

Abstract
Mixed leukocyte (white blood cells [WBCs]) trafficking using positron emission tomography (PET) is receiving growing interest to diagnose and monitor inflammatory conditions. PET, a high sensitivity molecular imaging technique, allows precise quantification of the signal produced from radiolabelled moieties. We have evaluated a new method for radiolabelling WBCs with either zirconium‐89 (89Zr) or copper‐64 (64Cu) for PET imaging. Chitosan nanoparticles (CNs) were produced by a process of ionotropic gelation and used to deliver radiometals into WBCs. Experiments were carried out using mixed WBCs freshly isolated from whole human blood. WBCs radiolabelling efficiency was higher with [89Zr]‐loaded CN (76.8 ± 9.6% (n = 12)) than with [64Cu]‐loaded CN (26.3 ± 7.0 % (n = 7)). [89Zr]‐WBCs showed an initial loss of 28.4 ± 5.8% (n = 2) of the radioactivity after 2 h. This loss was then followed by a plateau as 89Zr remains stable in the cells. [64Cu]‐WBCs showed a loss of 85 ± 6% (n = 3) of the radioactivity after 1 h, which increased to 96 ± 6% (n = 3) loss after 3 h. WBC labelling with [89Zr]‐loaded CN showed a fast kinetic of leukocyte association, high labelling efficiency and a relatively good retention of the radioactivity. This method using 89Zr has a potential application for PET imaging of inflammation.
Keywords: PET, Zr‐89, Cu‐64, inflammation imaging, white blood cell trafficking, chitosan nanoparticles
Introduction
Different imaging modalities have been applied for use in imaging inflammation. Magnetic resonance imaging (MRI) and ultrasound imaging have provided some promising methods of assessing synovitis in joints1 but have failed to provide quantitative and standardised methods of measuring inflammation.
Radiolabelled leukocyte scintigraphy with single‐photon emission computed tomography (SPECT) using technetium‐99m (t 1/2 = 6 h) and indium‐111 (t 1/2 = 67.9 h) chelates are the most widely used clinical procedures for the assessment of inflammatory diseases.2, 3 Molecular imaging technologies such as SPECT and positron emission tomography (PET) are well suited for tracking the migration of leukocytes to inflammatory foci in vivo. However, the sensitivity of PET (10−11–10−12 M) is at least 1–2 orders of magnitude higher than single photon imaging systems (10−10 M),4 and PET is more attractive for the quantification of the regional signal from migrated cells. Therefore, in vivo imaging of leukocyte trafficking using PET is receiving growing interest for applications in immunological studies to (i) track the selective recruitment of specific immune cells during pathogenesis, (ii) detect probable infectious/inflammatory foci and (iii) devise rational therapeutic strategies5 and carry out longitudinal studies.
Different approaches have been developed for tracking radiolabelled leukocytes in vivo with PET using the positron emitting isotopes copper‐64 (64Cu, t 1/2 = 12.7 h) and fluorine‐18 ([18F], t 1/2 = 109.7 min). Cationic [64Cu]2+ requires a chelate to transport it into cells; [64Cu]‐pyruvaldehyde‐bis(N4‐methylthiosemi‐carbazone) (PTSM), [64Cu]‐polyethylenimine (PEI) and [64Cu]‐loaded magnetic nanoparticles have been reported for white blood cell (WBC) labelling.4, 5, 6 [64Cu]‐PTSM proved to be superior to [18F]‐fluorodeoxyglucose‐labelled leukocytes with a higher and more reproducible labelling efficiency.7 The short physical half‐life of 18F is not suitable for long‐term observations, and non‐specific uptake was observed with [18F]‐fluorodeoxyglucose‐labelled leukocytes partly because of high efflux of 18F after injection.8 In addition, carbon‐11 (11C, t 1/2 = 20.3 min) has been utilised for macrophage imaging in rheumatoid synovitis9 using the PET tracer [11C]PK11195, which is a ligand for the translocator protein 18 kDa that is expressed on macrophages. The constraints of the short physical half‐life of 11C means that this method of imaging macrophages is restricted to static scans of joints and is not appropriate for trafficking of macrophages. Moreover, the presence of blood metabolites of [11C]PK11195 hampers the quantification of the PET signal.
The positron emitting isotope zirconium‐89 (89Zr, t 1/2 = 78.4 h) has emerged as a suitable radioisotope for imaging processes with longer pharmacokinetics with PET10, 11, 12, 13 because of its physical decay properties. 89Zr has had successful applications in antibody labelling for immuno‐PET imaging11, 12, 13, 14, 15, 16 and is ideally suited for cell trafficking over long periods of time.10 89Zr‐labelled dextran nanoparticles have been used for in vivo macrophage imaging.17 In comparison with 64Cu, 89Zr has a longer physical half‐life as well as a higher fraction of decays that occur by positron emission, with a branching ratio of 22.3% compared with 17.5% for 64Cu. Furthermore, the relatively low energy of the emitted positron (E aveβ+ = 396 keV) results in high‐resolution 89Zr images comparable with those observed with 64Cu with equivalent radioactive doses. Desferrioxamine B (DFO) is the most successfully employed chelator for [89Zr]4+ metal ions owing to its high affinity for the metal.12, 13 Nevertheless, the hydrophilic property of DFO makes it unsuitable for carrying 89Zr across the lipophilic membrane into cells. [89Zr]‐DFO‐NCS has however been used to randomly radiolabel primary amine groups at the surface of stem cells for PET‐based cell trafficking.18 More recently, [89Zr]‐oxinate4 has been used for in vivo cell trafficking with PET.19 This technique showed a radiolabelling yield comparable with that with indium‐111 SPECT, as well as a high retention of 89Zr in cells; however, further evaluation of its potential cytotoxicity effects is required.19
Chitosan, a biocompatible and non‐antigenic co‐polymer of glucosamine and N‐acetylglucosamine with metal ion chelating properties,20 is an appropriate carrier of 89Zr for cell labelling. Chitosan has been reported to show potential as a carrier for drug and gene delivery to cells,21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 and chitosan‐based formulations have been prepared for drug delivery systems. Chitosan is widely available, inexpensive and can be obtained in a wide variety of molecular weight distributions as well as at various degrees of deacetylation (DD).
The free amino (depending on DD) and hydroxyl groups of chitosan allows for conjugation with peptides and proteins or the incorporation of inorganic materials including metal ions.20 Both chitosan polymer and chitosan nanoparticles (CNs) have acquired great interest in various fields including wound dressing23, 33 and tissue engineering22 applications that are made possible by the biocompatibility, biodegradability and non‐toxicity offered by chitosan.23, 24, 34 Furthermore, various chitosan composites have been investigated for biomedical applications35, 36, 37, 38 and molecular imaging.39, 40 The preparation of chitosan quantum dot composites for drug or gene delivery with optical imaging utility has been reported,36, 41, 42 and biocompatible carboxymethyl chitosan‐coated super paramagnetic iron oxides have been prepared to visualise human stem cells with MRI.39, 43 Arginylglycylaspartate peptide delivery systems have been prepared from glycol CN for use as an antiangiogenic model drug in cancer therapy.39, 44
CN can be constructed from chitosan polymer by microemulsification or complex co‐acervation; however, both methods require harsh processing conditions and organic solvents or toxic reagents.45 Alternatively, ionotropic gelation27, 28, 29, 45, 46 can be used that involves cross‐linking chitosan polymer chains with a poly‐anion. This is carried out in mild and aqueous conditions that could potentially be exploited for delivery of PET radioisotopes into circulating leukocyte cells. CN have been shown to penetrate cells by a number of endocytosis and pinocytosis uptake pathways.21 Compared with other cationic polymers used for cell transfection such as PEI,47 chitosan is reported to display less cytotoxicity while maintaining cell viability.48
We describe here a new technique to radiolabel mixed human leukocytes with 89Zr and 64Cu using CN as a carrier. The radiometals are directly compared for their affinity for CN, leukocyte association and cell retention. Previously, 89Zr‐labelled dextran nanoparticles have been used for macrophage imaging17; however, the reported method uses toxic organic reagents and required the use of epichlorohydrin as a reticulating agent to form the nanoparticles. This was followed by coupling the nanoparticles to DFO in order to complex 89Zr and finally an amino end‐capping step. The method reported here is much less complex in terms of building the CN and requires neither the use of toxic organic reagents to construct the nanoparticles nor the coupling to DFO or any other complexing agent. This method for radiolabelling mixed leukocytes can be translated to preclinical and potentially clinical use for quantitative imaging of inflammatory disease.
Experimental
Materials and equipment
[89Zr]‐oxalate was purchased from Perkin Elmer (US)/BV cyclotron (Netherlands); [64Cu] copper chloride was purchased from Cambridge University; chitosan 15 kDa (>85% DD) was purchased from Tebu‐bio (France); chitosan 50–190 kDa (85% DD) and chitosan 190–310 kDa (85% DD) were purchased from Sigma‐Aldrich (UK). Acetic acid, sodium hydroxide and sodium tripolyphosphate (TPP) were all purchased from Sigma‐Aldrich (UK) and were used without any further purification. An Edwards Modulyo 4K freeze dryer (UK) was used to freeze dry CN.
Acid citrate dextrose (formula A) was purchased from Huddersfield Pharmacy Manufacturing Unit (UK), and hydroxyethyl starch 6% solution was purchased from Grifols (UK). Hank's balanced salt solution, trypan blue solution (0.4% w/v) and haemocytometer were all acquired from Sigma‐Aldrich (UK).
A PK121R multispeed refrigerated centrifuge from Thermo Scientific (UK) was used along with a Micro Centaur centrifuge (MSE, UK). For radiolabelling of CN, measurement of the association and retention of [89Zr]‐ or [64Cu]‐loaded CN into leukocyte cells, a Thermo Shaker PHMT (from Grant Instruments UK) was used to heat and agitate solutions. A Vortex Genie‐2, which was obtained from Scientific Instruments (USA), was used to re‐suspend solutions where necessary. The procedure for separation of mixed leukocytes from whole blood was performed in a SafeFlow 1.2 safety cabinet from Bioair Instruments (Italy). Measurements of radioactive samples were performed with an Isomed 2000 dose calibrator from MED (Germany) and a 1470 Wizard Automatic Gamma Counter from Perkin Elmer (UK). Scanning electron microscopy images were taken using a Phillips XL30 FEG SEM, and an Olympus BX51 microscope (Japan) was used for the cell viability assay.
Chitosan nanoparticle construct
Chitosan (15 kDa, >85% DD, 0.3 g) was solubilised in a 1% acetic acid solution (60 mL) and was left to stir for 1 h. The pH of the chitosan solution was then adjusted to 4.7 with the addition of sodium hydroxide (0.2 M). A solution of TPP (50 mg in 20 mL of deionised water) was added in a drop‐wise manner to the chitosan solution with stirring. CN formed instantaneously on addition of the poly‐anion. CNs were isolated by centrifugation at 3025g for 30 min at 4 °C; the supernatant was removed, and the nanoparticles were washed with deionised water, and centrifugation was repeated. The supernatant was removed, and CNs were isolated as a gel and placed in a desiccator for 3 h. CN where stored at 2–4 °C as this temperature was reported to have no effect on size and physical stability of the particles,49 and the particles were characterised via scanning electron microscopy. In order to assess the amount of dry chitosan in the CN gel, samples were freeze‐dried to remove water. Following the freeze‐dry process, the dry residue was weighed to determine the content of chitosan.
Isolation of mixed human leukocytes
Each experiment was carried out using mixed WBCs freshly isolated from whole blood following erythrocyte sedimentation according to a standard procedure.3, 50 Venous blood (51 mL) was taken from volunteers and was drawn into syringes containing acid citrate dextrose in a proportion of 1.5 parts to 8.5 parts of whole blood. The contents were mixed well and dispensed into two 50‐mL Falcon tubes. Next, 3 mL of 6% hydroxyethyl starch was added to each tube, and the contents mixed slowly to avoid the formation of bubbles. The Falcon tubes were maintained in an upright position for 60 min to allow red blood cells to settle. The leukocyte‐rich platelet‐rich plasma supernatant was then carefully removed and centrifuged at 150g for 5 min at room temperature to obtain a supernatant of platelet‐rich plasma (PRP) and a pellet of mixed leukocytes. The PRP supernatant was removed, and the mixed leukocyte pellet was re‐suspended in saline (2 mL). Next, the PRP was centrifuged at 1500g for 10 min at room temperature to obtain cell‐free plasma (CFP) that was removed and retained for washing and re‐suspending cells following the radiolabelling process.
Radiolabelling of chitosan nanoparticles with 89Zr and 64Cu
Approximately 15–20 mg of the chitosan nanoparticle gel was re‐suspended in deionised water (300 μL) in a 1.5‐mL Eppendorf vial. 89Zr (in oxalic acid) or 64Cu (as aqueous [64Cu]‐chloride ([64Cu]Cl2)) was added to the Eppendorf vial (approximately 400 kBq) before being placed in a thermo‐shaker and incubated at 1400 rpm at room temperature for up to 45 min. Following incubation, the mixture was centrifuged at 11 600g for 10 min to leave a supernatant containing free [89Zr]‐oxalate or [64Cu]Cl2 and a pellet containing [89Zr]‐ or [64Cu]‐loaded CN. Next, the [89Zr]‐ or [64Cu]‐loaded CNs were washed with water (300 μL), and centrifugation was repeated to remove any free 89Zr or 64Cu. The radioactivity in the supernatants and pellet was then counted in a gamma counter, and the radiolabelling efficiency is expressed as percentage of the ratio between radioactivity associated with the CN and total radioactivity.
Measure of leukocyte radiolabelling efficiency and retention of [89Zr]‐ or [64Cu]‐loaded chitosan nanoparticles into mixed human leukocytes
The [89Zr]‐ or [64Cu]‐loaded CNs were re‐suspended in saline (200 μL), and the re‐suspended mixed leukocytes (200 μL in saline) were added to the solution and vortexed for 30 s. The mixture was then incubated in a thermo‐shaker at 37 °C, at 1400 rpm for 10, 15, 20, 30 and 60 min (10, 20 and 30 min for [64Cu]‐loaded CN). Following incubation, leukocyte radiolabelling was terminated by the addition of CFP (1 mL). The radiolabelled leukocytes were separated from the mixture by centrifugation at 150g for 5 min to leave a pellet of [89Zr]‐ or [64Cu]‐labelled leukocytes (it should be noted that [89Zr]‐ and [64Cu]‐loaded CNs were studied using different blood draws). The radioactivity in the supernatant and the pellet was counted in a gamma counter. Radiolabelling efficiency is expressed as a percentage of the ratio between radioactivity associated with the leukocytes and total radioactivity.
To assess cell retention of the radioactivity, a sample of leukocytes labelled with [89Zr]‐ or [64Cu]‐loaded CN (after a 15‐min incubation time) were re‐suspended in CFP and allowed to incubate for 24 h at 37 °C. At intervals of 1, 2, 3, 4 and 24 h, the mixture was centrifuged at 150g for 5 min, the supernatant was removed and the pellet containing [89Zr]‐ or [64Cu]‐labelled leukocytes was re‐suspended in fresh CFP. Removed CFP fractions and leukocyte pellets were measured for radioactivity in a gamma counter and the radioactivity efflux rate determined as seen in Figure 33.
Figure 3.
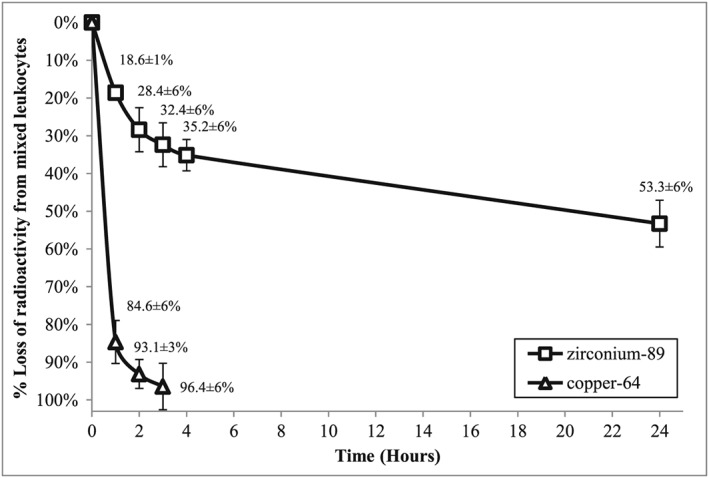
Retention of radioactivity in mixed leukocyte cells over a 24‐h period (all values are decay corrected to time = 0 h). Data expresses as mean ± SD.
Measurement of cell viability by trypan blue exclusion assay
To measure the viability of cells, a trypan blue exclusion assay was used. The cell pellet was initially re‐suspended in Hank's balanced salt solution (9.5 g/L, 500 μL). Next, 50 μL of the cell suspension was added to a falcon tube containing a further 4 mL of Hank's balanced salt solution, and 0.4% (w/v) trypan blue solution (500 μL) was added. The solution was mixed, and a Pasteur pipette was used to load the mixture onto a haemocytometer; viable and non‐viable cells were then counted in the haemocytometer under a microscope.
Results
Chitosan nanoparticle construct
Analysis by scanning electron microscope of CN generated from 15 kDa chitosan showed particles with spherical shape and a size distribution ranging from 57 to 153 nm in diameter (Figure 1).
Figure 1.

Scanning electron microscopy images of CN showing size (57–153 nm) and shape of nanoparticles.
CNs were formed from the process of ionotropic gelation. From freeze‐dried CN, we evaluated that only a fraction (26%, n = 3) of the chitosan polymer formed into nanoparticles. On assessment of the freeze‐dried CN generated from medium molecular weight (MMW) and low molecular weight chitosan, it was discovered that they were difficult to re‐suspend in saline solution. Nanoparticles with higher stability and water solubility were generated from the 15 kDa chitosan polymer. These CNs were less prone to agglomeration and were easily re‐suspended following centrifugation. As a result, nanoparticles formed from 15 kDa chitosan polymers were used in gel form in all further experiments.
Radiolabelling of chitosan nanoparticles with 89Zr and 64Cu
Results show that CN bind both 89Zr and 64Cu very efficiently averaging slightly more than 70% of radiometal uptake after 45 min (Tables 1 and 2 respectively). The highest labelling efficiency with 89Zr was observed with nanoparticles built from 190 to 310 kDa (MMW). The labelling efficiency was consistent over all size distributions of CN tested for 64Cu radiolabelling.
Table 1.
Binding efficiency of 89Zr to CN produced from various weight distributions of chitosan polymer
| CN | 15 kDa | LMW (50–190 kDa) | MMW (190–310 kDa) |
|---|---|---|---|
| 89Zr uptake (%) | 73.8 ± 18.8 (n = 30) | 72.8 ± 24.4 (n = 8) | 90.3 ± 14.8 (n = 8) |
Incubation was carried out in a thermo‐shaker at 1400 rpm at room temperature for 45 min.
Table 2.
Binding efficiency of 64Cu to CN produced from various weight distributions of chitosan polymer
| CN | 15 kDa | LMW (50–190 kDa) | MMW (190–310 kDa) |
|---|---|---|---|
| 64Cu uptake (%) | 70.7 ± 16.2 (n = 17) | 73.5 ± 5.3 (n = 3) | 71.9 ± 2.3 (n = 3) |
Incubation was carried out in a thermo‐shaker at 1400 rpm at room temperature for 45 min.
The content of dry CN in 20 mg of CN gel was determined to be approximately 0.6 mg after freeze‐drying of the gel and weighing the solid CN that remained. It was found that 5 mg of CN gel, corresponding to 0.15 mg of CN, was able to load 3.7 MBq of 89Zr, resulting in a specific radioactivity of 24.7 MBq/mg CN.
Measure of leukocyte radiolabelling efficiency and retention of [89Zr]‐ or [64Cu]‐loaded chitosan nanoparticles into mixed human leukocytes
Leukocyte‐associated 89Zr was measured as a function of incubation time (Figure 2). After 10‐min of incubation, [89Zr]‐leukocyte radiolabelling efficiency was observed at 82.7 ± 1.9% (n = 3); this was followed by a decrease and eventually a plateau at 65.5 ± 13.1% (n = 3) of cell‐associated 89Zr after 30 to 60‐min incubation (63% [89Zr]‐leukocyte radiolabelling efficiency). Retention of 89Zr by the mixed leukocytes after a 15‐min incubation of [89Zr]‐loaded CN with leukocytes was subsequently measured over a period of 24 h (Figure 3). The 15‐min incubation time was chosen as a balance of having a stable fraction of [89Zr]‐loaded CN bound to the leukocyte cell but still maintaining a method of radiolabelling cells in a timely manner for clinical translation.
Figure 2.
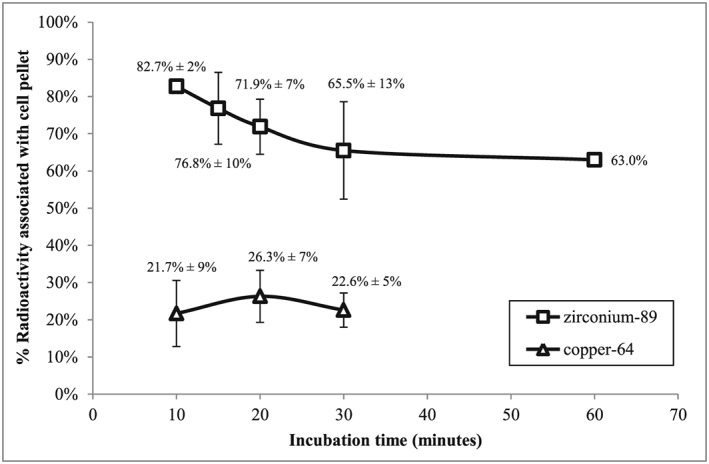
Leukocyte radiolabelling efficiency as a function of time. Data expresses as mean ± SD.
Similarly, [64Cu]‐leukocyte radiolabelling efficiency and the retention of 64Cu in leukocyte cells was investigated (Figures 2 and 3). [64Cu]‐leukocyte radiolabelling efficiency was significantly lower than was observed for its 89Zr counterpart as can be seen in Figure 2. A maximum of 26.3 ± 7.0% (n = 7) [64Cu]‐leukocyte radiolabelling efficiency was reached after 20‐min incubation that decreased to 22.6 ± 4.6% (n = 3) after 30 min.
Efflux of 64Cu from leukocytes was observed after 20‐min incubation of [64Cu]‐loaded CN with mixed leukocytes and was measured over a period of 3 h. Figure 3 shows that the 64Cu is removed from leukocyte cells to a much greater extent and at a much faster rate when compared with 89Zr. More than 90% of the radioactivity was lost from the cells after 2 h.
Viability of cells was studied using the trypan blue exclusion assay. The viability of the leukocytes exposed to 4.6 mg of CN (with no radioactivity) was 87%. After exposure to 10 mg of CN, cell viability decreased to 81%, and exposing cells to 20 mg of CN resulted in a further decrease in cell viability (70%). Viability was then tested with [89Zr]‐loaded CN only (20 mg). Following the radiolabelling of mixed leukocytes with [89Zr]‐loaded CN, 61% of the cells remained viable.
Discussion
Chitosan nanoparticle construct
CN formation by ionotropic gelation results from the electrostatic interaction between the protonated tertiary amine groups of chitosan and the charged groups of TPP. The size and surface charge of the nanoparticles formed is dependent upon the molecular weight and DD of chitosan, the weight ratio of chitosan to TPP as well as the pH of the solutions.27, 28, 51, 52, 53, 54 The fast kinetics of the formation of an opalescent solution containing CN slows the diffusion of the reactants into the solution and prevents the progress of further nanoparticle formation.
Separation of freeze‐dried CN and mixed leukocyte cells by centrifugation proved to be problematic in solutions that contained both materials, and co‐precipitation was observed. In order to make the nanoparticles more soluble, methylation of chitosan polymer at the amine functionality leading to quaternisation at that position was attempted using a method described by Kean et al.48 It was expected that the permanent quaternisation (completed with methyl iodide and under basic conditions) would increase the water solubility of chitosan and that the resulting increase in cationic character could ultimately lead to interactions with the negatively charged surface of cells. However, the decreased number of free amine moieties resulted in low binding affinities of the trimethylated chitosan to the radiometals (data not shown). CNs with higher water solubility were generated from 15 kDa chitosan polymer; these CNs were less prone to agglomeration and were easily re‐suspended following centrifugation.
Radiolabelling of chitosan nanoparticles with 89Zr and 64Cu
The metal binding efficiency of CN is not necessarily correlated with the molecular mass of chitosan used to prepare the nanoparticles as seen in Tables 1 and 2. Although CN that were prepared from MMW chitosan had the highest labelling efficiency, they were not selected for further experiments because of the difficulty in re‐suspending them in saline solution as well as the problems of separating them from mixed leukocytes by centrifugation that would often lead to co‐precipitation. As a result, CNs built from the 15 kDa chitosan polymer that showed an average of 73.8% uptake of 89Zr and 70.7% uptake of 64Cu were selected and used to radiolabel leukocyte cells.
Measurement of leukocyte radiolabelling efficiency and retention of [89Zr]‐ or [64Cu]‐loaded chitosan nanoparticles into mixed human leukocytes
The results shown in Figures 2 and 3 suggest a two‐step process for the mechanism of uptake of [89Zr]‐loaded CN through the cell membrane via pinocytosis21 or by phagocytosis.5 A similar mechanism to that reported by Lesniak et al. for the cell uptake of carboxylated polystyrene and silica nanoparticles55 may be occurring here. We postulate a two‐step mechanism, comprising an initial step in which the cationic nanoparticles adhere reversibly to the negatively charged cell membrane by electrostatic interaction, followed by internalisation into the cell via a slower process. The initial fast association of 89Zr into the cells (as seen in Figure 2) may be the result of an accumulation of the [89Zr]‐CN at the cell membrane through a weak adhesive contact after the initial mixing with the leukocytes using a vortex for 30 s followed by dissociation and dispersion until an equilibrium is reached. Leukocytes were radiolabelled in saline rather than a plasma medium to avoid any potential binding of the 89Zr or 64Cu to plasma proteins that would result in lower labelling efficiencies. The differences observed in the leukocyte radiolabelling efficiency and retention of radioactivity between 89Zr and 64Cu are a consequence of the differences of the initial cell membrane adhesion step. [64Cu]‐loaded CN might have a slower or weaker membrane binding that may in some part be as a result of a lower binding strength of copper to the nanoparticle.
Efflux of the radioactivity from the cells reached 28.4% (±5.8%) loss of 89Zr after 2 h and was followed by a slow release afterwards (Figure 3). After 24 h, around 50% of the radioactivity was still associated with the cells. We can infer from the results shown in Figures 2 and 3 that only a fraction of the [89Zr]‐loaded CN are internalised after 15‐min incubation and a fraction is still bound to the cell membrane. The loss of radioactivity from this point is likely to be because of competition between plasma proteins in the CFP and the cell membrane‐bound CN for 89Zr.
Because of the size (<0.5 µm) and shape of the CN and their ability to interact with the cell membrane resulting from their surface charge, it is likely that cellular internalisation involves an endocytosis56 or a phagocytosis pathway.5 These results reflect fast initial adhesion of [89Zr]‐ or [64Cu]‐loaded CN to the cell membrane followed by partial release leading to only a fraction of nanoparticles being internalised into the cell.
[64Cu]‐loaded CN showed a much poorer association into leukocyte cells compared with its 89Zr counterpart (Figure 2). In addition, the efflux of 64Cu from cells was much more rapid with more than 90% of the 64Cu being expelled from the cell or released from the membrane after 2‐h incubation in CFP (Figure 3). Similarly to the situation with the [89Zr]‐loaded CN, the loss could be attributed to the competition between components in the CFP and the leukocyte membrane‐bound [64Cu]‐loaded CN. The rapid loss of radioactivity from the cells might be a result of the greater affinity of components of the CFP for copper rather than zirconium. Human plasma contains specific copper transporter proteins such as ceruloplasmin or transcuprein57 that could be competing for the binding of 64Cu. Our results suggest that 89Zr is the superior radiometal to use for human inflammation scanning. The uptake mechanism of [89Zr]‐ or [64Cu]‐loaded CN into cells is unclear. There is a possibility of phagocytic engulfment5 or internalisation via an endocytosis or pinocytosis mechanism based on the size, shape and surface charge of the CN.56
The cytotoxicity effect of unlabelled CN on mixed leukocytes was also studied. Interestingly, we found that increased exposure to CN gel had a negative effect on the viability of leukocyte cells, suggesting a link between the amounts of CN and the viability of leukocyte cells, conversely to what was reported by Kean et al.48
Our method of radiolabelling WBCs with long‐lived PET isotopes is made more attractive because of its ease of use when compared with other reported methods using nanoparticles17 that require the addition of an organic reticulating agent and coupling of a metal complexing agent. Our technique requires neither the use of toxic organic reagents to construct the nanoparticles nor coupling to a complexing agent. Recently, a promising method using [89Zr]‐oxinate4 has been used to monitor cell traffic in vivo with PET,19 but further evaluation of the cytotoxicity of this method is required. In addition, [89Zr]‐DFO‐NCS has been used to radiolabel mesenchymal stem cells,18 but further investigation is required to asses if this method is translatable to leukocyte cell labelling.
From the measurements of leukocyte radiolabelling efficiency as well as the retention of 89Zr in the cell, we could estimate a time from blood draw to re‐administration of [89Zr]‐leukocytes to be up to 345 min (5.75 h) with this method. This takes into consideration (i) erythrocyte sedimentation from whole blood (60 min), (ii) isolation of leukocytes by centrifugation (15 min), (iii) radiolabelling of leukocyte cells (30 min), (iv) washing of the radiolabelled cells with CFP and (v) an interval to allow for the retention of 89Zr‐CN by the leukocytes to stabilise (240 min).
Conclusion
We report an improved and reproducible method for radiolabelling mixed human leukocytes with 89Zr. This new technique will allow for the quantitative imaging of inflammation using PET. With this method, which utilises CNs as a delivery system of PET isotopes into the cells, we have demonstrated a fast association of 89Zr into leukocyte cells with a slow rate of efflux thereafter.
This technique is suitable for measuring and monitoring the regional signal from migrated radiolabelled WBCs to pathological tissue in infectious and inflammatory diseases such as inflammatory arthritis. It offers the potential for superior quantification and sensitivity compared with current methods using SPECT. This represents the first essential steps towards the development of a generic PET technique for quantitative imaging of inflammation in the clinic with potential for applications to monitor other types of cell traffic such as stem cells.
Acknowledgements
This work was funded by the NIHR Manchester Musculoskeletal Biomedical Research Unit (Grant number: R114999). MF PhD studentship was funded by the Wolfson Molecular Imaging Centre, University of Manchester.
Fairclough, M. , Prenant, C. , Ellis, B. , Boutin, H. , McMahon, A. , Brown, G. , Locatelli, P. , and Jones, A. K. P. (2016) A new technique for the radiolabelling of mixed leukocytes with zirconium‐89 for inflammation imaging with positron emission tomography. J. Label Compd. Radiopharm, 59: 270–276. doi: 10.1002/jlcr.3392.
References
- 1. Karsdal M., Woodworth T., Henriksen K., Maksymowych W., Genant H., Vergnaud P., Christiansen C., Schubert T., Qvist P., Schett G., Platt A., Bay‐Jensen A.‐C., Arthritis Res. Ther. 2011, 13, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Palestro C. J., Love C., Bhargava K. K., Q. J. Nucl. Med. Mol. Imaging 2009, 53, 105. [PubMed] [Google Scholar]
- 3. Al‐Janabi M. A., Jones A. K., Solanki K., Sobnack R., Bomanji J., Al‐Nahhas A. A., Doyle D. V., Britton K. E., Huskisson E. C., Nucl. Med. Commun. 1988, 9, 987. [DOI] [PubMed] [Google Scholar]
- 4. Adonai N., Nguyen K. N., Walsh J., Iyer M., Toyokuni T., Phelps M. E., McCarthy T., McCarthy D. W., Gambhir S. S., Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pala A., Liberatore M., D'Elia P., Nepi F., Megna V., Mastantuono M., Al‐Nahhas A., Rubello D., Barteri M., Mol. Imaging Biol. 2012, 14, 593. [DOI] [PubMed] [Google Scholar]
- 6. Li Z.‐B., Chen K., Wu Z., Wang H., Niu G., Chen X., Mol. Imaging Biol. 2009, 11, 415. [DOI] [PubMed] [Google Scholar]
- 7. Bhargava K. K., Gupta R. K., Nichols K. J., Palestro C. J., Nucl. Med. Biol. 2009, 36, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu C., Ma G., Li J., Zheng K., Dang Y., Shi X., Sun Y., Li F., Zhu Z., Clin. Imaging 2013, 37, 28. [DOI] [PubMed] [Google Scholar]
- 9. van der Laken C. J., Elzinga E. H., Kropholler M. A., Molthoff C. F. M., van der Heijden J. W., Maruyama K., Boellaard R., Dijkmans B. A. C., Lammertsma A. A., Voskuyl A. E., Arthritis Rheum. 2008, 58, 3350. [DOI] [PubMed] [Google Scholar]
- 10. Fischer G., Seibold U., Schirrmacher R., Wängler B., Wängler C., Molecules 2013, 18, 6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deri M. A., Zeglis B. M., Lewis J. S., Nucl. Med. Biol. 2013, 40, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perk L., Vosjan M., Visser G., Budde M., Jurek P., Kiefer G., van Dongen G., Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vosjan M. J. W. D., Perk L. R., Visser G. W. M., Budde M., Jurek P., Kiefer G. E., van Dongen G. A. M. S., Nat. Protocols 2010, 5, 739. [DOI] [PubMed] [Google Scholar]
- 14. Borjesson P. K. E., Jauw Y. W. S., Boellaard R., de Bree R., Comans E. F. I., Roos J. C., Castelijns J. A., Vosjan M. J. W. D., Kummer J. A., Leemans C. R., Lammertsma A. A., van Dongen G. A. M. S., Clin. Cancer Res. 2006, 12, 2133. [DOI] [PubMed] [Google Scholar]
- 15. Nayak T. K., Brechbiel M. W., Bioconjug. Chem. 2009, 20, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Börjesson P. K. E., Jauw Y. W. S., de Bree R., Roos J. C., Castelijns J. A., Leemans C. R., van Dongen G. A. M. S., Boellaard R., J. Nucl. Med. 2009, 50, 1828. [DOI] [PubMed] [Google Scholar]
- 17. Keliher E. J., Yoo J., Nahrendorf M., Lewis J. S., Marinelli B., Newton A., Pittet M. J., Weissleder R., Bioconjug. Chem. 2011, 22, 2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bansal A., Pandey M. K., Demirhan Y. E., Nesbitt J. J., Crespo‐Diaz R. J., Terzic A., Behfar A., DeGrado T. R., EJNMMI Res. 2015, 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Charoenphun P., Meszaros L. K., Chuamsaamarkkee K., Sharif‐Paghaleh E., Ballinger J. R., Ferris T. J., Went M. J., Mullen G. E., Blower P. J., Eur. J. Nucl. Med. Mol. Imaging 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Varma A. J., Deshpande S. V., Kennedy J. F., Carbohydr. Polym. 2004, 55, 77. [Google Scholar]
- 21. Nam H. Y., Kwon S. M., Chung H., Lee S.‐Y., Kwon S.‐H., Jeon H., Kim Y., Park J. H., Kim J., Her S., Oh Y.‐K., Kwon I. C., Kim K., Jeong S. Y., J. Control. Release 2009, 135, 259. [DOI] [PubMed] [Google Scholar]
- 22. Kean T., Thanou M., Adv. Drug Deliv. Rev. 2010, 62, 3. [DOI] [PubMed] [Google Scholar]
- 23. Jayakumar R., Menon D., Manzoor K., Nair S. V., Tamura H., Carbohydr. Polym. 2010, 82, 227. [Google Scholar]
- 24. Peniche H., Peniche C., Polym. Int. 2011, 60, 883. [Google Scholar]
- 25. Garcia‐Fuentes M., Alonso M. J., J. Control. Release 2012, 161, 496. [DOI] [PubMed] [Google Scholar]
- 26. Chen M.‐C., Mi F.‐L., Liao Z.‐X., Hsiao C.‐W., Sonaje K., Chung M.‐F., Hsu L.‐W., Sung H.‐W., Adv. Drug Deliv. Rev. 2013, 65, 865. [DOI] [PubMed] [Google Scholar]
- 27. Zhang H.‐L., Wu S.‐H., Tao Y., Zang L.‐Q., Su Z.‐Q., J. Nanomater. 2010, 2010. [Google Scholar]
- 28. Calvo P., Remuñán‐López C., Vila‐Jato J. L., Alonso M. J., J. Appl. Polym. Sci. 1997, 63, 125. [Google Scholar]
- 29. Qi L., Xu Z., Jiang X., Hu C., Zou X., Carbohydr. Res. 2004, 339, 2693. [DOI] [PubMed] [Google Scholar]
- 30. Dodane V., Vilivalam V. D., Pharm. Sci. Technol. Today 1998, 1, 246. [Google Scholar]
- 31. Tiyaboonchai W., Naresuan Univ J. 2003, 11, 51. [Google Scholar]
- 32. Ishii T., Okahata Y., Sato T., Biochim. Biophys. Acta Biomembr. 2001, 1514, 51. [DOI] [PubMed] [Google Scholar]
- 33. Baldrick P., Regul. Toxicol. Pharmacol. 2010, 56, 290. [DOI] [PubMed] [Google Scholar]
- 34. Bagheri‐Khoulenjani S., Taghizadeh S. M., Mirzadeh H., Carbohydr. Polym. 2009, 78, 773. [Google Scholar]
- 35. Casettari L., Vllasaliu D., Lam J. K. W., Soliman M., Illum L., Biomaterials 2012, 33, 7565. [DOI] [PubMed] [Google Scholar]
- 36. Hein S., Wang K., Stevens W. F., Kjems J., Mater. Sci. Technol. 2008, 24, 1053. [Google Scholar]
- 37. Sashiwa H., Aiba S.‐I., Prog. Polym. Sci. 2004, 29, 887. [Google Scholar]
- 38. Yang W., Mou T., Guo W., Jing H., Peng C., Zhang X., Ma Y., Liu B., Bioorg. Med. Chem. Lett. 2010, 20, 4840. [DOI] [PubMed] [Google Scholar]
- 39. Agrawal P., Strijkers G. J., Nicolay K., Adv. Drug Deliv. Rev. 2010, 62, 42. [DOI] [PubMed] [Google Scholar]
- 40. Sonaje K., Chuang E.‐Y., Lin K.‐J., Yen T.‐C., Su F.‐Y., Tseng M. T., Sung H.‐W., Mol. Pharm. 2012, 9, 1271. [DOI] [PubMed] [Google Scholar]
- 41. Tan W. B., Jiang S., Zhang Y., Biomaterials 2007, 28, 1565. [DOI] [PubMed] [Google Scholar]
- 42. Linlin L., Dong C., Yanqi Z., Zhengtao D., Xiangling R., Xianwei M., Fangqiong T., Jun R., Lin Z., Nanotechnology 2007, 18, 405102. [Google Scholar]
- 43. Shi Z., Neoh K. G., Kang E. T., Shuter B., Wang S.‐C., Poh C., Wang W., ACS Appl. Mater. Interfaces 2008, 1, 328. [DOI] [PubMed] [Google Scholar]
- 44. Kim J.‐H., Kim Y.‐S., Park K., Kang E., Lee S., Nam H. Y., Kim K., Park J. H., Chi D. Y., Park R.‐W., Kim I.‐S., Choi K., Chan Kwon I., Biomaterials 2008, 29, 1920. [DOI] [PubMed] [Google Scholar]
- 45. Nagpal K., Singh S. K., Mishra D. N., Chem. Pharm. Bull. 2010, 58, 1423. [DOI] [PubMed] [Google Scholar]
- 46. Janes K. A., Calvo P., Alonso M. J., Adv. Drug Deliv. Rev. 2001, 47, 83. [DOI] [PubMed] [Google Scholar]
- 47. Fischer D., Li Y., Ahlemeyer B., Krieglstein J., Kissel T., Biomaterials 2003, 24, 1121. [DOI] [PubMed] [Google Scholar]
- 48. Kean T., Roth S., Thanou M., J. Control. Release 2005, 103, 643. [DOI] [PubMed] [Google Scholar]
- 49. Thakur M. L., Lavender J. P., Arnot R. N., Silvester D. J., Segal A. W., J. Nucl. Med. 1977, 18, 1014. [PubMed] [Google Scholar]
- 50. Ellis B., In Sampson's Book of Radiopharmacy (Ed.: Theobold T.), 2011, pp. 421. [Google Scholar]
- 51. Nasti A., Zaki N., Leonardis P., Ungphaiboon S., Sansongsak P., Rimoli M., Tirelli N., Pharm. Res. 2009, 26, 1918. [DOI] [PubMed] [Google Scholar]
- 52. Rampino A., Borgogna M., Blasi P., Bellich B., Cesàro A., Int. J. Pharm. 2013, 455, 219. [DOI] [PubMed] [Google Scholar]
- 53. Zhang H., Oh M., Allen C., Kumacheva E., Biomacromolecules 2004, 5, 2461. [DOI] [PubMed] [Google Scholar]
- 54. Gan Q., Wang T., Cochrane C., McCarron P., Colloids Surf. B Biointerfaces 2005, 44, 65. [DOI] [PubMed] [Google Scholar]
- 55. Lesniak A., Salvati A., Santos‐Martinez M. J., Radomski M. W., Dawson K. A., Åberg C., J. Am. Chem. Soc. 2013, 135, 1438. [DOI] [PubMed] [Google Scholar]
- 56. Canton I., Battaglia G., Chem. Soc. Rev. 2012, 41, 2718. [DOI] [PubMed] [Google Scholar]
- 57. Hueting R., J. Label. Compd. Radiopharm. 2014, 57, 231. [DOI] [PubMed] [Google Scholar]