

Abstract
A controlled balance between cell proliferation and differentiation is essential to maintain normal intestinal tissue renewal and physiology. Such regulation is powered by several intracellular pathways that are translated into the establishment of specific transcription programs, which influence intestinal cell fate along the crypt‐villus axis. One important check‐point in this process occurs in the transit amplifying zone of the intestinal crypts where different signaling pathways and transcription factors cooperate to manage cellular proliferation and differentiation, before secretory or absorptive cell lineage terminal differentiation. However, the importance of epigenetic modifications such as histone methylation and acetylation in the regulation of these processes is still incompletely understood. There have been recent advances in identifying the impact of histone modifications and chromatin remodelers on the proliferation and differentiation of normal intestinal crypt cells. In this review we discuss recent discoveries on the role of the cellular epigenome in intestinal cell fate, development, and tissue renewal. J. Cell. Physiol. 231: 2361–2367, 2016. © 2016 The Authors. Journal of Cellular Physiology Published by Wiley Periodicals, Inc.
One of the most rapidly renewing tissues in the human body is the epithelial lining of the intestine (Vermeulen and Snippert, 2014). Taking place along the crypt‐villus axis in the small intestine, this renewing process is characterized by a rapid and continuous proliferation in the crypt and general migration toward the tip of the villus where cells are released into the lumen (Bjerknes and Cheng, 2005; Crosnier et al., 2006; Scoville et al., 2008). The renewing process which maintains the dynamics of this system has been the subject of many seminal reviews (Cheng and Leblond, 1974; Barker et al., 2008; Potten et al., 2009; Li and Clevers, 2010; Shaker and Rubin, 2010). The stem cells which reside in the lower crypt supply the rapidly dividing progenitors that expand in the middle region of the crypt, referred to as the transit‐amplifying (TA) zone. Upon reaching the upper part of the crypt, corresponding to the terminal differentiation (TD) compartment, proliferating cells exit mitosis and acquire fully functional properties before reaching the base of the villus (Fig. 1).
Figure 1.
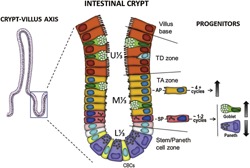
Human intestinal crypt architecture. The human intestinal crypt is subdivided into lower, middle, and upper thirds (L⅓, M⅓, U⅓) corresponding to the stem/Paneth cell compartment, the transit‐amplifying (TA) and terminal differentiation (TD) zones, respectively. TA undifferentiated progenitors arising from intestinal stem cell division undergo multiple rounds of mitosis prior to executing their differentiation program. Within the TA zone, absorptive progenitors (AP) divide approximately four times while secretory lineage progenitors (SP) will undergo one to two cycles before differentiating. APs, as well as goblet and enteroendocrine‐specific SPs are characterized by an upward migratory process in the crypt‐villus axis whereas Paneth‐determined SPs migrate downward.
It is noteworthy that several key events take place in the TA zone. Cell lineage specification to either secretory precursor (SP) cells that give rise to goblet, enteroendocrine, and Paneth cells or absorptive precursor (AP) cells occurs at the time of entry into the TA zone, under the control of the Notch pathway (Vooijs et al., 2011). Interestingly, a cell differentiation process is also ongoing in the TA zone as illustrated by the occurrence of relatively well‐differentiated cells of the SP lineages such as the goblet cells. Paneth cells are not seen here because, in contrast to other precursor cells, they migrate downward to complete their differentiation at the bottom of the crypts (Bjerknes and Cheng, 1981; van der Flier and Clevers, 2009). Intriguingly, AP cells only express a limited subset of differentiation markers in the TA zone while their full maturation occurs in the TD compartment in the human (Beaulieu, 1997; Benoit et al., 2012). In rodents, absorptive cell differentiation in the TA zone is even more obvious (Traber, 1999). Consistent with this phenomenon, it is noteworthy that AP cells undergo approximately four division cycles before undertaking their terminal differentiation program whilst SP cells divide only once or twice (Bjerknes and Cheng, 1999; Bjerknes and Cheng, 2005), explaining also why the majority of the cells on the villi are absorptive cells (Fig. 1).
However, key transcription factors involved in AP differentiation, such as CDX2, HNF1α, and GATA4 are expressed by the epithelial cells of the TA zone (Benoit et al., 2010). A question that needs to be addressed is what prevents spontaneous AP terminal differentiation in the presence of these factors. Some research groups have proposed the involvement of suppressive epigenetic mechanisms such as histone methylation/deacetylation for cell differentiation during development (Tou et al., 2004; Boyer et al., 2006). Indeed, epigenetic mechanisms have important implications in the transcriptional programming and regulation of embryonic and cancer stem cells (Munoz et al., 2012; Suva et al., 2013). Changes in the balance between different histone markers can lead to deregulation of gene transcription and can be related to cellular transformation and different cancers such as colorectal cancer (Fraga et al., 2005; Seligson et al., 2005; Mariadason, 2008; Hammoud et al., 2013).
In this review, we analyse recent advances in the detailed characterization of the epigenetic control of different aspects of intestinal cell physiology in general with a specific emphasis on the regulation of cell proliferation and differentiation in the intestinal TA zone. These mechanisms must be precisely tuned to establish proper tissue renewal and organism maintenance.
Epigenetics and Histone Modifications
Epigenetics is described as inherited alterations in gene expression or silencing that take place without changes in DNA sequence (Jaenisch and Bird, 2003; Bird, 2007). Most epigenetic studies are focused on the characterization of covalent and non‐covalent modifications of DNA and histones and the mechanisms by which such modifications influence chromatin architecture and subsequent gene expression (Goldberg et al., 2007). Thus, we sought to review the critical role of the Histone code in the context of intestinal physiology. The Histone code is defined by post‐translational modifications, such as acetylation and methylation of key residues of core histone tails, which can synergize or antagonize each other and influence chromatin accessibility (active vs. silenced) and transcription during cell fate determination and tissue renewal (Jenuwein and Allis, 2001) (Fig. 2). It is known that core histone N‐terminal chains can impact transcriptional activity via different types of multivalent modifications, including methylation and acetylation, but also ubiquitination, phosphorylation, and sumoylation (Kouzarides, 2007; Ruthenburg et al., 2007). In addition to their role as transcriptional regulators (Koch et al., 2007; Pandian et al., 2014), histone modifications can influence other DNA‐related processes, including DNA replication, double‐strand break reparation, and retrotransposable element silencing (Groth et al., 2007; Kinner et al., 2008; Di Giacomo et al., 2014).
Figure 2.
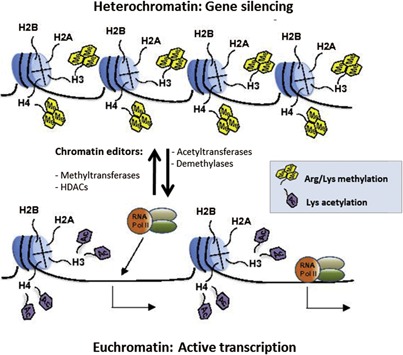
Histone tail modifications dictate chromatin organization and transcriptional activity. Methylation/acetylation of histone tails is one of the most important functional characteristics of euchromatin and heterochromatin patterning in the nucleus. Mono, di, and trimethylation of specific histone lysine and arginine residues are mostly associated with a tightly compacted and transcriptionally repressed form of chromatin called heterochromatin. Histone lysine acetylation however confers to DNA a relaxed and accessible (RNA Pol II, transcription factors) conformation called euchromatin. The concerted action of histone deacetylases (HDACs) and methyltransferases is generally responsible for heterochromatin domain organization, while the activity of histone acetyltransferases and demethylases is associated with transcriptionally active euchromatin regions.
Methylation and acetylation of histone lysine tails constitute the most important post‐translational modifications occurring in heterochromatin and euchromatin organization (Fig. 2) (Grunstein, 1997; Jaenisch and Bird, 2003; Kim and Kim, 2012). Due to their compact assembly, heterochromatin structures limit the access of transcriptional machinery to the more packaged DNA molecules which results in gene silencing (Li and Zhang, 2012). In contrast, euchromatin conformation is associated with the recruitment of transcription factors and RNA polymerase, resulting in gene expression (Fig. 2) (Kouzarides, 2007). Deregulation of the physiological balance between the formation of euchromatin and heterochromatin structures can result in different malignancies such as colorectal cancer (Chen et al., 2010). For example, histone H3 lysine‐27 trimethylation (H3K27me3), H4K16 methylation, or H4K20 deacetylation have all been associated with transcriptional aberrations observed in multiple human cancers, including colorectal tumors (Fraga et al., 2005; Hammoud et al., 2013). Interestingly, pharmacological strategies targeting key chromatin editors, including histone methyltransferases and histone deacetylases (HDAC) have recently been proposed to fix aberrant cell growth and survival in these human neoplasms (Benoit et al., 2013a,2013b; Carson et al., 2015).
Although epigenetic modifications have been widely studied in different biological contexts, there is still a lack of comprehensive understanding of the role played by epigenetics and chromatin remodeling on critical gene expression patterns regulating differentiation and proliferation of intestinal progenitors. So far, our group and others have highlighted that a tightly regulated cooperation between chromatin marks, such as histone methylation and acetylation have indispensable roles in the control of proliferation and differentiation of absorptive cells in the TA zone of the intestinal crypt (Suzuki et al., 2008; Benoit et al., 2012; Roostaee et al., 2015). In the next sections, we will focus on recent discoveries on the fundamental roles of chromatin regulation in human intestinal tissue homeostasis.
Histone Methylation and the Role of the PcG Complex in Intestinal Crypt Cells
Methylation of specific lysines or arginines on histone tails creates a platform for transcriptional repression or activation depending on the exact position of the lysine residue in the histone protein. For example, methylation of histone H3K4, H3K36, and H3K79 gives rise to a transcriptionally active region of chromatin while H3K9, H3K27, and H4K20 methylation are usually associated with gene silencing (Sims et al., 2003). The core catalytic subunit which controls histone lysine methylation is the SET domain, the active functional unit of many protein methyltransferases responsible for the addition of methyl groups to histones (Greer and Shi, 2012). One important example of SET‐containing proteins is the polycomb group (PcG) proteins which are a protein family implicated in transcriptional repression during tissue formation and development (Lewis, 1978). PRC1 and PRC2 are two distinct “PcG Repressive Complexes.” The core PRC2 ultra‐structure contains the methyltransferase unit Enhancer of Zeste homolog 2 (EZH2), the stabilization protein Suppressor of Zeste‐12 (SUZ12) (Margueron and Reinberg, 2011), as well as EED which is acting as a bridge between PRC2 and PRC1 (Cao et al., 2014). PcGs have been known to maintain pluripotence in embryonic stem cells (Boyer et al., 2006; Lee et al., 2006) to control somatic cell differentiation (Caretti et al., 2004; Ezhkova et al., 2009; Benoit et al., 2012) and contribute to cancer development (Piunti and Pasini, 2011). PcGs play a pivotal role in intestinal tissue development by controlling cell fate determination of absorptive cells, which are the predominant cell type in the intestinal epithelium with the important function of absorbing nutrients.
Specifically, recent characterization of the role of PRC2 in intestinal cells (Benoit et al., 2012) disclosed an important key concept in the regulation of intestinal cell differentiation. As aforementioned, the SUZ12 subunit is essential for proper functioning of the PRC2 complex which consists of the addition of three methyl groups to lysine 27 of histone H3 (H3K27) at target loci, leading to a transcriptionally silent chromatin state (Cao et al., 2002; Martinez‐Garcia and Licht, 2010; Margueron and Reinberg, 2011). In the TA zone of the intestinal crypt compartment, PcG proteins catalyse trimethylation of histone H3 (H3K27me3) and this process coincides with the repression of terminal differentiation genes such as sucrase‐isomaltase (SI), a specific marker of mature absorptive cells (Benoit et al., 2012). Incidentally, expression of the SI protein was found to be inversely correlated with the presence of SUZ12, supporting the inhibitory influence of PcGs on cell differentiation in the TA zone (Benoit et al., 2012). The possibility that the activity of PcG proteins could allow TA cells to preserve their proliferative state while repressing the expression of intestine‐specific differentiation genes was then tested using human cell models. Abolition of PRC2 activity in colon carcinoma (Caco‐2/15) and human normal non‐immortalized intestinal crypt (HIEC) cells resulted in the acceleration of absorptive cell terminal differentiation, manifested by a robust expression of the SI protein (Benoit et al., 2012). In these cell models, the PRC2‐catalyzed H3K27me3 mark (Martinez‐Garcia and Licht, 2010; Margueron and Reinberg, 2011) was significantly down‐regulated, confirming the essential implication of PcGs and histone methylation in the regulation of absorptive cell‐specific gene expression. Molecular mechanisms that control epigenetic modifications such as H3K27me3 are largely unknown. Our laboratory has recently found that inhibition of Src family kinases (SFKs) is associated with a decrease of H3K27me3 mark as well as a significant increase in intestinal cell differentiation markers. These data suggest that SFKs, which are negative regulators of the differentiation of intestinal cells, could act upstream of the PRC2 complex (Seltana et al., 2013). A detailed characterization of these SFK regulatory mechanisms needs further investigation. Thus, the elevated expression of PcGs in the TA zone of the crypt‐villus axis, as well as their impact on cell state‐specific gene expression provides compelling evidence on the role of PcG proteins in the maintenance of cellular proliferation and suppression of differentiation.
The PcG proteins are best known for their repressive action on the differentiation of various tissue‐specific stem cells such as mesenchymal stem cells, hematopoietic stem cells and skeletal muscle stem cells (Chen et al., 2012). Deregulation of PcG gene expression has also been reported in many cancers including colon cancer (Sauvageau and Sauvageau, 2010; Benoit et al., 2012). In human colon adenocarcinoma, an up‐regulation of SUZ12 expression and distribution throughout the tissue has been observed, confirming the implication of PcGs in the preservation of cancer progression (Benoit et al., 2012). Interestingly, pharmacological inhibition of the catalytic subunit of PRC2 (EZH2) has shown impaired growth and survival properties in human colorectal cancer cell lines and patient‐derived colon cancer stem cells (Benoit et al., 2013a,2013b). In each case, a knockdown of SUZ12 was sufficient to phenocopy the inhibition of EZH2, further supporting a critical involvement of the PRC2 in colorectal tumorigenesis.
Histone Acetylation and Differentiation of Intestinal Crypt Cells
Histone acetylation is another set of epigenetic mechanisms able to regulate cell proliferation and differentiation in the intestinal TA zone. Indeed, acetylation of histone tails counterbalances the histone net charge in order to induce a more relaxed chromatin conformation facilitating access of transcriptional machinery to the DNA promoter and resulting in de‐repression of gene expression associated with cellular differentiation (Fig. 2) (Marks et al., 2000). However, histone deacetylases (HDACs) counter this effect by removing acetyl groups from lysine residues within DNA bound core histones (Haberland et al., 2009). Histone deacetylation is a fundamental phenomenon that increases DNA interactions with histones, and as a result, down‐regulates gene expression by inhibiting the recruitment of RNA polymerase, transcription factors and cofactors at promoter regions and other regulatory elements (Fig. 2) (Marks et al., 2000; Glozak and Seto, 2007). Human HDACs are grouped into 4 classes: HDACI, HDACII, HDACIII, and HDACIV based on their sequence homology to the original yeast enzymes and domain organization. Class I includes HDAC1, −2, −3, −8, class II includes HDAC4, −5, −7, and −9, while HDAC6 and −10 constitute class III and HDAC11 is known as class IV (Marks, 2007; Witt et al., 2009). Out of the four HDAC classes, classes I and II have been show to play pivotal roles in the maintenance of normal intestinal physiology and are over‐expressed in colon tumors (Mariadason, 2008). In 2008, Suzuki et al. (2008) demonstrated that during differentiation of absorptive cells, the promoter of the sucrase‐isomaltase (SI) gene, a hallmark of terminal differentiation of absorptive cells (Beaulieu, 1997), was highly acetylated (Suzuki et al., 2008). Previous studies have tested HDAC inhibitors (HDACi) to directly investigate the role of histone acetylation in the regulation of the physiology of intestinal cells. For example, the HDACi sodium butyrate has been used to assess differentiation and proliferation of Caco‐2 cells (Mariadason et al., 2000, 2001). Although sodium butyrate has been shown to induce expression of some absorptive cell differentiation markers such as DPP4 and alkaline phosphatase, it does not up‐regulate SI expression (Mariadason et al., 2000). Newly confluent Caco‐2/15 cells represent a relevant model of absorptive cell progenitors in their last dividing cycles in the TA zone since their proliferation rate diminishes while a differentiation process begins (Beaulieu and Quaroni, 1991; Vachon and Beaulieu, 1992). Treatment of these cells with HDACi suberanilohydroxamic acid (SAHA) which suppresses the activity of both class I and II HDACs resulted in a dramatic up‐regulation of histone acetylation and SI expression at both mRNA and protein levels (Roostaee et al., 2015). In addition, in newly confluent Caco‐2/15 cells incubated with SAHA, the colonic enterocyte maturation marker SLC26A3 (Down Regulated in Adenoma, SLC26A3/DRA) was significantly up‐regulated. If histone acetylation accelerates intestinal cell maturation, HDAC inhibition should result in an early expression of intestine‐specific markers in the crypts as shown previously in mouse fetal gut explants (Tou et al., 2004). To test this possibility, mice were treated with SAHA and the expression of differentiation markers in intestinal crypts was investigated. The results showed that overall expression of SI and SLC26A3 was significantly enhanced while the SI and SLC26A3 proteins were detected in the upper part of ileal (Fig. 3A,B) and colonic crypts, respectively, in mice receiving SAHA, suggesting that histone acetylation participates in the repression of absorptive cell differentiation in the TA zone (Roostaee et al., 2015).
Figure 3.
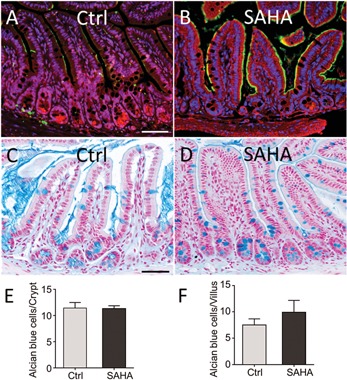
Selective effects of histone acetylation on the differentiation of intestinal cell progenitors. (A and B) Representative immunofluorescence illustrations showing that histone acetylation promotes expression of enterocyte specific differentiation protein SI (green) in the mouse ileum treated with SAHA (B) compared to the control (Ctrl) mouse (A). Blue (nuclei), Red (Evans blue). (C–F) SAHA treatment has no significant effect on the differentiation of goblet cells. (C and D) Alcian blue staining of mouse ileum for the detection of goblet cells in the Ctrl mice treated with DMSO (C) or SAHA (D). (E and F) Counting of goblet cells in the intestinal crypts (E) and villi (F) demonstrates that there is no significant difference in goblet cell number between mice treated with SAHA or control mice. Blue (goblet cells) and red (nuclei). Scale bar: 100 µm.
The use of genetically modified mouse models has also contributed to our understanding of the involvement of HDAC in intestinal homeostasis. Ablation of Hdac1 and Hdac2 in the mouse intestinal epithelium, early in development, was reported to promote expression of absorptive cell differentiation markers such as SI, intestinal alkaline phosphatase and fatty acid binding protein but this phenomenon was accompanied by a loss of differentiation of secretory lineages and a stimulation of cell proliferation (Turgeon et al., 2013). However, some of these observations were attributed to the only partial deletion of the target genes since a complete and simultaneous deletion of Hdac1 and Hdac2 in the adult intestine leads to a rapid loss of proliferative crypt cells in vivo and in intestinal organoid cultures (Gonneaud et al., 2015; Zimberlin et al., 2015), a phenomenon also observed in Caco‐2/15 cells and in the intestine of mice treated with the HDACi SAHA (Roostaee et al., 2015). Consistently, enhanced histone acetylation has been related to elevated p21WAF1 cellular levels, resulting in cell cycle arrest and apoptosis (Xiong et al., 1993; Chopin et al., 2004). Moreover, evaluation of goblet and Paneth cell number in the intestine of mice treated with SAHA suggests that HDAC inhibition does not influence SP cell differentiation (Fig. 3C–F). Further studies in murine models knocked down for either Hdac1 or Hdac2 as well as those expressing low levels of Hdac1 and in the absence of Hdac2 and vice versa disclosed the existence of dosage‐dependent specific and similar effects on intestinal epithelial cells (Gonneaud et al., 2015; Zimberlin et al., 2015). Further work is thus needed to more precisely assess the respective roles of these HDACs. The presence of other HDACs in intestinal epithelial cells that may participate to the phenomenon should also be considered in future studies. For instance, HDAC3 and HDAC4 which were both characterized as repressors of p21WAF1 expression in colorectal cancer cell lines have been reported to be predominantly expressed in the proliferative cells of the crypt in the normal intestine (Wilson et al., 2008, 2006).
Histone Methylation/Acetylation Transition in the Intestinal Crypt TA Zone
The data reported above indicate a role for both PcG proteins and HDACs for promoting cell proliferation and repressing absorptive cell differentiation in the TA zone of the intestinal crypt. Transition of cells from the proliferative to the terminal differentiation state requires global changes in histone modifications which influence the expression of multiple gene targets (Glozak and Seto, 2007). One example for such modifications is the inter‐conversion of histone methylation and acetylation. This process involves removal of methyl groups of H3K27me3 and subsequent acetylation of H3K27 to form an acetylated histone H3K27 (H3K27ac) complex (Ong and Corces, 2011). In this context, the study of Suzuki et al. (2008) showing that histone H3 modifications regulate SI expression is interesting. Indeed, di‐/tri‐methylation levels of histone H3 at lysine 9 on the promoter region of the SI gene were found to be associated with low SI expression in the crypt while its change to di‐acetylation at lysine 9/14 was linked to the up‐regulation of SI expression at the crypt‐villus junction (Suzuki et al., 2008). The transition of specific histone tail residues from a methylated to an acetylated state to impact proliferation rate versus differentiation and apoptosis is not limited to the intestine. For instance, human acute myeloid leukemia cells treated with the DNA methyltransferase inhibitors 5‐azacitidine and decitabine showed exchanges of methyl groups for acetyl functions on histone H3 lysine 27 that were associated with interleukin 3 expression (Buchi et al., 2014). Furthermore, Fiskus et al. (2009) reported that DZNep and Panobinostat, histone methyltransferase and HDAC inhibitors respectively, acted synergistically to induce anti‐proliferative and pro‐apoptotic gene expression in human acute myeloid cells.
Conclusions and Proposed Model
Cooperation between transcription factors, signaling pathways and epigenetic mechanisms is essential for the tight control of the constant renewal of intestinal tissue. Based on the recent data discussed in the previous sections, we propose an updated model (Fig. 4) to illustrate major mechanisms involved in the regulation of proliferation and differentiation of absorptive intestinal cells in the crypt. On one hand, the pro‐differentiation factors CDX2, HNF1α, and GATA4, which are necessary for the induction of SI expression and the control of absorptive cell proliferation and differentiation, are expressed in all epithelial cells of the TA zone and above (Escaffit et al., 2006; Benoit et al., 2010). On the other hand, epigenetic mechanisms act upstream of these pro‐differentiation transcription factors to maintain cell proliferation and transiently prevent differentiation in the TA zone. Interestingly, abolition of PRC2 activity or HDACs has no significant effect on the expression of CDX2 or HNF1α (Benoit et al., 2012; Roostaee et al., 2015). Therefore, transcription factors and histone modifications under the regulation by HDAC and PcG proteins appear as two sets of counteracting mechanisms that exceptionally integrate in the TA zone to maintain rapid and proper renewal of the intestinal epithelium. However, these mechanisms are transient so that after a few rounds of cell division, stimulation of cell proliferation and transcriptional repression of cell differentiation are halted through a yet unknown mechanism causing histone demethylation and acetylation both occurring in the upper crypt and corresponding to absorptive cell terminal differentiation (Fig. 4). This model provides a more detailed understanding of the crucial regulatory role of epigenetic mechanisms in the TA zone, in proper and constant maintenance of intestinal tissue renewal. This information may also have important implications in the comprehension of gastrointestinal disorders such as colorectal cancer in which perturbations in epigenetic mechanisms have been observed (Hammoud et al., 2013).
Figure 4.
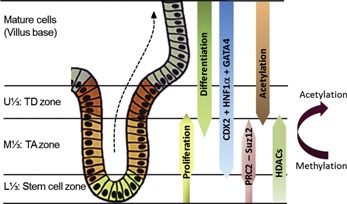
Integrative scheme of molecular mechanisms controlling intestinal tissue renewal. The intestinal crypt compartments are defined as the Stem cell, TA, and TD zones. The TA zone is composed of quickly dividing cells issued from the stem cell zone. For absorptive cells, experimental evidence suggests that the PRC2 complex and HDACs maintain cellular proliferation and restrain differentiation. Removal of methyl groups from H3K27me3 and acetylation of histones eliminates transcriptional repression, and the cell differentiation process starts (Benoit et al., 2012; Roostaee et al., 2015). Some studies have also reported a conversion of methylated‐to‐acetylated histones (Suzuki et al., 2008; Ong and Corces, 2011). Acetylation of histones is associated with robust expression of SI protein. The pro‐differentiation factors CDX2 and HNF1α are expressed by all epithelial cells including the cells residing in the TA zone and can up‐regulate expression of intestine‐specific genes such as SI.
Epigenetics is a fast‐growing field, which opens new avenues for fundamental research on the regulation of cellular proliferation, differentiation and subsequent generation of intestinal epithelial tissue under normal and diseased conditions. In this review, we discussed recent findings concerning epigenetic mechanisms that regulate intestinal cell fate in the TA zone. However, further research is still needed to explain how these epigenetic mechanisms are regulated at the cellular and molecular level under physiological and cancer conditions.
Acknowledgments
The authors thank Elizabeth Herring for reviewing the manuscript. This work was supported by Grant MOP 123415 from the Canadian Institute of Health Research (JFB). JFB is the recipient of the Canada Research Chair of Intestinal Physiopathology and a member of the Fonds de la recherche du Québec/Santé‐funded Centre de Recherche du Centre Hospitalier Universitaire de Sherbrooke.
Literature Cited
- Barker N, van de Wetering M, Clevers H. 2008. The intestinal stem cell. Genes Dev 22:1856–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JF. 1997. Extracellular matrix components and integrins in relationship to human intestinal epithelial cell differentiation. Prog Histochem Cytochem 31:1–78. [DOI] [PubMed] [Google Scholar]
- Beaulieu JF, Quaroni A. 1991. Clonal analysis of sucrase‐isomaltase expression in the human colon adenocarcinoma Caco‐2 cells. Biochem J 280:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit YD, Laursen KB, Witherspoon MS, Lipkin SM, Gudas LJ. 2013a. Inhibition of PRC2 histone methyltransferase activity increases TRAIL‐mediated apoptosis sensitivity in human colon cancer cells. J Cell Physiol 228:764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit YD, Witherspoon MS, Laursen KB, Guezguez A, Beausejour M, Beaulieu JF, Lipkin SM, Gudas LJ. 2013b. Pharmacological inhibition of polycomb repressive complex‐2 activity induces apoptosis in human colon cancer stem cells. Exp Cell Res 319:1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit YD, Lepage MB, Khalfaoui T, Tremblay E, Basora N, Carrier JC, Gudas LJ, Beaulieu JF. 2012. Polycomb repressive complex 2 impedes intestinal cell terminal differentiation. J Cell Sci 125:3454–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit YD, Pare F, Francoeur C, Jean D, Tremblay E, Boudreau F, Escaffit F, Beaulieu JF. 2010. Cooperation between HNF‐1alpha, Cdx2, and GATA‐4 in initiating an enterocytic differentiation program in a normal human intestinal epithelial progenitor cell line. Am J Physiol Gastrointest Liver Physiol 298:G504–G517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. 2007. Perceptions of epigenetics. Nature 447:396–398. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H. 1981. The stem‐cell zone of the small intestinal epithelium. I. Evidence from Paneth cells in the adult mouse. Am J Anat 160:51–63. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H. 1999. Clonal analysis of mouse intestinal epithelial progenitors. Gastroenterology 116:7–14. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, Cheng H. 2005. Gastrointestinal stem cells. II. Intestinal stem cells. Am J Physiol Gastrointest Liver Physiol 289:G381–G387. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. 2006. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441:349–353. [DOI] [PubMed] [Google Scholar]
- Buchi F, Masala E, Rossi A, Valencia A, Spinelli E, Sanna A, Gozzini A, Santini V. 2014. Redistribution of H3K27me3 and acetylated histone H4 upon exposure to azacitidine and decitabine results in de‐repression of the AML1/ETO target gene IL3. Epigenetics 9:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, Poliakov A, Yocum AK, Li Y, Chen W, Cao X, Jiang X, Dahiya A, Harris C, Feng FY, Kalantry S, Qin ZS, Dhanasekaran SM, Chinnaiyan AM. 2014. The central role of EED in the orchestration of polycomb group complexes. Nat Commun 5:3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument‐Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb‐group silencing. Science 298:1039–1043. [DOI] [PubMed] [Google Scholar]
- Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V. 2004. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 18:2627–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson R, Celtikci B, Fenning C, Javadi A, Crawford N, Perez‐Carbonell L, Lawler M, Longley DB, Johnston PG, Van Schaeybroeck S. 2015. HDAC inhibition overcomes acute resistance to MEK inhibition in BRAF‐mutant colorectal cancer by downregulation of c‐FLIPL. Clin Cancer Res 21:3230–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Zhang Y, Guo WH, Meng MB, Mo XM, Lu Y. 2010. Effects of heterochromatin in colorectal cancer stem cells on radiosensitivity. Chin J Cancer 29:270–276. [DOI] [PubMed] [Google Scholar]
- Chen YH, Hung MC, Li LY. 2012. EZH2: A pivotal regulator in controlling cell differentiation. Am J Transl Res 4:364–375. [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Leblond CP. 1974. Origin, differentiation and renewal of the four main epithelial cell types in the mouse small intestine. V. Unitarian Theory of the origin of the four epithelial cell types. Am J Anat 141:537–561. [DOI] [PubMed] [Google Scholar]
- Chopin V, Toillon RA, Jouy N, Le Bourhis X. 2004. P21(WAF1/CIP1) is dispensable for G1 arrest, but indispensable for apoptosis induced by sodium butyrate in MCF‐7 breast cancer cells. Oncogene 23:21–29. [DOI] [PubMed] [Google Scholar]
- Crosnier C, Stamataki D, Lewis J. 2006. Organizing cell renewal in the intestine: Stem cells, signals and combinatorial control. Nat Rev Genet 7:349–359. [DOI] [PubMed] [Google Scholar]
- Di Giacomo M, Comazzetto S, Sampath SC, Sampath SC, O'Carroll D. 2014. G9a co‐suppresses LINE1 elements in spermatogonia. Epigenetics Chromatin 7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escaffit F, Pare F, Gauthier R, Rivard N, Boudreau F, Beaulieu JF. 2006. Cdx2 modulates proliferation in normal human intestinal epithelial crypt cells. Biochem Biophys Res Commun 342:66–72. [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E. 2009. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue‐specific stem cells. Cell 136:1122–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, Balusu R, Koul S, Atadja P, Marquez VE, Bhalla KN. 2009. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3‐deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood 114:2733–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar‐Garea A, Boix‐Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez‐Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37:391–400. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Seto E. 2007. Histone deacetylases and cancer. Oncogene 26:5420–5432. [DOI] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. 2007. Epigenetics: A landscape takes shape. Cell 128:635–638. [DOI] [PubMed] [Google Scholar]
- Gonneaud A, Turgeon N, Boisvert FM, Boudreau F, Asselin C. 2015. Loss of histone deacetylase Hdac1 disrupts metabolic processes in intestinal epithelial cells. FEBS Lett 589:2776–2783. [DOI] [PubMed] [Google Scholar]
- Greer EL, Shi Y. 2012. Histone methylation: A dynamic mark in health, disease and inheritance. Nat Rev Genet 13:343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Rocha W, Verreault A, Almouzni G. 2007. Chromatin challenges during DNA replication and repair. Cell 128:721–733. [DOI] [PubMed] [Google Scholar]
- Grunstein M. 1997. Histone acetylation in chromatin structure and transcription. Nature 389:349–352. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. 2009. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet 10:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Cairns BR, Jones DA. 2013. Epigenetic regulation of colon cancer and intestinal stem cells. Curr Opin Cell Biol 25:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. 2003. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 33:245–254. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. 2001. Translating the histone code. Science 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim H. 2012. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J 53:232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinner A, Wu W, Staudt C, Iliakis G. 2008. Gamma‐H2AX in recognition and signaling of DNA double‐strand breaks in the context of chromatin. Nucleic Acids Res 36:5678–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, James KD, Lefebvre GC, Bruce AW, Dovey OM, Ellis PD, Dhami P, Langford CF, Weng Z, Birney E, Carter NP, Vetrie D, Dunham I. 2007. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res 17:691–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. 2007. Chromatin modifications and their function. Cell 128:693–705. [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, Koseki H, Fuchikami T, Abe K, Murray HL, Zucker JP, Yuan B, Bell GW, Herbolsheimer E, Hannett NM, Sun K, Odom DT, Otte AP, Volkert TL, Bartel DP, Melton DA, Gifford DK, Jaenisch R, Young RA. 2006. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125:301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB. 1978. A gene complex controlling segmentation in Drosophila. Nature 276:565–570. [DOI] [PubMed] [Google Scholar]
- Li L, Clevers H. 2010. Coexistence of quiescent and active adult stem cells in mammals. Science 327:542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhang Z. 2012. Linking DNA replication to heterochromatin silencing and epigenetic inheritance. Acta Biochim Biophys Sin (Shanghai) 44:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariadason JM. 2008. HDACs and HDAC inhibitors in colon cancer. Epigenetics 3:28–37. [DOI] [PubMed] [Google Scholar]
- Mariadason JM, Rickard KL, Barkla DH, Augenlicht LH, Gibson PR. 2000. Divergent phenotypic patterns and commitment to apoptosis of Caco‐2 cells during spontaneous and butyrate‐induced differentiation. J Cell Physiol 183:347–354. [DOI] [PubMed] [Google Scholar]
- Mariadason JM, Velcich A, Wilson AJ, Augenlicht LH, Gibson PR. 2001. Resistance to butyrate‐induced cell differentiation and apoptosis during spontaneous Caco‐2 cell differentiation. Gastroenterology 120:889–899. [DOI] [PubMed] [Google Scholar]
- Marks PA. 2007. Discovery and development of SAHA as an anticancer agent. Oncogene 26:1351–1356. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Rifkind RA. 2000. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst 92:1210–1216. [DOI] [PubMed] [Google Scholar]
- Martinez‐Garcia E, Licht JD. 2010. Deregulation of H3K27 methylation in cancer. Nat Genet 42:100–101. [DOI] [PubMed] [Google Scholar]
- Munoz P, Iliou MS, Esteller M. 2012. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol 6:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Corces VG. 2011. Enhancer function: New insights into the regulation of tissue‐specific gene expression. Nat Rev Genet 12:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandian GN, Taniguchi J, Junetha S, Sato S, Han L, Saha A, AnandhaKumar C, Bando T, Nagase H, Vaijayanthi T, Taylor RD, Sugiyama H. 2014. Distinct DNA‐based epigenetic switches trigger transcriptional activation of silent genes in human dermal fibroblasts. Sci Rep 4:3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Pasini D. 2011. Epigenetic factors in cancer development: Polycomb group proteins. Future Oncol 7:57–75. [DOI] [PubMed] [Google Scholar]
- Potten CS, Gandara R, Mahida YR, Loeffler M, Wright NA. 2009. The stem cells of small intestinal crypts: Where are they? Cell Prolif 42:731–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roostaee A, Guezguez A, Beausejour M, Simoneau A, Vachon PH, Levy E, Beaulieu JF. 2015. Histone deacetylase inhibition impairs normal intestinal cell proliferation and promotes specific gene expression. J Cell Biochem 116:2695–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. 2007. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 8:983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Sauvageau G. 2010. Polycomb group proteins: Multi‐faceted regulators of somatic stem cells and cancer. Cell Stem Cell 7:299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville DH, Sato T, He XC, Li L. 2008. Current view: Intestinal stem cells and signaling. Gastroenterology 134:849–864. [DOI] [PubMed] [Google Scholar]
- Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. 2005. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435:1262–1266. [DOI] [PubMed] [Google Scholar]
- Seltana A, Guezguez A, Lepage M, Basora N, Beaulieu JF. 2013. Src family kinase inhibitor PP2 accelerates differentiation in human intestinal epithelial cells. Biochem Biophys Res Commun 430:1195–1200. [DOI] [PubMed] [Google Scholar]
- Shaker A, Rubin DC. 2010. Intestinal stem cells and epithelial‐mesenchymal interactions in the crypt and stem cell niche. Transl Res 156:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, III , Nishioka K, Reinberg D. 2003. Histone lysine methylation: A signature for chromatin function. Trends Genet 19:629–639. [DOI] [PubMed] [Google Scholar]
- Suva ML, Riggi N, Bernstein BE. 2013. Epigenetic reprogramming in cancer. Science 339:1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Mochizuki K, Goda T. 2008. Histone H3 modifications and Cdx‐2 binding to the sucrase‐isomaltase (SI) gene is involved in induction of the gene in the transition from the crypt to villus in the small intestine of rats. Biochem Biophys Res Commun 369:788–793. [DOI] [PubMed] [Google Scholar]
- Tou L, Liu Q, Shivdasani RA. 2004. Regulation of mammalian epithelial differentiation and intestine development by class I histone deacetylases. Mol Cell Biol 24:3132–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traber PG. 1999. Carbohydrate assimilation In: Yamada T, editor. Textbook of gastroenterology. 3rd ed Philadelphia: J.B. Lippincott; pp 404–428. [Google Scholar]
- Turgeon N, Blais M, Gagne JM, Tardif V, Boudreau F, Perreault N, Asselin C. 2013. HDAC1 and HDAC2 restrain the intestinal inflammatory response by regulating intestinal epithelial cell differentiation. PLoS ONE 8:e73785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachon PH, Beaulieu JF. 1992. Transient mosaic patterns of morphological and functional differentiation in the Caco‐2 cell line. Gastroenterology 103:414–423. [DOI] [PubMed] [Google Scholar]
- van der Flier LG, Clevers H. 2009. Stem cells, self‐renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 71:241–260. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, Snippert HJ. 2014. Stem cell dynamics in homeostasis and cancer of the intestine. Nat Rev Cancer 14:468–480. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Liu Z, Kopan R. 2011. Notch: Architect, landscaper, and guardian of the intestine. Gastroenterology 141:448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AJ, Byun DS, Nasser S, Murray LB, Ayyanar K, Arango D, Figueroa M, Melnick A, Kao GD, Augenlicht LH, Mariadason JM. 2008. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell 19:4062–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AJ, Byun DS, Popova N, Murray LB, L'Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. 2006. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem 281:13548–13558. [DOI] [PubMed] [Google Scholar]
- Witt O, Deubzer HE, Milde T, Oehme I. 2009. HDAC family: What are the cancer relevant targets? Cancer Lett 277:8–21. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. 1993. P21 is a universal inhibitor of cyclin kinases. Nature 366:701–704. [DOI] [PubMed] [Google Scholar]
- Zimberlin CD, Lancini C, Sno R, Rosekrans SL, McLean CM, Vlaming H, van den Brink GR, Bots M, Medema JP, Dannenberg JH. 2015. HDAC1 and HDAC2 collectively regulate intestinal stem cell homeostasis. FASEB J 29:2070–2080. [DOI] [PubMed] [Google Scholar]