthy individuals have illustrated that but pathway predominates [18]. Further studies are needed to assess the impact of butyrate production through the Glutarate pathway on health of colonic cells, in particular in autism where C. bolteae is overabundant [2].
Identification of antibiotic resistance determinants
Drug resistance genes that had not been recognized by automated annotation were identified by homology sequence research on ARDB. Resistance genes were predicted on a value up to 40 % identity (50 % of positive substitutions) on 70 % of length above the cut-off value usually recommended (see list in Table 3). Then, the gene content and genetic organization of microbial resistance loci of the six C. bolteae and the six C. clostridioforme were compared to previous data obtained from C. clostridioforme CM201.1 in our laboratory.
Table 3.
Distribution of CDSs annotated as antimicrobial resistance genes
| ARDB | Number of clusters and orphans | C. bolteae 90A5 | C. bolteae 90A7 | C. bolteae 90A9 | C. bolteae 90B3 | C. bolteae 90B7 | C. bolteae 90B8 | C. clostridioforme 90A1 | C. clostridioforme 90A3 | C. clostridioforme 90A4 | C. clostridioforme 90A6 | C. clostridioforme 90A8 | C. clostridioforme 90B1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| class A beta-lactamase | blaCLO1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| class B beta-lactamase | metallo-enzymes | 4 | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 2 | 2 | 2 | 1 |
| class C beta-lactamase | Amp C and Penicillin Binding Protein | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| class D beta-lactamase | Amp D (oxa type) | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 |
| Teicoplanin, Vancomycin | VanRA | 4 | 2 | 1 | 1 | 1 | 1 | 3 | 1 | |||||
| VanSA | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||
| VanD | 2 | 2 | 2 | 2 | 2 | 2 | ||||||||
| VanSD | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||
| VanXD | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||
| VanRD | 2 | 1 | 1 | 1 | 1 | 1 | ||||||||
| Vancomycin | VanHB | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 |
| VanRB | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | |
| VanB | 1 | 1 | ||||||||||||
| VanSB | 1 | 1 | ||||||||||||
| VanWB | 1 | 1 | ||||||||||||
| VanXB | 1 | 1 | ||||||||||||
| VanYB | 2 | 2 | ||||||||||||
| VanSC | 1 | 1 | ||||||||||||
| VanRE | 1 | 1 | 1 | 1 | ||||||||||
| VanG | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| VanRG | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | |
| VanYG | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||||||
| VanUG | 3 | 2 | 1 | |||||||||||
| Teicoplanin | VanZ | 4 | 2 | 3 | 1 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| Lincomycin | LnuA | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 2 |
| Macrolides | ErmB | 1 | 1 | 1 | ||||||||||
| Macrolide-Lincosamide- Streptogramin (efflux pumps) | MacB | 1 | 6 | 8 | 7 | 7 | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 6 |
| MefA | 1 | 3 | 2 | 4 | 4 | 3 | 3 | 3 | 3 | 3 | 4 | 3 | 3 | |
| MsrA/MsrB | 5 | 3 | 3 | 3 | 3 | 3 | ||||||||
| Lsa | 1 | 1 | 1 | 1 | ||||||||||
| ABC transporter (efflux pumps) | VgaA | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Tirc | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| CcmA | 6 | 2 | 2 | 3 | 3 | 2 | 4 | 3 | ||||||
| Streptogramin_A Chloramphenicol | VatB | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| chloramphenicol | CAT | 3 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | ||||
| linezolide | Cfr | 2 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| Rifampicin | Arr | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||
| Metronidazole | Nim | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Trimethoprim | Dfra20 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Dfra | 1 | 1 | ||||||||||||
| Aminoglycosides | Aac6ie | 2 | 2 | |||||||||||
| Aad9ib | 1 | 1 | ||||||||||||
| Ant6ia | 3 | 1 | 1 | 2 | 1 | 2 | 1 | |||||||
| Aph3iiia | 1 | 2 | 1 | 1 | ||||||||||
| Apha | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | |
| Tetracycline | Tet40 | 1 | 1 | 1 | ||||||||||
| TetO | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||
| TetW | 1 | 1 | 1 | 1 | 1 | |||||||||
| Tet32 | 1 | 1 | 1 | |||||||||||
| Bacitracin | BacA | 3 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 2 |
| BcrA | 1 | 2 | 3 | 4 | 4 | 2 | 3 | 3 | 4 | 4 | 3 | 4 | 5 | |
| Multidrug resistance efflux pump | Blt | 1 | 1 | |||||||||||
| AcrB | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 3 | 3 | 3 | 3 |
aBroad annotation
Because sequence-based predictions might potentially identify determinants that do not lead to antimicrobial resistance, susceptibility testing was performed to obtain information on the predicted response of bacteria to antibiotics. The strains included in this study showed resistance patterns, including ampicillin, macrolides, lincomycin and quinolones, now common in anaerobes (Table 4). Both genomic data (CDSs and annotations) and phenotypic susceptibility tests were considered to identify antibiotic resistance determinants (Tables 3 and 4). Preliminary assays for cloning certain genes were also performed in order to check their capacity to confer antibiotic resistance (see below).
Table 4.
Antibiogram of C. clostridioforme and C. bolteae
| C. bolteae | C. bolteae | C. bolteae | C. bolteae | C. bolteae | C. bolteae | C. clostridioforme | C. clostridioforme | C. clostridioforme | C. clostridioforme | C. clostridioforme | C. clostridioforme | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Antimicrobial agent | 90A5 | 90A7 | 90A9 | 90B3 | 90B7 | 90B8 | 90A1 | 90A3 | 90A4 | 90A6 | 90A8 | 90B1 |
| Beta - lactams | ||||||||||||
| ampicillin | R | R | R | R | R | R | R | R | R | R | R | R |
| amox-clavulanic acid | S | S | S | S | S | S | S | S | S | S | S | S |
| cefoxitin | S | S | S | S | S | S | S | R | S | I | S | I |
| imipenem | S | S | S | S | S | S | S | S | S | S | S | S |
| Glycopeptides | ||||||||||||
| Vancomycin | S | S | S | S | S | S | S | S | S | S | Ra | S |
| Macrolides | ||||||||||||
| Erythromycin | R | R | I | R | R | R | R | R | R | R | R | R |
| Telithromycin (Ketolides) | R | R | S | R | R | R | R | R | R | R | R | R |
| Spiramycin | S | S | S | R | S | I | S | S | R | R | R | S |
| Lincosamides | ||||||||||||
| Lincomycin | I | R | R | R | R | R | S | I | R | R | R | R |
| Clindamycin | S | S | S | R | S | S | S | S | R | S | R | S |
| Streptogramins | ||||||||||||
| Pristinamycin | S | S | S | nd | I | S | S | S | S | S | S | S |
| Amphenicols | ||||||||||||
| Chloramphenicol | nd | S | S | R | S | R | S | S | S | S | S | S |
| Oxazolidinones | ||||||||||||
| Linezolid | S | S | S | Rb | S | S | S | S | S | S | S | S |
| Rifampin | S | Rc | S | S | S | S | S | S | S | S | S | S |
| Quinolones | ||||||||||||
| Ciprofloxacin | R | R | R | R | R | R | R | R | R | R | R | R |
| Moxifloxacin | R | R | R | R | R | R | R | R | R | R | R | R |
| Tetracyclines | ||||||||||||
| Doxycycline | S | R | R | R | R | S | R | R | R | R | R | R |
| Nitroimidazole | ||||||||||||
| Metronidazole | S | S | S | S | S | S | S | S | S | S | S | S |
| Polypeptides | ||||||||||||
| Colistin | R | R | R | R | R | R | R | R | R | R | R | R |
aMIC vancomycin > 250 mg/l
bMIC linezolid =16 mg/l
cMIC rifampin =32 mg/l
Genes of resistance to antibiotics used for treatment of anaerobic infections
A total of 76 clusters and 21 strain-specific genes potentially involved in antimicrobial resistance were identified (Table 3). It’s included from 42 to 50 CDSs in C. bolteae and 48 to 58 CDSs in C. clostridioforme. From 27 to 42 CDSs per genomes were related to drug resistance mechanisms to beta-lactams, glycopeptides, macrolides, lincosamides, and metronidazole.
Seven clusters involved in beta-lactam resistance are shared or part of the core genome of the two species (Fig. 3). Three types of beta-lactamases, including class A beta-lactamase, class C beta-lactamase, class D [Oxa type] beta-lactamase and several metallo-enzymes were recognized in the twelve genomes. All the strains studied, selected for their resistance to ampicillin, shared the gene blaCLO1, previously found in C. clostridioforme CM201.1 (unpublished), but the structure of integrative conjugative element (ICE) observed in CM201.1 was not found in the new genomes sequenced. The gene blaCLO1 confers resistance to aminopenicillins and carboxypenicillins in E. coli, and its activity is inhibited by clavulanate and sulbactam. Nine amino acid changes were observed in beta-lactamases of C. bolteae 90A9, 90B3 and 90A8. This closely related beta-lactamase was flanked by insertion sequences (IS66) and a putative gene for class D beta-lactamase (COG 2602) also described in Clostridium sp M62/1 from the human intestinal microflora (HMP project). Genes for class C beta-lactamases, previously found in the chromosomes of enteric bacteria (COG2680), were also present in C. bolteae and C. clostridioforme.
Fig. 3.
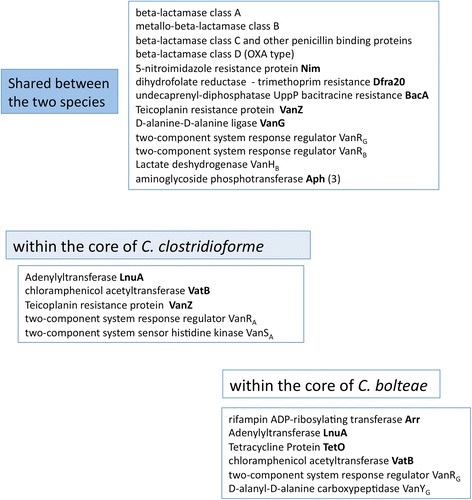
Distribution of antibiotic resistance genes shared between and within the core of C. bolteae and C. clostridioforme. Genes overlapping at least 90 % length and 90 % of similarity were considered homologs. Resistance genes were predicted on a value up to 40 % identity (50 % of positive substitutions) on 70 % of length by homology sequence research on ARDB. For all C. clostridioforme and some C. bolteae 23S rRNA methyltransferase Cfr-like
A high number of predicted genes (32 CDSs) were involved in resistance to glycopeptides (Additional file 6: Figure S2). C. clostridioforme 90A8 was the single strain with all genes required for glycopeptide resistance in agreement with the phenotype (MIC > 256 mg/l). Vancomycin resistance in this strain was attributed to a VanB-type operon borne by a Tn1549-like element. Unfortunately, a deletion of ten nucleotides within the relaxase gene of Tn1549 leads to the inability of 90A8 to transfer vancomycin resistance in vitro [19]. The other genomes of C. clostridioforme included part of the VanD-type vancomycin resistance operon, but the D-Ala-Lac ligase vanD gene was disrupted by a stop codon leading to a truncated protein (Additional file 6: Figure S2). In addition vanH and vanY, which encode a D-lactate dehydrogenase and a DD carboxypeptidase, respectively, were missing. Similarly, the genomes of C. bolteae harboured four CDSs, homolog of vanRG vanUG vanG vanYG, which formed an incomplete and non-functional operon due to the lack of a serine racemase gene. The high number of CDSs coding for glycopeptide resistance (including vanD or vanG known to be chromosomal and not transferable) found in the genomes of C. bolteae and C. clostridioforme, suggests that they are part of ancestral entire operons, which have evolved in the absence of antibiotic selective pressure [20]. Presence of incomplete van operon is intriguing, but similar observations in other anaerobes living in microbiomes such as Clostridium difficile 630 or Ruminococcus spp., have been reported [21, 22].
Homologues to the adenylyltransferase [lnuA] gene conferring resistance to the lincosamides, were present in the core genome of both species (Fig. 3). LnuA genes of C. clostridioforme and C. bolteae had 70 to 72 % identity with orthologs found in C. hathewayi and C. citroniae, respectively. C. clostridioforme 90B1 and 90A6 harboured an additional lnu gene (68 % identity with LnuA90A5), with no traces of mobile elements. Lincomycin resistance is common in C. bolteae and C. clostridioforme, often associated with resistance to clindamycin. LnuA proteins of the two species displayed 51 to 54 % of identity with LnuA from Staphylococcus suggesting common functionality, but the role of lnu genes is difficult to establish due to the presence of other putative mechanisms [23]. Similarly, all strains were resistant to erythromycin, whereas two genes homolog to the erythromycin ribosome methylase gene, ermB, were only predicted in the genomes of C. clostridioforme 90A4 and 90A8. Over-expression of multidrug efflux pumps and Macrolide-, and various Macrolide-Lincosamide-Streptogramin B—specific efflux systems, such as MacB, MefA, VgaA, MsrA/MsrB, and CcmA (Table 3, Additional file 7: Figure S3), found in the genomes studied, can lead to macrolide and lincosamide resistance [24]. In addition, two clusters of CDSs coding for xenobiotic acetyltransferases related to VatB (48 % identity) were found in the core genomes of each of the species (Fig. 3). VatB inactivates virginiamycin [25], but here, the resistance was not detected by anti-microbial susceptibility tests (Table 4).
A cluster of CDSs homologues to the metronidazole resistance (nim) genes was detected in the core genomes of both species, and metronidazole was very active on the species studied [26]. In Bacteroides fragilis it has been demonstrated that increased expression of nim genes when downstream from IS elements leads to metronidazole resistance [27]. In the lack of IS directly upstream the nim genes, the mechanism to confer metronidazole resistance to C. bolteae and C. clostridioforme remains to be established.
Unexpected observation of new genes of resistance
Regarding other drug resistances, genome data revealed genes related to chloramphenicol and rifampin resistance mechanisms (Table 3). In most genomes of each species, we found CDSs coding for a group A chloramphenicol acetyltransferase [cat] which can inactivate chloramphenicol. However, only C. bolteae 90B3 and 90B8 were resistant to chloramphenicol. The genome of strain 90B8 contained a second copy of the cat gene borne by a Tn4451-like transposon (96 and 90 % identity with Tn4451 and Tn4453, respectively). The 90B3 genome contained a CDS homolog to the gene cfr coding for a 23S rRNA methyl-transferase largely spread in Gram-positive bacteria. As expected, the strain 90B3 was also resistant to florfenicol, tiamulin, and linezolid (MIC = 16 mg/l). Other 23S rRNA methyltransferase (Cfr-like) CDSs were detected in an environment rich in transposable elements (Tn 6103-6110-CTn4 [fragments]) in the genomes of C. clostridioforme and C. bolteae 90A5 and 90B7, but the rRNA methylation did not appear to affect the susceptibility to chloramphenicol (Fig. 3).
The analysis of genomic data allowed to recognize CDSs homologues to rifampin-ADP-ribosyltransferase (arr) genes in C. bolteae but not in C. clostridioforme. No mobile elements or traces of mobile elements were found around the arr genes suggesting they were indigenous to this species. This new Arr sequences branched in the vicinity of Arr proteins from C. saccharoperbutylacetonicum and some Cyanobacteria on the phylogenetic tree (Additional file 8: Figure S4). They were distinct from Arr-2 proteins of Enterobacteriaceae and from Arr proteins of Mycobacterium and Streptomyces spp.. All strains, except 90A7, were susceptible to rifampin. In the absence of mutations in rpoB (known to be responsible for rifampin resistance), resistance of C. bolteae 90A7 (MIC: 32 mg/l) was likely due to positive selection of mutations in aar Cbol90A7. Susceptibility to rifampin of other C. bolteae was likely due to the lack of promoters upstream from arr (as predicted by in silico analysis) or to nucleotide substitutions within arr leading to amino acid replacement and functional inactivation (data not shown).
Concerning the resistance of all strains against moxifloxacin and ciprofloxacin, all strains of C. bolteae showed several substitutions in the “quinolone-resistance–determining region” (QRDR) of gyrB. We didn’t find any substitutions in this region for gyrA, nor described in the protein of the quinolone-resistant epidemic strain, C. difficile 027 [28]. Therefore, GyrB was likely the preferred target in acquisition of quinolone resistance in these two species. In addition, several CDSs coding for AcrB inner membrane transporter were present in all the strains. These transporters are part of a resistance-nodulation-division [RND] multidrug efflux pump, known to increase efflux of quinolones in some Gram-negative bacteria [29]. Further studies are needed to determine their influence in the loss of susceptibility of Clostridium spp. to fluoroquinolones.
Overall, similar resistance profiles against antibiotics in C. bolteae and C. clostridioforme can result from various mechanisms.
Genes of resistance to antibiotics less active or inactive against Clostridium spp
The genomes of C. bolteae and C.clostridioforme carried one or two copies of the undecaprenyl pyrophosphate phosphatase gene bacA, and 2 to 5 copies of the efflux pump genes, bcrA, involved in bacitracin resistance, in agreement with their low susceptibility [26]. We also found one cluster of CDSs homolog to the dihydrofolate reductase gene, dfrA20 (41 % identity / 97 % length) of Pasteurella multocida in the core genome of both species that could explain the poor activity of trimethoprim on our Clostridium spp. [30]. In addition, C. clostridioforme 90A6 harbored a CDS identical to dfrA from Enterococcus faecium. This gene detected in an environment rich in mobile elements is consistent with a new example of horizontal transfer between Enterococcus spp. and Clostridiales.
Interestingly, the genomes of C. bolteae or C. clostridioforme contained various resistance genes against antibiotics naturally inactive on these species. Five CDSs were homologs of genes that phosphorylate, acetylate or adenylylate aminoglycosides. Four of these putative resistance genes were detected in an environment of mobile elements. Three genes, aadE, sat4 and aph(3’)-III, conferring resistance to streptothricin, streptomycin and kanamycin, respectively, was found part of a transposon delineated by two IS1182 copies in C. clostridioforme 90A3 (two copies), 90A6 and 90B1. The aph(3’)-III detected was identical to the aph(3’)-III, part of the multidrug resistant plasmid PF856 from E. faecium [31], also related to an internal domain (99 % identity) of a SSCmec element of Staphylococcus aureus HT20040085. Similarly, the aminoglycoside 6-adenylyltransferase gene ant(6’)-Ia, conferring resistance to streptomycin was shared by C. clostridioforme 90A1, 90A3 (2 copies), 90A4, 90A6 (2 copies) and C. bolteae 90A7. The adenylyltransferase gene aad(9’)-b which mediates resistance to streptomycin/spectinomycin was found in C. bolteae 90B3. Two copies of the acetyl transferase aac(6’)-Im were also present in the genome of C. clostridioforme 90B1. Homologs of AAC(6’)-Im which confers resistance to tobramycin and amikacin resistance was also found in E. coli (96 % identity), Coprococcus sp, C. difficile and Enterococcus faecium (data not shown). In addition, three CDSs coding for an aminoglycoside kinase (APH), known to be widely distributed in Gram-positive bacteria, were also observed among all the strains [32].
Numerous tetracycline resistance genes were also detected in C. bolteae and C. clostridioforme. They include both efflux genes such as tet40, and ribosome protection determinants such as tetO, tetW, and tet32, previously reported as circulating in gut microflora among distantly related bacteria [33].
Conclusion
We studied the genomes of C. bolteae and C. clostridioforme, two species of the complex Clostridioforme, which can behave either as members of the human microbiota or as opportunistic pathogens.
We compared the genomes of six clinical isolates of C. bolteae and 6 strains of C. clostridioforme with available genome sequences in laboratory and international databanks. These data were used as a basis to reveal differences in functional patterns between the two species. Among them, differences in flagella coding genes and butyrate pathways can potentially influence host-gut microbiota interactions.
The patterns of resistance genes in the genomes were also of peculiar interest: (i) these bacterial species harbour specific and indigenous putative resistance genes, which included ABC transporters, antibiotic modifying enzymes, rRNA methyltransferases, (ii) Other resistance genes were acquired as shown by their location within mobile elements in the genomes studied. Some of them corresponded to genes mainly spread in gut bacteria. Others were new determinants, which remain to be analysed for their ability to confer antibiotic resistance. This study emphasizes the role of commensal bacteria of the digestive microbiota as reservoir for antibiotic resistances.
Future directions
As the costs of whole-genome sequencing continue to decline, it becomes increasingly available in routine diagnostic laboratories to detect antimicrobial resistance genes in genomes as substitute of traditional methods for resistance identification. However, the true challenge will remain to extract the relevant information from the large amount of data and to ensure the functionality of the genes detected.
Methods
Bacterial strains, molecular Identification and antimicrobial susceptibility testing
We studied retrospectively 6 strains of C. bolteae and 6 strains of C. clostridioforme, resistant to beta-lactams. Strains were isolated from intra-abdominal infections from patients (without links) over 2 years in two hospitals, Paris Saint Joseph and CHRU of Nancy, France.
Clostridium isolates were grown in Brain Heart Infusion agar or broth (Oxoid) in an anaerobic atmosphere (5 % CO2, 5 % H2 and 90 % N2) at 37 °C. Antibiotic susceptibility was tested by disk diffusion on Mueller-Hinton (MH) medium supplemented with 5 % defibrillated horse blood, according to the standards of the Comité de l’Antibiogramme de la Société Française de Microbiologie (http://www.sfm-microbiologie.org/). MICs of antimicrobial agents were determined on MH agar by Etest (bioMérieux).
Genomic DNA for sequencing was prepared from cells by lysis in lysozyme, incubation in proteinase K/SDS, followed by a standard phenol/chloroform extraction procedure. Prior to library preparation DNA quality was assessed by Nanodrop analysis (Thermo Scientific).
Molecular identification of strains was performed by sequencing a 1483-bp PCR fragment from 16S rRNA using universal primers B27F (5’-AGAGTTTGATCCTGGCTCAG) and U1492R (5’-GGTTACCTTGTTACGACTT) [34]. The taxonomic assignment of sequences was checked on the RDP classifier of the Ribosomal Database Project v11.1 [35]. Closely related 16S rRNA sequences found in the database were aligned with the newly determined sequences and a phylogenetic tree was re-constructed according to the neighbor-joining method of the Phylip package [36]. The stability of the groupings was estimated by bootstrap analysis (100 replications). The identification of C. bolteae 90A7, previously misnamed C. clostridioforme 90A7, was corrected (see Results and discussion).
Whole genome sequencing
This sequencing project was part of the Human Microbiome U54 initiative of the Broad Institute (broadinstitute.org). For each genome, paired end libraries were generated using Illumina’s Phusion-based library kits following the manufacturer’s protocols (Illumina, Hayward, CA, USA). Samples were multiplexed and sequenced on Illumina GAIIx machines and base-called following the manufacturer’s protocols. Individual samples of paired 90 nt reads generated 76 to 185-fold coverage of the genomes of C. bolteae (~6.37 Mb) and 82 to 264-fold coverage of the genomes of C. clostridioforme (~5.75 Mb), respectively (Table 1). Sequences were de novo assembled, yielding 1 to 21 contigs for genomes of C. bolteae and 10 to 48 contigs for those of C. clostridioforme.
Accession to our set of genomic sequences of Clostridium spp in public databases
All the genomes sequenced are available on the Broad Institute website (http://www.broadinstitute.org/data-software-and-tools) (Genbank Bioprojects : PRJNA64845, PRJNA64847, PRJNA64849, PRJNA64851, PRJNA64853, PRJNA64857, PRJNA659, PRJNA64861, PRJNA64863, PRJNA64865, PRJNA64867, PRJNA64869). The genomes of C. beijerinckii NCIMB 8052 (Clostridium sensu stricto), C. saccharolyticum WM1, C. bolteae BAA613 were retrieved from the PATRIC website (Genbank acc number: CP000721, CP002109; Bioproject: PRJNA18165). The genomes of Clostridium butyricum 60E.3.1, Clostridium colicanis 209318 (Clostridium sensu stricto), Clostridium bolteae WAL-14578, C. clostridioforme CM201.1, Clostridium clostridioforme 2149FAA.1, C. clostridioforme WAL-7855 (Clostridium cluster XIVa) and, C. innocuum 6_1_30.1 (Clostridium cluster XVIII) were selected from the Broad Institute (Genbank Bioprojects: PRJNA64855, PRJNA64873, PRJNA46383, PRJNA64871, PRJNA46389, PRJNA46391, PRJNA39359, respectively).
Annotation of genome sequences and ortholog groups
Automated genome annotation was generated at the Broad Institute. Additional information was obtained through a proteome analysis pipeline including protein domains search by RPS-Blast with the NCBI Conserved Domain Database [37] and, manual annotation. To improve annotation of the genomes, we determined ortholog groups accross our dataset by using the single-linkage algorithm of BlastClust (NCBI Blast package) at a high stringency level (S90-L0.9). If necessary, a protein sequence was (re)annotated with the ortholog group whose average Blast similarity was highest. In addition, CDSs named according to their position on contigs (ordered locus number) allowed to identify conserved synteny between genomes. Annotation of clusters was also conducted by homology search against the Clusters of Orthologous Groups (COG) protein database [38].
Detection of putative resistance genes
Antibiotic resistance genes were searched for by using BLASTp at various stringency levels against the Antibiotic Resistance Genes Database (ARDB) on a local machine [39]. Manual sorting of similarities and positive matches of the predicted protein sequences permitted to select manually the closely related AR proteins, and to assign functional annotation.
Putative extrachromosomal elements (e.g., ICEs or Transposons) were detected in the genomes of C. clostridioforme and C. bolteae by using BLASTp through the ICEberg database [40] upgraded with a personal collection of mobile elements from Firmicutes. A visual inspection of microsynteny (small scale of synteny) was also performed in regions surrounding resistance genes in order to detect mobile elements (e.g. transposase encoding genes) or their traces.
Acknowledgements
The authors thank Patrice Courvalin for the opportunity to sequence the 12 genomes of C. bolteae and C. clostridioforme at the Broad Institute, for helpful discussions and for comments regarding this manuscript. The authors thank all collaborators involved in the sequencing project. This research work has been funded in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No.:HHSN272200900018C and Grant U54HG0049 to the Broad Institute.
Availability of data and materials
Supporting files included within the article and its additional files contain all the information required to reproduce our results.
Authors’ contributions
PD performed the sequence analyses and helped to draft the manuscript. TL and JCM performed laboratory experiments. TL, JCM participated in the design of this study and helped to draft the manuscript. AA performed specialized analysis of whole genome assemblies. AME participated in the design of the study and oversaw sequencing and primary analysis of genomic data. All authors read and approved the final manuscript. CD conceived of the strategy of data analysis, participated in analysis and in coordination and drafted the manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Ethical approval was not required to use any of the isolates in this study. This analysis utilized open access data, exclusively.
Abbreviations
- ARDB
Antibiotic resistance gene database
- CDS
Coding DNA Sequence
- COG
Clusters of orthologous groups
- MIC
Minimum inhibitory concentration
- NGS
Next generation sequencing
Additional files
Lactose operons of C.bolteae and C. clostridioforme. (XLSX 10 kb)
Overview of the three flagellar loci found in C. bolteae. (XLSX 14 kb)
Overview of the flagellar locus found in C. clostridioforme. (XLSX 13 kb)
Metabolic pathways for butyrate. (XLSX 11 kb)
{kind=link}
Distance based phylogenetic tree of genes coding for butyrate kinase (buk). (PNG 721 kb)
{kind=link}
Schematic view of operons coding for glycopeptides antibiotic resistance. Canonical organisation and CDSs present in genomes of C. bolteae and C. clostridioforme were shown. Percent sequence identity and percent positive substitutions with the nearest homolog of each CDS were indicated. (PNG 409 kb)
{kind=link}
Distribution of genes coding for efflux pumps (potentially involved in antibiotic resistance) in the core genomes of the two species. (PNG 562 kb)
{kind=link}
Distance based phylogenetic tree of genes coding for rifampin resistance (Arr). (PNG 995 kb)
Contributor Information
Pierre Dehoux, Email: pdehoux@gmail.com.
Jean Christophe Marvaud, Email: jean-christophe.marvaud@u-psud.fr.
Amr Abouelleil, Email: amr@broadinstitute.org.
Ashlee M. Earl, Email: aearl@broadinstitute.org
Thierry Lambert, Email: thierry.lambert@u-psud.fr.
Catherine Dauga, Email: catherine.dauga@pasteur.fr.
References
- 1.Finegold SM, Molitoris D, Song Y, Liu C, Vaisanen ML, Bolte E, Mc Teague M, Sandler R, Wexler H, Marlowe EM, Collins MD, Lawson PA, Summanen P, Baysallar M, Tomzynski TJ, Read E, Johnson E, Rolfe R, Nasir P, Shah H, Haake DA, Manning P, Kaul A. Gastrointestinal microflora studies in late-onset autism. Clin Infect Dis. 2002;35(Suppl 1):S6–S16. doi: 10.1086/341914. [DOI] [PubMed] [Google Scholar]
- 2.Pequegnat B, Sagermann M, Valliani M, Toh M, Chow H, Allen-Vercoe E, Monteiro MA. A vaccine and diagnostic target for Clostridium bolteae, an autism-associated bacterium. Vaccine. 2013;31(26):2787–90. doi: 10.1016/j.vaccine.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 3.Martin Williams O, Brazier J, Peraino V, Goldstein EJC. A review of three cases of Clostridium aldenense bacteremia. Anaerobe. 2010;16(5):475–7. doi: 10.1016/j.anaerobe.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 4.Alexander CJ, Citron DM, Brazier JS, Goldstein EJ. Identification and antimicrobial resistance patterns of clinical isolates of Clostridium clostridioforme, Clostridium innocuum, and Clostridium ramosum compared with those of clinical isolates of Clostridium perfringens. J Clin Microbiol. 1995;33(12):3209–15. doi: 10.1128/jcm.33.12.3209-3215.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finegold SM, Song Y, Liu C, Hecht DW, Summanen P, Kononen E, Allen SD. Clostridium clostridioforme: a mixture of three clinically important species. Eur J Clin Microbiol Infect Dis. 2005;24:319–24. doi: 10.1007/s10096-005-1334-6. [DOI] [PubMed] [Google Scholar]
- 6.Warren YA, Tyrrell KL, Citron DM, Goldstein EJC. Clostridium aldenense sp nov and Clostridium citroniae sp nov Isolated from Human Clinical Infections. J Clin Microbiol. 2006;44(7):2416–22. doi: 10.1128/JCM.00116-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song Y, Liu C, Molitoris DR, Tomzynski TJ, Lawson PA, Collins MD, Finegold SM. Clostridium bolteae sp. nov., isolated from human sources system. Appl Microbiol. 2003;26:84–9. doi: 10.1078/072320203322337353. [DOI] [PubMed] [Google Scholar]
- 8.Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, Cai J, Hippe H, Farrow JA. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol. 1994;44(4):812–26. doi: 10.1099/00207713-44-4-812. [DOI] [PubMed] [Google Scholar]
- 9.Marshall BM, Ochieng DJ, Levy SB. Commensals: underappreciated reservoir of antibiotic resistance. Microbe. 2009;4(5):231–8. [Google Scholar]
- 10.Yu Y, Tangney M, Aass HC, Mitchell WJ. Analysis of the mechanism and regulation of lactose transport and metabolism in Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol. 2007;73:1842. doi: 10.1128/AEM.02082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aleksandrzak-Piekarczyk T, Kok J, Renault P, Bardowski J. Alternative lactose catabolic pathway in Lactococcus lactis IL1403. Appl Environ Microbiol. 2005;71(10):6060–9. doi: 10.1128/AEM.71.10.6060-6069.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christof Francke C, Robert Kerkhoven R, Michiel Wels M, Roland J, Siezen RJ. A generic approach to identify Transcription Factor-specific operator motifs; Inferences for LacI-family mediated regulation in Lactobacillus plantarum WCFS1. BMC Genomics. 2008;9(1):145. doi: 10.1186/1471-2164-9-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neville BA, Sheridan PO, Harris HMB, Coughlan S, Flint HJ, Duncan SH, Jeffery IB, Claesson MJ, Ross RP, Scott KP, O’Toole PW. Pro-inflammatory flagellin proteins of prevalent motile commensal bacteria are variably abundant in the intestinal microbiome of elderly humans. PLoS One. 2013;8(7):e68919. doi: 10.1371/journal.pone.0068919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barketi-Klai A, Monot M, Hoys S, Lambert-Bordes S, Kuehne SA, Minton N, Collignon A, Dupuy B, Kansau I. The flagellin FliC of Clostridium difficile is responsible for pleiotropic gene regulation during in vivo infection. PLoS One. 2014;9(5):e96876. doi: 10.1371/journal.pone.0096876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294(1):1–8. doi: 10.1111/j.1574-6968.2009.01514.x. [DOI] [PubMed] [Google Scholar]
- 16.Vital M, Howe AC, Tiedje JM. Revealing the bacterial butyrate synthesis pathways by analyzing (Meta)genomic data. MBio. 2014;5(2):e00889-14. doi: 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van den Abbeele P, Belzer C, Goossens M, Kleerebezem M, De Vos WM, Thas O, De Weirdt R, Kerckhof FM, Van de Wiele T. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. ISME J. 2013;7:949–61. doi: 10.1038/ismej.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vital M, Penton CR, Wang Q, Young VB, Antonopoulos DA, Sogin ML, Morrison HG, Raffals L, Chang EB, Huffnagle GB, Schmidt TM, Cole JR, Tiedje JM. A gene-targeted approach to investigate the intestinal butyrate-producing bacterial community. Microbiome. 2013;1(1):1–14. doi: 10.1186/2049-2618-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marvaud JC, Mory F, Lambert T. Clostridium clostridioforme and Atopobium minutum Clinical Isolates with VanB-Type Resistance in France. J Clin Microbiol. 2011;49(9):3436. doi: 10.1128/JCM.00308-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fajardo A, Martínez-Martín N, Mercadillo M, Galán JC, Ghysels B, Matthijs S, Cornelis P, Wiehlmann L, Tümmler B, Baquero F, Martínez JL. The neglected intrinsic resistome of bacterial pathogens. PLoS One. 2008;3(2):e1619. doi: 10.1371/journal.pone.0001619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Nicholas R, Thomson NR, Roberts AP, Cerdeño-Tárraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–86. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 22.Domingo MC, Huletsky A, Giroux R, Picard FJ, Bergeron MG. vanD and vanG-Like Gene Clusters in a Ruminococcus Species Isolated from Human Bowel Flora. Antimicrob Agents Chemother. 2007;51(11):4111–17. doi: 10.1128/AAC.00584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lozano C, Aspiroz C, Sáenz Y, Ruiz-García M, Royo-García G, Gómez-Sanz E, Ruiz-Larrea F, Zarazaga M, Torres C. Genetic environment and location of the lnu(A) and lnu(B) genes in methicillin-resistant Staphylococcus aureus and other staphylococci of animal and human origin. J Antimicrob Chemother. 2012;67(12):2804–8. doi: 10.1093/jac/dks320. [DOI] [PubMed] [Google Scholar]
- 24.Butaye P, Cloeckaert A, Schwarz S. Mobile genes coding for efflux-mediated antimicrobial resistance in Gram-positive and Gram-negative bacteria. Int J Antimicrob Agents. 2003;22(3):205–10. doi: 10.1016/S0924-8579(03)00202-4. [DOI] [PubMed] [Google Scholar]
- 25.Sugantino M, Roderick SL. Crystal structure of Vat(D): an acetyltransferase that inactivates streptogramin group A antibiotics. Biochemistry. 2002;41(7):2209–16. doi: 10.1021/bi011991b. [DOI] [PubMed] [Google Scholar]
- 26.Finegold SM, St John S, Vu AW, Li CM, Molitoris D, Song Y, Liu C, Wexler HM. In vitro activity of ramoplanin and comparator drugs against anaerobic intestinal bacteria from the perspective of potential utility in pathology involving bowel flora. Anaerobe. 2004;10(4):205–11. doi: 10.1016/j.anaerobe.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Löfmark S, Fang H, Hedberg M, Edlund C. Inducible metronidazole resistance and nim genes in clinical Bacteroides fragilis group isolates. Antimicrob Agents Chemother. 2005;49(3):1253–6. doi: 10.1128/AAC.49.3.1253-1256.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10(9):R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amaral L, Martins A, Spengler G, Molnar J. Efflux pumps of Gram-negative bacteria: what they do, how they do it, with what and how to deal with them. Front Pharmacol. 2014;4:1–11. doi: 10.3389/fphar.2013.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kehrenberg C, Schwarz S. dfrA20, A novel trimethoprim resistance gene from Pasteurella multocida. Antimicrob Agents Chemother. 2005;49(1):414–7. doi: 10.1128/AAC.49.1.414-417.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szakacs TA, Kalan L, Mcconnell MJ, Eshaghi A, Shahinas D, Mcgeer A, Wright GD, Low DE, Patel SN. Outbreak of vancomycin-susceptible enterococcus faecium containing the wild-type vanA gene. J Clin Microbiol. 2014;52(5):1682–86. doi: 10.1128/JCM.03563-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deutscher J, Saier MHJ. Ser/Thr/Tyr protein phosphrylation in bacteria – for long time neglected, now well established. J Mol Microbio Biotechnol. 2005;9:125–31. doi: 10.1159/000089641. [DOI] [PubMed] [Google Scholar]
- 33.de Vries LE, Valles Y, Agersø Y, Vaishampayan PA, Garcıa-Montaner A, Kuehl JV, Christensen H, Barlow M, Pilar Francino M. The gut as reservoir of antibiotic resistance: microbial diversity of tetracycline resistance in mother and infant. PLoS One. 2011;6(6):e21644. doi: 10.1371/journal.pone.0021644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner S, Pryer KM, Miao VPW, Palmer JD. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 1999;46:327–38. doi: 10.1111/j.1550-7408.1999.tb04612.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian Classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–67. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felsenstein J. PHYLIP - phylogeny inference package (version 3.2) Cladistics. 1989;5:164–6. [Google Scholar]
- 37.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41(Database issue):D348–52. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–7. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 39.Liu B, Pop M. ARDB--antibiotic resistance genes database. Nucleic Acids Res. 2009;37(Database issue):D443–D447. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bi D, Xu Z, Harrison EM, Tai C, Wei Y, He X, Jia S, Deng Z, Rajakumar K, Ou HY. ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res. 2012;40(Database issue):D621–626. doi: 10.1093/nar/gkr846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Supporting files included within the article and its additional files contain all the information required to reproduce our results.