

Abstract
The tissue distribution of teneligliptin, a dipeptidyl peptidase (DPP)‐4 inhibitor, was investigated in rats, and compared with tissue distributions previously reported for other DPP‐4 inhibitors. Following the oral administration of [14C]teneligliptin to Sprague–Dawley rats, it was predominantly distributed to the kidney and liver, followed by the lung, spleen and pituitary gland. The elimination half‐life (t 1/2) of [14C]teneligliptin was 68.3 and 69.0 h in the kidney and liver, respectively; these values were about 10 times greater than the plasma t 1/2. Of note, the elimination of [14C]teneligliptin from tissues with high DPP‐4 activity (kidney, liver and lung) was slower in wild‐type rats than in DPP‐4‐deficient rats, especially in the kidney. By contrast, in the heart and pancreas, which weakly express DPP‐4, no difference was observed in [14C]teneligliptin concentrations between the two animal strains. In the kidney, most radioactivity was attributable to unchanged teneligliptin from 0.5 to 72 h after administration. The marked difference in the distribution of [14C]teneligliptin between the two strains suggests that the high binding affinity of teneligliptin for DPP‐4 is involved in its tissue distribution. The currently marketed DPP‐4 inhibitors are highly distributed to the liver, kidney and lung, but the extent of tissue distribution varies greatly among the drugs. The differences in the tissue distributions of DPP‐4 inhibitors might be related to differences in their pleiotropic effects. © 2016 The Authors Biopharmaceutics & Drug Disposition Published by John Wiley & Sons Ltd.
Keywords: DPP‐4 inhibitor, tissue distribution, teneligliptin
Introduction
Glucagon‐like peptide‐1 (GLP‐1) is an incretin hormone secreted from the gastrointestinal tract in response to food intake. The active form of GLP‐1 stimulates insulin secretion in a blood glucose‐dependent manner while simultaneously suppressing glucagon secretion 1. The active form of GLP‐1 is rapidly degraded and inactivated by dipeptidyl peptidase‐4 (DPP‐4). DPP‐4 inhibitors prevent the degradation of GLP‐1, and thus increase the blood concentrations of the active form of GLP‐1.
Several DPP‐4 inhibitors are now prescribed in clinical practice for the treatment of type 2 diabetes mellitus. DPP‐4 inhibitors have a variety of chemical structures, and are classified as either peptide mimetic compounds, which are designed by modifying the structure of the peptide substrate (anagliptin 2, saxagliptin 3, teneligliptin 4 and vildagliptin 5), or non‐peptide mimetic compounds, which are designed by modifying the structures of compounds identified in random screening assays (alogliptin 6, linagliptin 7 and sitagliptin 8) 9 (Figure 1). Because of differences in their chemical structures, there are also differences in their binding modes 10, DPP‐4 inhibitory activity 2, 11 and physical properties, including ClogP, an index of lipophilicity (Table 1). It has been reported that DPP‐4 inhibitors vary greatly with respect to their plasma concentration–time profiles and elimination pathways in humans, and that these differences may necessitate dose adjustments owing to drug interactions and renal impairment 12, 13.
Figure 1.
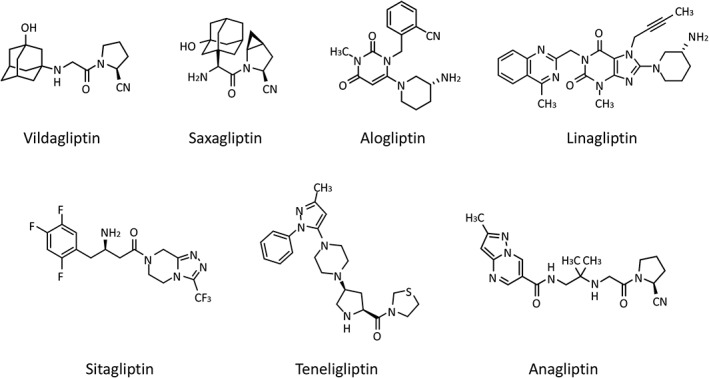
Chemical structures of DPP‐4 inhibitors
Table 1.
Characteristic features of DPP‐4 inhibitors
| Drug | Binding subsitesa | ClogP b | pKac | V ss (l/kg)d [Reference] | IC50 (nmol/l)e [Reference] |
|---|---|---|---|---|---|
| Alogliptin | S1, S2, S’1 | 0.99 | 8.5 | 3.5 19 | 4.9 11 |
| Anagliptin | – | 0.55 | 6.7 | 0.68 2 | 3.8 2 |
| Linagliptin | S1, S2, S’1, S’2 | 1.91 | 1.9, 8.6 | 50.3 23 | 0.6 11 |
| Saxagliptin | S1, S2 | −0.03 | 7.3 | 5.2 20 | 6.3 11 |
| Sitagliptin | S1, S2, S2 extensive | 0.69 | 7.7 | 8.8 25 | 10.3 11 |
| Teneligliptin | S1, S2, S2 extensive | 2.24 | 1.7, 3.8, 7.3 | 8.9 | 1.9 11 |
| Vildagliptin | S1, S2 | 0.69 | 7.6 | 8.6 26 | 29.2 11 |
These binding subsites were determined from the co‐crystal structures of the inhibitors with DPP‐4 by x‐ray crystallography 10.
These values were calculated using the PCModels in Daylight software version 4.93 (http://www.daylight.com/).
As stated on the manufacturer's interview form.
The volume of distribution at steady state (V ss) was calculated after intravenous administration to rats.
Median inhibitory concentration against DPP‐4.
Teneligliptin is a DPP‐4 inhibitor that was approved for the treatment of type 2 diabetes mellitus in 2012 in Japan and in 2014 in Korea 14, 15. In humans, teneligliptin is eliminated by hepatic metabolism mediated by cytochrome P450 3A4 or flavin‐containing monooxygenase 3, or excreted from the kidney as the unchanged drug 16. Teneligliptin is thus removed via several elimination routes. Variations in the exposure of patients to teneligliptin with renal and hepatic disorders were 1.68‐fold and 1.59‐fold higher than those in healthy subjects 17, 18; thus, a dose adjustment was not required, even for patients with renal or hepatic disorders.
This study investigated the tissue distribution of teneligliptin in rats during the preclinical development of teneligliptin, and compared the tissue distribution profiles of teneligliptin and other currently available DPP‐4 inhibitors to examine differences in their tissue distribution. The tissue distribution and physicochemical properties differ between the currently available DPP‐4 inhibitors, as shown in this study and other studies; therefore, their tissue distribution might be associated with their potential pleiotropic effects beyond their glucose‐lowering properties.
Materials and Methods
Objectives
Four independent preclinical studies were performed with the following objectives. Study 1: to determine the change in tissue concentration of teneligliptin in rats, and to quantify the distribution and the disappearance of teneligliptin in each tissue. Study 2: to obtain in vivo images visualizing the tissue distribution of teneligliptin and to verify the results of study 1. Study 3: to determine the role of DPP‐4 in the tissue distribution of teneligliptin. Study 4: to determine the relative concentrations of radiolabeled teneligliptin and its major metabolites in plasma and the kidney, which showed the highest concentration of teneligliptin in studies 1–3.
Materials
[14C]Teneligliptin (specific radioactivity 1.03–1.08 MBq/mg and radiochemical purity 98.5%–99.2%) was synthesized by Sekisui Medical Co., Ltd (Tokyo, Japan). The chemical structure of teneligliptin and the position of the radiolabel are shown in Figure 2. All other chemicals were of analytical grade or higher purity, and were obtained from commercial suppliers.
Figure 2.
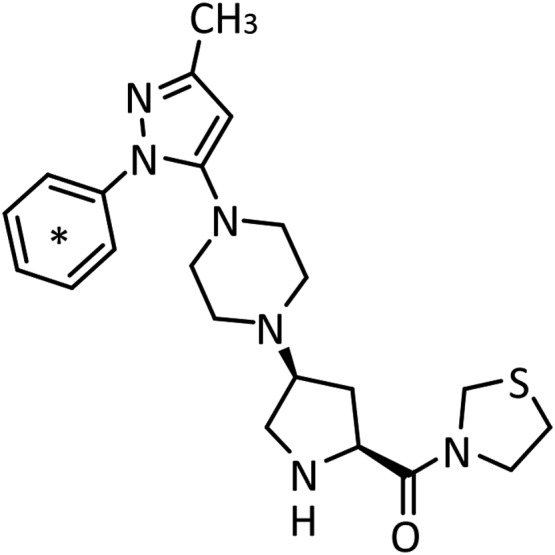
Chemical structure of [14C]teneligliptin. The position of the 14C radiolabel on teneligliptin is indicated by an asterisk
Animals
The study protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Mitsubishi Chemical Safety Institute Ltd (now LSI Medience Corporation, Tokyo, Japan). For studies 1, 2 and 4, male Sprague–Dawley rats (Crj:CD(SD)IGS; hereafter, SD rats) were purchased from Charles River Japan (Kanagawa, Japan). For study 3, male wild‐type Fischer rats (F344/Jcl; hereafter, wild‐type rats) and DPP‐4‐deficient male rats (F344/DuCrlCrlj; hereafter, DPP‐4‐deficient rats) were purchased from CLEA Japan (Shizuoka, Japan) and Charles River Japan, respectively. The rats were housed with a 12‐h light/dark cycle and controlled temperature and humidity. The rats were fasted from the evening before the dosing day to about 5 h after administration, but were given drinking water during this time.
Tissue distribution of radioactivity in SD rats (study 1)
Male SD rats were given a single oral dose of 1 mg/1.67 MBq/kg [14C]teneligliptin. Rats were euthanized at 0.5, 5, 12, 24, 72 and 168 h after administration (n = 4/time), and the following tissues were collected: blood, plasma, brain, pituitary, medulla oblongata, eyeball (bilateral), submaxillary gland, thyroid, spinal cord (neck), thymus, heart, lung, liver, adrenal (bilateral), kidney (bilateral), spleen, pancreas, prostate, testis (bilateral), epididymis, seminal vesicle, aorta (thoracic), skin (around the armpit), skeletal muscle (femoral), bone (femur), bone marrow (femoral), perirenal white adipose tissue, brown adipose tissue, mesenteric lymph nodes, stomach, small intestine, cecum and large intestine (except for the cecum). The entire blood sample was immediately transferred to a test tube with heparin sodium as an anticoagulant and gently inverted several times. One portion (200 μl) of the blood was set aside as a blood sample. The remaining blood was centrifuged (4 °C, 10 min at 1870 × g) and the supernatant (plasma) was retained. The other tissues were washed with physiological saline, wiped with pieces of filter paper to remove saline, and weighed. The samples were stored at −20 °C until used to measure radioactivity.
Whole‐body autoradiography of SD rats (study 2)
Male SD rats were given a single oral dose of 1 mg/1.67 MBq/kg [14C]teneligliptin and were euthanized at 0.5, 24 or 168 h after administration (n = 1/time). The animals were immediately frozen in a dry ice/acetone bath. The frozen body was embedded in about 4%–5% (w/v) carboxymethylcellulose solution and sliced using a cryomicrotome. The resulting frozen thin sections (40 μm thick) were collected on adhesive tape and freeze‐dried in the cryomicrotome. The sections were exposed for 72 h in a lead‐shielded box. The radiogram on the imaging plate was scanned using a bio‐imaging analyser (BAS‐2000II, Fujifilm Corporation, Tokyo, Japan) to obtain radioluminograms.
Whole‐body autoradiography of wild‐type or DPP‐4‐deficient rats (study 3)
Male wild‐type or DPP‐4‐deficient rats were given a single oral dose of 1 mg/1.60 MBq/kg [14C]teneligliptin. Tail vein blood samples were collected at 0.5, 24, 36 and 48 h after administration (n = 1/time). After the blood sampling, each rat was euthanized, frozen in a dry ice/acetone bath and embedded in about 4%–5% (w/v) carboxymethylcellulose solution together with the calibration blood samples of four different concentrations of [14C]teneligliptin. Tissue sections (40 μm thick) were prepared using a cryomicrotome, collected on adhesive tape and freeze‐dried in the cryomicrotome. Radioluminograms were obtained as described for study 2. The radioluminograms obtained in study 3 were analysed using an RLG analysing system (Multi Gauge Ver.2.2, Fujifilm Corporation, Tokyo, Japan) with the blood calibration standards to determine the radioactivity concentration in each tissue.
Metabolite profiles in plasma and kidney (study 4)
Male SD rats were given a single oral dose of 1 mg/1.60 or 1.67 MBq/kg [14C]teneligliptin. The rats were euthanized (n = 2 or 4/time) at 0.5 and 5 h after administration for plasma sampling or at 0.5, 24 and 72 h after administration for kidney sampling. The kidney was weighed and homogenized in a 4‐fold volume of phosphate‐buffered saline. The plasma or kidney homogenates were mixed in a 2‐fold volume of extraction solvent (acetonitrile for plasma, methanol for kidney homogenate), shaken and centrifuged (4 °C, 5 min at 1870 × g) to obtain the supernatant. The pellet was extracted with the same volume of extraction solvent, shaken and centrifuged (4 °C, 5 min at 1870 × g). The obtained supernatants were combined and evaporated under a nitrogen gas stream. Then, 5% acetonitrile was added to the dry residue and the mixture was injected into a high‐performance liquid chromatography (HPLC) system (LC‐10Avp HPLC system; Shimadzu, Kyoto, Japan) with an in‐line radioactivity detector (Flo‐One Radiomatic 610TR; PerkinElmer, Waltham, MA). Chromatographic separation was performed on an HPLC column (Capcell Pak C18 UG120, 5 μm, 4.6 mm × 250 mm; Shiseido, Tokyo, Japan) with gradient elution. The mobile phase consisted of 10 mm ammonium acetate/formic acid (100:0.5 [v/v], solvent A) and acetonitrile/10 mm ammonium acetate/formic acid (80:20:0.5 [v/v/v], solvent B). The gradient was started with 10% solvent B for 15 min, and the amount of solvent B was increased linearly to 20% over 20 min, 30% over 5 min and 70% over 10 min, followed by holding at 70% for 10 min. The elution sites of 14C‐labeled metabolites were confirmed by reference to HPLC‐ultraviolet chromatograms of unlabeled standard of teneligliptin and metabolites M1, M2, M3, M4 and M5 16.
Measurement of radioactivity
Radioactivity was determined using a liquid scintillation counter (LSC, Tri‐Carb 2300TR, PerkinElmer Life and Analytical Sciences, Waltham, MA) with quenching correction for the transformed spectral index of an external standard in accordance with the manufacturer's instructions. Each sample was measured once for 5 min. The net count was determined by subtracting the background count, which was obtained by measuring the scintillation cocktail alone for 5 or 10 min, from the gross count for each sample. The detection limit of radioactivity was set at twice the background counts per minute (cpm).
Data analysis
The radioactivity concentrations in tissues were analysed using WinNonlin (Ver. 4.1 or 5.2, Certara, St Louis, MO) and the elimination half‐life (t 1/2) was calculated using the values for the last three quantifiable time‐points after administration.
Reference values for marketed DPP‐4 inhibitors
The reference values for other marketed DPP‐4 inhibitors were obtained from previous studies 19, 20, 21 and the Japanese Common Technical Documents submitted to the Pharmaceuticals and Medical Devices Agency (PMDA) 22, 23, 24, 25, 26.
Results
Tissue distribution of radioactivity in SD rats (study 1)
Figure 3 shows the radioactivity concentration–time profiles in tissues, and Table 2 shows the tissue radioactivity concentrations in fasted SD rats given a single oral dose of 1 mg/kg [14C]teneligliptin. The absorbed radioactivity was rapidly distributed to tissues throughout the body, and the maximum tissue concentration was observed at 0.5 h after administration in all tissues except for the testis, epididymis, cecum and large intestine. The maximum tissue concentration was reached at 5 h in the testis, epididymis and cecum, and at 12 h in the large intestine. The plasma concentrations of radioactivity decreased over time with a t 1/2 of 6.5 h, and radioactivity was undetectable at 72 h after administration. High radioactivity concentrations were detected in the kidney and liver aside from the gastrointestinal tract. The t 1/2 was 68.3 and 69.0 h in the kidney and liver, respectively. At 168 h after administration, the radioactivity concentration was 13% of the maximum concentration in the kidney, compared with ≤ 3% of the maximum concentration in other tissues.
Figure 3.
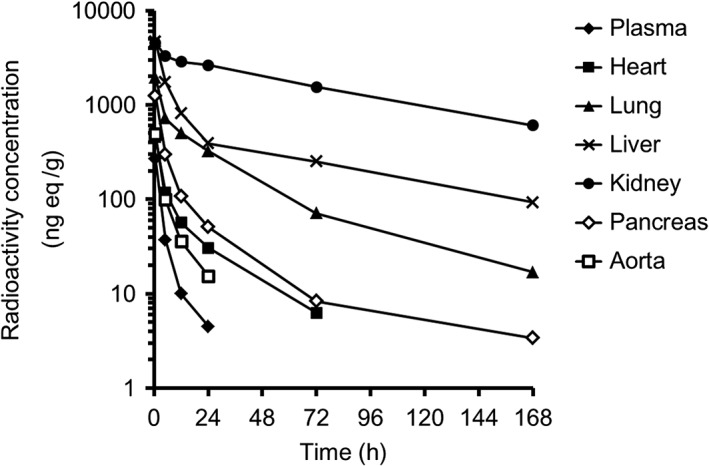
Tissue radioactivity concentration–time profiles (ng eq/g) in SD rats after a single oral dose of 1 mg/kg [14C]teneligliptin. Four SD rats were sampled at each time point
Table 2.
Concentration of radioactivity in tissues after a single oral dose of 1 mg/kg [14C]teneligliptin in SD rats
| Tissue | Concentration of radioactivity (ng eq/ml or g)a | t 1/2 (h)b | |||||
|---|---|---|---|---|---|---|---|
| 0.5 h | 5 h | 12 h | 24 h | 72 h | 168 h | ||
| Blood | 209.9 ± 57.1 (0.8) | 29.9 ± 13.7 (0.8) | 9.3 ± 2.1 (0.9) | 3.9 ± 0.4 (0.9) | N.D. (N.C.) | 0.6 ± 0.7 (N.C.) | 6.7 |
| Plasma | 266.5 ± 74.9 (1.0) | 37.2 ± 17.3 (1.0) | 10.0 ± 2.1 (1.0) | 4.5 ± 0.3 (1.0) | N.D. (N.C.) | N.D. (N.C.) | 6.5 |
| Brain | 24.4 ± 5.3 (0.1) | 7.7 ± 2.6 (0.2) | 3.3 ± 0.7 (0.3) | N.D. (N.C.) | N.D. (N.C.) | N.D. (N.C.) | 4.1 |
| Pituitary | 1412.4 ± 316.0 (5.3) | 406.6 ± 198.9 (10.9) | 149.8 ± 20.6 (15.0) | 68.6 ± 11.7 (15.2) | 13.0 ± 15.0 (N.C.) | N.D. (N.C.) | 17.8 |
| Medulla oblongata | 26.6 ± 6.0 (0.1) | 9.3 ± 4.5 (0.3) | 3.0 ± 0.3 (0.3) | N.D. (N.C.) | N.D. (N.C.) | N.D. (N.C.) | 3.7 |
| Eyeball | 107.9 ± 27.9 (0.4) | 39.0 ± 11.9 (1.0) | 17.4 ± 3.8 (1.7) | 8.4 ± 0.9 (1.9) | 1.5 ± 0.3 (N.C.) | N.D. (N.C.) | 17.6 |
| Submaxillary gland | 1009.9 ± 265.5 (3.8) | 309.9 ± 62.2 (8.3) | 123.7 ± 24.6 (12.4) | 78.5 ± 12.3 (17.4) | 18.0 ± 5.0 (N.C.) | 6.9 ± 2.5 (N.C.) | 43.6 |
| Thyroid | 826.4 ± 164.2 (3.1) | 279.2 ± 83.8 (7.5) | 124.3 ± 25.9 (12.4) | 55.2 ± 7.4 (12.3) | N.D. (N.C.) | N.D. (N.C.) | 8.3 |
| Spinal cord | 28.2 ± 7.8 (0.1) | 11.4 ± 5.8 (0.3) | 4.9 ± 2.4 (0.5) | 1.6 ± 1.1 (0.4) | N.D. (N.C.) | N.D. (N.C.) | 6.8 |
| Thymus | 538.8 ± 138.4 (2.0) | 298.8 ± 61.8 (8.0) | 181.8 ± 19.4 (18.2) | 132.1 ± 3.9 (29.4) | 37.5 ± 8.1 (N.C.) | 10.6 ± 3.3 (N.C.) | 41.0 |
| Heart | 454.0 ± 85.0 (1.7) | 118.7 ± 34.6 (3.2) | 56.6 ± 8.9 (5.7) | 30.6 ± 2.7 (6.8) | 6.3 ± 1.1 (N.C.) | N.D. (N.C.) | 19.5 |
| Lung | 1938.4 ± 456.7 (7.3) | 714.7 ± 116.7 (19.2) | 500.7 ± 40.2 (50.1) | 323.6 ± 46.7 (71.9) | 71.3 ± 12.9 (N.C.) | 16.9 ± 5.9 (N.C.) | 35.2 |
| Liver | 4686.7 ± 932.3 (17.6) | 1753.2 ± 130.4 (47.1) | 814.1 ± 68.7 (81.4) | 389.5 ± 49.5 (86.6) | 250.2 ± 21.8 (N.C.) | 92.4 ± 17.2 (N.C.) | 69.0 |
| Adrenal | 1221.7 ± 289.0 (4.6) | 323.7 ± 90.7 (8.7) | 177.0 ± 22.7 (17.7) | 100.4 ± 8.8 (22.3) | 20.5 ± 4.3 (N.C.) | 6.5 ± 4.5 (N.C.) | 38.5 |
| Kidney | 4568.2 ± 609.1 (17.1) | 3292.8 ± 76.8 (88.5) | 2872.1 ± 419.4 (287.2) | 2637.8 ± 410.3 (586.2) | 1548.5 ± 192.3 (N.C.) | 606.1 ± 203.2 (N.C.) | 68.3 |
| Spleen | 1377.2 ± 292.1 (5.2) | 510.7 ± 68.2 (13.7) | 312.9 ± 31.8 (31.3) | 217.7 ± 21.3 (48.4) | 62.4 ± 7.3 (N.C.) | 17.0 ± 4.3 (N.C.) | 40.5 |
| Pancreas | 1253.9 ± 341.0 (4.7) | 298.9 ± 83.1 (8.0) | 107.8 ± 22.6 (10.8) | 50.8 ± 1.2 (11.3) | 8.3 ± 1.8 (N.C.) | 3.4 ± 0.9 (N.C.) | 39.8 |
| Prostate | 478.6 ± 167.1 (1.8) | 169.0 ± 53.9 (4.5) | 76.7 ± 18.8 (7.7) | 38.6 ± 3.3 (8.6) | 6.1 ± 1.3 (N.C.) | 2.2 ± 0.6 (N.C.) | 37.3 |
| Testis | 68.5 ± 21.6 (0.3) | 128.2 ± 41.8 (3.4) | 49.6 ± 10.4 (5.0) | 18.7 ± 2.4 (4.2) | 8.3 ± 10.2 (N.C.) | N.D. (N.C.) | 26.5 |
| Epididymis | 232.5 ± 61.7 (0.9) | 352.5 ± 76.0 (9.5) | 259.9 ± 44.4 (26.0) | 167.6 ± 35.4 (37.2) | 44.9 ± 3.8 (N.C.) | 11.2 ± 3.3 (N.C.) | 38.1 |
| Seminal vesicle | 329.0 ± 105.4 (1.2) | 133.8 ± 57.4 (3.6) | 51.0 ± 6.3 (5.1) | 18.7 ± 4.4 (4.2) | 1.7 ± 2.0 (N.C.) | N.D. (N.C.) | 12.7 |
| Aorta | 488.7 ± 78.4 (1.8) | 99.0 ± 46.0 (2.7) | 35.7 ± 16.7 (3.6) | 15.4 ± 5.2 (3.4) | N.D. (N.C.) | N.D. (N.C.) | 7.3 |
| Skin | 366.3 ± 77.9 (1.4) | 199.1 ± 47.5 (5.4) | 99.2 ± 14.6 (9.9) | 55.2 ± 2.9 (12.3) | 18.9 ± 1.6 (N.C.) | 9.3 ± 3.3 (N.C.) | 59.5 |
| Skeletal muscle | 242.5 ± 66.9 (0.9) | 46.7 ± 20.1 (1.3) | 16.7 ± 2.4 (1.7) | 7.9 ± 1.1 (1.8) | N.D. (N.C.) | N.D. (N.C.) | 7.7 |
| Bone | 118.7 ± 22.2 (0.4) | 52.3 ± 19.9 (1.4) | 23.8 ± 7.5 (2.4) | 11.8 ± 3.1 (2.6) | 3.4 ± 0.9 (N.C.) | 0.6 ± 0.7 (N.C.) | 34.1 |
| Bone marrow | 880.0 ± 223.0 (3.3) | 289.9 ± 63.9 (7.8) | 159.2 ± 15.7 (15.9) | 98.6 ± 7.0 (21.9) | 30.7 ± 5.7 (N.C.) | 7.0 ± 4.9 (N.C.) | 38.6 |
| Fat | 120.4 ± 28.3 (0.5) | 33.3 ± 9.7 (0.9) | 17.2 ± 2.8 (1.7) | 7.8 ± 1.2 (1.7) | 2.2 ± 1.5 (N.C.) | N.D. (N.C.) | 21.7 |
| Brown fat | 501.8 ± 157.5 (1.9) | 134.2 ± 55.8 (3.6) | 57.6 ± 10.4 (5.8) | 26.9 ± 2.8 (6.0) | 7.5 ± 1.3 (N.C.) | 3.0 ± 1.1 (N.C.) | 48.1 |
| Mesenteric lymph nodes | 853.9 ± 176.8 (3.2) | 269.9 ± 46.4 (7.3) | 128.0 ± 21.0 (12.8) | 77.9 ± 9.6 (17.3) | 22.6 ± 3.3 (N.C.) | 9.2 ± 2.2 (N.C.) | 49.3 |
| Stomach | 2193.4 ± 953.1 (8.2) | 243.1 ± 117.9 (6.5) | 47.6 ± 12.0 (4.8) | 21.7 ± 1.7 (4.8) | 4.2 ± 1.5 (N.C.) | 1.3 ± 1.6 (N.C.) | 37.5 |
| Small intestine | 4476.6 ± 1778.8 (16.8) | 2688.4 ± 1240.2 (72.3) | 344.0 ± 52.7 (34.4) | 179.7 ± 29.3 (39.9) | 57.8 ± 11.4 (N.C.) | 34.8 ± 13.0 (N.C.) | 65.8 |
| Cecum | 332.4 ± 101.9 (1.2) | 1103.8 ± 713.4 (29.7) | 627.2 ± 294.5 (62.7) | 123.4 ± 23.7 (27.4) | 12.8 ± 2.7 (N.C.) | 5.6 ± 2.5 (N.C.) | 35.3 |
| Large intestine c | 416.0 ± 110.3 (1.6) | 342.6 ± 160.7 (9.2) | 475.8 ± 195.4 (47.6) | 120.7 ± 13.9 (26.8) | 19.6 ± 3.5 (N.C.) | 7.4 ± 3.3 (N.C.) | 38.4 |
Data are expressed as the mean ± standard deviation of four animals. The values in parentheses represent the ratio of the tissue concentration to the plasma concentration (K p) calculated from the mean value.
Average lower limit of quantification (plasma) ≈ 2 ng eq./ml.
The t 1/2 for the tissue concentration was calculated using the mean value at the last three quantifiable time‐points after administration.
Excluding the cecum.
N.D., not determined; N.C., not calculated.
Whole‐body autoradiography in SD rats (study 2)
Figure 4 shows a whole‐body autoradiogram of a fasted SD rat given a single oral dose of 1 mg/kg [14C]teneligliptin. At 0.5 h after administration, the radioactivity was rapidly distributed to tissues throughout the body, and the concentration was greatest in the kidney and liver, as well as the gastrointestinal tract. In the kidney, radioactivity was mainly detected in the outer layer of the medulla, with lower concentrations in the cortex and renal pelvis. At 168 h after administration, the kidney showed a relatively high level of radioactivity. A low level of radioactivity was detected in the liver, and negligible radioactivity was detected in the other tissues examined. In the kidney, the radioactivity was mainly detected in the outer layer of the medulla at all time‐points.
Figure 4.
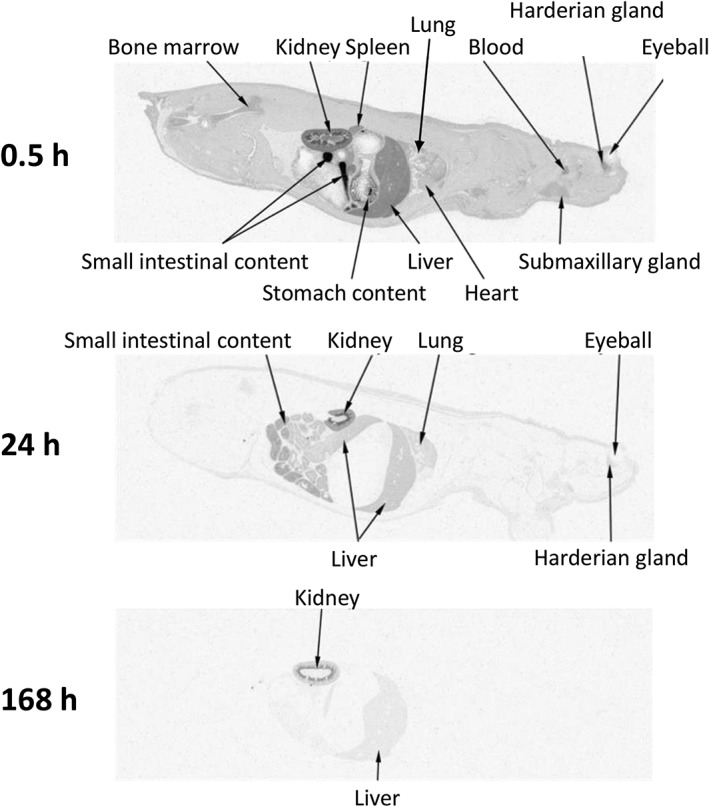
Whole‐body autoradiograms of one SD rat after a single oral dose of 1 mg/kg [14C]teneligliptin
Whole‐body autoradiography in wild‐type or DPP‐4‐deficient rats (study 3)
Figures 5, 6 show whole‐body autoradiograms after a single oral administration of [14C]teneligliptin at 1 mg/kg to fasted wild‐type (Figure 5) and DPP‐4‐deficient (Figure 6) rats. The radioactivity concentration–time profiles in the kidney, liver, lung, heart and pancreas are shown in Figure 7. At 0.5 h after administration, the radioactivity was rapidly distributed to most organs in both strains, especially the kidney and liver, in addition to the gastrointestinal tract. The tissue distribution was similar in both strains, except for the kidney, where the radioactivity concentration in the DPP‐4‐deficient rats was approximately 45% of that in wild‐type rats at 0.5 h after dosing. The radioactivity concentration–time profiles in the heart and pancreas were similar in both strains, whereas those in the kidney, liver and lung indicated greater concentrations for a longer duration in wild‐type rats than in DPP‐4‐deficient rats. The t 1/2 values for the radioactivity concentrations in the kidney, liver and lung were 257, 41.9 and 13.6 h, respectively, in wild‐type rats compared with 12.6 h, 13.4 h and undeterminable, respectively, in DPP‐4‐deficient rats. At 48 h after administration, however, the autoradiograms obtained for the DPP‐4‐deficient rat showed that the radioactivity concentrations in most tissues were low or undetectable, whereas radioactivity was detectable in almost all tissues, except for the pituitary gland, thyroid, heart and brown adipose tissue, in wild‐type rats. The autoradiograms of the kidney at all time‐points revealed the presence of radioactivity in the outer layer of the medulla in wild‐type rats, which was similar to that in SD rats, but not DPP‐4‐deficient rats.
Figure 5.
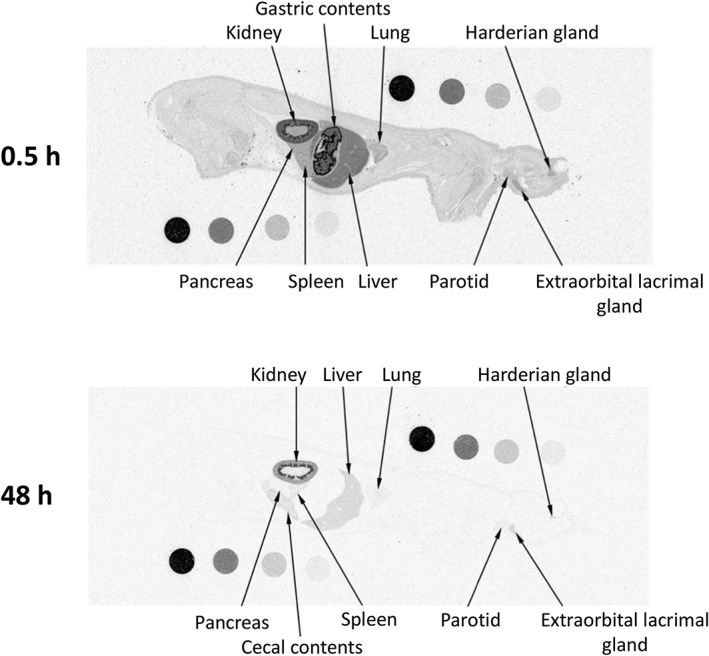
Whole‐body autoradiograms of one wild‐type Fischer rat after a single oral dose of 1 mg/kg [14C]teneligliptin
Figure 6.
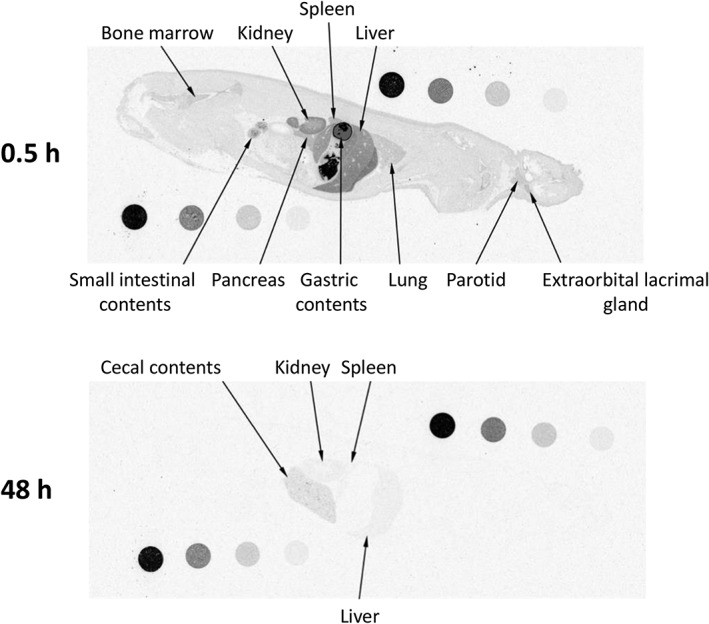
Whole‐body autoradiograms of one DPP‐4‐deficient rat after a single oral dose of 1 mg/kg [14C]teneligliptin
Figure 7.
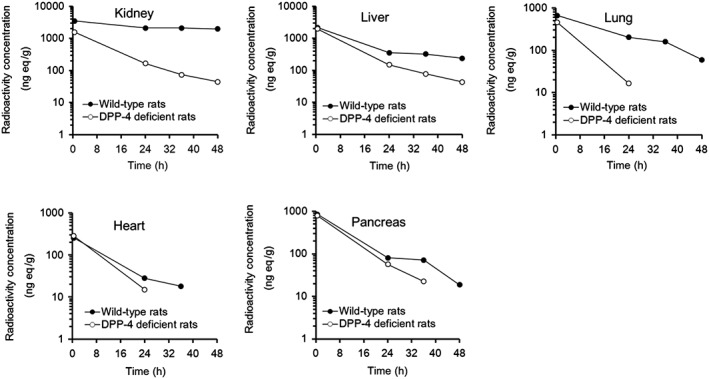
Tissue radioactivity concentration–time profiles (ng eq/g) in wild‐type Fischer rats or DPP‐4‐deficient rats after a single oral dose of 1 mg/kg [14C]teneligliptin. One SD rat was sampled at each time point
Metabolite profiles in plasma and kidney (study 4)
Figures 8, 9 show the representative radiochromatograms of plasma at 0.5 and 5 h and of the kidney at 0.5, 24 and 72 h after a single dose of 1 mg/kg [14C]teneligliptin in fasted SD rats. Teneligliptin and its metabolites in plasma or kidney were identified by comparing their HPLC‐ultraviolet chromatographic retention times with those of the reference standards. Teneligliptin was the most abundant radioactive component in plasma and kidney at all of the time‐points measured. M1 (thiazolidine‐1‐oxide metabolite) was detected at low levels in plasma at 0.5 h after administration. The other metabolites were not detected in plasma. Only M4 (hydroxymethyl metabolite) was detected in trace concentrations in the kidney.
Figure 8.
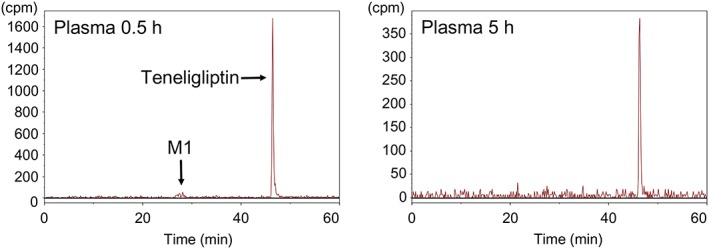
Representative HPLC radiochromatograms of plasma levels (cpm) from SD rats after a single oral dose of 1 mg/kg [14C]teneligliptin. Two or four SD rats were sampled at each time point
Figure 9.
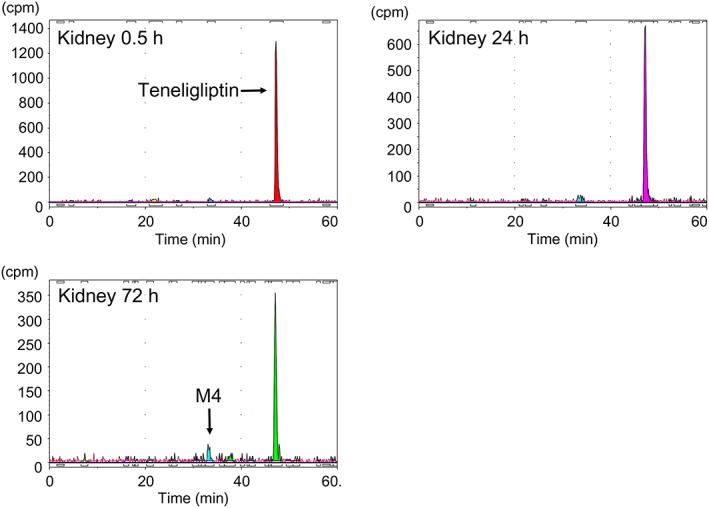
Representative HPLC radiochromatograms of kidney levels (cpm) from SD rats after a single oral dose of 1 mg/kg [14C]teneligliptin. Two or four SD rats were sampled at each time point
Discussion
This study showed that after an oral dose of [14C]teneligliptin, radioactivity corresponding to this molecule was rapidly distributed throughout the whole body of rats. Our findings suggest that most radioactivity distributed to these tissues corresponds to the unchanged drug. The volume of distribution (V ss: 8.9 l/kg, Table 1) based on the plasma concentration–time profile of the unchanged drug after the intravenous administration of teneligliptin to rats was greater than the extracellular fluid volume (approximately 0.3 l/kg) 27, consistent with a broad tissue distribution profile. Similar V ss values were obtained after intravenous administration of other DPP‐4 inhibitors (range, 3.5–50.3 l/kg), with the exception of anagliptin (0.68 l/kg), which indicated that most DPP‐4 inhibitors have a high tissue distribution (Table 1). After oral administration of [14C]teneligliptin to SD rats, it was predominantly distributed to the kidney and liver, in addition to the gastrointestinal tract, and high concentrations were also detected in the lung, spleen and pituitary gland. Of note, the t 1/2 values for radioactivity in the kidney and liver were approximately 10 times higher than the plasma t 1/2, which indicates that teneligliptin is slowly eliminated from the kidney and liver. Similar organ‐specific elimination patterns were also observed in wild‐type Fischer rats, which were used as a control for the DPP‐4‐deficient rats. Notably, the t 1/2 values for the elimination of [14C]teneligliptin radioactivity were much higher in the kidney and liver (257 and 41.9 h, respectively) than in other tissues (Figure 7). The t 1/2 values for the elimination of radioactivity from the kidney and liver were much lower in DPP‐4‐deficient rats (12.6 and 13.4 h, respectively). Additionally, there were no marked differences in the radioactivity concentration–time profile in the heart and pancreas between wild‐type and DPP‐4‐deficient rats. Mentlein previously reported that tissue DPP‐4 activity was greatest in the kidney, followed by the lung, adrenal gland, jejunum and liver, and was lowest in the heart and pancreas 28. These results suggest that tissue DPP‐4 is involved in the tissue distribution of teneligliptin.
In this study, the radioactivity in the kidney in wild‐type Fischer rats was greatest in the outer layer of the renal medulla followed by the renal cortex, and was lowest in the renal pelvis, consistent with the distribution detected in SD rats. By contrast, the radioactivity was not specifically distributed to the outer layer of the renal medulla in DPP‐4‐deficient rats. In a study by Fukasawa et al., immunostaining revealed that DPP‐4 expression was greatest around the outer layer of the renal medulla in the rat kidney 29. Thus, the distribution of DPP‐4 expression in the kidney was consistent with the distribution of radioactivity after administration of [14C]teneligliptin, and most radioactivity in the kidney was present as unchanged teneligliptin (Figure 9). These findings suggest that teneligliptin has a high affinity for DPP‐4 in the kidney.
The tissue‐to‐plasma ratio of radioactivity (K p) for all of the currently marketed DPP‐4 inhibitors tends to be greater in the kidney, liver and lung than in other organs, and linagliptin and teneligliptin had higher renal K p values (around 100) (Table 3). The renal K p values for saxagliptin and anagliptin were approximately 5 and 7, respectively. Although the V ss values suggest that DPP‐4 inhibitors show high tissue distribution, the extent of drug distribution in the kidney differed markedly among DPP‐4 inhibitors.
Table 3.
Tissue‐to‐plasma (or blood) ratio (K p) of radioactivity after an oral dose of radioisotope‐labeled DPP‐4 inhibitors to rats
| Drug [Reference] | Kidney | Lung | Liver | Heart | Pancreas | Dose (mg/kg) | Sampling time (h) |
|---|---|---|---|---|---|---|---|
| Alogliptin 22 | 29.7 | 8.6 | 17.3 | 2.36 | 4.94 | 3 | 4 |
| Anagliptin 21 | 6.71 | 1.50 | 4.36 | 0.74 | 0.81 | 10 | 6 |
| Linagliptin 23 | 113 | 11.1 | 42.3 | 4.78 | – | 2 | 4 |
| Saxagliptin 24 | 5.29 | 1.32 | 32.3 | 0.55 | 1.01 | 20 | 4 |
| Sitagliptin 25 | 14.7 | 9.09 | 22.0 | 2.73 | 5.69 | 5 | 4 |
| Teneligliptin | 88.5 | 19.2 | 47.1 | 3.20 | 8.00 | 1 | 5 |
| Vildagliptin 26 | 13.4 | 2.60 | 13.6 | 1.48 | 3.29 | 100 | 4 |
The distribution of radioactivity in the kidney after administration of [14C]linagliptin to rats in a prior study 30 was similar to that obtained with [14C]teneligliptin in our study. The authors of the earlier study proposed that binding to DPP‐4 is an important contributor to the high concentration in the kidney. Additionally, a non‐linear tissue distribution of linagliptin was observed after it was intravenously administered to rats at doses above 3 mg/kg 23. These results were attributed to the saturation of its binding to DPP‐4.
In general, the extent of tissue distribution for basic compounds is dependent on logP and pKa, in the absence of active uptake via transporters and specific intracellular binding 31. Because the pKa values of most DPP‐4 inhibitors are similar, it seems likely that logP values may be an important factor involved in their tissue distributions (Table 1). In fact, the ClogP was highest for teneligliptin (2.24) followed by linagliptin (1.91).
Nabeno et al. compared the binding modes of teneligliptin and linagliptin with DPP‐4 by x‐ray crystallography 10. As shown in Table 1, linagliptin and teneligliptin bind via distinct modes. Linagliptin binds to the S1, S2, S’1 and S’2 subsites, whereas teneligliptin binds to S1, S2 and S2 extensive subsites with high affinity owing to a considerably rigid structure. Because the physiological significance of this difference is unknown, further investigations are needed.
DPP‐4 is also expressed as a membrane‐bound form in many tissues, including the kidney and liver. Because DPP‐4 is also involved in inflammation, tissue injury and oxidative stress, DPP‐4 inhibitors may have protective effects in the kidney, liver and vascular tissue, in addition to their glucose‐lowering effects 32, 33, 34, 35, 36. The kidney has the highest DPP‐4 activity of all tissues, and DPP‐4 expression was up‐regulated in animal models of diabetic nephropathy and in human diabetes patients 36. Although the effects of teneligliptin on the kidney have not been clearly demonstrated, other DPP‐4 inhibitors were reported to show a renoprotective effect. Despite the weak glucose‐lowering effect of DPP‐4 inhibitors in a type 1 diabetes model, DPP‐4 inhibitors have generally improved renal function in this model. In clinical use, DPP‐4 inhibitors were reported to reduce albuminuria independent of the reduction in hemoglobin A1c (a marker of blood glucose control) 37. These results suggested that DPP‐4 inhibitors have a renoprotective effect independent of blood glucose control. Although the mechanism is not fully understood, inhibition of interactions between DPP‐4 and integrin β1 by DPP‐4 inhibitors is an important factor for its antifibrotic effect in diabetic kidneys 38. Regarding vascular protection, acetylcholine‐induced vascular relaxation in a ZDF rat model was significantly stronger with linagliptin compared with sitagliptin, although both drugs produced equivalent decreases in blood glucose. Vascular malondialdehyde, an oxidative stress marker, and vascular DPP‐4 activity were also attenuated by these drugs, with linagliptin producing significantly greater attenuation than sitagliptin. The author of this study suggested that the difference in effect between these drugs might be related to the vascular tissue distribution properties; thus, the vascular inhibition of DPP‐4 may be important for vascular protection 39. Teneligliptin attenuated endothelial dysfunction through the up‐regulation of eNOS and decreased TNF‐α expression in the aorta in an SHRcp model 40. Although the role of tissue DPP‐4 should be further investigated, differences in the tissue distributions of DPP‐4 inhibitors might be related to differences in their pleiotropic effects. In our study, teneligliptin showed a high affinity for the kidney and other organs with high DPP‐4 expression. Therefore, teneligliptin may have protective effects on these organs independent of its blood glucose‐lowering activity.
In conclusion, all DPP‐4 inhibitors are predominantly distributed to the kidney, liver and lung, all of which show high DPP‐4 expression. Marked differences were also found in the extent of tissue distribution for these DPP‐4 inhibitors. Teneligliptin showed greater distribution, especially in the kidney and the liver, than other DPP‐4 inhibitors. Further studies are needed to examine the relationship between the tissue distribution of DPP‐4 inhibitors and their pleiotropic effects by inhibiting DPP‐4.
Conflict of Interest
The authors are employees of Mitsubishi Tanabe Pharma Co., the manufacturer of teneligliptin.
Acknowledgements
This study was funded by Mitsubishi Tanabe Pharma Co. The authors thank Nicholas D. Smith, PhD, for editorial support, which was funded by Mitsubishi Tanabe Pharma Co.
Nakamaru, Y. , Akahoshi, F. , Iijima, H. , Hisanaga, N. , and Kume, T. (2016) Tissue distribution of teneligliptin in rats and comparisons with data reported for other dipeptidyl peptidase‐4 inhibitors. Biopharm. Drug Dispos., 37: 142–155. doi: 10.1002/bdd.2003.
References
- 1. Drucker DJ. The role of gut hormones in glucose homeostasis. J Clin Invest 2007; 117: 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kato N, Oka M, Murase T, et al. Discovery and pharmacological characterization of N‐[2‐({2‐[(2S)‐2‐cyanopyrrolidin‐1‐yl]‐2‐oxoethyl}amino)‐2‐methylpropyl]‐2‐methylpyrazolo[1,5‐a]pyrimidine‐6‐carboxamide hydrochloride (anagliptin hydrochloride salt) as a potent and selective DPP‐IV inhibitor. Bioorg Med Chem 2011; 19: 7221–7. doi:10.1016/j.bmc.2011.09.043. [DOI] [PubMed] [Google Scholar]
- 3. Augeri DJ, Robl JA, Betebenner DA, et al. Discovery and preclinical profile of saxagliptin (BMS‐477118): a highly potent, long‐acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem 2005; 48: 5025–37. [DOI] [PubMed] [Google Scholar]
- 4. Yoshida T, Akahoshi F, Sakashita H, et al. Discovery and preclinical profile of teneligliptin (3‐[(2S,4S)‐4‐[4‐(3‐methyl‐1‐phenyl‐1H‐pyrazol‐5‐yl)piperazin‐1‐yl] pyrrolidin‐2‐ylcarbonyl] thiazolidine): a highly potent, selective, long‐lasting and orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Bioorg Med Chem 2012; 20: 5705–19. doi:10.1016/j.bmc.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 5. Villhauer EB, Brinkman JA, Naderi GB, et al. 1‐[[(3‐Hydroxy‐1‐adamantyl)amino]acetyl]‐2‐cyano‐(S)‐pyrrolidine: a potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J Med Chem 2003; 46: 2774–89. [DOI] [PubMed] [Google Scholar]
- 6. Feng J, Zhang Z, Wallace MB, et al. Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J Med Chem 2007; 50: 2297–300. [DOI] [PubMed] [Google Scholar]
- 7. Eckhardt M, Langkopf E, Mark M, et al. 8‐(3‐I‐aminopiperidin‐1‐yl)‐7‐but‐2‐ynyl‐3‐methyl‐1‐(4‐methyl‐quinazolin‐2‐ylmethyl)‐3,7‐dihydropurine‐2,6‐dione (BI 1356), a highly potent, selective, long‐acting, and orally bioavailable DPP‐4 inhibitor for the treatment of type 2 diabetes. J Med Chem 2007; 50: 6450–3. [DOI] [PubMed] [Google Scholar]
- 8. Kim D, Wang L, Beconi M, et al. (2R)‐4‐Oxo‐4‐[3‐(trifluoromethyl)‐5,6‐dihydro[1,2,4]‐triazolo[4,3‐a] pyrazin‐7(8H)‐yl]‐1‐(2,4,5‐trifluorophenyl)butan‐2‐amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem 2005; 48: 141–51. [DOI] [PubMed] [Google Scholar]
- 9. Olson GL, Bolin DR, Bonner MP, et al. Concepts and progress in the development of peptide mimetics. J Med Chem 1993; 36: 3039–49. [DOI] [PubMed] [Google Scholar]
- 10. Nabeno M, Akahoshi F, Kishida H, et al. A comparative study of the binding modes of recently launched dipeptidyl peptidase IV inhibitors in the active site. Biochem Biophys Res Commun 2013; 434: 191–6. doi:10.1016/j.bbrc.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 11. Kimata H, Fukuda‐Tsuru S, Yoshida K, et al. Teneligliptin, a novel selective dipeptidyl peptidase‐4 inhibitor, improves postprandial hyperglycemia in a model of type 2 diabetes, Zucker diabetic fatty rats. Med Consult New Remedies 2012; 49: 9. [Google Scholar]
- 12. Golightly LK, Drayna CC, McDermott MT. Comparative clinical pharmacokinetics of dipeptidyl peptidase‐4 inhibitors. Clin Pharmacokinet 2012; 51: 501–14. doi:10.2165/11632930-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 13. Scheen AJ. Pharmacokinetics of dipeptidylpeptidase‐4 inhibitors. Diabetes Obes Metab 2010; 12: 648–58. doi:10.1111/j.1463-1326.2010.01212.x. [DOI] [PubMed] [Google Scholar]
- 14. Goda M, Kadowaki T. Teneligliptin for the treatment of type 2 diabetes. Drugs Today (Barc) 2013; 49: 615–29. doi:10.1358/dot.2013.49.10.2035882. [DOI] [PubMed] [Google Scholar]
- 15. Morishita R, Nakagami H. Teneligliptin: expectations for its pleiotropic action. Expert Opin Pharmacother 2015; 16: 417–26. doi:10.1517/14656566.2015.1000301. [DOI] [PubMed] [Google Scholar]
- 16. Nakamaru Y, Hayashi Y, Ikegawa R, et al. Metabolism and disposition of the dipeptidyl peptidase IV inhibitor teneligliptin in humans. Xenobiotica 2014; 44: 242–53. doi:10.3109/00498254.2013.816891. [DOI] [PubMed] [Google Scholar]
- 17. Halabi A, Maatouk H, Siegler KE, Faisst N, Lufft V, Klause N. Pharmacokinetics of teneligliptin in subjects with renal impairment. Clin Pharmacol Drug Develop 2013; 2: 246–54. doi:10.1002/cpdd.29. [DOI] [PubMed] [Google Scholar]
- 18. Halabi A, Maatouk H, Siegler KE, Faisst N, Holger H. Pharmacokinetics and safety of teneligliptin in subjects with hepatic impairment. Clin Pharmacol Drug Develop 2013; 3: 290–6. doi:10.1002/cpdd.89. [DOI] [PubMed] [Google Scholar]
- 19. Lee B, Shi L, Kassel DB, Asakawa T, Takeuchi K, Christopher RJ. Pharmacokinetic, pharmacodynamic, and efficacy profiles of alogliptin, a novel inhibitor of dipeptidyl peptidase‐4, in rats, dogs, and monkeys. Eur J Pharmacol 2008; 589: 306–14. doi:10.1016/j. [DOI] [PubMed] [Google Scholar]
- 20. Fura A, Khanna A, Vyas V, et al. Pharmacokinetics of the dipeptidyl peptidase 4 inhibitor saxagliptin in rats, dogs, and monkeys and clinical projections. Drug Metab Dispos 2009; 37: 1164–71. doi:10.1124/dmd.108.026088. [DOI] [PubMed] [Google Scholar]
- 21. Furuta S, Tamura M, Hirooka H, Mizuno Y, Miyoshi M, Furuta Y. Pharmacokinetic disposition of anagliptin, a novel dipeptidyl peptidase‐4 inhibitor, in rats and dogs. Eur J Drug Metab Pharmacokinet 2013; 38: 87–96. doi:10.1007/s13318-013-0119-z. [DOI] [PubMed] [Google Scholar]
- 22. Nesina® (alogliptin), Common Technical Document . Takeda Pharmaceutical Company 2010. Available at: http://www.pmda.go.jp/drugs/2010/P201000029/400256000_22200AMX00309_I100_2.pdf (in Japanese). Last accessed 26 June 2015.
- 23. Trazenta® (linagliptin), Common Technical Document 2011. Available at: http://www.pmda.go.jp/drugs/2011/P201100128/530353000_22300AMX00605_I100_1.pdf (in Japanese). Last accessed 26 June 2015.
- 24. Onglyza® (saxagliptin), Common Technical Document . Bristol‐Myers Squibb 2013. Available at: http://www.pmda.go.jp/drugs/2013/P201300036/180078000_22500AMX00877_I100_1.pdf (in Japanese). Last accessed 26 June 2015.
- 25. Januvia® (sitagliptin), Common Technical Document . Merck and Co. 2009. Available at: http://www.pmda.go.jp/drugs/2009/P200900043/63015300_22100AMX02257_I100_1.pdf (in Japanese). Last accessed 26 June 2015.
- 26. Equa® (vildagliptin), Common Technical Document . Novartis 2010. Available at: http://www.pmda.go.jp/drugs/2010/P201000008/30024200_22200AMX00233_I100_5.pdf (in Japanese). Last accessed 26 June 2015.
- 27. Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res 1993; 10: 1093–5. [DOI] [PubMed] [Google Scholar]
- 28. Mentlein R. Dipeptidyl‐peptidase IV (CD26)‐role in the inactivation of regulatory peptides. Regul Pept 1999; 85: 9–24. [DOI] [PubMed] [Google Scholar]
- 29. Fukasawa KM, Fukasawa K, Sahara N, Harada M, Kondo Y, Nagatsu I. Immunohistochemical localization of dipeptidyl aminopeptidase IV in rat kidney, liver, and salivary glands. J Histochem Cytochem 1981; 29: 337–43. [DOI] [PubMed] [Google Scholar]
- 30. Fuchs H, Binder R, Greischel A. Tissue distribution of the novel DPP‐4 inhibitor BI 1356 is dominated by saturable binding to its target in rats. Biopharm Drug Dispos 2009; 30: 229–40. doi:10.1002/bdd.662. [DOI] [PubMed] [Google Scholar]
- 31. Siebert GA, Hung DY, Chang P, Roberts MS. Ion‐trapping, microsomal binding, and unbound drug distribution in the hepatic retention of basic drugs. J Pharmacol Exp Ther 2004; 308: 228–35. [DOI] [PubMed] [Google Scholar]
- 32. Aroor AR, Sowers JR, Jia G, DeMarco VG. Pleiotropic effects of the dipeptidylpeptidase‐4 inhibitors on the cardiovascular system. Am J Physiol Heart Circ Physiol 2014; 307: H477–92. doi:10.1152/ajpheart.00209.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Avogaro A, Fadini GP. The effects of dipeptidyl peptidase‐4 inhibition on microvascular diabetes complications. Diabetes Care 2014; 37: 2884–94. doi:10.2337/dc14-0865. [DOI] [PubMed] [Google Scholar]
- 34. Itou M, Kawaguchi T, Taniguchi E, Sata M. Dipeptidyl peptidase‐4: a key player in chronic liver disease. World J Gastroenterol 2013; 19: 2298–306. doi:10.3748/wjg.v19.i15.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim NH, Yu T, Lee DH. The nonglycemic actions of dipeptidyl peptidase‐4 inhibitors. Biomed Res Int 2014; 2014: 368703. doi:10.1155/2014/368703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tanaka T, Higashijima Y, Wada T, Nangaku M. The potential for renoprotection with incretin‐based drugs. Kidney Int 2014; 86: 701–11. doi:10.1038/ki.2014.236. [DOI] [PubMed] [Google Scholar]
- 37. Groop PH, Cooper ME, Perkovic V, Emser A, Woerle HJ, von Eynatten M. Linagliptin lowers albuminuria on top of recommended standard treatment in patients with type 2 diabetes and renal dysfunction. Diabetes Care 2013; 36: 3460–8. doi:10.2337/dc13-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi S, Srivastava SP, Kanasaki M, et al. Interactions of DPP‐4 and integrin β1 influences endothelial‐to‐mesenchymal transition. Kidney Int. 2015; 88: 479–89. doi:10.1038/ki.2015.103. [DOI] [PubMed] [Google Scholar]
- 39. Takai S, Sakonjo H, Jin D. Significance of vascular dipeptidyl peptidase‐4 inhibition on vascular protection in Zucker diabetic fatty rats. J Pharmacol Sci 2014; 125: 386–93. [DOI] [PubMed] [Google Scholar]
- 40. Nakagami H, Pang Z, Shimosato T, et al. The dipeptidyl peptidase‐4 inhibitor teneligliptin improved endothelial dysfunction and insulin resistance in the SHR/NDmcr‐cp rat model of metabolic syndrome. Hypertens Res 2014; 37: 629–35. doi:10.1038/hr.2014.53. [DOI] [PubMed] [Google Scholar]