

Summary
While proteasome inhibition is a validated therapeutic approach for multiple myeloma (MM), inhibition of individual constitutive proteasome (c20S) and immunoproteasome (i20S) subunits has not been fully explored owing to a lack of effective tools. We utilized the novel proteasome constitutive/immunoproteasome subunit enzyme‐linked immunosorbent (ProCISE) assay to quantify proteasome subunit occupancy in samples from five phase I/II and II trials before and after treatment with the proteasome inhibitor carfilzomib. Following the first carfilzomib dose (15–56 mg/m2), dose‐dependent inhibition of c20S and i20S chymotrypsin‐like active sites was observed [whole blood: ≥67%; peripheral blood mononuclear cells (PBMCs): ≥75%]. A similar inhibition profile was observed in bone marrow–derived CD138+ tumour cells. Carfilzomib‐induced proteasome inhibition was durable, with minimal recovery in PBMCs after 24 h but near‐complete recovery between cycles. Importantly, the ProCISE assay can be used to quantify occupancy of individual c20S and i20S subunits. We observed a relationship between MM patient response (n = 29), carfilzomib dose and occupancy of multiple i20S subunits, where greater occupancy was associated with an increased likelihood of achieving a clinical response at higher doses. ProCISE represents a new tool for measuring proteasome inhibitor activity in clinical trials and relating drug action to patient outcomes.
Keywords: molecular analysis, multiple myeloma, myeloma therapy, pharmacology, trials
The 26S proteasome is a ubiquitously expressed protein complex responsible for the homeostatic control of protein turnover and regulated degradation of proteins involved in most cellular functions (Wilk & Orlowski, 1983; Coux et al, 1996; Ciechanover, 2012). Each proteasome has a 20S core, each containing two copies of three distinct proteolytic enzymes. The proteasome exists in two forms: the constitutive proteasome (c20S), expressed ubiquitously throughout the body, and the immunoproteasome (i20S), expressed primarily in haematopoietic cells or in cytokine‐exposed non‐haematopoietic cells (Martinez & Monaco 1991; Glynne et al, 1991, Nandi et al, 1996). In the c20S, proteolytic activities are encoded in the β5, β1 and β2 subunits and are characterized on the basis of substrate specificity as chymotrypsin‐like (CT‐L), caspase‐like and trypsin‐like, respectively. In the i20S, low‐molecular mass polypeptide 7 (LMP7), LMP2 and multicatalytic endopeptidase complex‐like 1 (MECL1) replace β5, β1 and β2, respectively.
Approval of the dipeptide boronic acid proteasome inhibitor, bortezomib, for the treatment of relapsed multiple myeloma (MM; Richardson et al, 2005) and relapsed or refractory mantle cell lymphoma (Fisher et al, 2006) has spurred the development of other agents that target proteasome activity (Bennett & Kirk, 2008; Dick & Fleming, 2010; Kuhn et al, 2011). These agents vary from targeting individual active sites to targeting all three catalytic activities (Chauhan et al, 2005; Muchamuel et al, 2009). Carfilzomib, a tetrapeptide epoxyketone proteasome inhibitor, has recently been approved in the US for the treatment of relapsed and refractory MM (Kuhn et al, 2007; Jain et al, 2011). While both carfilzomib and bortezomib primarily target the CT‐L activity of the c20S and i20S, carfilzomib displays greater specificity than bortezomib for CT‐L activity relative to caspase‐like and trypsin‐like activities (Demo et al, 2007).
Accurate and relevant markers to monitor pharmacodynamics are important in the development of targeted agents for cancer, in which narrow therapeutic margins may limit drug effectiveness (Kummar et al, 2010; Yap et al, 2010). Ideally, direct measurement of target inhibition in malignant cells should be related to patient outcome. This approach is challenging given the complexity of the target (particularly multi‐subunit intracellular protein complexes), the need for serial sampling, and the current lack of ready access to tumour cells. Instead, surrogate tissues (e.g. cells isolated from blood) and/or markers (e.g. changes in downstream signalling pathways) are used to approximate the clinical profile of an agent. This method may result in either equivocal results or the incorporation of potentially suboptimal dosing strategies into later clinical trials.
The standard method for determining proteasome inhibition uses biochemical substrates, such as Leu‐Leu‐Val‐Tyr‐7‐amino‐4‐methylcoumarin (LLVY‐AMC) for determining CT‐L activity (Lightcap et al, 2000); however, this approach cannot differentiate between c20S and i20S activity. MM cells express both proteasome types, and the role of individual subunits in the anti‐tumour effect of clinically relevant proteasome inhibitors remains an area of active research (Kuhn et al, 2009; Parlati et al, 2009; Wehenkel et al, 2012). Meanwhile, the role of the non–CT‐L active sites and their inhibition by proteasome inhibitors remain poorly understood, mainly due to a lack of suitable substrates for measuring enzymatic activity. Substrates for LMP2 and MECL1 are poorly defined and, similar to LLVY‐AMC, do not differentiate between immunoproteasome subunit activity and the corresponding constitutive proteasome subunits (Groettrup et al, 2010). Studies utilizing inhibitors that specifically target the caspase‐like or trypsin‐like active sites suggest that inhibition of non–CT‐L active sites alone may sensitize cancer cells to CT‐L inhibition but are not sufficient to induce cytotoxicity (Britton et al, 2009; Mirabella et al, 2011). Other assessment tools are needed to fully characterize the role of c20S and i20S active sites in the anti‐cancer activity of proteasome inhibitors. Considering these limitations, we developed the proteasome constitutive/immunoproteasome subunit enzyme‐linked immunosorbent (ProCISE) assay to measure the amounts and occupancy of proteasome active sites in samples with heterogeneous proteasome composition (Parlati et al, 2009). We previously reported the use of the ProCISE assay in quantifying c20S and i20S subunit levels in primary cells and cell lines derived from haematopoietic and solid tumours (ST) (Parlati et al, 2009). Herein, we present the first clinical application of ProCISE, demonstrating its utility in determining the level of occupancy of all six proteasome active sites in samples derived from MM and ST patients treated with carfilzomib from five phase I/II and phase II clinical trials.
Methods
Reagents
Proteasome active site‐binding probe (PABP) (Bennett et al, 2006) and carfilzomib (Smyth & Laidig, 2006) were synthesized as previously described (Smyth & Laidig, 2006). The fluorogenic substrate succinyl‐LLVY‐AMC and human c20S and i20S were purchased from Boston Biochem (Boston, MA, USA). Human whole blood and peripheral blood mononuclear cell (PBMC)‐enriched leukapheresis‐derived samples from healthy volunteers were purchased from AllCells (Berkeley, CA, USA). Monoclonal antibodies specific for β1, β2, LMP7 and LMP2 were purchased from Enzo Life Sciences (Farmingdale, NY, USA). Antibodies specific to MECL1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). A rabbit polyclonal anti‐β5‐antibody was generated as previously described (Muchamuel et al, 2009). Horseradish peroxidase (HRP)–conjugated goat anti‐rabbit and goat anti‐mouse were purchased from Jackson ImmunoResearch (West Grove, PA, USA). HRP‐conjugated rabbit anti‐goat was purchased from Invitrogen (Carlsbad, CA, USA).
Sample collection
Samples for analysis were derived from phase I/II and phase II trials (Table 1). Trial details can be found in the Supplementary Information. Patients received carfilzomib as an intravenous infusion over 2–10 min or, as in the case of PX‐171‐007, over 30 min on days 1, 2, 8, 9, 15 and 16 of a 28‐day cycle at doses from 15 to 56 mg/m2.
Table 1.
Clinical Samples used for Pharmacodynamic Analysis
| Blood and PBMC pharmacodynamic samples from MM patientsa | ||||
| Trialb | Phase | N | 2‐ to 10‐min infusion dose (initial/escalation mg/m2) | 30‐min infusion dose (initial/escalation mg/m2) |
|
PX‐171‐005 (NCT00721734) |
II | 31 | 15/20 (N = 31) | – |
|
PX‐171‐006 (NCT00603447) |
Ib/II | 16 |
15 only (n = 8) 20/27 (n = 8) |
– |
|
PX‐171‐007 (NCT00531284) |
I/II | 24 |
20/36 (n = 3) 20/45 (n = 3) 20/56 (n = 19) |
|
| Bone marrow pharmacodynamic samples from MM patientsc | ||||
| Trialb | Phase | N | 2‐ to 10‐min infusion dose (mg/m2) | 30‐min infusion dose (mg/m2) |
|
PX‐171‐003‐A0 and ‐A1 (NCT00511238) |
II | 7 |
20 only (N = 7; predose, n = 3; C1D2, n = 4) |
— |
|
PX‐171‐004 (NCT00530816) |
II | 11 |
20 only (N = 11; predose, n = 6; C1D2, n = 5) |
— |
|
PX‐171‐005 (NCT00721734) |
II | 15 |
Dose N/A (MM, N = 15; predose, n = 15; C1D2, n = 0) |
— |
| Blood and PBMC pharmacodynamic samples from ST patientsa | ||||
|
PX‐171‐007 (NCT00531284) |
I/II | 36 |
20 only (n = 3) 20/27 (n = 3) 20/36 (n = 18) |
20/45 (n = 2) 36 only (n = 6) 45 only (n = 6) |
C, cycle; D, day; MM, multiple myeloma; N/A, not applicable; PBMC, peripheral blood mononuclear cell; ST, solid tumour.
cycle 1, multiple days, and cycle 2, day 1 only.
Blood and/or bone marrow aspirates were collected from patients in five clinical trials prior to and after carfilzomib treatment and at the doses listed. Samples were analysed for proteasome activity and subunit occupancy as described in Fig 1.
Predose and C1, D2 only.
In PX‐171‐003‐A0/A1, PX‐171‐004 and PX‐171‐005, bone marrow samples were collected during screening and on day 2 of cycle 1 (approximately 4 h post‐dose). In PX‐171‐005, PX‐171‐006 and PX‐171‐007, whole blood and isolated PBMC samples were collected prior to dosing and 1 h post‐dose on days 1, 2 and 8 of cycle 1 and on day 1 of cycle 2 (no samples were collected on days 2 or 8 in cycle 1 for patients with ST receiving carfilzomib over 2–10 min). All samples were obtained after informed consent was given and in accordance with the Helsinki Declaration.
Sample preparation
Bone marrow aspirates were enriched for CD138+ tumour cells via immunomagnetic bead separation (RoboSep, Stem Cell Technologies, Vancouver, Canada) as per the manufacturer's protocol. Cells were lysed in lysis buffer (20 mM Tris pH 8∙0, 5 mM ethylenediaminetetraacetic acid, EDTA) and cleared of cellular debris by centrifugation prior to analysis.
CT‐L activity assay
CT‐L activity using succinyl‐LLVY‐AMC was determined as previously described (Lightcap et al, 2000; Demo et al, 2007). Protocol modifications for the current study included a blood quality control (QC) matrix, generation of an aminomethylcoumarin standard curve and QC samples. Details are provided in the Supplementary Information.
ProCISE assay
We previously published the methodology of the ProCISE assay for evaluation of samples from primary cells and tumour cell lines (Parlati et al, 2009). The clinical application of the ProCISE assay involves, in brief, the following steps: (i) incubation of PABP with whole blood, PBMC or bone marrow lysates; (ii) denaturation with 5∙6 M guanidine hydrochloride; (iii) addition of streptavidin‐coated beads; (iv) extensive bead washing; (v) addition of proteasome subunit‐specific primary antibody followed by a secondary HRP‐conjugated antibody; and (vi) luminescence‐based detection. Additional details on the clinical application of the ProCISE assay can be found in the Supplementary Information section.
Statistical analysis
One‐tailed Welch–Satterthwaite t‐tests, or fixed, or mixed‐effects analysis of variance models followed by Tukey's studentized range post‐hoc tests were used, as appropriate, to determine statistically significant differences. The presence of a linear trend was tested using an F‐test for linear contrast. Statistical analyses were performed using GraphPad Prism (version 5.03; GraphPad Prism Software Inc., San Diego, CA, USA) or SAS software (version 9.1.3; SAS Institute, Inc., Cary, NC, USA). Statistical significance was achieved when P < 0∙05.
Results
ProCISE clinical validation
ProCISE quantitatively measures the levels of proteasome subunit active sites within cell lysates via the addition of a biotinylated probe that covalently binds to all active sites. Probe‐bound active sites are captured on streptavidin‐sepharose beads and quantified with subunit‐specific antibodies compared with a standard curve of purified proteasomes (Fig 1A). We determined that ProCISE accurately measures c20S and i20S subunits within a large dynamic range (2–167% of c20S subunits and 3–250% of i20S subunits found within typical whole blood and PBMC samples, respectively; Table SI). In addition, when known concentrations of c20S and i20S were spiked into whole blood or PBMC samples, respectively, we observed good correlation between the measured and expected values (Figure S1).
Figure 1.
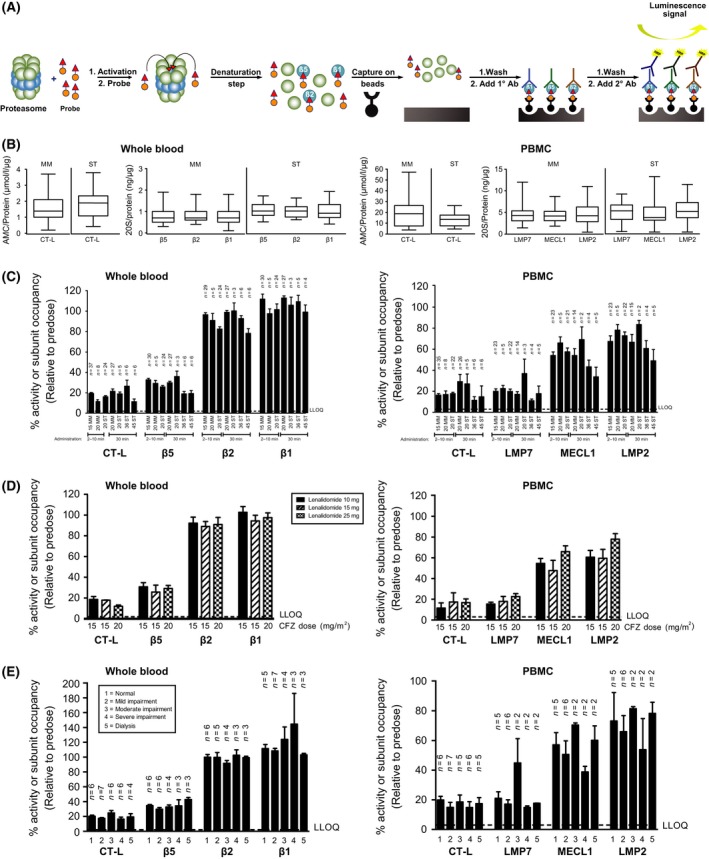
Carfilzomib administration results in potent inhibition of multiple proteasome subunits. (A) ProCISE assay schematic. PABP is added to purified 20S proteasomes (activated with 0∙03% SDS‐AB) or cell lysates and incubated for 2 h. Denaturation with guanidine and capture on beads is then done simultaneously for 1 h followed by five washes in PBS + 1% BSA + 0∙05% Tween. Primary antibodies for specific proteasome active site subunits are then added overnight, followed by another five washes. A secondary HRP antibody is then added for 2 h, followed by five washes. A chemiluminescent substrate is then added for 5–10 min, and luminescence is determined. (B) Proteasome subunit levels were measured by ProCISE in 62 whole blood and 54 PBMC samples from MM patients and 27 whole blood and 24 PBMC samples from ST patients. CT‐L activity was measured in the same samples using the LLVY‐AMC substrate. Data are presented as box and whisker plots with subunit concentration (ProCISE) or specific activity (LLVY‐AMC). Mean specific activity [AMC/protein (μM/μg)] and subunit concentrations [subunit/protein (ng/μg)] are represented by a horizontal line, and error bars represent minimum and maximum values. (C) Whole blood and PBMC samples taken 1 h after the first administration of carfilzomib in MM and ST patients (doses: 15–45 mg/m2) were analysed for proteasome subunit activity by LLVY‐AMC and occupancy by ProCISE. Values were normalized to predose values, and data are presented as mean (±SEM) relative activity or occupancy. The dotted line represents the LLOQ for whole blood. (D) MM patients were treated with carfilzomib 15 or 20 mg/m2, dexamethasone 40 mg and lenalidomide 10, 15, or 25 mg on day 1. Whole blood and PBMC were isolated predose and 1 h after the first dose of carfilzomib dose was administered. Samples were analysed for proteasome activity and subunit occupancy, as described in panel A. Values are normalized to predose values, and data are presented as mean (±SEM) specific activity or occupancy (N = 2–8 per cohort). LLVY‐AMC data are reproduced from Badros et al (2013) with permission from Nature Publishing Group. (E) MM patients with varying degrees of renal function were treated with 15 mg/m2 of carfilzomib on day 1. Whole blood and PBMC were isolated predose 1 h after carfilzomib dose administration. Samples were analysed for proteasome activity and subunit occupancy as described in panel B. Values are normalized to predose values, and data are presented as mean (±SEM) relative activity or occupancy (N = 2–7 per cohort). 1°, primary; 2°, secondary; Ab, antibody; AMC, aminomethylcoumarin; BSA, bovine serum albumin; CFZ, carfilzomib; CT‐L, chymotrypsin‐like; D, day; HRP, horseradish peroxidase; LLOQ, lower limit of quantitation; LLVY, Leu‐Leu‐Val‐Tyr‐7‐amino‐4‐methylcoumarin; LMP, low‐molecular mass polypeptide; MECL, multicatalytic endopeptidase complex‐like; MM, multiple myeloma; PABP, proteasome active site‐binding probe; PBMC, peripheral blood mononuclear cell; PBS, phosphate buffer solution; SDS‐AB, sodium dodecyl sulfate in assay buffer; SEM, standard error of the mean; ST, solid tumour.
To validate ProCISE as a measure of drug activity, we treated whole blood for 1 hwith varying concentrations of carfilzomib and analysed both whole blood and isolated PBMCs (Figure S2). The ex vivo dose response of carfilzomib‐mediated β5 and LMP7 occupancy measured by ProCISE mirrored that of the enzymatic activity assessed using LLVY‐AMC in the same samples. Carfilzomib showed more than 10‐fold selectivity for CT‐L subunits relative to other c20S and i20S subunits, findings that were similar to previously reported results from purified proteasomes (Demo et al, 2007). Finally, baseline levels of proteasome active sites from MM and ST patient samples collected from multiple studies were analysed by ProCISE (Fig 1B). Stoichiometric amounts of the c20S subunits and the i20S subunits were present in whole blood and PBMCs, respectively, independent of tumour type. Additionally, levels of CT‐L activity inhibition and subunit occupancy in patients receiving carfilzomib were similar using the LLVY‐AMC or ProCISE assays in whole blood or PBMCs, suggesting that measurement of subunit occupancy accurately reflects proteolytic activity (Fig 1C; Table SII). Taken together, these data validate ProCISE for the quantitative measurement of proteasome inhibition, active site occupancy and levels of active site subunits in patient samples.
Inhibition of proteasome subunits by carfilzomib in patients
Carfilzomib has demonstrated anti‐tumour activity in patients with MM at doses ranging from 15 to 56 mg/m2 (Siegel et al, 2012; Vij et al, 2012a,b; Badros et al, 2013; Papadopoulos et al, 2013, 2015). In phase I and II trials, only the LLVY‐AMC assay was used in a limited number of patient samples to measure CT‐L activity alone (O'Connor et al, 2009; Badros et al, 2013; Niesvizky et al, 2013; Papadopoulos et al, 2013). To obtain the full active‐site inhibition profile of carfilzomib, we utilized ProCISE to quantify active site activity and occupancy in whole blood and PBMC samples from patients in multiple studies (n = 41 ST and n = 73 MM) 1 h after the first dose of carfilzomib (15–45 mg/m2) was administered. In patients receiving 15 or 20 mg/m2 of carfilzomib as a 2‐ to 10‐min infusion, rates of inhibition of β5 and LMP7 occupancy in whole blood and PBMC samples were 67–74% and 77–80%, respectively (see Fig 1C). At 20 mg/m2, no significant difference in the level of proteasome inhibition was detected between patients receiving a 2‐ to 10‐min infusion and those receiving a 30‐min infusion. Inhibition of the other i20S subunits (MECL1 and LMP2) was also observed in PBMCs at doses of 15 and 20 mg/m2, albeit at lower levels than LMP7 inhibition (31–46% inhibition of MECL1 and 22–33% inhibition of LMP2). At the same doses, there was less than 18% inhibition of the c20S subunits β2 and β1 in whole blood samples. At a higher dose (45 mg/m2 administered as a 30‐min infusion), we noted increased occupancy for almost all subunit sites assessed (Fig 1C). The effect was most prominent for MECL1 and LMP2; occupancy of β1, however, was not observed. A statistically significant linear dose‐response effect was seen for occupancy of β5, β2 and MECL1 (P values were 0∙0044, 0∙0137 and 0∙0432 for the linear trend, respectively).
Carfilzomib has been used in combination with lenalidomide and dexamethasone and as a single agent in patients with relapsed and/or refractory MM and varying degrees of renal function (Badros et al, 2013; Niesvizky et al, 2013; Siegel et al, 2013). Whole blood and PBMC samples from patients receiving 15 or 20 mg/m2 of carfilzomib with 40 mg of dexamethasone and 10, 15 or 25 mg of lenalidomide (study PX‐171‐006) were taken 1 h after the first dose of carfilzomib and analysed for active site activity and occupancy. Levels of active site occupancy were similar in patients receiving carfilzomib in combination with varying doses of lenalidomide (Fig 1D). No effect from the co‐administration of lenalidomide was noted at later time points in cycle 1 or at the beginning of cycle 2 (data not shown). In addition, active site occupancy was similar when patients treated with 15 mg/m2 of carfilzomib were stratified based on renal function status (study PX‐171‐005) and was consistent with results previously reported using the LLVY‐AMC assay (Fig 1E) (Badros et al, 2013). Taken together, these data demonstrate that carfilzomib induces consistent and potent occupancy of multiple proteasomal active sites in MM patients, regardless of concomitant drug administration or renal function.
Sustained proteasome inhibition after repeated carfilzomib administration
As carfilzomib selectively and irreversibly binds to its target (Demo et al, 2007), we assessed the duration of subunit occupancy and recovery of proteasome activity following its use. Whole blood and PBMC samples were analysed from patients from multiple studies both prior to and 1 h after receiving carfilzomib on days 1, 2 and 8 of cycle 1 and on day 1 of cycle 2. Consistent with results from animal studies (Demo et al, 2007), we observed cumulative and sustained subunit occupancy in whole blood samples that was most evident for β2 and β5 (Fig 2A). In PBMCs, modest recovery was seen 24 h after the first dose (day 2, predose), but unbound subunit levels were lower than pretreatment values (see Fig 2A). Following the second dose of carfilzomib, a slight increase in occupancy was observed across all i20S subunits (day 2, 1 h). Recovery following a 5‐day non‐dosing period (day 8, predose) was more prominent than after a 24‐h period (day 2, predose), but full recovery was not achieved. By the start of cycle 2 (cycle 2, predose), complete or near‐complete recovery was noted in PBMCs for all three i20S active sites at doses below 36 mg/m2. These results, in which data for each dose level were pooled across studies, are similar to those previously reported using the LLVY‐AMC assay in patients treated with carfilzomib, lenalidomide and dexamethasone from PX‐171‐006 (Niesvizky et al, 2013). Our findings suggest that the addition of lenalidomide and dexamethasone to carfilzomib treatment does not impact the pharmacodynamics of carfilzomib‐mediated proteasome inhibition. In patients receiving 45 mg/m2 of carfilzomib, minimal recovery was observed between day 8 and the start of cycle 2; it should be noted, however, that relatively few patients (range, 3–9) were analysed at this dose level.
Figure 2.
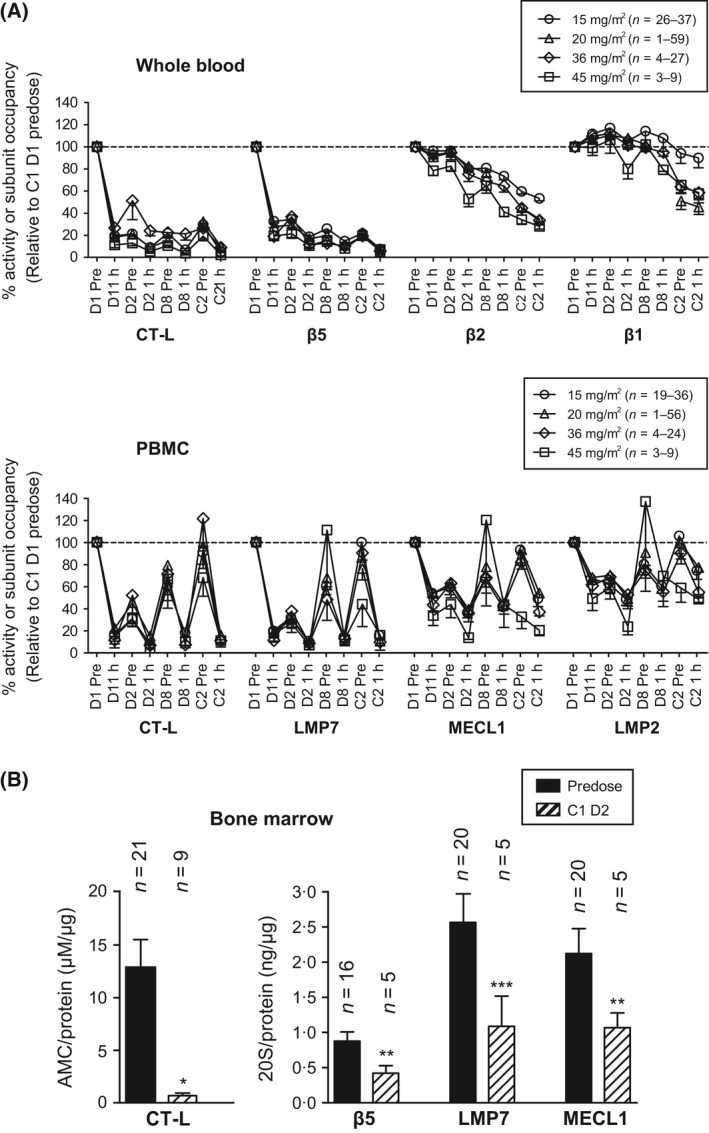
Repeat administration of carfilzomib results in sustained proteasome inhibition. (A) Patients received carfilzomib at 15, 20, 36 or 45 mg/m2 on days 1, 2, 8, D9, 15 and 16 of a 28‐day cycle. Whole blood and PBMC samples were taken prior to and 1 h after administration on days 1, 2 and 8 of cycle 1 and day 1 of cycle 2. Samples were analysed for proteasome activity and subunit occupancy as described in Fig 1 and were normalized to cycle 1, day 1 predose values. Data are presented as mean (±SEM) relative specific activity or occupancy. (B) Bone marrow samples from MM patients were taken during baseline screening and/or on cycle 1, day 2, 1 to 4 h after the second dose of carfilzomib. CD138+ cells and samples were analysed for proteasome activity and subunit occupancy. Data are presented as mean (±SEM) relative activity or subunit concentration. *P < 0∙001; **P < 0∙01; ***P < 0∙05 by a t‐test. C, cycle; CT‐L, chymotrypsin‐like; D, day; AMC, aminomethylcoumarin; LMP, low‐molecular mass polypeptide; MECL, multicatalytic endopeptidase complex‐like; MM, multiple myeloma; PBMC, peripheral blood mononuclear cell; SEM, standard error of the mean.
In study PX‐171‐007, patients were dose‐escalated from 20 mg/m2 to a target dose that ranged from 27 to 56 mg/m2 beginning on day 8 of cycle 1. As near complete recovery of proteasome activity and subunit levels in PBMC was observed by the beginning of cycle 2 (cycle 2, predose), we compared the level of individual i20S subunit occupancy on cycle 2, day 1 with pretreatment values. Results from ProCISE showed that all doses resulted in similar and potent increases in LMP7 occupancy, regardless of whether carfilzomib was administered as a 2‐ to 10‐min infusion or as a 30‐min infusion, while a dose‐dependent increase in MECL1 and LMP2 occupancy was observed at doses of up to 45 mg/m2 (Figure S3). At a dose of 56 mg/m2 (the maximum tolerated dose in patients with MM when infused over 30 min), carfilzomib produces near‐complete LMP7 inhibition (i.e. at the limit of quantitation of the assay) and was bound to, on average, more than 79% of all i20S active sites.
We next determined whether the levels of proteasome inhibition detected in whole blood and PBMC samples were representative of target inhibition in tumour cells. As an optional correlative study, CD138+ tumour cells were isolated from bone marrow aspirates taken during screening and on cycle 1, day 2, and levels of β5, LMP7 and MECL1 were assessed. Following administration of carfilzomib at a dose of 15 or 20 mg/m2, we observed a statistically significant (>95%) reduction in CT‐L activity as assessed by LLVY‐AMC (P < 0∙001). More moderate but still statistically significant reductions were seen at the β5, LMP7 and MECL1 active sites [reductions of 52% (P < 0∙05), 58% (P < 0∙01) and 50% (P < 0∙05), respectively] (Fig 2B). Intriguingly, we also observed that the i20S accounts for nearly 75% of the proteasome activity in these MM tumour cells (Table 2). Not all samples could be tested in both assays because of the limited sample volume, which may account for discrepancies in the inhibition levels measured by LLVY‐AMC and by ProCISE. Despite the limitations of this analysis, these data suggest that the use of carfilzomib results in inhibition lasting more than 48 h and that proteasome inhibition in tumour cells is similar to that observed in PBMC samples.
Table 2.
CT‐L Activity and Proteasome Subunit Levels in Baseline Primary Myeloma Cells and Multiple Myeloma Cell Lines
| Baseline CD138+ bone marrow proteasome activity and subunit composition | |||||
|---|---|---|---|---|---|
| Trialsa | Average number of prior regimens (range) | CT‐L (AMC/protein [μM/μg]) | β5 (ng/μg protein) | LMP7 (ng/μg protein) | LMP7 (%) |
| PX‐171‐003, PX‐171‐004, PX‐171‐005 | 5 (1–13) | 11∙3 ± 2∙8 | 0∙9 ± 0∙1 | 2∙6 ± 0∙4 | 74∙3 |
| Number of samples | 29 | 18 | 22 | ||
AMC, 7‐amino‐4‐methylcoumarin; CT‐L, chymotrypsin‐like; LMP, low‐molecular mass protein.
Bone marrow aspirates were collected from patients in trials PX‐171‐003, PX‐171‐004 and PX‐171‐005 prior to carfilzomib dosing and sorted into CD138+ and CD138− populations. CD138+ samples were analysed for proteasome activity and subunit occupancy as described in Fig. 1.
Correlation between proteasome inhibition and response
To determine whether the level of proteasome inhibition achieved with carfilzomib treatment corresponded to clinical response rates, samples from 29 patients with MM who received the highest tolerated dose (20/56 mg/m2 in PX‐171‐007) and the lowest administered dose of carfilzomib (15 or 15/20 mg/m2 in PX‐171‐005) were compared (Fig 3A; Table 3). During the first 2 days of carfilzomib treatment (15–20 mg/m2), no statistically significant differences in levels of proteasome inhibition were observed. When the dose was escalated to 56 mg/m2 on cycle 1, day 8 for patients in PX‐171‐007, significantly greater active site occupancy was observed across all three i20S subunits in patients who received the higher dose compared with patients in PX‐171‐005 who continued to receive the lower dose. In patients receiving 56 mg/m2, carfilzomib binding to LMP7, MECL1 and LMP2 was increased by 7∙9%, 23∙1% and 33∙6%, respectively, relative to patients receiving 15 or 20 mg/m2 (see Fig 3A and B; P < 0∙05, P <0∙001 and P < 0∙01, respectively). Increased occupancy of LMP2 at the beginning of C2 was also noted in the high‐dose group. Strikingly, 64% of patients in this analysis who received the higher dose had a clinical response compared with only 11% of patients receiving a lower dose (see Table 3). These data suggest a relationship between carfilzomib dose, proteasome inhibition and patient response, in which greater occupancy of multiple immunoproteasome subunits is correlated with an increased likelihood of achieving a clinical response.
Figure 3.
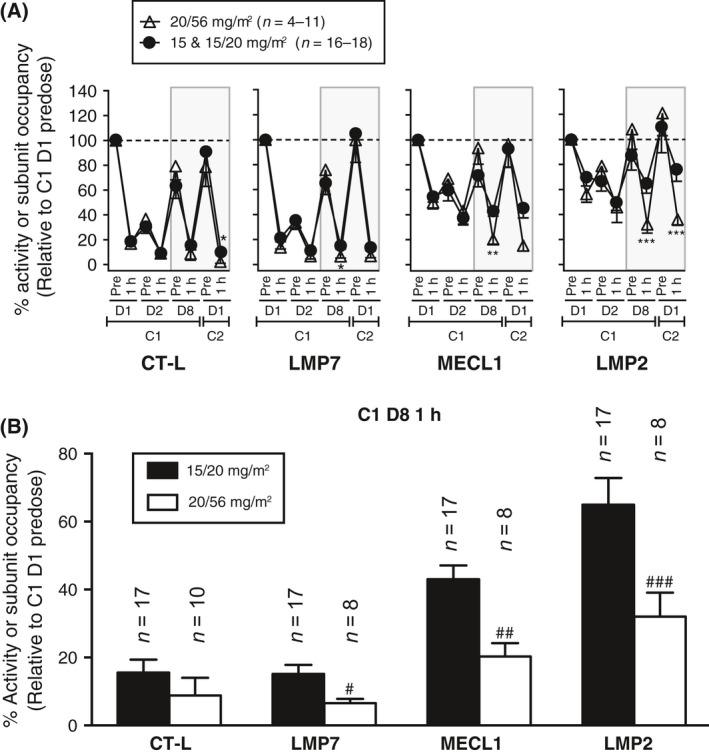
Immunoproteasome subunit inhibition is correlated with activity in multiple myeloma. (A) Proteasome inhibition in PBMC samples was measured as described in Fig 1. Patients were stratified according to carfilzomib dose: either 20/56 mg/m2 from PX‐171‐007 or 15 and 15/20 mg/m2 from PX‐171‐005. Values (relative to cycle 1, day 1 predose) are presented as mean (±SEM) relative subunit concentration or specific activity. The shaded area represents the time points in which the dose was stepped up to 56 mg/m2 for PX‐171‐007 patients or 20 mg/m2 for some PX‐171‐005 patients. *P < 0∙05; **P < 0∙01; ***P < 0∙001 by a t‐test. (B) Data from panel A at cycle 1, day 8 1‐h time point. # P < 0∙05; ## P < 0∙001; ### P < 0∙01; by a t‐test. C, cycle; CT‐L, chymotrypsin‐like; D, day; LMP, low‐molecular mass polypeptide; MECL, multicatalytic endopeptidase complex‐like; PBMC, peripheral blood mononuclear cell; SEM, standard error of the mean.
Table 3.
Clinical response in patients receiving the highest tolerated dose and the lowest administered dose of carfilzomib.a
| 20/56 mg/m2 (PX‐171‐007) (N = 11) | 15 or 15/20 mg/m2 (PX‐171‐005) (N = 18) | |
|---|---|---|
|
Responders (≥ PR) |
64% (n = 7) |
11% (n = 2) |
|
Non‐responders (SD + PD) |
36% (n = 4) |
89% (n = 16) |
PD, progressive disease; PR, partial response; SD, stable disease.
Patients received carfilzomib at a dose of 15 or 20 mg/m2 on days 1 and 2 and a target dose of 56 or 20 mg/m2 on days 8, 9, 15 and 16 in trials PX‐171‐007 and PX‐171‐005, respectively. Some patients in PX‐171‐005 did not step up to their target dose (e.g. from 15 to 20 mg/m2) and continued to receive 15 mg/m2. Patient responses were determined using International Myeloma Working Group criteria (Badros et al, 2013; Papadopoulos et al, 2013).
Discussion
Proteasome inhibition is a validated strategy for the treatment of B‐cell neoplasms, but limitations in conventional assays have prevented pharmacodynamic‐based correlative studies of drug action. We describe here the clinical validation and application of ProCISE, a novel assay for monitoring proteasome inhibition via active site occupancy in patient samples that circumvents these limitations and enables a more complete profile of drug action. ProCISE allows for quantitative monitoring of drug binding to all six proteasomal catalytic subunits in cells derived from patient samples, such as PBMC and primary tumour cells. By measuring the binding of each individual subunit, we are able to monitor specific active site inhibition in tumour cells (CD138+ myeloma cells) that express the c20S and i20S. This enables monitoring of the pharmacodynamic response to compounds that are selective for individual 20S active sites (Muchamuel et al, 2009). As existing substrate‐based assays are unable to differentiate between c20S and i20S enzymatic activities for any of the subunits, ProCISE currently represents the only known method to monitor the inhibition of individual subunits.
Previously, the pharmacodynamic profile of carfilzomib was assessed in vitro using the ProCISE assay (Parlati et al, 2009). In tumour cell lines, carfilzomib has been shown to have equivalent and potent activity against both β5 and LMP7 and to be at least 10‐fold less potent against the other subunits of the c20S and i20S (Parlati et al, 2009). In the current analysis, we verified the potency of carfilzomib for CT‐L active sites ex vivo and have extended previous findings in samples derived from carfilzomib‐treated patients. In patients treated with doses ≥15 mg/m2, on average an inhibition rate of greater than 83% was achieved in β5 and LMP7. As in the in vitro studies, there was a correspondence between values derived with ProCISE and those derived from the enzymatic assay for CT‐L activity. Slightly greater inhibition was observed in whole blood with the enzymatic assay compared with ProCISE, probably due to a quenching effect of the blood matrix – possibly haemoglobin – on the enzymatic activity and the presence of LMP7 in erythrocytes and PBMCs (whole blood samples were not depleted of PBMCs prior to the assay). However, there was a significant correlation between inhibition of CT‐L activity and ProCISE in whole blood and PBMCs for the aggregate of paired samples (Table SII). Inhibition levels were also equivalent in patients with MM receiving 20 mg/m2 carfilzomib as a 2‐ to 10‐min or 30‐min infusion, which is consistent with results reported in animals (Yang et al, 2011). It is noteworthy that the overall level of CT‐L inhibition seen with carfilzomib is higher than the 65–70% inhibition rate reported in bortezomib‐treated patients (Orlowski et al, 2002; Papandreou et al, 2004; Moreau et al, 2008). Using ProCISE, we also demonstrated that the administration of carfilzomib inhibits proteasome subunit occupancy in bone marrow–derived tumour cells. It is possible that the small sample size (n = 5) accounts for discrepancies seen between inhibition levels of CT‐L activity assessed by LLVY‐AMC and by β5 and LMP7 occupancy assessed by ProCISE; however, we cannot rule out altered binding properties within tumour cells. Nevertheless, these data represent the first known direct correlation between proteasome inhibition in blood samples to that in tumour cells in patients with MM.
Clinical trials have demonstrated that carfilzomib and bortezomib are clinically active regardless of the degree of renal insufficiency in patients (Jagannath et al, 2005; San‐Miguel et al, 2008; Badros et al, 2013). Consistent with this, we found that carfilzomib induces consistent and potent inhibition of multiple proteasomal active sites in MM patients, regardless of patient renal function. In addition, we found that concomitant drug administration did not affect levels of active site occupancy, as assessed in patients receiving carfilzomib in combination with varying doses of lenalidomide. To our knowledge, this is the first time the subunit‐specific pharmacodynamic activity of a proteasome inhibitor has been characterized in MM patients with renal impairment or concomitant drug administration, providing additional information on the activity of this class of agents in a variety of clinical settings.
In rodents, proteasome activity following treatment with carfilzomib recovers with a half‐life of approximately 24 h in tissues (e.g. adrenal and bone marrow), but prolonged inhibition was observed in whole blood for which recovery is dependent on red blood cell turnover (Demo et al, 2007). Little to no recovery of proteasome activity was observed in whole blood following the administration of carfilzomib, which is consistent with our preclinical results. Somewhat surprisingly, recovery of activity in PBMCs was slower than predicted based on animal data. Minimal recovery of proteasome activity was observed 24 h after the first dose, and incomplete recovery was seen after the 5‐day non‐dosing period during the first week of treatment. This effect is unlikely to be due to prolonged exposure, because carfilzomib is rapidly cleared from the plasma (O'Connor et al, 2009). Rather, these data demonstrate that carfilzomib results in proteasome inhibition that lasts at least 48 h and suggests that PBMCs have an altered rate of proteasome recovery relative to other cell types. In contrast, a 50% recovery of activity was seen 12 h after the administration of bortezomib, with full recovery occurring 36 h after dosing (Hellmann et al, 2011). We hypothesize that differences in proteasome recovery between the two drugs may be due to the irreversible binding of carfilzomib in contrast to the reversible binding of bortezomib to the proteasome (Demo et al, 2007).
Interestingly, administration of carfilzomib resulted in increased occupancy of MECL1 and LMP2 in PBMCs but not of β1 and β2 in whole blood. This finding was unexpected given the weak potency of carfilzomib ex vivo against non–CT‐L subunits in whole blood and PBMCs. Levels of MECL1 and LMP2 inhibition have a statistically significant linear dose effect in which higher doses of carfilzomib lead to greater inhibition. While inhibition of LMP2 in samples from bortezomib‐treated patients has previously been reported (Kraus et al, 2007; Arastu‐Kapur et al, 2011), this is the first known report of MECL1 inhibition by a proteasome inhibitor in patients. The mechanism of selective LMP2 and MECL1 inhibition by carfilzomib relative to β1 and β2 inhibition remains to be defined. Although data from our group demonstrated that combined inhibition of β5 and LMP7 was necessary and sufficient to induce apoptosis in myeloma cells in vitro (Parlati et al, 2009), several studies have postulated a role for the other active sites of the i20S in anti‐myeloma responses. Kuhn et al (2009) have shown that LMP2‐selective inhibitors can induce myeloma cell death, and multiple reports have demonstrated synergistic tumour cell killing with carfilzomib or bortezomib and either an LMP2/β1 or MECL1/β2 selective inhibitor (Britton et al, 2009; Mirabella et al, 2011). It is possible that the simultaneous inhibition of multiple i20S subunits contributes to the potent anti‐tumour activity of carfilzomib in patients with MM, including patients that are refractory to bortezomib (Siegel et al, 2012; Vij et al, 2012a), particularly given that approximately 75% of the proteasomes in isolated myeloma cells are i20S.
Importantly, we observed a relationship between carfilzomib dose, i20S inhibition, and clinical response in patients with MM. Patients receiving a dose of 56 mg/m2 of carfilzomib beginning on day 8 showed significantly greater inhibition of all three i20S subunits compared with patients who received 15 or 20 mg/m2 of carfilzomib. Given that near‐complete inhibition of LMP7 was noted even at 15 mg/m2, higher doses of carfilzomib resulted in small differences in LMP7 inhibition. However, significantly greater levels of inhibition of both LMP2 and MECL1 were noted in patients receiving 56 mg/m2 of carfilzomib compared with those receiving 15 or 20 mg/m2 (i.e. cycle 1, day 8). At cycle 2, day 1, LMP2 inhibition remained significantly different between the two groups, reaffirming that higher doses of carfilzomib led to greater i20S inhibition. The difference observed in MECL1 at cycle 2, day 1 is probably not statistically significant because of the small sample size (n = 4) in the 20/56‐mg/m2 cohort. Of the patients analysed for pharmacodynamics, 64% of patients receiving 20/56 mg/m2 achieved a clinical response compared with only 11% of patients receiving 15 or 20 mg/m2. While these samples were derived from different patient populations, both trials enrolled patients with relapsed and/or refractory MM who had been treated with at least two prior regimens. Overall, these data suggest that higher doses of carfilzomib lead to greater levels of i20S inhibition, resulting in improved clinical responses in patients with MM. Although these data are derived from small numbers of patients and should be verified in larger, randomized trials, they do support previous findings of a dose‐response relationship seen with earlier phase II trials of single‐agent carfilzomib (Squifflet et al, 2011).
In summary, we present the first clinical validation and application of the ProCISE assay. Administration of carfilzomib results in potent and prolonged inhibition of multiple proteasome active sites in patients with MM. In patients with MM, there is a dose‐dependent relationship between pharmacodynamic inhibition and anti‐tumour response following carfilzomib treatment. It is worth noting that the ProCISE assay can also be used to assess and compare the activity of other proteasome inhibitors besides carfilzomib (Parlati et al, 2009), as ProCISE is not treatment‐specific. Therefore, ProCISE represents a potentially valuable new tool for measuring the activity of proteasome inhibitors in clinical trials and can be used to relate drug action to patient outcomes.
Author contributions
SJL, KL and FP designed and performed research, collected data, and analysed and interpreted data. MKB, SAK and CJK designed research and analysed and interpreted data. LK performed statistical analysis. TFW and AFW collected data. KPP, RN, AZB, RV, SJ, DS, MW and GJA collected data and provided clinical samples. All authors contributed to the writing of this manuscript and approved it for submission.
Conflict of interest
SJL, KL, FP, MKB, SA‐K, LK, TFW, AFW and CJK were employees of Onyx Pharmaceuticals at the time of this work. RV has received research funding from Onyx Pharmaceuticals and Celgene, has participated in speaker bureaus for Celgene, Millennium and Onyx Pharmaceuticals, and has received honoraria from Eli Lily and Bristol‐Myers Squibb. DS has served as a consultant, participated in advisory boards and received honoraria from Millennium and Celgene. MW has received research funding from Onyx Pharmaceuticals, Celgene, Millennium, Pharmacyclics, Janssen‐Cilag and Novartis, and received honoraria from Onyx Pharmaceuticals and Celgene. SJ has received honoraria from Onyx Pharmaceuticals, Millennium, Celgene and Merck, and is on the board of or advisory committees for Ortho Biotech, Imedex, Medicom WorldWide, OptumHealth Education and PER Group. KPP has served as a consultant for Proteolix and has received research funding from Onyx Pharmaceuticals and Proteolix. RN has served as a consultant and received research funding from Onyx Pharmaceuticals, Celgene and Millennium, and has participated in speakers bureaus for Celgene and Millennium. GJA and AZB have no conflicts of interest to disclose.
Supporting information
Data S1: Methods and Results
Table SI. ProCISE precision and interday variability.
Table SII. Correlation of the relative change of enzymatic inhibition of chymotrypsin‐like active site occupancy and β5/LMP7 active site occupancy in whole blood and PBMC samples.
Fig. S1. Validation of the ProCISE assay.
Fig. S2. Potency of carfilzomib in ex vivo whole blood and PBMC.
Fig. S3. Dose‐dependent immunoproteasome inhibition.
Acknowledgements
Critical review of the manuscript for scientific accuracy was undertaken by Thomas Renau, PhD and A. Peter Morello III, PhD, CMPP (Onyx Pharmaceuticals, an Amgen subsidiary, Inc.). Medical writing and editorial assistance were provided by Cheryl Chun, PhD, CMPP (BlueMomentum, an Ashfield Company, part of UDG Healthcare plc, San Bruno, CA) and funded by Onyx Pharmaceuticals, Inc. (South San Francisco, CA) , an Amgen subsidiary.
References
- Arastu‐Kapur, S. , Anderl, J.L. , Kraus, M. , Parlati, F. , Shenk, K.D. , Lee, S.J. , Muchamuel, T. , Bennett, M.K. , Driessen, C. , Ball, A.J. & Kirk, C.J. (2011) Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clinical Cancer Research, 17, 2734–2743. [DOI] [PubMed] [Google Scholar]
- Badros, A.Z. , Vij, R. , Martin, T. , Zonder, J.A. , Kunkel, L. , Wang, Z. , Lee, S. , Wong, A.F. & Niesvizky, R. (2013) Carfilzomib in multiple myeloma patients with renal impairment: pharmacokinetics and safety. Leukemia, 27, 1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M.K. & Kirk, C.J. (2008) Development of proteasome inhibitors in oncology and autoimmune diseases. Current Opinion in Drug Discovery and Development, 11, 616–625. [PubMed] [Google Scholar]
- Bennett, M.K. , Buchholz, T.J. , Demo, S.D. , Laidig, G.J. , Lewis, E.R. & Smyth, M.S. (2006) Compound for enzyme inhibition (US Patent 2006/0088471 A1), April 27, 2006. http://patents.com/us-20060088471.html. Accessed January 16, 2015
- Britton, M. , Lucas, M.M. , Downey, S.L. , Screen, M. , Pletnev, A.A. , Verdoes, M. , Tokhunts, R.A. , Amir, O. , Goddard, A.L. , Pelphrey, P.M. , Wright, D.L. , Overkleeft, H.S. & Kisselev, A.F. (2009) Selective inhibitor of proteasome's caspase‐like sites sensitizes cells to specific inhibition of chymotrypsin‐like sites. Chemistry and Biology, 16, 1278–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan, D. , Catley, L. , Li, G. , Podar, K. , Hideshima, T. , Velankar, M. , Mitsiades, C. , Mitsiades, N. , Yasui, H. , Letai, A. , Ovaa, H. , Berkers, C. , Nicholson, B.l , Chao, T.‐H. , Neuteboom, S.T.C. , Richardson, P. , Palladino, M.A. & Anderson, K.C. (2005) A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib. Cancer Cell, 8, 407–419. [DOI] [PubMed] [Google Scholar]
- Ciechanover, A. (2012) Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin‐proteasome system and onto human diseases and drug targeting. Biochimica et Biophysica Acta, 1824, 3–13. [DOI] [PubMed] [Google Scholar]
- Coux, O. , Tanaka, K. & Goldberg, A.L. (1996) Structure and functions of the 20S and 26S proteasomes. Annual Review of Biochemistry, 65, 801–847. [DOI] [PubMed] [Google Scholar]
- Demo, S.D. , Kirk, C.J. , Aujay, M.A. , Buchholz, T.J. , Dajee, M. , Ho, M.N. , Jiang, J. , Laidig, G.J. , Lewis, E.R. , Parlati, F. , Shenk, K.D. , Smyth, M.S. , Sun, C.M. , Vallone, M.K. , Woo, T.M. , Molineaux, C.J. & Bennett, M.K. (2007) Antitumor activity of PR‐171, a novel irreversible inhibitor of the proteasome. Cancer Research, 67, 6383–6391. [DOI] [PubMed] [Google Scholar]
- Dick, L.R. & Fleming, P.E. (2010) Building on bortezomib: second‐generation proteasome inhibitors as anti‐cancer therapy. Drug Discovery Today, 15, 243–249. [DOI] [PubMed] [Google Scholar]
- Fisher, R.I. , Bernstein, S.H. , Kahl, B.S. , Djulbegovic, B. , Robertson, M.J. , de Vos, S. , Epner, E. , Krishnan, A. , Leonard, J.P. , Lonial, S. , Stadtmauer, E.A. , O'Connor, O.A. , Shi, H. , Boral, A.L. & Goy, A. (2006) Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Journal of Clinical Oncology, 24, 4867–4874. [DOI] [PubMed] [Google Scholar]
- Glynne, R. , Powis, S.H. , Beck, S. , Kelly, A. , Kerr, L.A. & Trowsdale, J. (1991) A proteasome‐related gene between the two ABC transporter loci in the class II region of the human MHC. Nature, 353, 357–360. [DOI] [PubMed] [Google Scholar]
- Groettrup, M. , Kirk, C.J. & Basler, M. (2010) Proteasomes in immune cells: more than peptide producers? Nature Reviews Immunology, 10, 73–78. [DOI] [PubMed] [Google Scholar]
- Hellmann, A. , Rule, S. , Walewski, J. , Shpilberg, O. , Feng, H. , van de Velde, H. , Patel, H. , Skee, D.M. , Girgis, S. & Louw, V.J. (2011) Effect of cytochrome P450 3A4 inducers on the pharmacokinetic, pharmacodynamic and safety profiles of bortezomib in patients with multiple myeloma or non‐Hodgkin's lymphoma. Clinical Pharmacokinetics, 50, 781–791. [DOI] [PubMed] [Google Scholar]
- Jagannath, S. , Barlogie, B. , Berenson, J.R. , Singhal, S. , Alexanian, R. , Srkalovic, G. , Orlowski, R.Z. , Richardson, P.G. , Anderson, J.A. , Nix, D. , Esseltine, D.L. & Anderson, K.C. (2005) Bortezomib in recurrent and/or refractory multiple myeloma. Initial clinical experience in patients with impaired renal function. Cancer, 103, 1195–1200. [DOI] [PubMed] [Google Scholar]
- Jain, S. , Diefenbach, C. , Zain, J. & O'Connor, O.A. (2011) Emerging role of carfilzomib in treatment of relapsed and refractory lymphoid neoplasms and multiple myeloma. Core Evidence, 6, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus, M. , Rückrich, T. , Reich, M. , Gogel, J. , Beck, A. , Kammer, W. , Berkers, C.R. , Burg, D. , Overkleeft, H. , Ovaa, H. & Driessen, C. (2007) Activity patterns of proteasome subunits reflect bortezomib sensitivity of hematologic malignancies and are variable in primary human leukemia cells. Leukemia, 21, 84–92. [DOI] [PubMed] [Google Scholar]
- Kuhn, D.J. , Chen, Q. , Voorhees, P.M. , Strader, J.S. , Shenk, K.D. , Sun, C.M. , Demo, S.D. , Bennett, M.K. , van Leeuwen, F.W.B. , Chanan‐Khan, A.A. & Orlowski, R.Z. (2007) Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin‐proteasome pathway, against preclinical models of multiple myeloma. Blood, 110, 3281–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, D.J. , Hunsucker, S.A. , Chen, Q. , Voorhees, P.M. , Orlowski, M. & Orlowski, R.Z. (2009) Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood, 113, 4667–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, D.J. , Orlowski, R.Z. & Bjorklund, C.C. (2011) Second generation proteasome inhibitors: carfilzomib and immunoproteasome‐specific inhibitors (IPSIs). Current Cancer Drug Targets, 11, 285–295. [DOI] [PubMed] [Google Scholar]
- Kummar, S. , Chen, H.X. , Wright, J. , Holbeck, S. , Millin, M.D. , Tomaszewski, J. , Zweibel, J. , Collins, J. & Doroshow, J.H. (2010) Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nature Reviews Drug Discovery, 9, 843–856. [DOI] [PubMed] [Google Scholar]
- Lightcap, E.S. , McCormack, T.A. , Pien, C.S. , Chau, V. , Adams, J. & Elliott, P.J. (2000) Proteasome inhibition measurements: clinical application. Clinical Chemistry, 46, 673–683. [PubMed] [Google Scholar]
- Martinez, C.K. & Monaco, J.J. (1991) Homology of proteasome subunits to a major histocompatibility complex‐linked LMP gene. Nature 353, 664–667. [DOI] [PubMed] [Google Scholar]
- Mirabella, A.C. , Pletnev, A.A. , Downey, S.L. , Florea, B.I. , Shabaneh, T.B. , Britton, M. , Verdoes, M. , Filippov, D.V. , Overkleeft, H.S. & Kisselev, A.F. (2011) Specific cell‐permeable inhibitor of proteasome trypsin‐like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chemistry and Biology, 18, 608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, P. , Coiteux, V. , Hulin, C. , Leleu, X. , van de Velde, H. , Acharya, M. & Harousseau, J.‐L. (2008) Prospective comparison of subcutaneous versus intravenous administration of bortezomib in patients with multiple myeloma. Haematologica, 93, 1908–1911. [DOI] [PubMed] [Google Scholar]
- Muchamuel, T. , Basler, M. , Aujay, M.A. , Suzuki, E. , Kalim, K.W. , Lauer, C. , Sylvain, C. , Ring, E.R. , Shields, J. , Jiang, J. , Shwonek, Pl , Parlati, F. , Demo, S.D. , Bennett, M.K. , Kirk, C.J. & Groettrup, M. (2009) A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nature Medicine, 15, 781–787. [DOI] [PubMed] [Google Scholar]
- Nandi, D. , Jiang, H. & Monaco, J.J. (1996) Identification of MECL‐1 (LMP‐10) as the third IFN‐gamma‐inducible proteasome subunit. Journal of Immunology, 156, 2361–2364. [PubMed] [Google Scholar]
- Niesvizky, R. , Martin, T.G. III , Bensinger, W.I. , Alsina, M. , Siegel, D.S. , Kunkel, L.A. , Wong, A.F. , Lee, S. , Orlowski, R.Z. & Wang, M. (2013) Phase Ib dose‐escalation study (PX‐171‐006) of carfilzomib, lenalidomide, and low‐dose dexamethasone in relapsed or progressive multiple myeloma. Clinical Cancer Research, 19, 2248–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, O.A. , Stewart, A.K. , Vallone, M. , Molineaux, C.J. , Kunkel, L.A. , Gerecitano, J.F. & Orlowski, R.Z. (2009) A phase 1 dose escalation study of the safety and pharmacokinetics of the novel proteasome inhibitor carfilzomib (PR‐171) in patients with hematologic malignancies. Clinical Cancer Research, 15, 7085–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski, R.Z. , Stinchcombe, T.E. , Mitchell, B.S. , Shea, T.C. , Baldwin, A.S. , Stahl, S. , Adams, J. , Esseltine, D.‐L. , Elliott, P.J. , Pien, C.S. , Guerciolini, R. , Anderson, J.K. , Depcik‐Smith, N.D. , Bhagat, R. , Lehman, M.J. , Novick, S.C. , O'Connor, O.A. & Soignet, S.L. (2002) Phase I trial of the proteasome inhibitor PS‐341 in patients with refractory hematologic malignancies. Journal of Clinical Oncology, 20, 4420–4427. [DOI] [PubMed] [Google Scholar]
- Papadopoulos, K.P. , Burris, H.A. III , Gordon, M. , Lee, P. , Sausville, E.A. , Rosen, P.J. , Patnaik, A. , Cutler, R.E., Jr. , Wang, Z. , Lee, S. , Jones, S.F. & Infante, J.R. (2013) A phase I/II study of carfilzomib 2‐10‐min infusion in patients with advanced solid tumors. Cancer Chemotherapy and Pharmacology, 72, 861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, K.P. , Siegel, D.S. , Vesole, D.H. , Lee, P. , Rosen, S.T. , Zojwalla, N. , Holahan, J.R. , Lee, S. , Wang, Z. & Badros, A. (2015) Phase I study of a 30‐minute infusion of carfilzomib as single agent or in combination with low‐dose dexamethasone in patients with relapsed and/or refractory multiple myeloma. Journal of Clinical Oncology, 33, 732–739. [DOI] [PubMed] [Google Scholar]
- Papandreou, C.N. , Daliani, D.D. , Nix, D. , Yang, H. , Madden, T. , Wang, X. , Pien, C.S. , Millikan, R.E. , Tu, S.M. , Pagliaro, L. , Kim, J. , Adams, J. , Elliott, P. , Esseltine, D. , Petrusich, A. , Dieringer, P. , Perez, C. & Logothetis, C.J. (2004) Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen‐independent prostate cancer. Journal of Clinical Oncology, 22, 108–121. [DOI] [PubMed] [Google Scholar]
- Parlati, F. , Lee, S.J. , Aujay, M. , Suzuki, E. , Levitsky, K. , Lorens, J.B. , Micklem, D.R. , Ruurs, P. , Sylvain, C. , Lu, Y. , Shenk, K.D. & Bennett, M.K. (2009) Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin‐like activity of the proteasome. Blood, 114, 3439–3447. [DOI] [PubMed] [Google Scholar]
- Richardson, P.G. , Sonneveld, P. , Schuster, M.W. , Irwin, D. , Stadtmauer, E.A. , Facon, T. , Harousseau, J.L. , Ben‐Yehuda, D. , Lonial, S. , Goldschmidt, H. , Reece, D. , San‐Miguel, J.F. , Bladé, J. , Boccadoro, M. , Cavenagh, J. , Dalton, W.S. , Boral, A.L. , Esseltine, D.L. , Porter, J.B. , Schenkein, D. & Anderson, K.C. ; for the Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators (2005) Bortezomib or high‐dose dexamethasone for relapsed multiple myeloma. New England Journal of Medicine, 352, 2487–2498. [DOI] [PubMed] [Google Scholar]
- San‐Miguel, J.F. , Richardson, P.G. , Sonneveld, P. , Schuster, M.W. , Irwin, D. , Stadtmauer, E.A. , Facon, T. , Harousseau, J.L. , Ben‐Yehuda, D. , Lonial, S. , Goldschmidt, H. , Reece, D. , Blade, J. , Boccadoro, M. , Cavenagh, J.D. , Neuwirth, R. , Boral, A.L. , Esseltine, D.L. & Anderson, K.C. (2008) Efficacy and safety of bortezomib in patients with renal impairment: results from the APEX phase 3 study. Leukemia, 22, 842–849. [DOI] [PubMed] [Google Scholar]
- Siegel, D.S. , Martin, T. , Wang, M. , Vij, R. , Jakubowiak, A.J. , Lonial, S. , Trudel, S. , Kukreti, V. , Bahlis, N. , Alsina, M. , Chanan‐Khan, A. , Buadi, F. , Reu, F.J. , Somlo, G. , Zonder, J. , Song, K. , Stewart, A.K. , Stadtmauer, E. , Kunkel, L. , Wear, S. , Wong, A.F. , Orlowski, R.Z. & Jagannath, S. (2012) A phase 2 study of single‐agent carfilzomib (PX‐171‐003‐A1) in patients with relapsed and refractory multiple myeloma. Blood, 120, 2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, D. , Martin, T. , Nooka, A. , Harvey, R.D. , Vij, R. , Niesvizky, R. , Badros, A.Z. , Jagannath, S. , McCulloch, L. , Rajangam, K. & Lonial, S. (2013) Integrated safety profile of single‐agent carfilzomib: experience from 526 patients enrolled in 4 phase 2 clinical studies. Haematologica, 98, 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, M.S. & Laidig, G.J. (2006) Compounds for enzyme inhibition (US 2006/0030533 A1), February 9, 2006. http://patents.com/us-20060030533.html. Accessed January 16, 2015.
- Squifflet, P. , Michiels, S. , Siegel, D.S. , Vij, R. , Ro, S. & Buyse, M.E. (2011) Multivariate modelling reveals evidence of a dose‐response relationship in phase 2 studies of single‐agent carfilzomib [abstract]. Blood, 118, Abstract1877.21680794 [Google Scholar]
- Vij, R. , Siegel, D.S. , Jagannath, S. , Jakubowiak, A.J. , Stewart, A.K. , McDonagh, K. , Bahlis, N. , Belch, A. , Kunkel, L.A. , Wear, S. , Wong, A.F. , Orlowski, R.Z. & Wang, W. (2012a) An open‐label, single‐arm, phase 2 study of single‐agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. British Journal of Haematology, 158, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vij, R. , Wang, M. , Kaufman, J.L. , Lonial, S. , Jakubowiak, A.J. , Stewart, A.K. , Kukreti, V. , Jagannath, S. , McDonagh, K.T. , Alsina, M. , Bahlis, N.J. , Reu, F.J. , Gabrail, N.Y. , Belch, A. , Matous, J.V. , Lee, P. , Rosen, P. , Sebag, M. , Vesole, D.H. , Kunkel, L.A. , Wear, S.M. , Wong, A.F. , Orlowski, R.Z. & Siegel, D.S. (2012b) An open‐label, single‐arm, phase 2 (PX‐171‐004) study of single‐agent carfilzomib in bortezomib‐naive patients with relapsed and/or refractory multiple myeloma. Blood, 119, 5661–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehenkel, M. , Ban, J.O. , Ho, Y.K. , Carmony, K.C. , Hong, J.T. & Kim, K.B. (2012) A selective inhibitor of the immunoproteasome subunit LMP2 induces apoptosis in PC‐3 cells and suppresses tumour growth in nude mice. British Journal of Cancer, 107, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk, S. & Orlowski, M. (1983) Evidence that pituitary cation‐sensitive neutral endopeptidase is a multicatalytic protease complex. Journal of Neurochemistry, 40, 842–849. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Wang, Z. , Fang, Y. , Jiang, J. , Zhao, F. , Wong, H. , Bennett, M.K. , Molineaux, C.J. & Kirk, C.J. (2011) Pharmacokinetics, pharmacodynamics, metabolism, distribution and excretion of carfilzomib in rats. Drug Metabolism and Disposition, 39, 1873–1882. [DOI] [PubMed] [Google Scholar]
- Yap, T.A. , Sandhu, S.K. , Workman, P. & de Bono, J.S. (2010) Envisioning the future of early anticancer drug development. Nature Reviews Cancer, 10, 514–523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Methods and Results
Table SI. ProCISE precision and interday variability.
Table SII. Correlation of the relative change of enzymatic inhibition of chymotrypsin‐like active site occupancy and β5/LMP7 active site occupancy in whole blood and PBMC samples.
Fig. S1. Validation of the ProCISE assay.
Fig. S2. Potency of carfilzomib in ex vivo whole blood and PBMC.
Fig. S3. Dose‐dependent immunoproteasome inhibition.