

Abstract
Taliglucerase alfa is an intravenous enzyme replacement therapy approved for treatment of type 1 Gaucher disease (GD), and is the first available plant cell–expressed recombinant therapeutic protein. Herein, we report long‐term safety and efficacy results of taliglucerase alfa in treatment‐naïve adult patients with GD. Patients were randomized to receive taliglucerase alfa 30 or 60 U/kg every other week, and 23 patients completed 36 months of treatment. Taliglucerase alfa (30 U/kg; 60 U/kg, respectively) resulted in mean decreases in spleen volume (50.1%; 64.6%) and liver volume (25.6%; 24.4%) with mean increases in hemoglobin concentration (16.0%; 35.8%) and platelet count (45.7%; 114.0%), and mean decreases in chitotriosidase activity (71.5%; 82.2%). All treatment‐related adverse events were mild to moderate in intensity and transient. The most common adverse events were nasopharyngitis, arthralgia, upper respiratory tract infection, headache, pain in extremity, and hypertension. These 36‐month results of taliglucerase alfa in treatment‐naïve adult patients with GD demonstrate continued improvement in disease parameters with no new safety concerns. These findings extend the taliglucerase alfa clinical safety and efficacy dataset. www.clinicaltrials.gov identifier NCT00705939. Am. J. Hematol. 91:656–660, 2016. © 2016 Wiley Periodicals, Inc.
Introduction
For more than two decades, the standard of care for patients with type 1 Gaucher disease (GD) has been enzyme replacement therapy (ERT). The most recently approved ERT for GD is taliglucerase alfa (ELELYSO®; Pfizer, New York, NY; Protalix BioTherapeutics, Carmiel, Israel). This agent was first approved in the United States in 2012 1, and it is the first plant cell–expressed human recombinant therapeutic protein to receive market authorization in the United States 1, 2. Taliglucerase alfa is indicated for treatment of adults with type 1 GD in the United States, Israel, Brazil, Australia, Canada, Chile, and other countries, and is approved for treatment of pediatric patients in the United States, Australia, Canada, and Israel, Mexico, other countries, and for hematologic manifestations of type 3 GD in pediatric patients in Canada.
The efficacy and safety of taliglucerase alfa at two biweekly doses of 30 U/kg and 60 U/kg in treatment‐naïve patients with GD were assessed in a pivotal, phase III, 9‐month, double‐blind, parallel‐group, randomized trial (NCT00376168; Study PB‐06‐001) 3, 4. Patients achieved significant reductions in spleen volume, liver volume, and chitotriosidase activity along with significant increases in hemoglobin concentration following treatment with taliglucerase alfa 30 U/kg and 60 U/kg. Platelet counts increased in both dose groups. All treatment‐related adverse events (AEs) were mild or moderate in intensity and no serious AEs were reported 4.
Patients who completed pivotal Study PB‐06‐001 were eligible to participate in extension Study PB‐06‐003 (NCT00705939) at the same dose as in Study PB‐06‐001 4, 5. The purpose of this report is to present the safety and efficacy results of treatment‐naïve adult patients with GD receiving taliglucerase alfa 30 U/kg or 60 U/kg for up to 36 total months of treatment in Studies PB‐06‐001 and PB‐06‐003. Patients who completed Study PB‐06‐003 were eligible to continue treatment in an additional extension study, PB‐06‐007 (NCT01422187), for a total duration of taliglucerase alfa treatment of up to 5 years 5.
PATIENTS AND METHODS
Study design
The design of the 9‐month pivotal study (PB‐06‐001) has been previously reported; it was a phase III, multicenter, double‐blind, randomized, parallel‐dose (30 U/kg and 60 U/kg) study of efficacy and safety in adult ERT‐naïve patients 4. Study PB‐06‐003 was designed to be an extension study for patients completing trials PB‐06‐001 and PB‐06‐002 (a 9‐month multicenter, open‐label study of efficacy and safety in adults and children switching from imiglucerase to taliglucerase alfa; NCT00712348). Patients from Study PB‐06‐001 continued double‐blind treatment for an additional 15 months in Study PB‐06‐003 for a total of 24 months of double‐blind treatment, at which time the investigator could request unblinding of the patient's dose. In the present report, data were analyzed from patients who completed Study PB‐06‐001 (9 months of treatment) and continued in Study PB‐06‐003 (27 months of treatment) for a total duration of up to 36 months of taliglucerase alfa treatment.
The ethics committees/institutional review boards (IRBs) associated with each study site reviewed and approved the protocol and informed consent forms. Protocol amendments were approved by the IRBs before implementation and included extension of the potential treatment period from 15 to 30 months (24–39 total months of taliglucerase alfa treatment) in Study PB‐06‐003 or until marketing approval was granted by the appropriate regulatory authority, whichever was earlier, and to remove pregnancy or nursing as an exclusion criterion and as a reason for study discontinuation. The study was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice guidelines and all local regulations. Written informed consent was obtained for all patients.
Patients
The main inclusion criteria for patients in Study PB‐06‐003 were completion of Study PB‐06‐001 and written informed consent to continue in Study PB‐06‐003. To be eligible for Study PB‐06‐001, patients were required to have a diagnosis of GD based on low glucocerebrosidase leukocyte activity level, splenomegaly (organ volume more than eight times the normal volume expected based on body weight), and thrombocytopenia (platelet count < 120,000/mm3). The main exclusion criteria from Study PB‐06‐003 were current use of another experimental drug (for any disease) and presence of any medical, emotional, behavioral, or psychological condition that in the judgment of the investigator would interfere with the patient's compliance with the requirements of the study.
Study treatment
In Study PB‐06‐001, patients were randomized to receive taliglucerase alfa 30 U/kg body weight/infusion or 60 U/kg body weight/infusion administered intravenously at 2‐week intervals. In Study PB‐06‐003, patients continued to receive the dose of taliglucerase alfa that they had been assigned at randomization for Study PB‐06‐001. After 24 months, patients who had been receiving 30 U/kg could have their dose increased to 60 U/kg, if the investigator deemed it clinically necessary.
Assessments
Efficacy evaluations included spleen volume, liver volume, hemoglobin concentration, platelet count, and biomarkers (chitotriosidase activity and chemokine [C‐C motif] ligand 18 [CCL18] concentration). Organ volumes were measured by magnetic resonance imaging using a single validated system and blinded central readers, as described previously 4, 6. Safety assessments included the incidence and severity of AEs, the incidence of serious AEs, presence of anti‐taliglucerase alfa antibodies, physical examination, clinical laboratory values, electrocardiogram, echocardiogram, and pulmonary function tests.
Statistical analysis
For each parameter, efficacy analyses included patients for whom data were available at each time point through 36 months. The safety analysis population included all patients from Study PB‐06‐001 who entered Study PB‐06‐003 and received study drug in PB‐06‐003. The full analysis population was defined as patients who enrolled in Study PB‐06‐003 and received at least one dose of study drug. The completer population was defined as patients who completed 36 months of treatment based on their study duration calculated from the first infusion date in Study PB‐06‐001 and the last infusion date in Study PB‐06‐003. Spleen and liver volumes were calculated as multiples of normal (MN) where normal spleen volume is 2 mL/kg and normal liver volume is 25 mL/kg 7. For categorical variables, counts and percentages are presented. Descriptive statistics for continuous variables, sample size (n), mean and its standard error (SE), standard deviation, median, and range were calculated for spleen volume, liver volume, hemoglobin concentration, platelet count, and change in biomarker activity. The time points were counted continuously from Study PB‐06‐001 through Study PB‐06‐003; “baseline” in this analysis denotes baseline from Study PB‐06‐001 4. No inferential statistics were performed for testing the change from baseline and/or for comparing between or among treatment groups. Analysis of AEs was limited to events that occurred during Study PB‐06‐003.
Results
Study patients
The patient disposition is provided in Fig. S1 Supporting Information. A total of 29 patients completed Study PB‐06‐001 4. Twenty‐six of these patients enrolled in extension Study PB‐06‐003, and all received treatment; of these, 23 completed ≥36 months of taliglucerase alfa treatment. Three patients discontinued Study PB‐06‐003 (all were in the 60 U/kg dose group): One patient (22‐030) was unable to continue for logistical reasons and withdrew voluntarily. Another patient (10‐013) was discontinued by the study investigator to assess possible allergic reaction of itching and rash on the arms associated with infusions 37 and 38. One patient (12‐024) stopped receiving the study drug after a total duration of 24 months because the study protocol in that patient's country ended at that time point; the patient continued to receive taliglucerase alfa in a compassionate use program.
Baseline patient demographics and disease characteristics are shown in Table SI Supporting Information for the 23 patients who completed 36 months of treatment. All but one patient (in the 60 U/kg group) were Caucasian. The ratio of male to female patients was 13:10. The mean baseline spleen volumes for both dose groups were >15 MN (range, 7.7–54.2 MN), and the mean liver volumes for both groups were ≥1.5 MN. Similar baseline patient demographics and disease characteristics were observed in the population of 26 patients who entered the study.
Efficacy
In the full analysis population (n = 26), mean spleen volume (measured in MN), liver volume (measured in MN), platelet count, chitotriosidase activity, and CCL18 concentration improved continuously through 36 months of treatment for patients receiving taliglucerase alfa 30 U/kg and 60 U/kg. Hemoglobin concentration reached normal levels after approximately 9 to 12 months of treatment and stayed stable through the study duration (Figs. 1, 2, 3). Individual patient results are shown in Tables SII–SIV Supporting Information.
Figure 1.
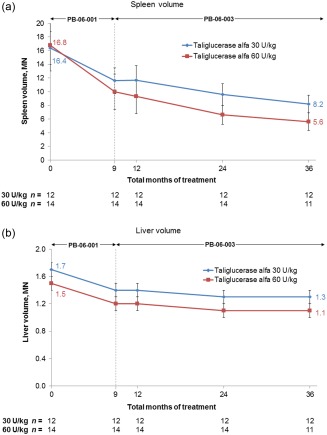
Spleen volume, expressed as multiples of normal (MN), where normal spleen volume is 2 mL/kg times body weight (kg), and liver volume, expressed as MN, where normal liver volume is 25 mL/kg times body weight (kg), through 36 total months of taliglucerase alfa treatment. At each time point, values represent the mean based on all patients with available data. Error bars represent standard error. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2.
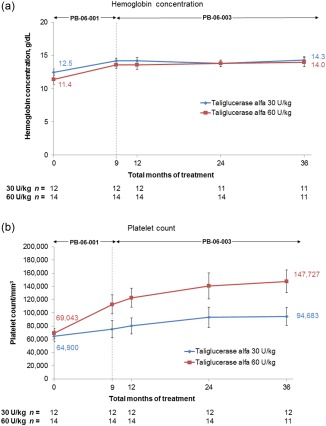
Hemoglobin concentration and platelet count through 36 total months of taliglucerase alfa treatment. One patient did not have hemoglobin concentration values at 36 months and was excluded from the analysis. At each time point, values represent the mean based on all patients with available data. Error bars represent standard error. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 3.

Mean percent change in chitotriosidase activity and CCL18 concentration mean percent change from baseline through 36 total months of taliglucerase alfa treatment. One patient in the 60 U/kg dose group (30–111) was found to have no chitotriosidase activity at baseline; therefore, no chitotriosidase activity data were available during the study, and the patient was excluded from chitotriosidase analysis but was followed via CCL18. At each time point, values represent the mean based on all patients with available data. Error bars represent standard error. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Analysis of the subset of only those patients who completed 36 total months of taliglucerase alfa treatment (n = 23) revealed the following: In the 30 U/kg dose group, results were identical to the full analysis population because there were no discontinuations; in the 60 U/kg dose group, curves for the patients who completed 36 total months of treatment (not shown) were similar to those shown in Fig. 1 through Fig. 3 for the full analysis population. The 23 patients who completed 36 months of taliglucerase alfa treatment demonstrated substantial percent changes in visceral and hematologic parameters and in biomarkers from baseline. For the 30 U/kg and 60 U/kg dose groups, respectively, mean (±SE) percent changes were as follows: spleen volume (measured in MN) decreased by 50.1% (±2.4%; n = 12) and 64.6% (±3.3%; n = 11), liver volume (measured in MN) decreased by 25.6% (±1.5%; n = 12) and 24.4% (±4.6%; n = 11), hemoglobin concentration increased by 16.0% (±4.4%; n = 11) and 35.8% (±13.2%; n = 11), platelet count increased by 45.7% (±12.7%; n = 12) and 114.0% (±28.8%; n = 11), chitotriosidase activity decreased by 71.5% (±4.3%; n = 12) and 82.2% (±7.2%; n = 10), and CCL18 concentration decreased by 58.1% (±4.8%; n = 12) and 71.0% (±6.8%; n = 11).
Three patients discontinued the study prior to completion of 36 months of treatment and they, too, demonstrated improvements in disease parameters from baseline to last observation. One patient (12‐024; 60 U/kg) discontinued after 15 months of treatment in Study PB‐06‐003 (24 total months of therapy) because IRB approval for treatment beyond the original study duration of 15 months (for a total of 24 months) was not received in time in that patient's country. From baseline to 24 months, this patient showed improvements in spleen volume (−49.5%; measured in MN), liver volume (−29.5%; measured in MN), hemoglobin concentration (+7.3%), platelet count (+35.4%), chitotriosidase activity (−73.0%), and CCL18 concentration (−40.9%). The second patient (22‐030; 60 U/kg) discontinued because of logistical reasons; this patient experienced fatigue and sinusitis, which were considered to be AEs that were not related to treatment. This patient received taliglucerase alfa for a total of 24 months; from baseline to last observation, the patient showed improvements in spleen volume (−43.5%; measured in MN), hemoglobin concentration (+13.3%), platelet count (+115.2%), chitotriosidase activity (−70.3%), and CCL18 concentration (−58.7%), while the liver volume remained in the normal range at 0.98 MN. The third patient (10‐013; 60 U/kg) discontinued at the investigator's request to investigate itching and rash on the arms, which were considered possibly related to treatment after the 38th infusion. This patient also experienced a rash under the adhesive that held the infusion catheter in place, nervousness, general body pain, and non‐related head trauma with bleeding that occurred during a seizure (epilepsy) and necessitated hospitalization, none of which was considered treatment related. This patient received taliglucerase alfa for a total of 26 months; improvements were observed from baseline to last observation in spleen volume (−62.9%; measured in MN), liver volume (−24.2%; measured in MN), hemoglobin concentration (+13.5%), platelet count (+56.5%), chitotriosidase activity (−94.4%), and CCL18 concentration (−71.8%).
Safety
Table 1 lists the safety findings for all 26 patients who entered Study PB‐06‐003 during the extension period. The most common all‐causality AEs were arthralgia (30 U/kg, n = 4; 60 U/kg, n = 4), headache (30 U/kg, n = 4; 60 U/kg, n = 3), upper respiratory tract infection (30 U/kg, n = 3; 60 U/kg, n = 3), pain in extremity (30 U/kg, n = 2; 60 U/kg, n = 4), nasopharyngitis (30 U/kg, n = 2; 60 U/kg, n = 3), and hypertension (30 U/kg, n = 2; 60 U/kg, n = 3). Most AEs (98.5%) were mild to moderate in severity and transient in nature, as were all treatment‐related AEs. Ten patients experienced a total of 36 AEs considered to be related to treatment. All of these events resolved and the patients continued the study with no change in dose. Four severe AEs, considered unrelated to treatment, occurred in three patients during the study: pain from a hemangioma in the left knee and surgical treatment for the hemangioma resulting in a pulmonary embolism (Patient 41‐022), immune thrombocytopenia (Patient 11‐007), and diabetes mellitus (Patient 40‐018). There were no safety issues that evolved as a consequence of low platelet count in the patient with the diagnosis of immune thrombocytopenia. Three patients experienced four serious AEs during the study: head injury in a patient with epilepsy (Patient 10‐013), immune thrombocytopenia (noted above), hemangioma, and pulmonary embolism (noted above), all of which were considered not related to treatment and all of which resolved.
Table 1.
AEs Occurring During the Extension Period (Safety Population; n = 26)
| No. of AEs (No. of patients) | % of total AEs | |
|---|---|---|
| Total AEs | 275 (24) | 100 |
| Mild or moderate | 271 (23) | 98.5 |
| Severe or very severe | 4 (3) | 1.1 |
| 30 U/kg | 2 (1) | |
| 60 U/kg | 2 (2) | |
| Non–treatment‐relateda | 239 (24) | 86.9 |
| Treatment‐relatedb | 36 (10) | 13.1 |
Probably or definitely not related to treatment.
Possibly, probably, or definitely related to study treatment.
AEs: adverse events.
Antidrug antibodies
Thirteen patients (30 U/kg dose group, n = 5; 60 U/kg dose group, n = 8) were found to have anti‐taliglucerase alfa IgG antibodies on at least one post‐baseline visit, and two of these patients (Patient 30‐008, 30 U/kg; Patient 15‐016, 60 U/kg) were found to have neutralizing antibodies based on an in vitro enzymatic activity assay but were negative in a cell‐based neutralizing antibody assay. Efficacy did not appear to be impaired by development of neutralizing antibodies, as noted in the Discussion. One of the 13 patients (Patient 30‐010) was found to have a very low anti‐taliglucerase alfa IgG titer present at baseline (before exposure). IgG titers in this patient did not increase during treatment, and this patient was negative for anti‐taliglucerase alfa antibodies from month 12 until the end of the study. Treatment‐related AEs in patients with a positive (post‐treatment) anti‐taliglucerase alfa antibody titer included a fixed drug eruption (Patient 10‐001), arthralgia (Patient 17‐032), infusion‐related reaction and infusion site pain (Patient 10‐005), pruritus and rash (Patient 10‐013), gynecomastia and pulmonary hypertension (Patient 15‐016), and hypersensitivity (Patient 30‐009).
Discussion
This multinational, multicenter, two‐dose study of taliglucerase alfa (30 U/kg and 60 U/kg) in treatment‐naïve adult patients with GD demonstrated improvement in visceral and hematologic disease parameters as well as biomarkers. All treatment‐related AEs were mild or moderate in severity and resolved.
Dose increases occurred in three patients: One patient (10‐001, 30 U/kg), with a baseline platelet count of 28,000/mm3, received blinded treatment through 30 total months after which the blind was opened and the dose increased to 60 U/kg for the remainder of the study. This patient's platelet counts were 28,000/mm3 at 30 months and 31,000/mm3 at 36 months. From baseline to 36 months, the patient's platelet count had risen by 10.7% and the patient also showed improvements in spleen volume (−45.7%; measured in MN), liver volume (−23.8%; measured in MN), hemoglobin concentration (+17.5%), chitotriosidase activity (−54.6%), and CCL18 concentration (−39.7%). The second patient (11‐014) received 30 U/kg through approximately 26 months of treatment, at which time a bone crisis occurred. This patient's dose was increased to 45 U/kg until the end of the study. The patient was treated with nonsteroidal anti‐inflammatory drugs, and the AE resolved after 1 month without recurrence of bone crisis throughout the remainder of the study. From baseline through 36 months, this patient experienced improvements in spleen volume (−47.5%; measured in MN), liver volume (−23.0%; measured in MN), hemoglobin concentration (+7.5%), platelet count (+42.0%), chitotriosidase activity (−88.3%), and CCL18 concentration (−77.7%). The third patient (42‐025) received blinded treatment through 24 total months at 30 U/kg. Due to a low baseline platelet count of 27,000/mm3 and small increase to only 33,000/mm3 (22.2%) at 24 months, the patient's dose was increased to 60 U/kg through the end of the study. At 36 months, the platelet count was 30,000/mm3. From baseline through 36 months, improvements were observed in spleen volume (−43.6%; measured in MN), liver volume (−17.9%; measured in MN), hemoglobin concentration (+23.3%), platelet count (+11.1%), chitotriosidase activity (−66.4%), and CCL18 concentration (−51.6%).
Although the dose increase in the patient experiencing bone crisis (11‐014) had a positive impact on the resolution of the bone symptoms, dose escalations did not make a clinically meaningful difference in the platelet counts of the other two patients. This poor platelet response is presently not understood. While focal splenic lesions are associated with refractory thrombocytopenia 8, magnetic resonance imaging scans of both patients' spleens did not reveal evidence of intra‐splenic lesions.
Development of neutralizing antibodies did not appear to result in a loss of efficacy, and no changes in treatment were initiated for either patient (15‐016 and 30‐008) with neutralizing antibodies; both patients completed the study. From baseline through 36 months, two patients (15‐016 and 30‐008, respectively) experienced improvements in spleen volume (−41.1 and −37.1%; measured in MN), liver volume (−15.1 and −25.9%; measured in MN), chitotriosidase activity (−80.9 and −60.5%), and CCL18 concentration (−63.7 and −27.2%). Hemoglobin concentration decreased nominally by 0.8% from 13.2 g/dL at baseline in Patient 15‐016 but remained in the normal range and increased by 10.3% in Patient 30‐008, who was anemic at baseline. Platelets increased by 71.8% in Patient 15‐016 and showed a clinically insignificant decrease from baseline in Patient 30‐008 (−1.2% from 86,000/mm3 at baseline). The numbers of patients reported as developing anti‐taliglucerase alfa IgG antibodies in Study PB‐06‐003 are higher than those seen in previous, shorter‐term studies 1, 4, and are likely due to increased sensitivity as part of assay modifications resulting in different antibody sample positivity reporting. These modifications were instituted to establish statistically based cut point definitions consistent with current industry practices.
In conclusion, these long‐term results of taliglucerase alfa demonstrated continued improvement in the clinical parameters with no new safety concerns through 36 months of treatment in treatment‐naïve adult patients with GD. These results extend the clinical safety and efficacy profile of taliglucerase alfa in patients with GD.
Supporting information
Supporting Information
Acknowledgments
Pfizer and Protalix entered into an agreement in November 2009 to develop and commercialize taliglucerase alfa. Statistical analyses were performed by Target Health, who provides clinical research services to Pfizer and Protalix BioTherapeutics. None of the authors received compensation for their contributions to this manuscript.
Conflicts of interest: AZ is a consultant for Protalix BioTherapeutics and is a member of the Speakers' Bureau for Genzyme/Sanofi, Protalix BioTherapeutics, and Shire; has options in Protalix BioTherapeutics; and has received honoraria from Genzyme/Sanofi, Pfizer, and Shire. In addition, the Gaucher Clinic receives grant/research support from Genzyme/Sanofi and Shire. PG, AM, GD, HR, FG, DJA, MP, ETM, PAC, SV, and SES‐M are study investigators. RC and EB‐A are employees of Protalix BioTherapeutics.
References
- 1. Elelyso [package insert]. New York, NY: Pfizer Labs; 2015. [Google Scholar]
- 2. Fox JL. First plant‐made biologic approved. Nat Biotechnol 2012;30:472 [Google Scholar]
- 3.National Institutes of Health. A Phase III Trial to Assess the Safety and Efficacy of Plant Cell Expressed GCD in Patients with Gaucher Disease NCT00376168; 2012. Available at: http://www.clinicaltrials.gov/ct2/show/NCT00376168, accessed on June 25, 2015.
- 4. Zimran A, Brill‐Almon E, Chertkoff R, et al. Pivotal trial with plant cell‐expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2011;118:5767–5773. [DOI] [PubMed] [Google Scholar]
- 5. Clinicaltrials.gov. A Multicenter Extension Study of Taliglucerase Alfa in Adult Subjects with Gaucher Disease NCT01422187; 2013; Available at: http://clinicaltrials.gov/ct2/show/NCT01422187?term=taliglucerase+alfa&rank=3, accessed on June 25, 2015.
- 6. Bracoud L, Ahmad H, Brill‐Almon E, Chertkoff R. Improving the accuracy of MRI spleen and liver volume measurements: A phase III Gaucher disease clinical trial setting as a model. Blood Cells Mol Dis 2011;46:47–52. [DOI] [PubMed] [Google Scholar]
- 7. Pastores GM, Weinreb NJ, Aerts H, et al. Therapeutic goals in the treatment of Gaucher disease. Semin Hematol 2004;41:4–14. [DOI] [PubMed] [Google Scholar]
- 8. Stein P, Malhotra A, Haims A, et al. Focal splenic lesions in type I Gaucher disease are associated with poor platelet and splenic response to macrophage‐targeted enzyme replacement therapy. J Inherit Metab Dis 2010;33:769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information