

Abstract
Cancer metastasis is a multistep process that involves a series of tumor‐stromal interaction, including extracellular matrix (ECM) remodeling, which requires a concerted action of multiple proteolytic enzymes and their endogenous inhibitors. This study investigated the role of tissue inhibitor of metalloproteinases (TIMP) 2 in the context of hepatocellular carcinoma (HCC) metastasis. We found that TIMP2 was the most significantly down‐regulated member among the TIMP family in human HCCs. Moreover, TIMP2 underexpression was frequent (41.8%; 23 of 55) in human HCCs and was significantly associated with liver invasion and poorer survival outcomes of HCC patients. Furthermore, stable silencing of TIMP2 in HCC cell lines enhanced cell invasive ability and ECM degradation associated with formation of invadopodia‐like feature, suggesting that TIMP2 is a negative regulator of HCC metastasis. Using an orthotopic tumor xenograft model, we demonstrated that ectopic expression of TIMP2 open reading frame in the highly metastatic HCC cell line, MHCC‐97L, significantly reduced HCC progression as well as pulmonary metastasis. Mechanistically, TIMP2 suppression, in a hypoxic environment, was induced through a regulatory feedback circuit consisting of hypoxia‐inducible factor (HIF) 1 alpha, microRNA‐210 (miR‐210), and HIF‐3α. Conclusion: TIMP2 is frequently down‐regulated in human HCCs and its down‐regulation is associated with aggressive tumor behavior and poorer patient outcome. Its suppression is under the regulation of a novel feedback circuit consisting of HIF‐1α/miR‐210/HIF‐3α. TIMP2 is an important regulator of ECM degradation and HCC metastasis. (Hepatology 2016;64:473‐487)
Abbreviations
- ECM
extracellular matrix
- EV
empty vector
- FFPE
formalin‐fixed paraffin‐embedded
- HCC
hepatocellular carcinoma
- HIFs
hypoxia‐inducible factors
- HPRT
hypoxanthine‐guanine phosphoribosyltransferase
- HREs
hypoxia‐responsive elements
- IHC
immunohistochemical
- LNA
locked nucleic acid
- miRNAs
microRNAs
- MMPs
matrix metalloproteinases
- mRNA
messenger RNA
- NTC
nontarget control
- NTLs
nontumorous livers
- ORF
open reading frame
- qPCR
quantitative polymerase chain reaction
- shRNA
short hairpin RNA
- T
tumorous
- TACE
transarterial chemo‐embolization
- TIMPs
tissue inhibitor of metalloproteinases
- VMP1
vacuole membrane protein 1
Liver cancer (hepatocellular carcinoma; HCC) is one of the most common fatal cancers worldwide,1 being the third‐leading cause of cancer deaths in the world and the second‐leading cause of cancer deaths in China and Southeast Asia.2 The poor prognosis of this disease is mainly attributable to the high rate of tumor recurrence or metastasis, contributing to around 90% of cancer‐related mortality.3
Hypoxia is a prevalent tumor microenvironment in HCC, attributed to the insufficient vascular networks to support the rapidly growing tumor. Hypoxia‐inducible factors (HIFs), consisting of an oxygen‐sensitive α subunit (HIFα) and a constitutively expressed β‐unit (HIFβ), are one of the most important transcriptional regulators that facilitate cellular response to hypoxia. In humans, three distinct forms of HIF‐α have been identified (HIF‐1α, HIF‐2α, and HIF‐3α), and all of them are involved in regulating transcriptional programs in response to hypoxia.
Cancer metastasis is a complicated process that involves concerted actions of many transcriptional regulators and proteolytic enzymes, some of which have been demonstrated to be regulated by hypoxia and HIFs in solid tumors. Invasion of the extracellular matrix (ECM) is an early and essential step of the metastatic cascade. Matrix metalloproteinases (MMPs) are extracellular endopeptidases primarily responsible for degradation of ECM proteins, preparing the path for tumor cells to invade and migrate across the stroma for distant metastasis.4, 5 Therefore, inhibition of MMPs may reduce ECM degradation and suppress dissemination of primary tumor cells. Apart from activation of zymogen forms and transcriptional control, activities of MMP family members are also regulated by their endogenous inhibitors, namely, the tissue inhibitors of metalloproteinases (TIMPs).6 Currently, four different members have been identified in the TIMP family (TIMP1, TIMP2, TIMP3, and TIMP4), and their primary function is to provide extracellular regulation of the proteolytic activities of MMPs.7 Despite potentially inhibiting all the MMPs, each TIMP member has different efficacies against different MMP members.
Deregulation of microRNAs (miRNAs) has been implicated in both promotion and suppression of cancer metastasis by functioning as posttranscriptional regulators of oncogenes or tumor‐suppressor genes.8, 9, 10, 11 Under this study, we sought to explore the contribution of the miRNA/HIF‐1α feedback loop in promoting the invasive abilities of HCC cells as well as its interaction with TIMP2. Here, we showed that TIMP2 suppression induced invadopodia‐like features in HCC cells, which, in turn, enhanced ECM degradation. Under hypoxia, TIMP2 was suppressed through a HIF‐1α‐dependent mechanism and the suppression was under regulation of a novel feedback circuit composed of HIF‐1α, miR‐210, and HIF‐3α. Conversely, inhibition of miR‐210 perturbed the feedback circuit, attenuated the suppression of TIMP2 under hypoxia, and eventually reduced HCC cell invasion. Our findings have provided evidence to show that TIMP2 is an important negative regulator of cell invasion, and thus metastasis, and is under regulation of a novel feedback circuit consisting of HIF‐1α/miR‐210/HIF‐3α.
Materials and Methods
CELL LINES AND HUMAN SAMPLES
Human HCC cell lines SMMC‐7721, PLC/PRF/5, MHCC‐97L, and BEL‐7402 were used in the present study. SMMC‐7721 was obtained from the Shanghai Institute of Cell Biology (Shanghai, China). PLC/PRF/5 was obtained from the American Type Culture Collection (Manassas, VA). MHCC‐97L was obtained from Prof. Z.Y. Tang of the Fudan University in Shanghai. All HCC specimens and their corresponding nontumorous liver tissues were resected from Chinese patients between 1991 and 2007 at Queen Mary Hospital, Hong Kong. Thirty‐nine patients were male and 16 were female. Ages ranged from 24 to 82 years (mean = 54.25 years). Overall, 44 of 55 (80%) of the patients had chronic hepatitis B viral infection with positive serum hepatitis B surface antigen status. None of these patients received any other therapies, including chemoembolization or chemotherapy, before hepatic tumor resection. After surgical resection, all specimens were either snap‐frozen immediately in liquid nitrogen and stored at ‐80°C or fixed in buffered 10% formalin for paraffin embedding. Use of clinical specimens was approved by the institutional review board of the University of Hong Kong/Hospital Authority.
ESTABLISHMENT OF STABLE TIMP2 KNOCKDOWN AND OVEREXPRESSING CELLS
TIMP2 was stably knocked down in HCC cell lines by means of lentiviral approach as described.12 In brief, short hairpin RNA (shRNA) against TIMP2 (shTIMP2‐33 or shTIMP2‐34) or nontarget control (shNTC) was cotransfected with the packaging mix into 293FT cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) to produce the lentiviral particles. HCC cells were transduced with the shRNA‐containing or shNTC‐containing recombinant lentivirus, and positive transduced cells were selected for at least two passages by puromycin treatment at a concentration of 1‐2 μg/mL. Successful knockdown or ectopic expression of TIMP2 was confirmed by means of western blotting. Stable TIMP2‐overexpressing HCC cells were similarly generated. TIMP2 knockdown vectors were purchased from Sigma‐Aldrich (St. Louis, MO), and TIMP2‐overexpressing vector was purchased from GeneCopoeia (Rockville, MD). HIF‐1α and HIF‐2α knockdown (shHIF1α and shHIF2α) HCC cells were generated in our previous study.13
RNA EXTRACTION AND QUANTITATIVE POLYMERASE CHAIN REACTION
Total RNA from HCC cell lines or frozen HCC clinical tissues was extracted using TRIzol reagent (Invitrogen, Grand Island, NY). Complementary DNA was synthesized from 1μg of total RNA using the GeneAmp RNA PCR Kit (Applied Biosystems, Foster City, CA). Taqman probes for TIMP1, TIMP2, TIMP3, TIMP4, hypoxanthine‐guanine phosphoribosyltransferase (HPRT), and 18s were purchased from Applied Biosystems. Reverse transcription of miRNAs was performed using the Taqman MicroRNA Reverse Transcription Kit with the microRNA‐specific primers. All quantitative polymerase chain reaction (qPCR) reactions were performed in triplicate using the Taqman Universal PCR Master Mix Kit and 7900 HT Fast Real Time PCR System (Applied Biosystems).
microRNA EXTRACTION AND PROFILING
microRNA (miRNA) from SMMC‐7721, MHCC‐97L‐luc, and PLC/PRF/5 after 48 hours of incubation at normoxic (20% O2) or hypoxic (0.1% O2) condition was extracted using TRIzol reagent (Invitrogen) and subsequently subject to Megaplex reverse transcription (Applied Biosystems). miRNA expression profiles were examined using the Taqman human MiRNA Low‐Density Array Set (v2.0). Expression levels were normalized against endogenous U6 controls. For individual miRNA assay on formalin‐fixed paraffin‐embedded (FFPE) HCC clinical samples, miRNA extraction was performed as described.11 In brief, hypoxic HCC cells in perinecrotic areas, aided by immunohistochemistry (IHC) using the hypoxia marker, CA9, were microdissected by a 27‐gauge needle under a dissecting microscope. miRNA extraction was performed with the miRNeasy FFPE kit (Qiagen, Valencia, CA).
PLASMIDS AND REAGENTS FOR LUCIFERASE REPORTER ASSAYS
Transcriptional activity of HIF‐1α by luciferase reporter assay was examined by luciferase reporter assay as described.14 In brief, a 60‐base‐pair minimal fragment of the cytomegalovirus promoter fused with six copies of consensus sequence of hypoxia‐responsive elements (HREs), tgcatACGTGggctccaa, was subcloned into pGL3 basic luciferase reporter construct. pRL‐SV40 was used as an internal control to normalize transfection efficiency. Twenty‐four hours after transfection of the construct, transfected HCC cells were subject to either normoxia or hypoxia condition for another 24 hours. Firefly and Renilla luciferase signals were determined by dual‐luciferase reporter assay (Promega, Madison, WI), according to the manufacturer's protocol. To examine the negative regulatory role of miR‐210 on HIF‐3α by luciferase reporter assay, two copies of either putative wild‐type or mutated miR‐210 binding sites of the HIF‐3α 3′UTR (untranslated region) were subcloned into the dual‐luciferase miRNA target expression vector, pmirGLO (Promega). Next, 15 pmole of miR‐210 precursor was transfected into BEL‐7402 cells using X‐tremeGene (Roche, Basel, Switzerland). Twenty‐four hours later, 0.5 μg of the pmirGLO construct containing the wild‐type or mutated HIF‐3α 3′UTR‐binding sequence was transfected into BEL‐7402 cells using Fugene 9 (Roche, Indianapolis, IN). Forty‐eight hours after the second transfection, firefly and Renilla luciferase signals were determined by dual‐luciferase reporter assay as described. Transfection efficiency was normalized with Renilla luciferase activity. Three independent experiments were performed for each group.
INVASION ASSAYS
In vitro cell invasive ability was examined by transwell assays as described.15 In brief, 5 × 104 cells were added evenly onto the upper chamber of the transwell, uncoated for migration assay, or coated with a thin layer of BD Matrigel Matrix (BD Biocsciences, Sparks, MD) for invasion assay. After incubation, migrated and invaded cells were fixed with 100% methanol and subsequently stained with 0.1% crystal for assessment. For each experimental group, a minimum of three random fields of stained cells photographed by a camera connected to a phase contrast microscope were counted.
IN VIVO ORTHOTOPIC LIVER INJECTION MODEL
Six‐ to 8‐week‐old BALB/c‐nu/nu athymic male mice were used for in vivo experiments in this study. Orthotopic liver injection was performed using a 25‐μL syringe with a cemented 22‐gauge needle. HCC cells (1 × 106) were resuspended in 20 μL of matrigel diluted with serum‐free cell culture medium in a 1:1 ratio. Mice were anesthetized with 80 mg/kg of pentobarbitol and a small incision was made in the abdomen. The left lobe of the liver was gently exposed and the resuspended cells were carefully injected into the lobe. After injection, the abdominal wound was sutured.
MHCC‐97L cells used for injection were stably transduced with firefly luciferase gene and enabled regular in vivo monitoring of tumor growth by bioluminescent imaging. For in vivo signal detection, 100 mg/kg of d‐luciferin (Xenogen, Hopkinton, MA) was injected intraperitoneally into tumor‐bearing mice and representative bioluminescent images were captured using an IVIS 100 Imaging System (Xenogen). Ex vivo imaging of lung was also performed to examine extrahepatic metastasis of HCC xenografts. All animal experiments were performed according to the Animals (Control of Experiments) Ordinance (Hong Kong) and the institution guidance on animal work.
INVADOPODIA AND IMMUNOFLUORESCENCE
HCC cells were plated on green fluorescent gelatin‐coated coverslips prepared according to the manufacturer's instruction (Millipore), and invadopodia formation was examined by confocal microscopy. After 24‐48 hours of culture, cells were fixed in 4% paraformaldehyde for 30 minutes, followed by immunostaining with phalloidin. Areas of degradation mediated by HCC clones were examined using a laser scanning confocal microscope and images were processed using Photoshop software (Adobe Systems, Newton, MA) and ImageJ (National Institutes of Health, Bethesda, MD). To quantitate the percentage of cells associated with gelatin degradation, a minimum of 200 cells were scored for each experimental group. Cells were scored as positive if the F‐actin‐rich structures colocalized with an area devoid of green fluorescent gelatin.
CLINICOPATHOLOGICAL CORRELATION AND SURVIVAL ANALYSIS
Clinicopathological features were correlated with the TIMP2 IHC protein expression using SPSS 20 for Windows (SPSS, Inc., Chicago, IL). The independent t test was used for continuous parametric data, the Mann‐Whitney U test for continuous nonparametric data, and the Fisher's exact test for continuous parametric data. Survival data were analyzed using Kaplan‐Meier's method and compared between groups by the log‐rank test. Differences were considered statistically significant when the P values were less than 0.05.
Results
TIMP2 WAS FREQUENTLY DOWN‐REGULATED IN HUMAN HCCs
From our whole‐transcriptome sequencing data on 16 pairs of primary human HCCs and their corresponding nontumorous livers (NTLs), we observed that TIMP2 was most substantially underexpressed among all four TIMP family members in HCC tissues (Fig. 1A). We further examined the expression pattern of TIMP2 by qPCR in an expanded cohort of 48 pairs of HCCs and their corresponding NTLs (Fig. 1B). Consistently, TIMP2 messenger RNA (mRNA) levels were found to be significantly underexpressed in human HCCs (P < 0.001, Mann‐Whitney U test). Furthermore, with IHC (Fig. 1C), TIMP2 protein underexpression was frequently 41.8% (23 of 55) observed in HCCs. Upon clinicopathological correlation, TIMP2 protein underexpression was significantly associated with direct tumor invasion into the adjacent liver parenchyma (P = 0.035, Fisher's exact test; Table 1). There was a trend of a positive association of TIMP2 underexpression and tumor microsatellite formation (P = 0.067, Fisher's exact test; Table 1), although it did not reach statistical significance. Furthermore, TIMP2 protein underexpression in HCCs was associated with significantly poorer overall survival rates (median survival rate of 79.1 months vs. 29.5 months; P = 0.002) as well as disease‐free survival rates of HCC patients (median survival rate of 41.3 months vs 15.6 months; P = 0.016; Fig. 1D).
Figure 1.
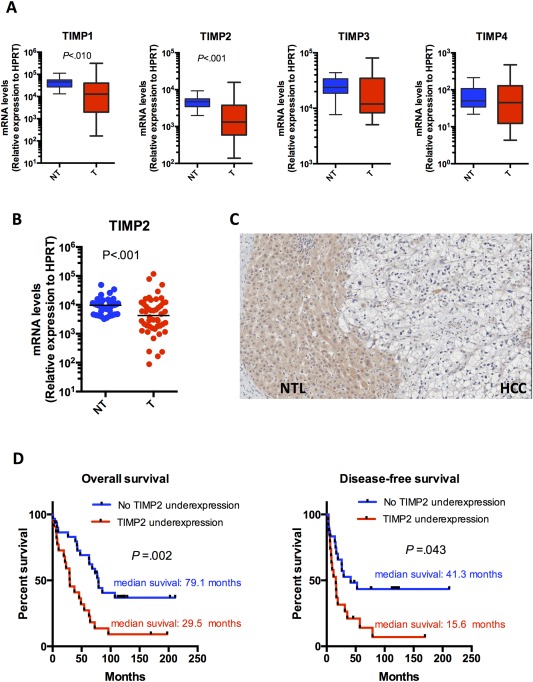
TIMP2 was the most frequently down‐regulated TIMP member in human HCCs. (A) mRNA levels of four TIMP members (TIMP1, −2, −3, and −4) normalized with HPRT in an initial cohort of 16 pairs of tumorous (T) and corresponding nontumorous (NT) liver tissues from HCC patients (Mann‐Whitney U test). (B) mRNA levels of TIMP2 normalized with HPRT in an expanded cohort of 48 pairs of T and corresponding NT tissues (Mann‐Whitney U test; P < 0.001). (C) IHC staining using antibody against TIMP2 on human HCCs and their corresponding NT livers. (D) Underexpression of TIMP2 in human HCCs significantly correlated with both overall and disease‐free survival rates of patients. HCC patients were classified into two groups according to underexpression of TIMP2. Survival data were analyzed using Kaplan‐Meier's method.
Table 1.
Clinicopathological Features of 55 HCC Patients Analyzed
| Clinicopathological Features | No. of Cases Without TIMP2 Underexpression | No. of Cases With TIMP2 Underexpression | P Value |
|---|---|---|---|
| Sex | |||
| Male | 23 | 16 | 0.543 |
| Female | 9 | 7 | |
| Tumor size, cm | |||
| >5 | 16 | 11 | 0.565 |
| ≤5 | 15 | 11 | |
| Liver invasion | |||
| Absent | 21 | 9 | 0.035a |
| Present | 7 | 11 | |
| Tumor microsatellite formation | |||
| Absent | 18 | 7 | 0.067 |
| Present | 14 | 15 | |
| Tumor encapsulation | |||
| Absent | 20 | 18 | 0.142 |
| Present | 11 | 4 | |
| Venous invasion | |||
| Absent | 13 | 9 | 0.602 |
| Present | 19 | 13 | |
| Cellular differentiation | |||
| Edmondson grade I‐II | 15 | 10 | 0.570 |
| Edmondson grade III‐IV | 17 | 12 | |
| TNM staging | |||
| I‐II | 11 | 6 | 0.416 |
| III‐IV | 20 | 15 |
Fisher's exact test.
Abbreviation: TNM, tumor node metastasis.
KNOCKDOWN OF TIMP2 PROMOTED THE INVASIVE ABILITY OF HCC CELLS IN VITRO
Given that down‐regulation of TIMP2 was frequently observed in patient HCCs and associated with aggressive behavior and poorer patient survival, we examined whether knockdown of TIMP2 in HCC cells would enhance their invasive abilities. We observed that TIMP2 was differentially expressed across a panel of HCC cell lines (Supporting Fig. S1). By lentiviral transduction of shRNA, we established stable knockdown TIMP2 clones in HCC cell lines (SMMC‐7721 and PLC/PRF/5), which are lowly metastatic and have high TIMP2 expression (Fig. 2A). By transwell Matrigel cell invasion assay, the number of cells invading the Matrigel chamber was significantly higher in the two independent TIMP2 knockdown clones, as compared to shNTC. Similar results were observed in both HCC cell lines (Fig. 2A,B), suggesting that the proinvasive ability of TIMP2 knockdown was not cell‐line specific. In addition, the regulatory role of TIMP1 was also examined, given that levels of TIMP1 were also underexpressed in primary HCC tissues. Knockdown of TIMP1 did not enhance invasive abilities of both SMMC‐7721 and PLC/PRF/5 (Supporting Fig. S2), suggesting that the preventive role of TIMP2 in HCC cell invasion is specific to TIMP2, but not TIMP1.
Figure 2.
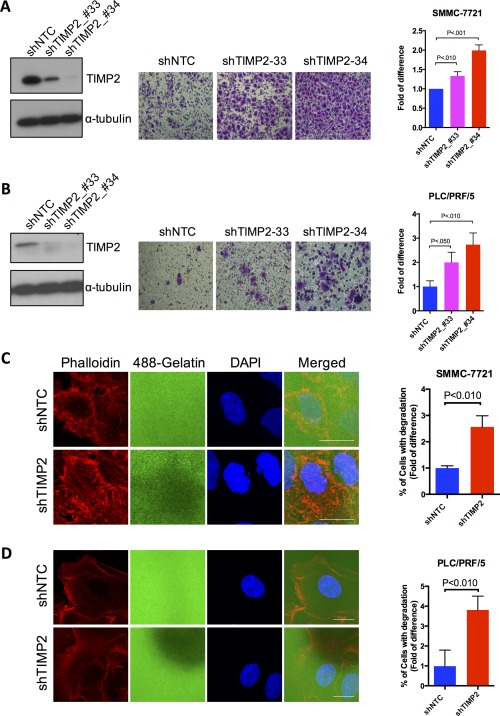
Knockdown of TIMP2 enhanced cell invasion and invadopodia formation in HCC cells. (A,B) Western blotting analysis showed successful knockdown of TIMP2. Knockdown of TIMP2 resulted in increased cell‐invasive abilities. Transwell cell invasion assays of (A) SMMC‐7721 and (B) PLC/PRF/5 were performed after transduction with shNTC and shTIMP2 lentivirus. Quantitative data are presented as mean ± SD, and statistical significance was determined by t test (* P < 0.05; ** P < 0.01; *** P < 0.001). Results were from three independent experiments. (C,D) Representative immunofluorescence images of (C) SMMC‐7721 and (D) PLC/PRF/5 plated on fluorescein‐isosthiocyanate‐conjugated gelatin and stained phalloidin. Scale bar, 20 μm. Invadopodia are defined as gelatin degradation colocalized with F‐actin‐rich structures. Percentage of cells associated with gelatin degradation was calculated as the number of cells with invadopodia normalized to the total number of cells scored (at least 200 cells were scored per experimental group). Quantitative data are presented as mean ± SD, and statistical significance was determined by t test (* P < 0.05; ** P < 0.01; *** P < 0.001). Results were from three independent experiments. Abbreviation: DAPI, 4′,6‐diamidino‐2‐phenylindole.
Invadopodia are highly regulated and transient subcellular protrusions that represent sites of ECM attachment as well as degradation.16 Accumulating evidence suggests that invadopodia highly correlate with cancer cell invasiveness by promoting ECM degradation and cell motility. Therefore, we examined whether TIMP2 expression level affected invadopodia formation and the associated ECM degradation. We observed microscopically that gelatin degradation, reminiscent of ECM degradation, was significantly enhanced in TIMP2 knockdown cells (Fig. 2C,D). Of note, the sites of gelatin degradation appeared to colocalize with cells with F‐actin‐rich structures, suggesting that degradation was mediated by invadopodia‐like features (Supporting Fig. S3A,B and Fig. 2C). Again, the effect of TIMP2 suppression in triggering ECM degradation associated with invadopodia was not cell‐line specific, given that similar observations were consistently found in the other HCC cell line, PLC/PRF/5 (Fig. 2D). These findings, in line with the Matrigel invasion assay, emphasized that suppression of TIMP2 promoted invasive abilities of HCC cells through enhancing ECM degradation association with invadopodia‐like features.
ECTOPIC EXPRESSION OF TIMP2 SUPPRESSED PULMONARY METASTASIS OF HCC IN VIVO
Next, we sought to determine whether ectopic expression of TIMP2 suppressed in vivo distant metastasis of HCC cells by orthotopic injection of luciferase‐labeled, TIMP2‐overexpressing MHCC‐97L cells (TIMP2) and its corresponding empty vector (EV) control cells. Xenogen imaging clearly revealed a significant reduction in volumes of the TIMP2‐overexpressing tumors as compared to those of the EV control group (Fig. 3A). Moreover, irregular, invasive tumor borders at the tumor growth front were observed in all (5 of 5) of the EV group, as compared to 60% (3 of 5) of the TIMP2 group. Of note, tumor microsatellite formation and venous invasion, both being recognized features of HCC metastasis, were not observed in any of the tumor xenografts derived from TIMP2‐overexpressing cells, whereas these pathological features were observed in most of the tumor xenografts derived from the EV control group (Fig. 3B,C). More importantly, lungs harvested from these tumor‐bearing mice with EV tumor xenografts had distinct metastasis formation in 5 of 5 (100%) mice, as compared to only 1 of 5 (20%) mice with TIMP2‐overexpressing tumor xenografts (Fig. 3D). Formation of tumor foci in lung tissues was further confirmed by histological analysis (Fig. 3E).
Figure 3.
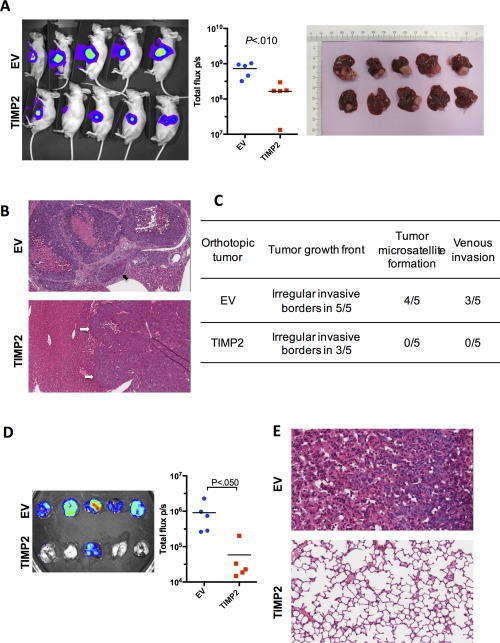
Ectopic expression of TIMP2 suppressed HCC progression and pulmonary metastasis in vivo. (A) Xenogen imaging of nude mice subject to orthotopic liver injection of EV and the open reading frame of TIMP2 in MHCC‐97L cells are shown. MHCC‐97L cells stably overexpressing luciferase were used for the orthotopic injection model. (B) Representative H&E sections of hepatic tumor xenografts are shown. Tumor microsatellite formation around primary tumor xenografts and multiple foci of venous invasion were frequently observed in the EV control group, but not in the TIMP2‐overexpressing group. Black arrow indicates venous invasion and white arrows indicate the tumor boundary. (C) Summary of pathological analysis on hepatic tumor xenografts derived from EV control and TIMP2‐overexpressing MHCC‐97L cells in nude mice. (D) Ex vivo bioluminescent imaging of lung tissues harvested from tumor‐bearing mice is shown. Presence of pulmonary metastasis was detected by luciferase signals. Formation of pulmonary metastases was significantly reduced in nude mice bearing hepatic tumor xenografts with TIMP2 overexpression, as compared to the EV control group. (E) Representative H&E sections of lung tissues confirmed the presence of metastases in the EV control group, as compared to none in the TIMP2‐overexpressing group. Abbreviation: H&E, hematoxylin and eosin.
SUPPRESSION OF TIMP2 BY HYPOXIA WAS MEDIATED BY HIF‐1α/miR‐210/HIF‐3α REGULATORY CIRCUIT
We observed that the expression of TIMP2 was significantly reduced in HCC cells under hypoxia (Supporting Fig. S5A and Fig. 4A). However, suppression of TIMP2 expression was significantly abolished upon knockdown of HIF‐1α, but not HIF‐2α, indicating that suppression of TIMP2 by hypoxia was HIF‐1α dependent.
Figure 4.
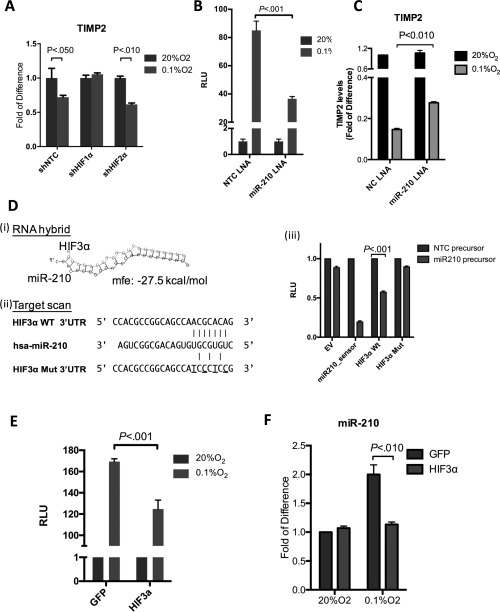
Suppression of TIMP2 by hypoxia was mediated by HIF‐1α/miR‐210/HIF‐3α regulatory circuit in human HCCs. (A) Hypoxic suppression of TIMP2 was abolished upon HIF‐1α knockdown, but not HIF‐2α knockdown. mRNA levels of TIMP2 in shNTC, shHIF‐1α, and shHIF‐2α MHCC‐97L clones were determined by qPCR and normalized to HPRT (P < 0.050, t test; mean ± SD; n = 3). (B) miR‐210 inhibition suppressed the transcriptional activity of HIF‐1α. Luciferase reporter assay demonstrated that miR‐210 inhibition suppressed the activity of HRE‐luciferase reporter construct under hypoxia (P < 0.001, t test; mean ± SD). (C) qPCR analysis of TIMP2 levels in SMMC‐7721 cells, subject to transfection with negative control (NC) LNA inhibitor or miR‐210 LNA inhibitor and incubated under normoxia (20% O2) or hypoxia (0.1% O2; P < 0.010, t test). (D) In silico analysis (i) RNA hybrid and (ii) TargetScan 5.2 revealed a putative miR‐210 target sequence in the 3′UTR of HIF‐3α. This fragment was fused with the pmiRGLO vector to examine the binding by luciferase reporter assay. The mutated seed sequence is underlined. (iii) miRNA luciferase reporter assay. pmiRGLO luciferase reporter fused with two copies of wild‐type miR‐210 target sequences were transfected into BEL‐7402 cells after transfection with NC precursor or miR‐210 precursor. miR‐210 precursor significant suppressed luciferase activity of wild‐type 3′UTR of HIF‐3α (P < 0.001, t test; mean ± SD; n = 5). (E) Luciferase reporter assay demonstrated that ectopic expression of HIF‐3α ORF suppressed activity of HRE‐luciferase reporter construct under hypoxia (P < 0.001, t test; mean ± SD; n = 5). (F) Ectopic expression of HIF‐3α ORF suppressed hypoxic induction of mR‐210. Transcript levels of miR‐210 in SMMC7721 stably expressing the empty vector GFP or HIF‐3α ORF are shown. U6 was used as a housekeeping gene for data normalization (P < 0.010, t test; mean ± SD; n = 3). Abbreviations: GFP, green fluorescent protein; ORF, open reading frame; RLU, relative light units.
Recently, the importance of miRNA transcription factor feedback loops has been highlighted in various human malignancies.17, 18, 19 Therefore, we further investigated whether specific miRNA(s) played a regulatory role in fine tuning the transcriptional activity of HIF‐1α, which, in turn, down‐regulated the expression of TIMP2 under hypoxia. To this end, we examined the microRNA expression profiles with TaqMan human microRNA Low‐Density Array on three HCC cell lines (SMMC‐7721, Huh‐7, and MHCC‐97L) under normoxic and hypoxic conditions and compared them with the microRNA profile of primary HCC tissues we previously reported on.20 From the 664 microRNAs examined, miR‐210 was commonly overexpressed in all three HCC cell lines under 0.1% O2 hypoxic condition (Supporting Fig. S4A). Hypoxic induction of miR‐210 was further validated in four other HCC cell lines (Hep3B, PLC/PRF/5, HLE, and BEL‐7402; Supporting Fig. S4B), as well as in hypoxic regions, as depicted by CA9 staining, in human HCC specimens (Supporting Fig. S4C).
Next, we further investigated the regulatory role of miR‐210 on HIF‐1α. We inhibited miR‐210 using miR‐210‐specific locked nucleic acid (LNA) and examined HIF‐1α transcriptional activities by luciferase reporter assay. Whereas cells transfected with (NTC) LNA expectedly displayed a dramatic increase of HIF‐1α‐dependent luciferase activity under hypoxia, induction of the luciferase signal mediated by hypoxia treatment was significantly abolished upon miR‐210 inhibition (Fig. 4B), revealing that miR‐210, as a microRNA induced by HIF‐1α (Supporting Fig. S5B), also regulated HIF‐1α transcriptional activity through a feedback circuit. In line with this observation, the mRNA and protein levels of TIMP2 were down‐regulated by hypoxia and the down‐regulation was significantly abolished upon miR‐210 inhibition (Fig. 4C and Supporting Fig. S5C), implicating that hypoxic induction of miR‐210 is an important mediator of TIMP2 suppression in HCC during hypoxia.
By in silico analysis using TargetScan 5.2 and RNA hybrid miRNA target prediction algorithms, we found a putative binding site for hsa‐miR‐210 in the 3′UTR of HIF‐3α (Fig. 4D), a classical dominant‐negative regulator of HIF‐1α.21, 22 With luciferase reporter assay to test whether miR‐210 induction modulated HIF‐1α signaling by a feedback mechanism by repressing HIF‐3α, transfection of the miR‐210 precursors significantly suppressed the signal of the wild‐type HIF‐3α 3′UTR‐coupled luciferase reporter construct by ∼40%, as compared to the NTC control (Fig. 4D). This suppressive effect was substantially abolished when the putative miR‐210‐binding site was mutated, revealing that miR‐210 bound to the 3′UTR of HIF‐3α in HCC cells and that HIF‐3α is a bona‐fide target of miR‐210 in human HCCs.
We postulated that ectopic overexpression of HIF‐3α with no 3′UTR might disturb the regulatory circuit and modulate HIF‐1α transcriptional activity. As revealed by luciferase reporter assays, overexpression of HIF‐3α mimicked the effect of miR‐210 inhibition and resulted in a significant decrease in HIF‐1α transcriptional activity (Supporting Fig. S5D and Fig. 4E). In line with this observation, the level of miR‐210 under hypoxia was also significantly reduced in HIF‐3α‐overexpressing cells compared to those in EV control cells (Fig. 4F). Taken altogether, the data suggest that TIMP2 is down‐regulated by hypoxia through a HIF‐1α/miR‐210/HIF‐3α regulatory feedback circuit.
ENHANCED CELL INVASION CONFERRED BY SUPPRESSION OF TIMP2 WAS PERTURBED BY miR‐210 INHIBITION IN HYPOXIC HCC CELLS
Given that HIF‐1α signaling is tightly regulated by miR‐210 and its direct target HIF‐3α in hypoxic HCC cells, we postulated that suppression of TIMP2 under hypoxia was mediated by a feedback circuit that might involve HIF‐1α/miR‐210/HIF‐3α. Therefore, we sought to investigate whether miR‐210 inhibition would attenuate suppression of TIMP2 mediated by hypoxia and thus reduce HCC cell‐invasive ability.
First, we observed that overexpression of miR‐210 was prevalent in HCC patients (34.0% [18 of 53]; P < 0.001) and significantly associated with poorer survival with shorter overall and disease‐free survival rates (P < 0.001 and P = 0.011, respectively; Fig. 5A,B). Overexpression of miR‐210 in human HCC tissues was significantly correlated with TIMP2 protein underexpression (P = 0.018), further supporting that miR‐210 plays a regulatory role in TIMP2 expression in human HCC (Table 2). Clinicopathological analysis revealed that miR‐210 overexpression was significantly associated with aggressive behavior, including advanced tumor stages (P = 0.009), presence of venous invasion (P = 0.022), and absence of tumor encapsulation (P = 0.002; Fig. 5C). The prometastatic role of miR‐210 was further examined by Matrigel transwell cell invasion assay. In addition, the SMMC‐7721 and BEL‐7402 HCC clones stably overexpressing miR‐210 by lentiviral infection showed significantly enhanced cell‐invasive abilities in these cells even under the normoxic condition (Fig. 5D), suggesting that miR‐210 plays a stimulatory role in HCC metastasis.
Figure 5.
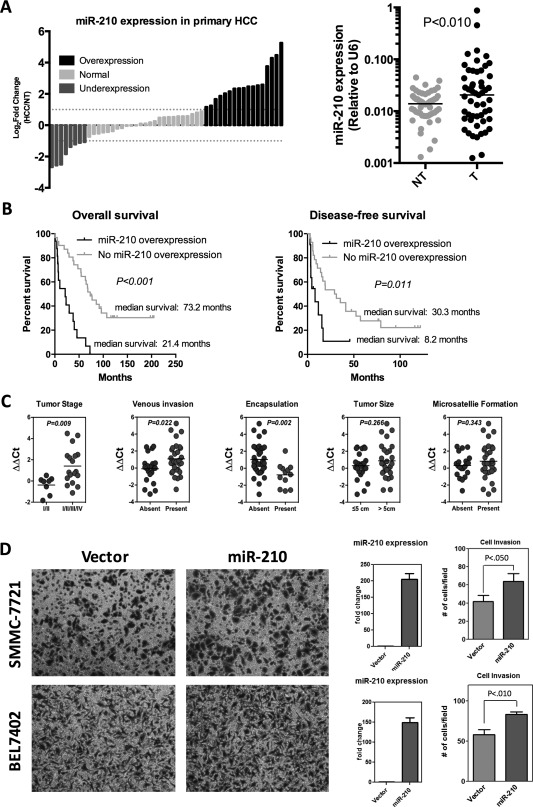
Overexpression of miR‐210 in human HCCs was associated with poorer survival rates and aggressive tumor behavior. (A) Overexpression of miR‐210 was frequent in human HCCs (34.0%; 18 of 53). Expression of mature miR‐210 in a cohort of 53 HCC patients was determined by qPCR and normalized against endogenous control U6. (B) Overexpression of miR‐210 in human HCCs was associated with both shorter overall and disease‐free survival rates. HCC patients were classified into two groups according to the expression of miR‐210. Overexpression indicates a log2 fold change ≥+1. Survival data were analyzed using Kaplan‐Meier's method. (C) Clinicopathological correlation of miR‐210 in human HCCs. miR‐210 overexpression significantly correlated with aggressive features of human HCCs, including more‐advanced tumor stage, presence of venous invasion, and absence of tumor encapsulation. miR‐210 expression levels (ΔΔCT) were presented as the log2 ratio of miR‐210 expression in HCCs, as compared to their corresponding nontumorous livers (log2 ratio HCC/NT). Statistical significance was determined by t test. (D) Transwell cell invasion assays of SMMC7721 and BEL‐7402 were performed after transduction with miR‐210 or empty vector lentivirus. Quantitative data are presented as mean ± SD, and statistical significance was determined by t test (* P < 0.05; ** P < 0.01; *** P < 0.001). Results were from three independent experiments. Successful overexpression of miR‐210 was confirmed by qPCR. U6 was used as housekeeping gene for data normalization. Abbreviation: NT, nontumorous.
Table 2.
Correlation of miR210, mRNA, and TIMP2 Protein Levels in Human HCC
| No. of Cases Without TIMP2 Underexpression | No. of Cases With TIMP2 Underexpression | P Value | |
|---|---|---|---|
| miR210 overexpression | |||
| No | 26 | 11 | 0.018 |
| Yes | 6 | 12 |
* Fisher's exact test.
It has been well documented that metastatic potential of various human cancers is greatly enhanced under hypoxic conditions. Invasive abilities of both SMMC‐7721 cells and PLC/PRF/5 were significantly enhanced under hypoxic conditions. However, when we transiently repressed miR‐210 levels using the specific LNA inhibitor, enhancement of cell‐invasive abilities by hypoxia was remarkably abolished in each of these cell lines, and the suppressive effect of miR‐210 inhibition was not observed in TIMP2 knockdown clones (Fig. 6A‐C), highlighting that TIMP2 is a crucial mediator for suppressing cell invasion conferred by miR‐210 inhibition. In line with these observations, we also demonstrated that the ECM degradation associated with invadopodia formation was also significantly enhanced in hypoxic HCC cells (Supporting Fig. S6). Similarly, miR‐210 inhibition significantly attenuated enhancement of ECM degradation conferred by hypoxia, and knockdown of TIMP2 evidently reversed the suppressive effect of miR‐210 inhibition.
Figure 6.
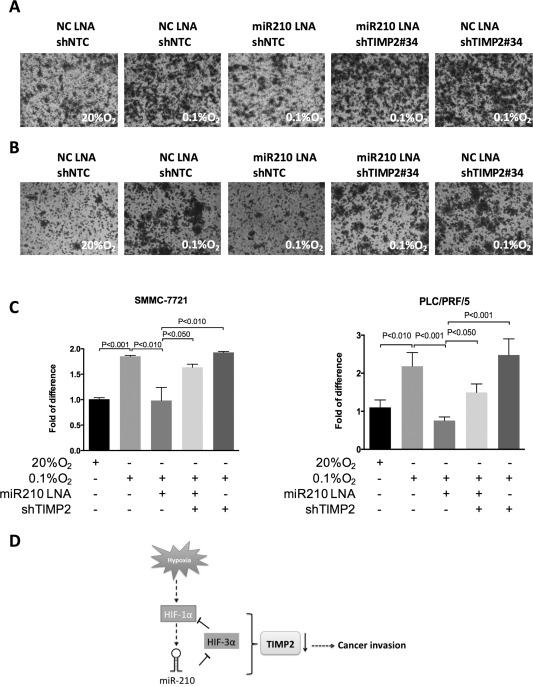
Enhanced cell invasive ability conferred by TIMP2 suppression in hypoxia was perturbed by miR‐210 inhibition. Transwell cell invasion assays of (A) SMMC7721 and (B) PLC/PRF/5 were performed after transfection with miR‐210 LNA inhibitor or negative control (NC) LNA inhibitor under normoxia (20% O2) or hypoxia conditions (0.1% O2). (C) Quantitative data are presented as mean ± SD, and statistical significance was determined by t test (* P < 0.05; ** P < 0.01; *** P < 0.001). Results were from three independent experiments. (D) Schematic representation of the relationship between TIMP2 and the HIF‐1α/miR‐210/HIF‐3α regulatory feedback circuit in HCC metastasis. Abbreviation: NC, negative control.
Discussion
In this study, we found that TIMP2 was frequently and significantly down‐regulated in human HCCs as compared to corresponding NTLs. Moreover, down‐regulation of TIMP2 significantly correlated with direct liver invasion into the adjacent liver parenchyma by the tumor. In line with this observation, we showed that knockdown of TIMP2 promoted HCC cell invasion in vitro as well as ECM degradation mediated by formation of invadopodia‐like features. Furthermore, our in vivo orthotopic tumor cell injection model demonstrated that ectopic expression of TIMP2 in highly metastatic HCC cells not only reduced the incidence of tumor microsatellite formation and venous invasion in the primary hepatic tumor xenografts, but also pulmonary metastasis, suggesting that both extrahepatic and intrahepatic metastasis in HCC was affected by TIMP2 expression. Taken together, our study has provided solid in vitro and in vivo evidence that TIMP2 is an important regulator of HCC metastasis.
ECM degradation is an early and essential step in cancer metastasis, which is a key factor for determining patient survival that contributes to around 90% of mortality associated with different types of solid tumor.3 The MMP/TIMP system is a critical regulator of ECM remodeling in the context of both normal physiological and pathological tissue compositions. It is well known that the imbalance between different members of MMP and TIMP can facilitate ECM degradation in malignant cells.5 In line with this phenomenon, we found that down‐regulation of TIMP2 protein significantly correlated with poorer survival of HCC patients. This clinical association implicates that further understanding of how TIMP2 was down‐regulated may provide therapeutic strategy for improving patient survival by antagonizing HCC cell invasion. In the present study, we demonstrated that TIMP2 was suppressed by hypoxia, and this is likely to be an important mechanism enhancing metastatic potential of HCC cells. Indeed, we observed that suppression of TIMP2 was contributed to by a novel feedback signaling circuit that involves HIF‐1α, miR‐210, and HIF‐3α.
HIF‐1α and/or HIF‐2α are frequently elevated in human malignancies and their up‐regulation is associated with poor prognosis of patients. On the contrary, HIF‐3α was found to be frequently down‐regulated in renal cell carcinomas.23, 24 Given its ability to inhibit the transactivation of HRE‐driven genes under hypoxic conditions, HIF‐3α has been classified as a dominant‐negative regulator of HIF‐1α/HIF‐2α. HIF‐3α can form an inactive complex with HIF‐1α or HIF‐2α and thus prevents their interaction with other transcriptional components.22, 24 In the present study, we found that TIMP2 down‐regulation was under the regulation of HIF‐1α and miR‐210 was directly up‐regulated by HIF‐1α under hypoxic conditions in HCC. Significantly, we further validated HIF‐3α as a direct target of miR‐210. Hence, the hypoxic induction of miR‐210 might mediate positive feedback signal on HIF‐1α by reducing expression of HIF‐3α. Importantly, the transcriptional activity of HIF‐1α was substantially abolished upon miR‐210 inhibition, implicating that hypoxic induction of miR‐210 plays a key role in enhancing the activity of HIF‐1α under hypoxic conditions, possibly by targeting HIF‐3α. Furthermore, we found that forced expression of HIF‐3α without 3′UTR significantly reduced the abundance of miR‐210.
In addition, we observed that miR‐210 enhanced the invasive ability of HCC cells. Similar observation was reported in the previous study of Liang et al., who demonstrated that miR‐210 plays a prometastatic role in HCC, and they attributed this effect to suppression of vacuole membrane protein 1 (VMP1).25 Nevertheless, the underlying biological mechanism by which suppression of VMP1 promotes cancer cell invasion remains poorly understood, given that VMP1 was well established as a regulator of autophagy and apoptosis rather than a suppressor of metastasis.26, 27 Therefore, whether miR‐210 enhances cancer metastasis through targeting VMP1 remains unclear in HCC.
Currently, little information is available to address the underlying mechanism by which hypoxia triggers invadopodia‐mediated ECM degradation in cancer cells. One recent report demonstrated that the HIF system contributed to invadopodia‐mediated ECM remodeling through activation of proto‐oncogene tyrosine‐protein kinase SRC to promote melanoma metastasis. This provided the first evidence supporting the regulatory role for HIF in this aspect.17 On the surface of invasive cancer cells and tumor‐associated endothelium, MMP2 is known to interact with αvβ3 integrin receptor, and this interaction facilitates cancer cell invasion by promoting collagen degradation in the ECM.28 TIMPs are the most important physiological inhibitors of MMP activity. Among all family members of TIMPs, TIMP2 is more effective in regulating the activity of MMP2.29 TIMP2 was previously demonstrated to be targeted by miR519d in HCC.30 However, in the present study, we did not observe miR519d up‐regulation in hypoxic HCC cells. Instead, our study provided mechanistic evidence that the HIF‐1α/miR‐210/HIF‐3α feedback circuit plays a regulatory role in TIMP2 suppression. Therefore, it is conceivable to believe that this feedback circuit represents a novel mechanism linking hypoxia to invadopodia‐mediated ECM remodeling through stimulation of MMP2 in hypoxic HCC cells.
Our findings have provided evidence that, under hypoxic conditions, TIMP2 is a critical mediator of HCC cell invasion and is under regulation of the dynamic HIF‐1a/miR‐210/HIF‐3a signaling circuit (Fig. 6D). These findings have clinical relevance to the transarterial chemoembolization (TACE) therapy employed to block the hepatic arterial blood supply as a treatment for HCC patients. Although most tumor cells are starved to death under the nutrient‐deprived and oxygen‐deprived conditions during TACE, it has been suggested that the hypoxic insult might provide a strong selection for the most metastatic tumor cells, resulting in unsatisfactory long‐term survival benefits in some patients.31, 32 From a recent study using a preclinical HCC model, it has been demonstrated that combination of TAE with antisense oligonucleotides against HIF‐1α inhibited tumor angiogenesis and further promoted the tumor shrinkage effect.33 Our data in this study demonstrated that perturbation of the dynamic HIF‐1a signaling circuit by miR‐210 inhibitor abolished TIMP2 down‐regulation and suppressed HCC cell invasion. These findings support the notion that targeting against HIF‐1α signaling is a promising direction to tackle the hypoxic responses in HCC elicited by TACE. Further investigation is warranted to examine the feasibility of miR‐210 inhibition as a novel therapeutic strategy in combination with TACE to suppress metastasis in hypoxic HCC cells.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28577/suppinfo
Supporting Information
Potential conflict of interest: Nothing to report.
The study was funded by the S.K. Yee Medical Research Fund 2011, University Development Fund of The University of Hong Kong, and Lee Shiu Family Foundation. I.O.L. Ng is Loke Yew Professor in Pathology.
REFERENCES
- 1. Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer 2001;94:153‐156. [DOI] [PubMed] [Google Scholar]
- 2. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005;55:74‐108. [DOI] [PubMed] [Google Scholar]
- 3. Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet 2007;8:341‐352. [DOI] [PubMed] [Google Scholar]
- 4. Stetler‐Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase‐independent biological activities. Sci Signal 2008;1:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010;141:52‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci 2002;115:3719‐3727. [DOI] [PubMed] [Google Scholar]
- 7. Stetler‐Stevenson WG. The tumor microenvironment: regulation by MMP‐independent effects of tissue inhibitor of metalloproteinases‐2. Cancer Metastasis Rev 2008;27:57‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White NM, Fatoohi E, Metias M, Jung K, Stephan C, Yousef GM. Metastamirs: a stepping stone towards improved cancer management. Nat Rev Clin Oncol 2011;8:75‐84. [DOI] [PubMed] [Google Scholar]
- 9. Ma L, Teruya‐Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA‐10b in breast cancer. Nature 2007;449:682‐688. [DOI] [PubMed] [Google Scholar]
- 10. Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008;451:147‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong CC, Wong CM, Tung EK, Au SL, Lee JM, Poon RT, et al. The microRNA miR‐139 suppresses metastasis and progression of hepatocellular carcinoma by down‐regulating Rho‐kinase 2. Gastroenterology 2011;140:322‐331. [DOI] [PubMed] [Google Scholar]
- 12. Ma W, Wong CC, Tung EK, Wong CM, Ng IO. RhoE is frequently down‐regulated in hepatocellular carcinoma (HCC) and suppresses HCC invasion through antagonizing the Rho/Rho‐kinase/myosin phosphatase target pathway. Hepatology 2013;57:152‐161. [DOI] [PubMed] [Google Scholar]
- 13. Wong CC, Tse AP, Huang YP, Zhu YT, Chiu DK, Lai RK, et al. Lysyl oxidase‐like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology 2014;60:1645‐1658. [DOI] [PubMed] [Google Scholar]
- 14. Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE 2005;2005:re12. [DOI] [PubMed] [Google Scholar]
- 15. Sze KM, Chu GK, Lee JM, Ng IO. C‐terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C‐Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013;57:131‐139. [DOI] [PubMed] [Google Scholar]
- 16. Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol 2011;12:413‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, et al. An HNF4alpha‐miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011;147:1233‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kent OA, Chivukula RR, Mullendore M, Wentzel EA, Feldmann G, Lee KH, et al. Repression of the miR‐143/145 cluster by oncogenic Ras initiates a tumor‐promoting feed‐forward pathway. Genes Dev 2010;24:2754‐2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR‐21 and miR‐181b‐1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010;39:493‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wong CM, Wong CC, Lee JM, Fan DN, Au SL, Ng IO. Sequential alterations of miRNA expression in hepatocellular carcinoma development and venous metastasis. Hepatology 2012;55:1453‐1461. [DOI] [PubMed] [Google Scholar]
- 21. Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA. Molecular characterization and chromosomal localization of a third alpha‐class hypoxia inducible factor subunit, HIF3alpha. Gene Expr 1998;7:205‐213. [PMC free article] [PubMed] [Google Scholar]
- 22. Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, et al. Inhibitory PAS domain protein is a negative regulator of hypoxia‐inducible gene expression. Nature 2001;414:550‐554. [DOI] [PubMed] [Google Scholar]
- 23. Maynard MA, Evans AJ, Hosomi T, Hara S, Jewett MA, Ohh M. Human HIF‐3alpha4 is a dominant‐negative regulator of HIF‐1 and is down‐regulated in renal cell carcinoma. FASEB J 2005;19:1396‐1406. [DOI] [PubMed] [Google Scholar]
- 24. Maynard MA, Evans AJ, Shi W, Kim WY, Liu FF, Ohh M. Dominant‐negative HIF‐3 alpha 4 suppresses VHL‐null renal cell carcinoma progression. Cell Cycle 2007;6:2810‐2816. [DOI] [PubMed] [Google Scholar]
- 25. Ying Q, Liang L, Guo W, Zha R, Tian Q, Huang S, et al. Hypoxia‐inducible microRNA‐210 augments the metastatic potential of tumor cells by targeting vacuole membrane protein 1 in hepatocellular carcinoma. Hepatology 2011;54:2064‐2075. [DOI] [PubMed] [Google Scholar]
- 26. Jiang PH, Motoo Y, Vaccaro MI, Iovanna JL, Okada G, Sawabu N. Expression of vacuole membrane protein 1 (VMP1) in spontaneous chronic pancreatitis in the WBN/Kob rat. Pancreas 2004;29:225‐230. [DOI] [PubMed] [Google Scholar]
- 27. Vaccaro MI, Ropolo A, Grasso D, Iovanna JL. A novel mammalian trans‐membrane protein reveals an alternative initiation pathway for autophagy. Autophagy 2008;4:388‐390. [DOI] [PubMed] [Google Scholar]
- 28. Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler‐Stevenson WG, et al. Localization of matrix metalloproteinase MMP‐2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 1996;85:683‐693. [DOI] [PubMed] [Google Scholar]
- 29. Bourboulia D, Stetler‐Stevenson WG. Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): positive and negative regulators in tumor cell adhesion. Semin Cancer Biol 2010;20:161‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fornari F, Milazzo M, Chieco P, Negrini M, Marasco E, Capranico G, et al. In hepatocellular carcinoma miR‐519d is up‐regulated by p53 and DNA hypomethylation and targets CDKN1A/p21, PTEN, AKT3 and TIMP2. J Pathol 2012;227:275‐285. [DOI] [PubMed] [Google Scholar]
- 31. Llovet JM, Real MI, Montana X, Planas R, Coll S, Aponte J, et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 2002;359:1734‐1739. [DOI] [PubMed] [Google Scholar]
- 32. Lo CM, Ngan H, Tso WK, Liu CL, Lam CM, Poon RT, et al. Randomized controlled trial of transarterial lipiodol chemoembolization for unresectable hepatocellular carcinoma. Hepatology 2002;35:1164‐1171. [DOI] [PubMed] [Google Scholar]
- 33. Sun X, Jiang H, Jiang X, Tan H, Meng Q, Sun B, et al. Antisense hypoxia‐inducible factor‐1alpha augments transcatheter arterial embolization in the treatment of hepatocellular carcinomas in rats. Hum Gene Ther 2009;20:314‐324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28577/suppinfo
Supporting Information