

Abstract
The trans‐o‐hydroxybenzylidene pyruvate aldolase‐catalysed reactions between fluoropyruvate and many (hetero)aromatic aldehydes yield aldol adducts without subsequent dehydration. Treatment of the reaction products with hydrogen peroxide yields the corresponding syn‐configured α‐fluoro β‐hydroxy carboxylic acids which have >98 % ee. The overall chemoenzymatic approach, in which fluoropyruvate serves as a fluoroacetate equivalent, may be exploited in the synthesis of polar building blocks and fragments with potential value in drug discovery.
Keywords: aldolase, asymmetric catalysis, chemoenzymatic synthesis, organofluorine compounds
Fluorination can profoundly affect the conformation, bioavailability, metabolism, pharmacokinetics and pharmacodynamics of bioactive small molecules.1 The introduction of fluorine is therefore widely used to tune biological function, and around 20 % of leading drugs contain at least one fluorine atom2 (10 of the top 50 drugs in 2013 by US prescription3). Examples of leading fluorinated pharmaceuticals include the cholesterol‐lowering drug Rosuvastatin4 and the antidiabetic drug Sitagliptin.4
The controlled formation of fluorine‐substituted stereocentres, however, presents a significant challenge. Most usually, the challenge is addressed by stereoselective C−F bond formation. For example, the fluorination of allylic silanes is often diastereoselective,5 and organo‐6 and Pd‐7 catalysed methods enable the enantioselective α‐fluorination of carbonyl compounds.
Stereoselective C−C bond formation could provide a complementary approach for controlling fluorine‐bearing stereocentres (Scheme 1). However, aldol (and related) reactions of esters of fluoroacetic acid generally exhibit poor diastereoselectivity (e.g. reactions of lithium enolates,8 Reformatsky reactions9 and Mukayama aldol reactions10). In any case, the reactants typically used in such aldol reactions (ethyl fluoroacetate and sodium fluoroacetate) are toxic.11 Very recently, an enantioselective organocatalysed reaction of fluoromalonic acid halfthioesters has been developed that yields the corresponding anti‐configured α‐fluoro thioester aldol adducts.12 We therefore envisaged a complementary approach in which fluoropyruvic acid (4) would serve as an alternative synthetic equivalent for fluoroacetate in an aldolase‐catalysed aldol reaction.13 Aldolase‐catalysed reaction of fluoropyruvate (4) and aldehydes 1 would result in the formation of α‐keto acids 5 which might then be decarboxylated14 to give the corresponding α‐fluoro β‐hydroxy carboxylic acids 6. The approach would complement organocatalysed reactions that yield other classes of α‐fluoro carbonyl compounds.12, 15
Scheme 1.
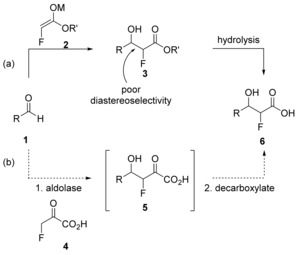
Alternative approaches for the conversion of aldehydes 1 into α‐fluoro β‐hydroxy carboxylic acids 6. a) Reaction with an enolate of an ester of fluoroacetic acid 2, followed by hydrolysis. b) Aldolase‐catalysed reaction with fluoropyruvic acid (4), followed by decarboxylation.
The Class I (lysine‐dependent) aldolase, N‐acetyl neuraminic acid lyase (NAL), has been shown to accept fluoropyruvate as an alternative donor substrate to pyruvate.16 The value of NAL in the synthesis of fluorinated products is, however, limited by poor stereocontrol and narrow demonstrated substrate scope. Moreover, NAL accepts only polyhydroxylated substrates which often yield products as complex anomeric mixtures of both pyranose and furanose forms. We therefore sought another Class I aldolase that would also accept fluoropyruvate as a donor, but would have more value in the synthesis of building blocks17 for drug discovery.
We selected trans‐o‐hydroxybenzylidene pyruvate hydratase‐aldolase (HBPA; EC 4.1.2.45) which catalyses the aldol reaction between salicylaldehyde (1 b) and pyruvate, and a subsequent dehydration.18 This enzyme is known to accept several aromatic aldehydes as substrates19 in reactions with pyruvate. Accordingly, we expressed a synthetic gene encoding the Pseudomonas putida enzyme and purified the gene product in His‐tagged form (Supporting Information).
Initially, we screened for catalysis of the reaction between fluoropyruvate (4) and a range of aromatic and heterocyclic aldehyde substrates 1 (Supporting Information). Accordingly, each alternative aldehyde (final concentration: 50 mm), fluoropyruvate (final concentration: 25 mm) and HBPA (final concentration: 0.1 mol %) was dissolved in 25 mm pH 6.0 MES buffer in an NMR tube. In each case, the conversion into products, and the disappearance of fluoropyruvate, was followed by 282 MHz 19F NMR spectroscopy (Figure 1 A–C and Supporting Information).
Figure 1.

Reaction between fluoropyruvate and 4‐pyridinecarboxaldehyde catalysed by HBPA. A) Overview of the reaction. B) 282 MHz 19F spectrum of fluoropyruvate in its keto and hydrated forms in 25 mm pH 6.0 MES buffer. C) Addition of 0.1 mol % HBPA results, after 24 h, in the appearance of double doublets corresponding to the keto and hydrated forms of the major (93 %) and minor (7 %) diastereomers 5 f. D) Selected examples of accepted aldehyde substrates, together with percentage conversions.
The HBPA‐catalysed reaction involving fluoropyruvate (4) as the donor was successful with a wide range of aromatic and heteroaromatic aldehydes 1 (Figure 1 D and Supporting Information). Unfortunately, no aliphatic aldehydes were found to be good substrates. The products were generally obtained with good diastereoselectivity as mixtures of keto and hydrated forms. Remarkably, dehydration of the products was not observed, perhaps because the presence of the fluorine atom precludes enzyme‐catalysed elimination. It was observed that the best aldehyde substrates typically had a hydrogen bond acceptor at the 2‐ or 4‐position of the (hetero)aromatic ring.
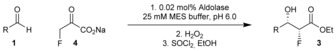
The HBPA‐catalysed reaction between fluoropyruvate (4) and an excellent substrate, 4‐pyridinecarboxaldehyde (1 f), was optimised for preparative application. First, it was shown that conversion into product was not changed significantly when a smaller excess of aldehyde substrate was used (92 % conversion with both 50 mm and 27.5 mm aldehyde). Second, it was shown that the conversion remained high with five‐fold less enzyme (89 % conversion with 0.02 mol % enzyme), though was compromised significantly at even lower loading (40 % conversion with 0.005 mol % enzyme). Third, it was shown that HPBA was tolerant of up to 20 % v/v DMSO as co‐solvent; however, most aldehydes were soluble in the buffer alone and those aldehydes that were not, typically, only required addition of 5–10 % v/v DMSO.
A wide range of aldehyde substrates was explored to determine the scope of our synthetic approach. Typically, an aldehyde 1 (27.5 mm), fluoropyruvate (4) (25 mm) and 0.02 mol % HBPA were dissolved in 25 mm pH 6.0 MES buffer. After 24 hr, the crude β‐fluoro γ‐hydroxy α‐keto acid products 5 were efficiently decarboxylated by treatment with H2O2 (resulting in dramatic simplification of the 19F NMR spectrum, see Supporting Information). Finally, to facilitate isolation and purification, the crude acid products were evaporated to dryness before esterification by treatment with SOCl2 in ethanol. The enantiomeric excesses of the products were determined by integration of the 282 MHz 19F spectra of the corresponding (R)‐ and (S)‐3,3,3‐trifluoro‐2‐methoxy‐2‐phenylpropanoic esters (Supporting Information).20 Our results are summarized in Table 1.
Table 1.
Chemoenzymatic synthesis of α‐fluoro β‐hydroxy esters.
| Entry | Product | Yield[a] | syn:anti [b] | ee [c] |
|---|---|---|---|---|
| 1 |
|
71 % | >98:<2 | >98 % |
| 2 |

|
71 % | >98:<2 | >98 % |
| 3[d] |
|
60 % | >98:<2 | >98 % |
| 4 |
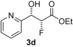
|
51 % | 93:7 | >98 % |
| 5 |

|
29 % | 83:17 | >98 % |
| 6 |
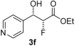
|
57 % | 90:10 | >98 % |
| 7[d] |

|
76 % | 94:6 | >98 % |
| 8[d] |

|
38 % | 92:8 | >98 % |
| 9[d] |

|
25 % | >98:<2 | >98 % |
| 10 |
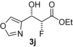
|
43 % | 94:6 | >98 % |
| 11 |
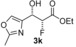
|
45 % | 93:7 | >98 % |
| 12[d] |

|
70 % | >98:<2 | >98 % |
| 13 |
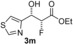
|
41 % | 94:6 | >98 % |
| 14 |
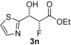
|
57 % | 93:7 | >98 % |
[a] Yield of purified product. The absolute and relative configuration of the major diastereoisomer is as drawn. [b] Diastereomeric ratio of purified products. [c] The enantiomeric excess of the major product was determined by conversion of the purified product into (R)‐ and (S)‐3,3,3‐trifluoro‐2‐methoxy‐2‐phenylpropanic esters. [d] Biotransformations were performed in solutions of MES buffer with 10 % DMSO as a co‐solvent to solubilise aldehydes.
Our chemoenzymatic synthesis of α‐fluoro β‐hydroxy esters was successful with a wide range of heteraromatic and aromatic aldehydes (Table 1). The HBPA‐catalysed reaction was generally highly diastereoselective, and, after purification, a single diastereomeric product was often observed. In all cases, the major diastereomeric product was shown to have >98 % ee. The absolute and relative configuration of 3 f was determined by X‐ray crystallography, and that of 3 g determined by X‐ray crystallographic analysis of the corresponding camphanic ester 7 g (Figure 2). The relative configuration of the other esters 3 was determined by correlation of the 19F NMR spectra (Supporting Information). Specifically, the 3 J HF values were diagnostic9b of the relative configuration of the α‐fluoro β‐hydroxy esters 3 (syn: ≈24 Hz; anti: ≈18 Hz). These data are consistent with the syn‐configured esters adopting a conformation in which the fluorine and hydroxy groups adopt a gauche arrangement.21
Figure 2.
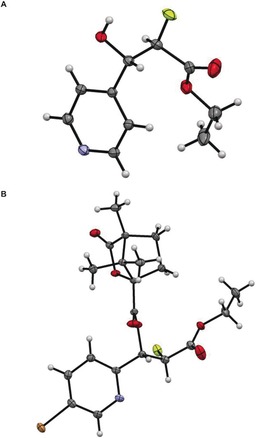
X‐ray crystal structures of α‐fluoro β‐hydroxy acid derivatives. A) X‐ray structure of ester 3 f. B) X‐ray crystal structure of 7 g. The thermal ellipsoids are set at 50 % probability. Color code: C gray, H white, red O, yellow F, blue N, brown Br.
Our synthetic approach yields small, polar molecules that may have value in drug discovery applications. To understand this value, we determined the molecular properties of the α‐fluoro β‐hydroxy esters 3 a–n; the intermediate carboxylic acids 6 a–n; and the corresponding 2‐fluoro 1,3‐diols 8 a–n (see Supporting Information for the synthesis of an exemplar 1,3‐diol) (Figure 3). Most of these compounds meet guidelines that have been established both for high‐quality fragments22 and building blocks17 for drug discovery.
Figure 3.
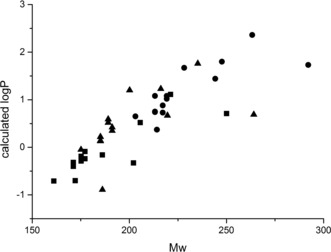
Molecular properties of the α‐fluoro β‐hydroxy esters 3 a–n (circles), the α‐fluoro β‐hydroxy acids 6 a–n (triangles) and the corresponding 2‐fluoro 1,3‐diols 8 a–n (squares).
In conclusion, we have developed a robust chemoenzymatic synthesis of α‐fluoro β‐hydroxy esters. The scope of the approach is broad, enabling the conversion of many aromatic and heteroaromatic aldehydes into chiral products with >98 % ee and high diastereoselectivity. Crucially, the syn diasteroselectivity of the process complements the anti selectivity of a recent organocatalysed synthesis of α‐fluoro thioesters.12 Moreover, the distinctive products have molecular properties suitable for application as high‐quality fragments and building blocks for drug discovery.
Experimental Section
General procedure: β‐fluoropyruvic acid sodium salt (0.50 mmol in 25 mm aq. MES buffer, final conc. 25 mm) followed by aldehyde (0.55 mmol in 25 mm aq. MES buffer, final conc. 27.5 mm) was added to HBPA (250 μg mL−1 enzyme in 25 mm MES buffer, pH 6.0, final vol. 20 mL), and the reaction mixture was left to stand at RT for 24 h before the addition of solution of H2O2 (30 % aq. solution, 150 μL). After 30 min of vigorous stirring, the reaction mixture was cooled to 0 °C and Na2S2O5 (solid) was slowly added, before the water was removed in vacuo to reveal a crude product. SOCl2 (200 μL, 1.03 mmol) was slowly added to a suspension of the crude product in EtOH (10 mL) at 0 °C and stirred at RT for 4 h before being cooled to 0 °C and made alkaline with NaHCO3 (sat. aq. sol). The resulting mixture was extracted with EtOAc (3×10 mL), the combined organic layers were washed with H2O (2×30 mL), dried (MgSO4), filtered and concentrated in vacuo to reveal a crude product.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The research leading to these results has received funding from the Innovative Medicines Initiative Joint Undertaking project CHEM21 under grant agreement no. 115360, resources of which are composed of financial contribution from the European Union's Seventh Framework Programme (FP7/2007‐2013) and EFPIA companies’ in kind contribution. We thank Dr. Chris Pask for obtaining X‐ray crystal structures.
J. K. Howard, M. Müller, A. Berry, A. Nelson, Angew. Chem. Int. Ed. 2016, 55, 6767.
Contributor Information
Prof. Alan Berry, Email: a.berry@leeds.ac.uk.
Prof. Adam Nelson, Email: a.s.nelson@leeds.ac.uk.
References
- 1.
- 1a. Ismail F. M. D., J. Fluorine Chem. 2002, 118, 27–33; [Google Scholar]
- 1b. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]
- 1c. Hagmann W. K., J. Med. Chem. 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]
- 2. O'Hagan D., J. Fluorine Chem. 2010, 131, 1071–1081. [Google Scholar]
- 3. McGrath N. A., Brichacek M., Njardarson J. T., J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar]
- 4. Wang J., Sánchez-Roselló M., Aceña J. L., del Pozo C., Sorochinsky A. E., Fustero S., Soloshonok V. A., Liu H., Chem. Rev. 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Greedy B., Paris J.-M., Vidal T., Gouverneur V., Angew. Chem. Int. Ed. 2003, 42, 3291–3294; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 3413–3416; [Google Scholar]
- 5b. Giuffredi G. T., Purser S., Sawicki M., Thompson A. L., Gouverneur V., Tetrahedron: Asymmetry 2009, 20, 910–920. [Google Scholar]
- 6.
- 6a. Beeson T. D., MacMillan D. W. C., J. Am. Chem. Soc. 2005, 127, 8826–8828; [DOI] [PubMed] [Google Scholar]
- 6b. Marigo M., Fielenbach D., Braunton A., Kjærsgaard A., Jørgensen K. A., Angew. Chem. Int. Ed. 2005, 44, 3703–3706; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3769–3772. [Google Scholar]
- 7. Kim S. M., Kim H. R., Kim D. Y., Org. Lett. 2005, 7, 2309–2311. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Welch J. T., Seper K., Eswarakrishnan S., Samartino J., J. Org. Chem. 1984, 49, 4720–4721; [Google Scholar]
- 8b. Welch J. T., Eswarakrishnan S., J. Chem. Soc. Chem. Commun. 1985, 186–188. [Google Scholar]
- 9.
- 9a. Linderman R. J., Graves D. M., J. Org. Chem. 1989, 54, 661–668; [Google Scholar]
- 9b. Ocampo R., W. R. Dolbier, Jr. , Abboud K. A., Zuluaga F., J. Org. Chem. 2002, 67, 72–78. [DOI] [PubMed] [Google Scholar]
- 10. Huang X.-T., Chen Q.-Y., J. Org. Chem. 2002, 67, 3231–3234. [DOI] [PubMed] [Google Scholar]
- 11. Proudfoot A. T., Bradberry S. M., Vale J. A., Toxicol. Rev. 2006, 25, 213–219. [DOI] [PubMed] [Google Scholar]
- 12. Saadi J., Wennemers H., Nat. Chem. 2016, 8, 276–280. [DOI] [PubMed] [Google Scholar]
- 13.For reviews, see:
- 13a. Whitesides G. M., Wong C.-H., Angew. Chem. Int. Ed. Engl. 1985, 24, 617–638; [Google Scholar]; Angew. Chem. 1985, 97, 617–638; [Google Scholar]
- 13b. Dean S. M., Greenberg W. A., Wong C.-H., Adv. Synth. Catal. 2007, 349, 1308–1320; [Google Scholar]
- 13c. Windle C. L., Müller M., Nelson A., Berry A., Curr. Opin. Chem. Biol. 2014, 19, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. W. B. Wright, Jr. , Collins K. H., J. Am. Chem. Soc. 1956, 78, 221–224. [Google Scholar]
- 15.
- 15a. Zhong G., Fan J., C. F. Barbas III , Tetrahedron Lett. 2004, 45, 5681–5684; [Google Scholar]
- 15b. Xu X.-Y., Wang Y.-Z., Gong L.-Z., Org. Lett. 2007, 9, 4247–4249; [DOI] [PubMed] [Google Scholar]
- 15c. Xu X.-Y., Wang Y.-Z., Cun L.-F., Gong L.-Z., Tetrahedron: Asymmetry 2007, 18, 237–242. [Google Scholar]
- 16.
- 16a. Chokhawala H. A., Cao H., Yu H., Chen X., J. Am. Chem. Soc. 2007, 129, 10630–10631; [DOI] [PubMed] [Google Scholar]
- 16b. Beliczey J., Kragl U., Liese A., Wandrey C., Hamacher K., Coenen H. H., Tierling T., US Patent, 635543, 2002;
- 16c. Watts A. G., Withers S. G., Can. J. Chem. 2004, 82, 1581–1588; [Google Scholar]
- 16d. Stockwell J., Daniels A. D., Windle C. L., Harman T. A., Woodhall T., Lebl T., Trinh C. H., Mulholland K., Pearson A. R., Berry A., Nelson A., Org. Biomol. Chem. 2016, 14, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldberg F. W., Kettle J. G., Kogej T., Perry M. W. D., Tomkinson N. P., Drug Discovery Today 2015, 20, 11–17. [DOI] [PubMed] [Google Scholar]
- 18. Kuhm A. E., Knackmuss H.-J., Stolz A., J. Biol. Chem. 1993, 268, 9484–9489. [PubMed] [Google Scholar]
- 19. Eaton R. W., Appl. Environ. Microbiol. 2000, 66, 2668–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoye T. R., Jeffrey C. S., Shao F., Nat. Protoc. 2007, 2, 2451–2458. [DOI] [PubMed] [Google Scholar]
- 21. O'Hagan D., J. Org. Chem. 2012, 77, 3689–3699. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Congreve M., Carr R., Murray C., Jhoti H., Drug Discovery Today 2003, 8, 876–877; [DOI] [PubMed] [Google Scholar]
- 22b. Murray C. W., Rees D. C., Angew. Chem. Int. Ed. 2016, 55, 488–492; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 498–503. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary