

Abstract
G-protein coupled receptors (GPCRs) initiate a variety of signaling cascades depending on effector coupling. β-arrestins, which were initially characterized by their ability to “arrest” GPCR signaling by uncoupling receptor and G protein, have recently emerged as important signaling effectors for GPCRs. β-arrestins engage signaling pathways that are distinct from those mediated by G-protein. As such, arrestin-dependent signaling can play a unique role in regulating cell function, but whether neuromodulatory GPCRs utilize β-arrestin-dependent signaling to regulate neuronal excitability remains unclear. Here, we find that D3 dopamine receptors (D3R) regulate axon initial segment (AIS) excitability through β-arrestin-dependent signaling, modifying CaV3 voltage dependence to suppress high frequency action potential generation. This non-canonical D3R signaling thereby gates AIS excitability via pathways distinct from classical GPCR signaling pathways.
ETOC BLURB
The neuronal site of action potential initiation, the axon initial segment (AIS), has recently gained recognition as a site of neuromodulation and plasticity. Here, Yang et al. demonstrate that D3 dopamine receptors regulate AIS CaV3 channels through a non-canonical, arrestin-dependent mechanism, suppressing AIS excitability by hyperpolarizing CaV3 voltage-dependent inactivation.
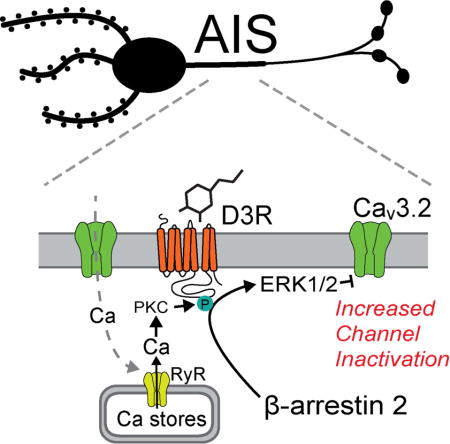
Introduction
G-protein coupled receptor (GPCR) signaling is regulated by β-arrestin, a scaffolding protein that both arrests G-protein dependent signaling and initiates GPCR trafficking following ligand binding (Reiter et al., 2012). In recent years, β-arrestins have emerged as an important downstream effector of GPCRs, supporting signaling through protein kinases including ERK, Akt and Src. Dopamine receptors are a major class of neuronal GPCR involved in various neuronal processes, including movement, cognition, and reward (Schultz, 2007), and are characterized by their coupling to Gs (D1, D5) or Gi/o (D2, D3, D4) G-proteins. While G-protein-dependent signaling supports various actions of dopaminergic signaling in the brain, there is emerging evidence that β-arrestin-dependent signaling plays a role in dopaminergic function in health and disease (Beaulieu et al., 2009). And yet, whether dopaminergic systems utilize β-arrestin-dependent signaling in neurons to regulate neuronal activity remains unexplored.
Recently, we discovered a mechanism by which D3 dopamine receptors (D3R) regulate neuronal activity. The axon initial segment (AIS), which is the site of action potential initiation in most neurons, is enriched with Na, K and Ca channels that each contribute to different aspects of action potential generation (Bender and Trussell, 2012; Kole and Stuart, 2012). We found that D3Rs selectively modulate Ca influx through AIS-localized low-voltage activated (CaV3, T-type) Ca channels, reducing spike-evoked Ca influx in the AIS. Because these channels contribute to the generation of high-frequency action potential bursts, dopaminergic regulation of these channels suppresses burst output (Bender et al., 2010; Bender et al., 2012).
Gi/o-coupled receptors, including D3R, are thought to signal through Gi/o to suppress cyclic AMP and protein kinase A (PKA) activity (Beaulieu et al., 2009). Initial studies identified no role for PKA in D3R-mediated inhibition of AIS Ca channels, suggesting that D3R may act through non-canonical signaling pathways (Bender et al., 2010). Here, we used a combination of electrophysiology and 2-photon imaging in acute brain slice and heterologous expression systems to probe the cellular mechanisms that allow for D3R-dependent modulation of AIS Ca channels. We found that β-arrestin-dependent signaling is required for D3R-dependent modulation at the AIS, and that dopamine signaling hyperpolarizes CaV3 steady state inactivation, resulting in a marked loss of AIS Ca channels available to support spike initiation. These results thus identify an arrestin-dependent mechanism for dopaminergic regulation of neuronal excitability at the AIS.
Results
Whole-cell recordings were made from cartwheel cells, a class of glycinergic interneuron in the auditory brainstem dorsal cochlear nucleus. Cartwheel cells were ideal for these studies, as their spike-evoked Ca dynamics are well understood (Kim and Trussell, 2007; Molitor and Manis, 2003; Roberts et al., 2008), and their spiking activity is readily modified by D3R signaling (Bender et al., 2010; Bender et al., 2012). Cartwheel cells were filled with Alexa 594 (red) and Fluo 5F (green) for simultaneous 2-photon imaging of neuronal morphology and intracellular Ca dynamics, and were identified based on their laminar position, dendritic morphology, and ability to generate high-frequency spike bursts in response to somatic current injection (Fig. 1A, B) (Wouterlood and Mugnaini, 1984). Consistent with previous results, the D3R agonist quinpirole (2 μM) suppressed spike-evoked AIS Ca influx when compared to within-cell baselines (Fig. 1C, H, normalized ΔG/Gsat, quinpirole: 0.76 ± 0.01, n = 18).
Fig 1. D3R-dependent regulation of AIS Ca is β-arrestin dependent.
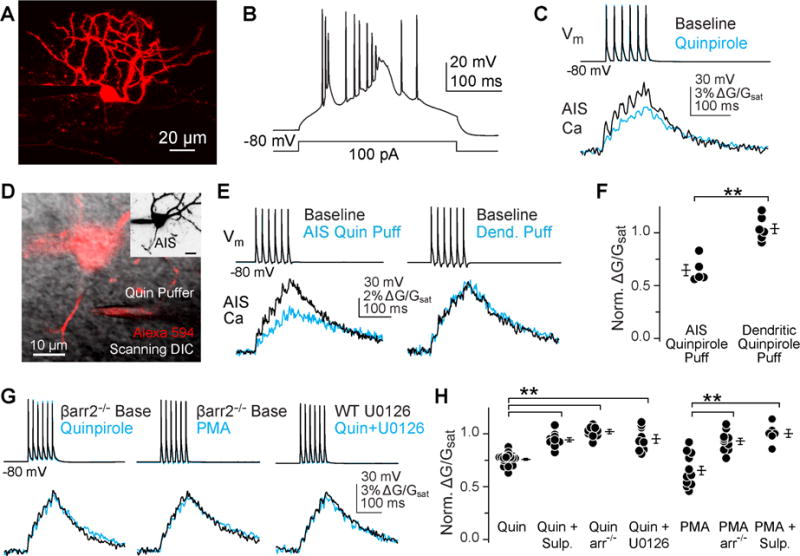
A) 2-photon z-stack of cartwheel cell, visualized with Alexa 594.
B) Characteristic burst spiking of cartwheel cells evoked with somatic current injection.
C) Quinpirole (2 μM) suppresses spike-evoked Ca influx in the AIS, as seen previously (Bender et al., 2010).
D) Simultaneous 2P fluorescence and scanning-DIC was used to position a puffer pipette near the AIS. Inset: maximum intensity z-stack of neuron, highlighting that the AIS was well separated from dendrites.
E) AIS Ca modulation occurred only when quinpirole was applied near the AIS. Somatic voltage and AIS Ca transients are shown before (black) and following (cyan) local application of quinpirole.
F) Summary of local quinpirole application across cells. Circles are single cells, bars are mean ± SEM. Data are normalized to pre-drug conditions within each experiment. ** p < 0.01, Mann-Whitney.
G) AIS Ca modulation was not observed in β-arrestin-2 knockouts, or when blocking ERK1/2 signaling with U0126. Drugs were applied to ACSF.
H) Summary of β-arrestin-dependent signaling in the AIS. Format as in panel f, ** p < 0.01, ANOVA, Newman-Keuls. Sulpiride is abbreviated “sulp.”
Spike burst suppression mediated by D3R activation requires several minutes to modulate AIS Ca influx and spike output (Bender et al., 2010), suggesting that either D3Rs are physically separated from AIS CaV channels and must transduce signals over long distances, or that signaling pathway kinetics are slow. To test for the location of D3Rs, a puffer pipette filled with quinpirole (10 μM) was placed in close proximity to the AIS or a dendritic branch under 2-photon guidance (Fig. 1D). Pipettes were co-filled with 10 μM Alexa 594 to visualize application. Under these conditions, we found that spike-evoked AIS Ca was modulated only when quinpirole was applied to the AIS (norm. ΔG/Gsat, 0.65 ± 0.05, n = 5), and not the dendrite (norm. AIS ΔG/Gsat, 1.05 ± 0.06, n = 5, p < 0.001 vs AIS puff), suggesting that D3Rs are localized in or near the axon and signal locally to AIS CaV3 channels (Fig. 1E–F).
We found previously that AIS Ca modulation required PKC, but not PKA activity (Bender et al., 2010). Interestingly, β-arrestin-dependent endocytosis of D3R occurs only if D3R is phosphorylated by PKC (Cho et al., 2007; Thompson and Whistler, 2011). Together, these observations suggest that signaling to CaV3 could be arrestin mediated. In support of this hypothesis, we found that quinpirole did not modulate AIS Ca in slices from β-arrestin-2−/− mice (Fig. 1G–H; norm. ΔG/Gsat, 1.02 ± 0.02, p < 0.001 vs. WT). β-arrestin recruitment was still Gβγ-dependent, as preventing Gβγ hydrolysis with GDPβs blocked signaling (Pitcher et al., 1992) (0.3 mM in internal in lieu of tris-GTP; Fig. 2, norm. ΔG/Gsat, 0.94 ± 0.02, n = 11, p < 0.01 vs quinpirole).
Fig 2. AIS Ca modulation requires intracellular store signaling.
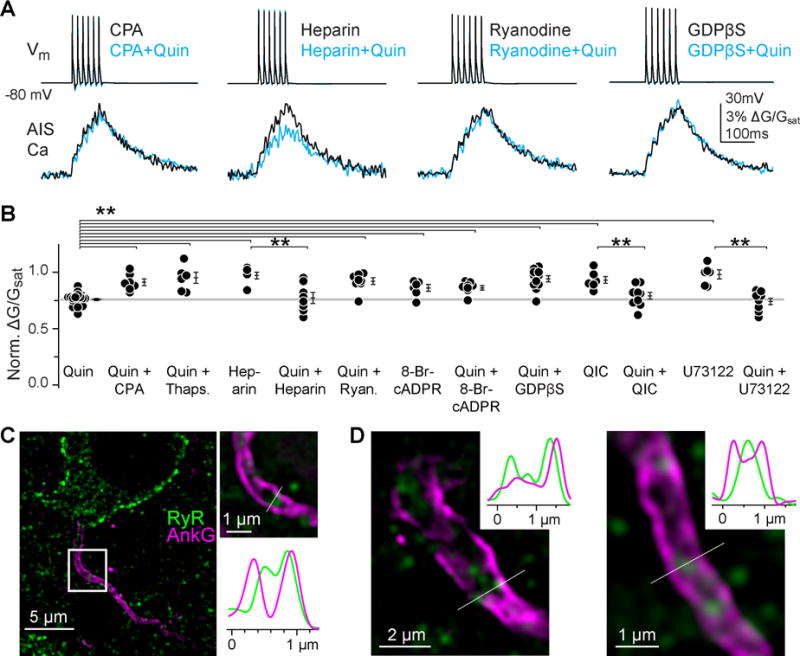
A) Ca modulation is ryanodine-store dependent in cartwheel AIS. Data displayed as in Fig. 1E.
B) Summary of Ca store- and G-protein-dependent signaling in the AIS. Format as in Fig. 1F.
C) Ryanodine receptor (RyR) and the AIS scaffolding protein ankyrin-G (AnkG) immunofluorescence in cartwheel cells, visualized with structured illumination superresolution microscopy. Initial segments localized to the middle of the DCN molecular layer were chosen for analysis, as cartwheel cells are the only DCN cell class with somata and initial segments in this region. Often, initial segments were connected to RyR outlined somata with a teardrop shape, a common feature of cartwheel cells (see Fig. 1A for comparison). Left, 3.4 μm-thick maximum intensity z-stack of cartwheel AIS. Top right, single optical section of boxed region in z-stack. Bottom right, RyR and AnkG fluorescence along a line transecting the AIS is plotted below.
D) Single optical sections from other putative cartwheel cell initial segments, with corresponding RyR and AnkG fluorescence along a line transecting the AIS.
In this model, PKC is required to phosphorylate D3R, not CaV3. And yet, PKC activation alone using the direct PKC activator phorbol 12-myristate 13-acetate (PMA, 10 μM) was sufficient to modulate AIS Ca (Fig. 1G, H; norm. ΔG/Gsat, 0.65 ± 0.04, n = 12). This suggests that either PKC is downstream of arrestin, or that PMA has additional effects in slice that allow for activation of D3R. In other systems, phorbol esters can drive reverse transport at the dopamine transporter (Cowell et al., 2000; Kantor and Gnegy, 1998), raising the possibility that PMA is acting both to activate PKC that phosphorylates D3R and to promote release of dopamine. To test this, we applied PMA to cells preincubated in the D3R antagonist sulpiride (1 μM). AIS Ca was not modulated in these conditions (1.01 ± 0.04, n = 6, p < 0.001 vs PMA alone). PKC activation was still required in cartwheel cells, as we have shown previously that AIS Ca modulation was blocked with intracellular inhibitors of PKC (Bender et al., 2010). Furthermore, PMA application in the absence of sulpiride had no effect in arrestin−/− neurons (0.93 ± 0.03, n = 10, p < 0.001 vs PMA in WT), indicating that PKC activation is upstream of β-arrestin. These data therefore indicate that CaV3 channels are modulated by an effector other than PKC, downstream of β-arrestin. β-arrestins mediate G protein-independent signaling by forming scaffolds with other kinases, including the mitogen-activated protein kinase ERK1/2 (Shenoy et al., 2006). Consistent with this, AIS Ca modulation was blocked in the presence of the ERK1/2 inhibitor U0126 (10 μM), suggesting that arrestin-dependent scaffolding of ERK1/2 is important for D3R modulation of CaV3 (Fig. 1G–H; norm. ΔG/Gsat, 0.95 ± 0.04, p < 0.001 vs WT).
Ca release from intracellular stores is required for activation of conventional forms of PKC. Cartwheel cell initial segments contain endoplasmic reticula (Wouterlood and Mugnaini, 1984), though its role in AIS function is unclear. To test whether these stores are required for D3R signaling, we first depleted all stores with the Ca-ATPase inhibitors cyclopiazonic acid (CPA, 20 μM) or thapsigargin (10 μM). Both inhibitors blocked quinpirole-induced AIS Ca modulation (Fig. 2A–B, ΔG/Gsat, CPA, 0.91 ± 0.03, n = 7, p < 0.001; thapsigargin, 0.95 ± 0.05, n = 6, p < 0.001 vs. quinpirole). Ca can be released from stores by activating IP3 receptors or by ryanodine receptors (RyR), which support Ca-induced-Ca-release. IP3 receptors have not been observed in cartwheel cell initial segments (Ryugo et al., 1995). Correspondingly, we found that blockade of various components of the canonical IP3 signaling cascade, including Gαq, phospholipase C, and IP3 receptors themselves, had no effect on AIS Ca modulation [Fig. 2A–B; QIC (Gaq inhibitor), 5 μM in pipette: norm. ΔG/Gsat, 0.79 ± 0.03, n = 6, p = 0.14 vs. quinpirole; U73122 (phospholipase C inhibitor), 10 μM: norm. ΔG/Gsat, 0.74 ± 0.03, n = 8, p = 0.23 vs. quinpirole; heparin (IP3 receptor inhibitor), 1 mg/1 ml in pipette: norm. ΔG/Gsat, 0.77 ± 0.05, n = 8, p = 0.75 vs. quinpirole]. In contrast, depleting ryanodine-dependent stores with high concentrations of ryanodine (100 μM in pipette), or blocking the endogenous activator of RyRs, cyclic ADP-ribose (cADPR), with 8-Br-cyclic ADPR (100 μM in pipette), attenuated quinpirole-mediated effects on AIS Ca2+ transients (Fig. 2A–B, ryanodine: norm. ΔG/Gsat, 0.92 ± 0.03, n = 8, p < 0.05 vs. quinpirole; 8-Br-cyclic ADPR: norm. ΔG/Gsat, 0.86 ± 0.02, n = 8, p < 0.05 vs. quinpirole). Furthermore, structured illumination super-resolution microscopy revealed RyRs puncta in cartwheel cell initial segments (Fig. 2C–D). Thus, RyRs provide a source of local store Ca that is required for D3R-dependent modulation.
CaV3 channels are expressed as one of 3 isoforms (3.1, 3.2, 3.3), each with different kinetics and sensitivity to neuromodulators. We found previously that AIS Ca influx was sensitive to low concentrations of Ni and ascorbate (Bender et al., 2010), both of which are preferential CaV3.2 antagonists (Nelson et al., 2007; Perez-Reyes, 2003). To test whether CaV3.2 channels are the target of D3R signaling, we made recordings from CaV3.2−/− mice and found that quinpirole had no effect on AIS Ca transients (Fig. 3B, norm. ΔG/Gsat, 0.91 ± 0.02, n = 10, p = 0.24 vs. WT quinpirole + sulpiride). Furthermore, this isoform appeared to be a major contributor to the generation of spike bursts at the onset of step depolarizations (Kim and Trussell, 2007), as the instantaneous frequency of onset spikes was reduced in CaV3.2−/− cells (Fig. 3A, WT: 188 ± 13 Hz, n = 18; KO: 121 ± 18 Hz, n = 11; p < 0.01, Mann-Whitney). This suggests that CaV3.2 channels contribute to subthreshold Ca influx in cartwheel cell dendrites and initial segments.
Fig 3. CaV3.2 channels are the target of D3Rs in the AIS.
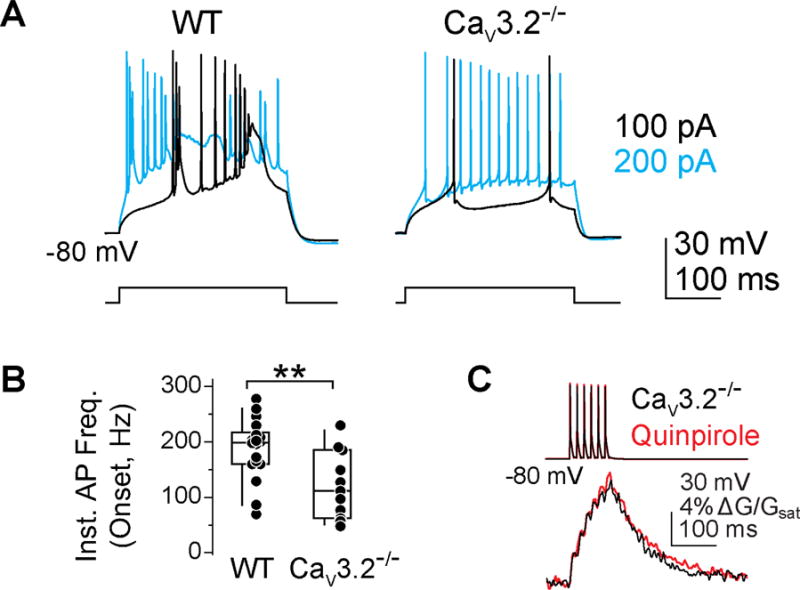
A) Cartwheel cell burst firing is suppressed in CaV3.2−/− animals. Spike trains are color coded to current injection amplitude.
B) Instantaneous frequency of first two spikes in a train was lower in CaV3.2−/− animals. Box plot indicates median, quartiles and 10/90 percentiles. ** p < 0.01, Mann Whitney.
C) Quinpirole did not alter AIS Ca in CaV3.2−/−.
D3R regulates AIS, but not dendritic, CaV3 channels. Since dendritic CaV3 current dominates whole-cell voltage clamp recordings in cartwheel cells, modulation occurring exclusively at the AIS is masked in somatic whole-cell recordings (Bender et al., 2010). We therefore used a two-step electrical/optical approach to determine how D3R alters CaV3.2 biophysical properties. First, we recapitulated this neuromodulatory pathway in HEK293 cells by transiently expressing D3R and CaV3.2. This allowed us to assess the modulation of CaV3.2 biophysical properties with high-quality voltage clamp. HEK293 cell stores are different than those found in the AIS: HEK293 cells appear to lack endogenous RyRs (Rossi et al., 2002); however they express M3 muscarinic receptors (mAChRs) endogenously, which allow for activation of PKC via IP3 (Atwood et al., 2011). Consistent with this, we found that neither quinpirole alone nor quinpirole paired with ryanodine concentrations that activate RyR-dependent stores (5 μM in ACSF) had an effect on CaV3.2 biophysics. Instead, modulation occurred when D3R agonists were paired with the mAChR agonist carbachol (10 μM). PKC/D3R co-activation hyperpolarized half-maximal steady state inactivation by 8.0 mV relative to cells co-treated in the presence of the D3R antagonist sulpiride (CCh+Quin: −90.7 ± 1.0 mV, n = 11, CCh+Quin+Sulp: −82.7 ± 1.0 mV, n = 9, p < 0.001). More modest hyperpolarizations in voltage-dependent activation were also observed in some conditions (Fig. 4). These data support results in the AIS that coincident activation of D3R and PKC are required to engage β-arrestin. Further, effects were blocked by sulpiride, pertussis toxin, and U0126, indicating that D3R receptor activation, mobilization of beta-gamma following Gi/o activation, and ERK1/2 signaling were all required in HEK cells as in the AIS.
Fig 4. D3R-dependent regulation of CaV3.2 currents in HEK 293 cells.
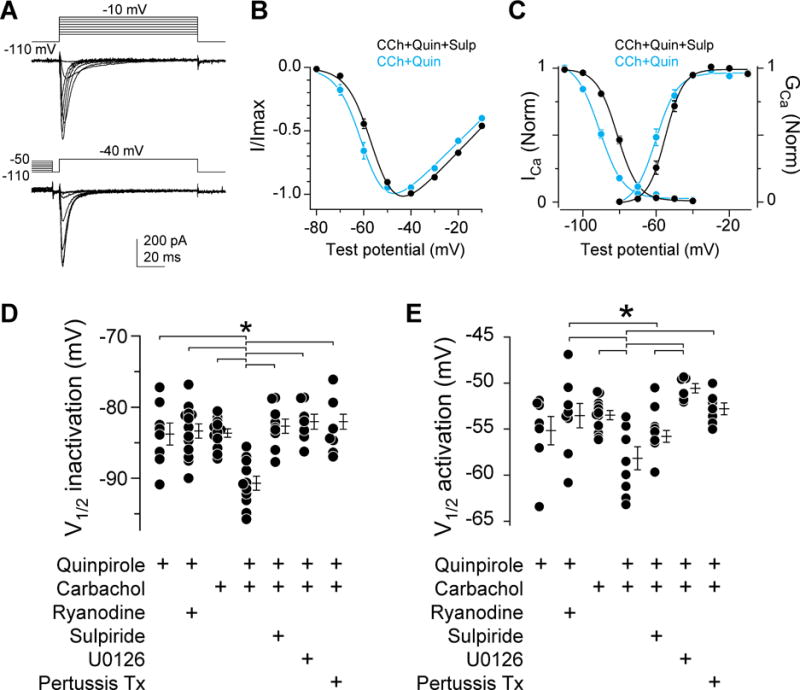
A) CaV3.2 steady-state activation and inactivation in HEK293 cells co-expressing D3R. Data are from untreated cells.
B) Peak activation current for CCh+quinpirole and CCh+quinpirole+sulpiride conditions. Data are fit to a Boltzmann equation. n = 8 each. Bars are SEM.
C) Activation and inactivation curves for CCh+quinpirole and CCh+quinpirole+sulpiride conditions. Activation: n = 8 each. Inactivation: n = 11 CCh+quinpirole, n = 9 CCh+quinpirole+sulpiride. Bars are SEM.
D) Half inactivation voltage is hyperpolarized by co-application of CCh and quinpirole. A concentration of ryanodine (5 μM) that activates RyR-dependent stores is ineffective in HEK cells. CCh+Quin effects are blocked by inhibiting D3R signaling with sulpiride, blocking ERK1/2 signaling with U0126, or preventing Gβγ-dependent recruitment of β-arrestin to D3R with pertussis toxin. Circles are single cells. Bars are mean ± SEM. **, p < 0.01, ANOVA, Newman-Keuls.
E) Half-activation voltage is hyperpolarized by co-application of CCh and quinpirole vs carbachol alone, but not quinpirole or quinpirole/CCh/sulpiride. Dots are single cells. Bars are mean ± SEM. *, p < 0.05, ANOVA, Newman-Keuls.
The relative channel availability at a resting Vm of −80 mV was reduced by 62% in HEK cells. This reduction may account for changes in spike-evoked Ca influx at the AIS. Here, CaV3-mediated Ca influx accounts for ~45% of total spike-evoked Ca influx (Bender et al., 2012). Thus, roughly half of the Ca influx mediated by CaV3.2 channels would need to be suppressed for quinpirole to modulate total spike-evoked Ca influx by 25% (Fig. 1). To test whether D3R alters CaV3.2 inactivation in neurons, we isolated CaV3 currents in voltage clamp and imaged AIS Ca with Fluo-4, as this allows one to monitor Ca channel activity local to the AIS. Cells were held at membrane potentials that deinactivate (−110 mV) or half inactivate CaV3 (−80 mV) and then depolarized to −50 mV to activate CaV3. AIS Ca transients evoked from −110 mV were reduced to 18 ± 6% of baseline by the CaV3 selective antagonist TTA-P2 (2 μM, Reger et al., 2011). As observed previously, quinpirole did not alter whole-cell currents at either voltage (norm. I−110: 1.05 ± 0.03, n = 8, norm. I−80: 1.01 ± 0.04, n = 8). However, modulation of AIS Ca was observed when cells were held at V1/2 inactivation for CaV3 (norm. G/Gsat: 0.46 ± 0.07, n = 8). AIS Ca influx was not altered if the cell was held at −110 mV, a voltage where CaV3 currents are deinactivated (Fig. 5; norm. G/Gsat: 1.00 ± 0.03, n = 8). This is consistent with a change in voltage dependent inactivation, and not with an overall reduction of CaV3 current (i.e., channel endocytosis). Thus, a relatively subtle modulation in the voltage dependence of inactivation can account for the marked reduction in the number of channels available to flux Ca during a spike (Bender et al., 2010; Bender et al., 2012).
Fig. 5. D3R signaling regulates AIS Ca by hyperpolarizing CaV3.2 inactivation.
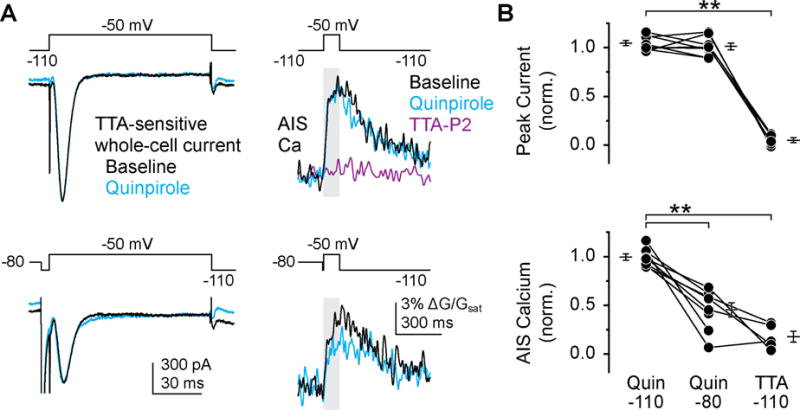
A) TTA-P2 sensitive CaV3 whole-cell currents (left, Residual current following application of the selective CaV3 antagonist TTA-P2 [2 μM] have been subtracted) and coincident AIS Ca transients (right) in cartwheel cells voltage clamped to fully deinactivate CaV3 (top) or half inactivate CaV3 (bottom). Whole cell current, which is dominated by non-modulated dendritic CaV3, is not altered (Bender et al., 2010); AIS Ca is suppressed only in half-inactivated condition. TTA-P2 (magenta) was added following quinpirole and its block was assessed with steps from −110 mV.
B) Summary of peak current amplitude and AIS Ca (integrated over step to −50 mV; grey shadow in g), normalized to pre-quinpirole baseline at each voltage. Bars and symbols as in Fig. 1f. ** p < 0.01, Newman-Keuls.
Discussion
The AIS is gaining recognition as an important site for neuromodulation and plasticity (Grubb and Burrone, 2010; Grubb et al., 2011; Kole and Stuart, 2012; Kuba et al., 2010). Spike-evoked AIS Ca influx has been observed in many central and peripheral neurons (Apostolides et al., 2016; Bender and Trussell, 2009; Callewaert et al., 1996; Francois et al., 2015; Grundemann and Clark, 2015; Luscher et al., 1996; Martinello et al., 2015; Schiller et al., 1995; Yu et al., 2010). AIS Ca influx is often mediated by a mix of Ca channels that includes CaV3 (Bender and Trussell, 2009; Francois et al., 2015; Grundemann and Clark, 2015; Martinello et al., 2015), suggesting that these channels play a role in initial segment excitability throughout the brain. Here, we found that D3Rs altered CaV3 voltage dependent inactivation, effectively blocking a major fraction of CaV3 channels in the AIS at resting membrane potentials. These effects were mediated at least in part by the CaV3.2 isoform, as quinpirole did not alter AIS Ca influx in CaV3.2−/− animals; however, additional experiments would be required to establish definitively whether other CaV3 isoforms are also present at the AIS. Interestingly, the opposite has been observed in hippocampal granule cells. There, M1 muscarinic receptors have been shown to alter the voltage dependent activation properties of AIS-localized CaV3 channels. This increased neuronal excitability by inhibiting Kv7 in a Ca-dependent manner (Martinello et al., 2015). AIS CaV3 channels can therefore be targets for bidirectional modulation, controlling overall neuronal activity by regulating the excitability of the spike initiation zone.
Intracellular Ca cisternae are localized to the AIS of many neuron classes, including cartwheel cells (Benedeczky et al., 1994; Wouterlood and Mugnaini, 1984). More recently, ryanodine receptors were identified at the AIS of pyramidal cells and striatal spiny neurons (King et al., 2014; Mandikian et al., 2014). In these cells, RyRs co-localize in ankyrin-G deficient clusters with GABAA receptors and KV2.1 channels. Here, we found that that dopaminergic regulation at the AIS required ryanodine-dependent stores. We hypothesize that these RyRs are sensitive to spike-evoked Ca influx, and that store Ca mobilization is necessary for PKC activation in the AIS. Indeed, PKC is enriched along the plasma membrane of the AIS in Purkinje neurons (Cardell et al., 1998), potentially in close proximity to RyRs. This may suggest that dopaminergic regulation is sensitive to cartwheel cell activity levels, as D3Rs would require both bound agonist and PKC-dependent phosphorylation to engage β-arrestin.
Dorsal cochlear nucleus circuits are regulated by multiple neuromodulatory pathways, including cholinergic, serotonergic (Tang and Trussell, 2015; Thompson and Thompson, 2001), noradrenergic (Kuo and Trussell, 2011), and dopaminergic systems (Cransac et al., 1995). Here, we found that D3R-dependent regulation of cartwheel cell excitability is β-arrestin-mediated. β-arrestin not only engages signaling cascades that are distinct from canonical GPCR pathways, but also signals over longer timescales than traditional G-protein dependent signaling (Reiter et al., 2012). This slow timecourse is consistent with previous results, where D3R-dependent suppression of spike bursts occurred over minutes in cartwheel cells (Bender et al., 2010). Cartwheel cell bursts have been hypothesized to contribute to adaptive noise filtering in dorsal cochlear nucleus by providing feedforward and lateral inhibition to principal cells and cartwheel cells, respectively (Oertel and Young, 2004; Roberts and Trussell, 2010; Roberts and Portfors, 2008; Sedlacek et al., 2011; Shore, 2005). Moreover, bursts generate dendritic Ca spikes that may be important for short- and long-term plasticity at cartwheel cell synapses (Sedlacek et al., 2011; Tzounopoulos et al., 2004). Long-lasting burst suppression may therefore be useful when transitioning to new environments that have new forms of background noise. Dopaminergic input, perhaps related to entering novel environments (Schultz, 2007), may aid the acquisition of new noise filtering rules by suppressing bursting associated with older environmental conditions.
D3Rs are localized to multiple brain regions beyond the dorsal cochlear nucleus, including limbic regions that are sites for therapeutic intervention in neuropsychiatric disease (Sokoloff et al., 1992). Indeed, many antipsychotic drugs have high affinity for D3R (Joyce and Millan, 2005), though how they interact with D3R to regulate neuronal function is relatively unclear. Recently discovered β-arrestin-biased dopamine receptor agonists show potent antipsychotic properties, indicating that β-arrestin-dependent signaling may underlie some of the beneficial actions of dopaminergic therapeutics (Beaulieu et al., 2009; Chen et al., 2012). Because dopamine system dysfunction is central to a variety of neuronal disorders, and because dopamine receptor pharmacology can differentially engage G-protein and β-arrestin-dependent pathways, it is critical to understand how these different signaling pathways regulate neuronal excitability. Whether D3Rs regulate AIS excitability in other cell classes, and whether these actions are affected in psychiatric disease, remain to be explored.
Experimental Procedures
Slice Electrophysiology and Imaging
All procedures were pre-approved by UCSF IACUC. Coronal brainstem slices (210 μm) were made from postnatal day (P)16-24 C57BL/6NJ, B6N(Cg)-Arrb2tm1.1(KOMP)Vlcg/J (RRID:IMSR_KOMP:VG17027), or B6;129-Cacna1htm1Kcam/J (RRID:IMSR_JAX:013770) mice of either sex. Dissection solution contained (in mM) 87 NaCl, 25 NaHCO3, 25 glucose, 75 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, and 7 MgCl2. Slices were incubated in the same solution for 30 min at 32.5 °C, then at room temperature for 0.5 –3 hrs before recording. Recording solution contained (in mM): 129 NaCl, 3 KCl, 1 CaCl2, 2.7 MgSO4, 1.2 KH2PO4, 20 NaHCO3, 3 Na-HEPES, 10 glucose; bubbled with 5% CO2/95% O2; 32–34 °C. 10 μM NBQX, 0.5 μM strychnine, and 10 μM SR95531 were included in all experiments to block AMPA, glycine and GABA receptors, respectively. For current-clamp recordings, patch electrodes with 3–5 MΩ tip resistance were filled with a solution containing (in mM): 113 K-Gluconate, 9 HEPES, 4.5 MgCl2, 14 Tris2-phosphocreatine, 4 Na2-ATP, 0.3 tris-GTP, 0.25 Fluo-5F and 0.02 Alexa 594. For voltage-clamp recordings, the recording solution was supplemented with 2 mM CaCl2 (total), 1 mM CsCl, 250 nM tetrodotoxin, and 10 μM nifedipine; patch electrodes were filled with a solution containing (in mM): 110 CsMeSO3, 40 HEPES, 1 KCl, 4 NaCl, 4 Mg-ATP, 10 Na-phosphocreatine, 0.4 Na2-GTP, 0.25 Fluo-4, 0.02 Alexa 594; ~290 mOsm (pH 7.22). 2-photon imaging and electrophysiological acquisition was performed as previously described (Bender et al., 2010). Baseline and post-drug measurements of resting membrane potential (Vm) and input resistance (Rin) were reported as the average of 5 minute epochs immediately before and 10 minutes following drug application. For experiments in which drugs were introduced via the pipette, time-locked controls were performed to account for potential drug-induced rundown of AIS Ca if quinpirole-dependent modulation was observed. Recordings were excluded if Rin changed by more than 15%. Data were corrected for a measured junction potential of 10 and 12 mV in voltage and current-clamp conditions, respectively.
For localized puffing experiments, 10 μM quinpirole was dissolved in the following solution (in mM): 142 NaCl, 3 KCl, 1 CaCl2, 2.7 MgSO4, 1.2 KH2PO4, 10 Na-HEPES, 10 glucose, and 0.1 Alexa-594 (pH, 7.2; 305 mOsm). Baseline AIS Ca transients were assessed before positioning the puffer pipette within the slice. Pipettes were positioned within 5 μm of the target neurite, and solution was expelled 5× (20 ms duration, 1 min interval, 8–10 psi). The pipette was then withdrawn, and post-drug AIS Ca transients were assessed 5–10 min later. During puffs, Alexa 594 fluorescence radiated ~25 μm from the pipette. Therefore, dendritic branches 40–50 μm from the soma were selected to minimize quinpirole spillover to the AIS.
To calculate onset spike frequency in wild type and CaV3.2 knockout animals, 300 ms current steps (100–400 pA, incremented at 20–50 pA) were injected through the somatic pipette. The maximum instantaneous spike frequency between the first two spikes was noted, regardless of injected current amplitude. Maximal frequency occurred at similar current injection amplitudes in these two populations (WT: 207.8 ± 19.1 pA, n = 18; KO: 263.4 ± 16.6 pA, n = 11; p = 0.2, U = 71.5, Mann-Whitney).
Immunohistochemistry
P20-P35 C57BL/6NJ and were perfused and fixed with 1% paraformaldehyde. Coronal sections (30 μm) were collected on a cryostat and rinsed in PBS. Sections were rinsed with PBST (PBS with 3% TX-100) (3X), then blocked with 10% normal goat serum with 3% TX-100 for 2h at room temperature (RT). Sections were incubated overnight at room temperature using validated primary antibodies (King et al., 2014) (mouse IgG1 anti-RyR, 1:100, Pierce MA3-925, RRID:AB_2254138; mouse IgG2a anti-AnkyrinG, 1:200, NeuroMab N106/36, RRID:AB_10697718). Sections were rinsed with PBS (3X) and incubated with AlexaFluor 568 goat anti-mouse IgG1 (1:1000) and AlexaFluor488 goat anti-mouse IgG2a (1:1000) for 2h at room temperature (Invitrogen). Sections were washed and mounted with Vectashield with DAPI (Vector Labs). Images were acquired with a Nikon N-SIM structured illumination microscope (100X, 1.49 NA) at UCSF’s Nikon Imaging Center. 4 separate sections were obtained from one animal. Results were identical across sections.
Heterologous expression and electrophysiology
cDNA encoding human D3R (Missouri S&T cDNA Resource Center) was cloned into the pIRES2/AcGFP vector (Clontech) using NheI and XhoI restriction sites. HEK293 cells were maintained in a culture medium of 90% DMEM (Invitrogen) and 10% fetal bovine serum (HyClone), incubated at 37°C. Cells were transfecte d with Lipofectamine 2000 (I10 μL/500 mL), a D3R/GFP plasmid (500 ng/500 mL), and pcDNA3.1 encoding the rat Cav3.2 alpha subunit (4 μg/500 mL) (Lee et al., 1999) and incubated for 16–24 hours at 37°C. Cells were then plated on 25 mm coverslips and recorded within 4–48 hours. Only isolated, GFP-positive cells with peak currents >200 pA were selected for analysis. Internal solution contained (in mM): 110 CsMeSO3, 40 HEPES, 1 KCl, 4 NaCl, 4 Mg-ATP, 10 Na-phosphocreatine, 0.4 Na2-GTP, 0.1 EGTA; ~290 mOsm (pH 7.2). Recording solution contained (in mM): 148 TEA-Cl, 10 HEPES and 2 CaCl ~305 mOsm (pH 7.4 with TEA-OH). Recordings were made at 32–34°C, and Rseries was compensated 75%. Observed current in CaV3.2-expressing cells was −880 ± 88 pA (n = 32, including carbachol, quinpirole, and carbachol + quinpirole + sulpiride groups). A small current was observed in untransfected cells (−9 ± 9 pA, n = 3, range: −28 to 0.6 pA), presumably mediated by native nonselective cation channels (Zhu et al., 1998).
CaV3.2 activation was assessed with current steps from −110 to a range of voltages from −80 to −10 mV (10 mV increments, 100 ms duration). Peak currents were fit to a Boltzmann equation to calculate conductance (G = 1/(1 + exp [−(V − V1/2/s]), G: norm. conductance, V1/2: half-maximal activation, s: slope). Inactivation was assessed with a test pulse from −110 to −40 mV proceeded by 2s voltage steps from −100 to 0 mV (10 mV increments, offset 5 ms before test pulse). Cells were incubated in quinpirole and/or carbachol for >15 min before recording. U0126, ryanodine, and sulpiride were applied >10 min before carbachol and/or quinpirole to ensure that they reached equilibrium before agonizing D3R signaling. Cells were incubated in 1 μg/mL pertussis toxin for 16–24 hrs before recording in pertussis toxin-free solution. Recording sessions were limited to 1 hr following removal of pertussis toxin.
Chemicals
Fluo-5F and Fluo-4 pentapotassium salt and Alexa Fluor 594 hydrazide Na salt were from Molecular Probes. UBO-QIC (Gαq-inhibiting compound) was from the Institute of Pharmaceutical Biology, University of Bonn, Germany. 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo[f]quinoxaline-2,3-dione (NBQX), gabazine (SR95531), (-)-Quinpirole hydrochloride, (S)-(-)-sulpiride, phorbol 12-myristate 13-acetate (PMA), U0126, cyclopiazonic acid (CPA), thapsigargin, heparin sodium salt, ryanodine, U73122, 8-Br-cyclic ADP ribose, tetrodotoxin-citrate, pertussis toxin, and nifedipine were from Tocris. All others were from Sigma. All drugs were introduced to the artificial cerebrospinal fluid unless otherwise noted.
Statistics
An ANOVA followed by a Newman-Keuls posthoc test was used, unless otherwise stated. Significance was set at 0.05. Data were presented as mean ± SEM.
Highlights.
D3 dopamine receptors utilize non-canonical signaling pathways to regulate AIS CaV3
D3Rs signal via β-arrestin, ryanodine-dependent calcium stores, and ERK
Arrestin-dependent signaling hyperpolarizes voltage dependent inactivation of CaV3
D3R signaling results in a loss of AIS CaV3 channels available for spike initiation
Acknowledgments
We thank Dr. Edward Perez-Reyes for providing CaV3.2 cDNA, Drs. Fei Li and Gabriel Mercado Besserer for cell culture support, and Dr. Chris Ford and members of the Bender lab for valuable comments. This work was supported by funds from the NIH to RBS (F32NS095580), RMvR (R00AA020539), JLW (R01DA037963), KJB (R00DC011080, R01DA035913, P50AA017072), and the UCSF Open Access Publishing Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions: SY, RBS, and KJB performed electrophysiology and imaging experiments. SY, MA and ATL performed immunohistochemistry. RBS, GMB, RMvR and JLW performed heterologous expression. All authors contributed to experimental design and manuscript preparation.
References
- Apostolides PF, Milstein AD, Grienberger C, Bittner KC, Magee JC. Axonal Filtering Allows Reliable Output during Dendritic Plateau-Driven Complex Spiking in CA1 Neurons. Neuron. 2016;89:770–783. doi: 10.1016/j.neuron.2015.12.040. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics. 2011;12:14. doi: 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Ford CP, Trussell LO. Dopaminergic modulation of axon initial segment calcium channels regulates action potential initiation. Neuron. 2010;68:500–511. doi: 10.1016/j.neuron.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Trussell LO. Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron. 2009;61:259–271. doi: 10.1016/j.neuron.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Trussell LO. The physiology of the axon initial segment. Annu Rev Neurosci. 2012;35:249–265. doi: 10.1146/annurev-neuro-062111-150339. [DOI] [PubMed] [Google Scholar]
- Bender KJ, Uebele VN, Renger JJ, Trussell LO. Control of firing patterns through modulation of axon initial segment T-type calcium channels. J Physiol. 2012;590:109–118. doi: 10.1113/jphysiol.2011.218768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedeczky I, Molnar E, Somogyi P. The cisternal organelle as a Ca(2+)-storing compartment associated with GABAergic synapses in the axon initial segment of hippocampal pyramidal neurones. Exp Brain Res. 1994;101:216–230. doi: 10.1007/BF00228742. [DOI] [PubMed] [Google Scholar]
- Callewaert G, Eilers J, Konnerth A. Axonal calcium entry during fast ‘sodium’ action potentials in rat cerebellar Purkinje neurones. J Physiol. 1996;495:641–647. doi: 10.1113/jphysiol.1996.sp021622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardell M, Landsend AS, Eidet J, Wieloch T, Blackstad TW, Ottersen OP. High resolution immunogold analysis reveals distinct subcellular compartmentation of protein kinase C gamma and delta in rat Purkinje cells. Neuroscience. 1998;82:709–725. doi: 10.1016/s0306-4522(97)00305-9. [DOI] [PubMed] [Google Scholar]
- Chen X, Sassano MF, Zheng L, Setola V, Chen M, Bai X, Frye SV, Wetsel WC, Roth BL, Jin J. Structure-functional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. J Med Chem. 2012;55:7141–7153. doi: 10.1021/jm300603y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EY, Cho DI, Park JH, Kurose H, Caron MG, Kim KM. Roles of protein kinase C and actin-binding protein 280 in the regulation of intracellular trafficking of dopamine D3 receptor. Mol Endocrinol. 2007;21:2242–2254. doi: 10.1210/me.2007-0202. [DOI] [PubMed] [Google Scholar]
- Cowell RM, Kantor L, Hewlett GH, Frey KA, Gnegy ME. Dopamine transporter antagonists block phorbol ester-induced dopamine release and dopamine transporter phosphorylation in striatal synaptosomes. Eur J Pharmacol. 2000;389:59–65. doi: 10.1016/s0014-2999(99)00828-6. [DOI] [PubMed] [Google Scholar]
- Cransac H, Cottet-Emard JM, Pequignot JM, Peyrin L. Monoamines (noradrenaline, dopamine, serotonin) in the rat cochlear nuclei: endogenous levels and turnover. Hearing Res. 1995;90:65–71. doi: 10.1016/0378-5955(95)00147-x. [DOI] [PubMed] [Google Scholar]
- Francois A, Schuetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, Mantilleri A, Nargeot J, Noel J, Wood JN, et al. The Low-Threshold Calcium Channel Cav3.2 Determines Low-Threshold Mechanoreceptor Function. Cell Reports. 2015;10:370–382. doi: 10.1016/j.celrep.2014.12.042. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb MS, Shu Y, Kuba H, Rasband MN, Wimmer VC, Bender KJ. Short-and long-term plasticity at the axon initial segment. J Neurosci. 2011;31:16049–16055. doi: 10.1523/JNEUROSCI.4064-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundemann J, Clark BA. Calcium-Activated Potassium Channels at Nodes of Ranvier Secure Axonal Spike Propagation. Cell Reports. 2015;12:1715–1722. doi: 10.1016/j.celrep.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JN, Millan MJ. Dopamine D3 receptor antagonists as therapeutic agents. Drug Discov Today. 2005;10:917–925. doi: 10.1016/S1359-6446(05)03491-4. [DOI] [PubMed] [Google Scholar]
- Kantor L, Gnegy ME. Protein kinase C inhibitors block amphetamine-mediated dopamine release in rat striatal slices. J Pharmacol Exp Ther. 1998;284:592–598. [PubMed] [Google Scholar]
- Kim Y, Trussell LO. Ion channels generating complex spikes in cartwheel cells of the dorsal cochlear nucleus. J Physiol. 2007;97:1705–1725. doi: 10.1152/jn.00536.2006. [DOI] [PubMed] [Google Scholar]
- King AN, Manning CF, Trimmer JS. A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J Comp Neurol. 2014;522:2594–2608. doi: 10.1002/cne.23551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Stuart GJ. Signal processing in the axon initial segment. Neuron. 2012;73:235–247. doi: 10.1016/j.neuron.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates Na(+) channel distribution at the axon initial segment. Nature. 2010;465:1075–1078. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- Kuo SP, Trussell LO. Spontaneous spiking and synaptic depression underlie noradrenergic control of feed-forward inhibition. Neuron. 2011;71:306–318. doi: 10.1016/j.neuron.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Lipp P, Luscher HR, Niggli E. Control of action potential propagation by intracellular Ca2+ in cultured rat dorsal root ganglion cells. J Physiol. 1996;490(Pt 2):319–324. doi: 10.1113/jphysiol.1996.sp021146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandikian D, Bocksteins E, Parajuli LK, Bishop HI, Cerda O, Shigemoto R, Trimmer JS. Cell type-specific spatial and functional coupling between mammalian brain Kv2.1 K(+) channels and ryanodine receptors. J Comp Neurol. 2014;522:3555–3574. doi: 10.1002/cne.23641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinello K, Huang Z, Lujan R, Tran B, Watanabe M, Cooper EC, Brown DA, Shah MM. Cholinergic afferent stimulation induces axonal function plasticity in adult hippocampal granule cells. Neuron. 2015;85:346–363. doi: 10.1016/j.neuron.2014.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molitor SC, Manis PB. Dendritic Ca2+ transients evoked by action potentials in rat dorsal cochlear nucleus pyramidal and cartwheel neurons. J Neurophysiol. 2003;89:2225–2237. doi: 10.1152/jn.00709.2002. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Su P, Kang HW, Van Deusen A, Baumgart JP, David LS, Snutch TP, Barrett PQ, Lee JH, et al. Molecular mechanisms of subtype-specific inhibition of neuronal T-type calcium channels by ascorbate. J Neurosci. 2007;27:12577–12583. doi: 10.1523/JNEUROSCI.2206-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel D, Young ED. What’s a cerebellar circuit doing in the auditory system? Trends Neurosci. 2004;27:104–110. doi: 10.1016/j.tins.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ. Role of beta gamma subunits of G proteins in targeting the beta-adrenergic receptor kinase to membrane-bound receptors. Science. 1992;257:1264–1267. doi: 10.1126/science.1325672. [DOI] [PubMed] [Google Scholar]
- Reger TS, Yang ZQ, Schlegel KA, Shu Y, Mattern C, Cube R, Rittle KE, McGaughey GB, Hartman GD, Tang C, et al. Pyridyl amides as potent inhibitors of T-type calcium channels. Bioorg Med Chem Lett. 2011;21:1692–1696. doi: 10.1016/j.bmcl.2011.01.089. [DOI] [PubMed] [Google Scholar]
- Reiter E, Ahn S, Shukla AK, Lefkowitz RJ. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu Rev Pharm. 2012;52:179–197. doi: 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MT, Bender KJ, Trussell LO. Fidelity of complex spike-mediated synaptic transmission between inhibitory interneurons. J Neurosci. 2008;28:9440–9450. doi: 10.1523/JNEUROSCI.2226-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MT, Trussell LO. Molecular layer inhibitory interneurons provide feedforward and lateral inhibition in the dorsal cochlear nucleus. J Neurophysiol. 2010;104:2462–2473. doi: 10.1152/jn.00312.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PD, Portfors CV. Design principles of sensory processing in cerebellum-like structures. Early stage processing of electrosensory and auditory objects. Biol Cybern. 2008;98:491–507. doi: 10.1007/s00422-008-0217-1. [DOI] [PubMed] [Google Scholar]
- Rossi D, Simeoni I, Micheli M, Bootman M, Lipp P, Allen PD, Sorrentino V. RyR1 and RyR3 isoforms provide distinct intracellular Ca2+ signals in HEK 293 cells. J Cell Sci. 2002;115:2497–2504. doi: 10.1242/jcs.115.12.2497. [DOI] [PubMed] [Google Scholar]
- Ryugo DK, Pongstaporn T, Wright DD, Sharp AH. Inositol 1,4,5-trisphosphate receptors: immunocytochemical localization in the dorsal cochlear nucleus. J Comp Neurol. 1995;358:102–118. doi: 10.1002/cne.903580107. [DOI] [PubMed] [Google Scholar]
- Schiller J, Helmchen F, Sakmann B. Spatial profile of dendritic calcium transients evoked by action potentials in rat neocortical pyramidal neurones. J Physiol. 1995;487:583–600. doi: 10.1113/jphysiol.1995.sp020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci. 2007;30:259–288. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- Sedlacek M, Tipton PW, Brenowitz SD. Sustained firing of cartwheel cells in the dorsal cochlear nucleus evokes endocannabinoid release and retrograde suppression of parallel fiber synapses. J Neurosci. 2011;31:15807–15817. doi: 10.1523/JNEUROSCI.4088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Shore SE. Multisensory integration in the dorsal cochlear nucleus: unit responses to acoustic and trigeminal ganglion stimulation. Eur J Neurosci. 2005;21:3334–3348. doi: 10.1111/j.1460-9568.2005.04142.x. [DOI] [PubMed] [Google Scholar]
- Sokoloff P, Giros B, Martres MP, Andrieux M, Besancon R, Pilon C, Bouthenet ML, Souil E, Schwartz JC. Localization and function of the D3 dopamine receptor. Arznei-Forschung. 1992;42:224–230. [PubMed] [Google Scholar]
- Tang ZQ, Trussell LO. Serotonergic regulation of excitability of principal cells of the dorsal cochlear nucleus. J Neurosci. 2015;35:4540–4551. doi: 10.1523/JNEUROSCI.4825-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AM, Thompson GC. Serotonin projection patterns to the cochlear nucleus. Brain Res. 2001;907:195–207. doi: 10.1016/s0006-8993(01)02483-0. [DOI] [PubMed] [Google Scholar]
- Thompson D, Whistler JL. Dopamine D(3) receptors are down-regulated following heterologous endocytosis by a specific interaction with G protein-coupled receptor-associated sorting protein-1. J Biol Chem. 2011;286:1598–1608. doi: 10.1074/jbc.M110.158345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Wouterlood FG, Mugnaini E. Cartwheel neurons of the dorsal cochlear nucleus: a Golgi-electron microscopic study in rat. J Comp Neurol. 1984;227:136–157. doi: 10.1002/cne.902270114. [DOI] [PubMed] [Google Scholar]
- Yu Y, Maureira C, Liu X, McCormick D. P/Q and N channels control baseline and spike-triggered calcium levels in neocortical axons and synaptic boutons. J Neurosci. 2010;30:11858–11869. doi: 10.1523/JNEUROSCI.2651-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Zhang Y, Xu H, Jiang C. Identification of endogenous outward currents in the human embryonic kidney (HEK 293) cell line. J Neurosci Methods. 1998;81:73–83. doi: 10.1016/s0165-0270(98)00019-3. [DOI] [PubMed] [Google Scholar]