

Abstract
Human microbiome research is an actively developing area of inquiry, with ramifications for our lifestyles, our interactions with microbes, and how we treat disease. Advances depend on carefully executed, controlled, and reproducible studies. Here, we provide a Primer for researchers from diverse disciplines interested in conducting microbiome research. We discuss factors to be considered in the design, execution, and data analysis of microbiome studies. These recommendations should help researchers to enter and contribute to this rapidly developing field.
Introduction
Many studies have documented differences in the composition of host-associated microbial communities between healthy and disease states (Clemente et al., 2012; Karlsson et al., 2013; Knights et al., 2013). For a growing number of diseases, an altered microbiome is not just a marker of disease, but also actively contributes to pathology (Chassaing et al., 2012). The best empirical direct evidence that microbiomes can drive disease comes from experiments in which the microbiota from diseased donors and controls are “transplanted” into healthy germ-free hosts: if recipients of the disease-associated microbiome display the disease phenotype, the microbiome is considered causal. This approach, pioneered by Jeffrey Gordon and his group (Turnbaugh et al., 2006), has directly demonstrated that the composition of gut microbial communities can alter host metabolism (Koren et al., 2012; Vijay-Kumar et al., 2010), transmit colitis (Garrett et al., 2007), and modulate type I diabetes (Wen et al., 2008). The range of conditions with a host-microbiome interaction component continues to grow and has recently started to include neurological conditions (Collins et al., 2012). Consequently, researchers from a wide array of disciplines are interested in testing whether microbes, and especially gut microbes, are associated with various pathologies, whether they actively participate in disease, and ultimately whether they can present novel targets for therapies. This Primer is intended for non-experts who are considering their first microbiome project and summarizes lessons learned from past successful and unsuccessful projects.
Mammalian microbiome research has a long history (Savage, 1977), recently marked by dramatic increases in scale and scope due to advances in DNA-sequencing technologies and in associated computational methods. Anecdotal descriptions of community composition that set the standard in the recent past have given way to study designs that allow for repeated measurements, error estimates, correlations of microbiota with covariates, and increasingly sophisticated statistical tests (Knight et al., 2012). Today, microbiome data are obtained predominantly in three forms: (1) 16S rRNA gene sequence surveys that provide a view of microbiome membership, (2) metagenomic data used to portray functional potential, and (3) metatranscriptomic data to describe active gene expression. Here, we focus primarily on 16S rRNA gene surveys because they are economical and therefore scale to larger projects. 16S rRNA gene sequence data provide a relatively unbiased characterization of bacterial and archaeal diversity (Box 1 provides a brief overview of methods for characterizing the diversity of microbial eukaryotes and viruses). Regardless of the types of microorganisms targeted or the methodology used to characterize them, choices made at every step, from study design to analysis, can impact results. This Primer highlights resources that address specific technical questions and provides general advice stemming from our collective experience working in the field. Although we focus mainly on the mammalian gut microbiota, many of the same issues apply to microbial communities of other habitats. We have structured the Primer to answer questions that are commonly raised by researchers entering the field (Figure 1).
Box 1. Archaeal, Viral, and Eukaryotic Diversity.
Most studies of the human microbiota describe bacterial diversity, which typically dominates the cellular fraction of the microbiota; but other taxa, including Archaea, fungi, and other microbial eukaryotes, and viruses can be present.
Archaea
Archaeal diversity can be characterized using the commonly employed 515F/806R primer set (and others), and their diversity can be analyzed in the same way as bacterial diversity. The 16S rRNA gene is the most widely used marker gene for the Archaea, and their diversity is represented in reference data sets commonly used for Bacteria.
Microbial Eukaryotes
Characterization of fungal communities, in particular, is an active research area. In principle, the bioinformatics pipeline is the same for eukaryotic marker genes as for bacterial marker genes (Iliev et al., 2012). However, the lack of a standard marker gene and reference database means that the bioinformatics protocols are not as standardized as for 16S rRNA gene analysis. For fungi, although several marker gene options exist, the internal transcribed spacer (ITS) region of the 16S rRNA gene is generally preferred for obtaining high taxonomic resolution. The UNITE database (Abarenkov et al., 2010) is often used for ITS sequence-based analyses of fungal sequences. However, the ITS region is not amenable to alignments across distinct fungal taxa, so ITS-based fungal community studies frequently do not make use of phylogenetic metrics for alpha- and beta-diversity comparisons. One strategy that is being explored is using the 18S rRNA gene and ITS in conjunction to define fungal phylogenetic trees. Moreover, the 18S rRNA gene can, in principle, be used to analyze eukaryotic communities in the same manner that 16S rRNA genes are used. A reference database containing many eukaryotic sequences, such as SILVA (Quast et al., 2013), should be used for such analyses. One should confirm that the region of the 18S gene amplified discriminates between the taxa studied and should be aware that the 18S rRNA gene is not sufficient to characterize the eukaryotic phylogeny: trees built from 18S sequence alone will likely be of questionable utility.
Viruses
Characterizing the human virome requires a different approach because, unlike for cellular life, no gene or genomic region is homologous across all viruses. The current approach for studying these communities is to isolate virus-like particles (VLPs) using size fractionation and to sequence those using metagenomics (Caporaso et al., 2011a; Handley et al., 2012; Hurwitz et al., 2013; Reyes et al., 2010). Alternatively, viruses can be characterized using DNA microarrays (Jack et al., 2009; Palacios et al., 2007).
Figure 1. Conducting a Microbiome Study.
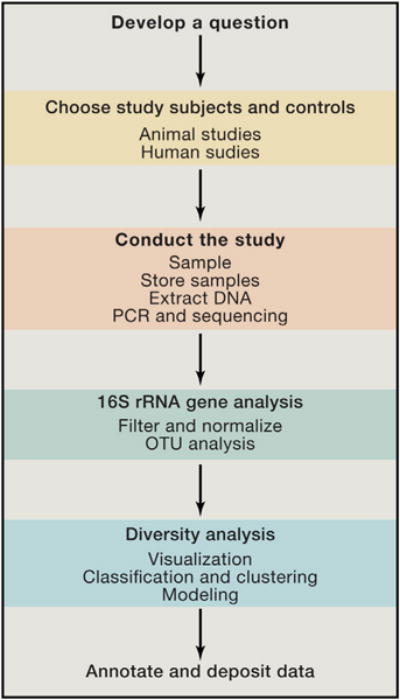
The sequential steps of conducting a microbiome study are diagramed, mirroring the sections of this Primer.
Animal Studies
The Maternal Effect
A large fraction of microbiome studies are conducted in animals, particularly rodents, as they offer attractive models for human biology and their environmental conditions can be tightly controlled. How animals are bred and raised is the most important source of confounding factors in microbiome studies conducted in animals. Inoculation of mice at birth (the maternal effect) is a major factor shaping the composition of the microbiota and leads to a sharing of suites of bacteria between littermates and their mothers that differentiates them from members of other families and can persist over several generations (Ley et al., 2005). The maternal effect determines the specific suite of microbes available to colonize a host. Subsequently, the individual host and host diet shapes the relative abundances of these taxa (Ley et al., 2005; Rawls et al., 2006).
Mitigating the Maternal Effect
The maternal effect is particularly problematic when it confounds the experimental effect (see Figure 2 for an example). Because littermates and even co-caged unrelated animals can share microbiotas due to coprophagy and other modes of transmission, randomization of treatments across litters/cages becomes an important aspect of experimental design. When the goal is to compare the effect of different genotypes on the microbiome, the options range from the use of germ-free mice gavaged with the same inoculum to the use of mixed-genotype litters. When these options are not available, alternate approaches include embryo transfers so that mixed genotypes are born together, cross-fostering, and cohousing post weaning. The last two options may be the least effective, as microbiotas will be at least partly assembled. In large studies with multiple litter/cage replicates, the variance in the data that is attributable to the maternal effect can be accounted for in statistical models (Benson et al., 2010).
Figure 2. The Maternal Effect Can Confound the Experimental Effect.
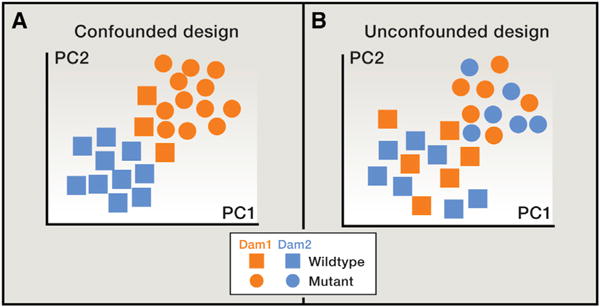
(A and B) In this mock example, each point represents a gut microbial community as characterized by a set of 16S rRNA gene sequences from a single mouse sample. In principal coordinates analysis (PCoA), points that are closer together represent microbial communities that are more similar in sequence composition. Samples from two different mouse genotypes are represented, and the mice are derived from two different dams. In all panels, squares indicate wild-type, and circles indicate mutant mouse genotypes. In (A), the effect of genotype is confounded by the effect of a shared dam, whereas in (B), the effect of dam is randomized across the two genotypes.
Using the Maternal Effect to Maximize a Phenotype
In some cases, animals of different genotypes are maintained separately in order to maximize the maternal effect and obtain a strong microbial phenotype (Vaishnava et al., 2011; Vijay-Kumar et al., 2010). Separately maintained mice can then be cohoused to demonstrate the spread of a microbiota between adults (Lawley et al., 2012; Ridaura et al., 2013). Conversely, mice can be housed individually to minimize cross-contamination and maintain individual microbiotas (Ley et al., 2005).
Environment Matters: Microbiotas Vary Greatly among Facilities
Mouse microbiotas can differ significantly between facilities even if they have identical genotypes (Friswell et al., 2010). Environmental conditions can differ between facilities—for instance, the water acidity, food, bedding, and so on can differ. But it appears that different colonies harbor their own populations of microbes as well. One striking example of this facility effect is that of the segmented filamentous bacteria (SFB), which have been reported more common in mice obtained from one common vendor (JAX) than another (Taconic) (Ivanov et al., 2009). Because their impact on the murine immune system is substantial, their presence or absence may confound experimental treatments, particularly in immunological studies. Less is known about the effects of housing regimen (e.g., specific pathogen free [SPF] versus conventional corridors—and note that definitions of SPF differ between facilities). For instance, the microbiota can differ enough between the SPF and conventional areas within facilities to impact the prevalence of certain microbially mediated phenotypes, such as type 1 diabetes in NOD mice (Wen et al., 2008).
Human Studies
Commonly Applied Exclusion Criteria
In human studies, antibiotics, diet, body mass index, age, pregnancy, and ethnicity all have been reported in the literature to have varying degrees of influence on the microbiota composition of the gut and of other body sites (Costello et al., 2012). Some of these factors, notably antibiotic use, have effects strong enough that they are often exclusion criteria (Cho et al., 2012; Dethlefsen et al., 2008; Dethlefsen and Relman, 2011; Ubeda et al., 2010). The exclusion criteria list from the NIH Human Microbiome Project (HMP, see dbGAP) includes use of systemic antibiotics, antifungals, antivirals, or antiparasitics within 6 months of sampling. Each antibiotic can affect the microbiome differently, and microbiome responses to a single antibiotic can also vary substantially between individuals (Dethlefsen et al., 2008; Dethlefsen and Relman, 2011; Maurice et al., 2013). The 6 month window of exclusion is somewhat arbitrary, however, and in some cases, a shorter window has been applied based on results (Koren et al., 2012).
Controls
As in any well-controlled study, factors impacting the microbiota should be balanced across the experimental groups. Selection of subjects can be limited by the demographics of the population available for study. Of course, a carefully selected control group, such as case-matched controls, is preferable to the use of “normal” individuals from another study. In time-series studies, individuals can be treated as their own control by collecting baseline samples before and during treatment (Dethlefsen et al., 2008; Ley et al., 2006). In studies that do not include a time series, the best-matched controls are monozygotic twins, in which genotype is constant within twin pairs: for instance, the effect of obesity on the microbiome has been studied in monozygotic co-twins discordant for this phenotype (Smith et al., 2013). Comparisons between monozygotic and dizygotic twin pairs are used to differentiate between environmental and genetic contributions to the microbiome, with the assumption that early environmental influences are similar for both twin types.
The Human Microbiome Project (HMP) generated the largest human metagenomic study to date (Consortium, 2012a, b), with the aim of providing a healthy reference set to be used for comparison in future studies. This reference set, however, introduces questions on how to best use the HMP data in new studies. Combining new data sets with the HMP is most effective when the protocols are compatible and the effect size (i.e., differences between controls and subjects) of the study is large. For example, in the Koren et al. (2012) study, the average between-subject microbial diversity was higher in the third trimester of pregnancy compared to the first trimester, but from the study’s data alone it was not possible to tell which was most similar to a non-pregnant state. Comparison with the HMP data set suggested that the first-trimester pattern was typical of a non-pregnant state and that the third trimester was aberrant (Koren et al., 2012). Another observation that emerged from this comparison was a difference in overall diversity between the American (HMP) and Finnish (pregnancy study) samples. Because the data were generated separately, the source of this difference could be technical artifacts or something more interesting such as culture, diet, or geography. Indeed, for studies with subtle effects on the microbiome such as those associated with obesity or colon cancer, technical effects such as PCR primer choice and DNA extraction method (see below) can greatly outweigh the biological effects. Therefore, most studies will require a carefully matched control group and cannot rely on the HMP data set as a “universal” control group, just as epidemiological studies cannot rely on a single “universal” reference population.
Before Sampling: Study Design Elements
Pilot Studies
The number of samples required for a microbiome study depends on the effect size. A tool called “Evident” has been developed to aid in estimating the study size required based on the anticipated effect size and similar previous studies (https://github.com/biocore/Evident). However, because there is no standardized way of reporting effect sizes and similar studies may not exist, a pilot study may be necessary to define the effect size.
Document Everything and Be Consistent
Before, during, and after sample collection, all information about the sample and experimental procedures should be recorded. This information will constitute the “metadata” (covariates) surrounding the sample and will later be used in analyzing the data. Furthermore, such analyses will be simplified when procedures are as consistent as possible across groups and extraneous variables are minimized.
Repeated Sampling of Individuals
One question that comes up in study design is whether to resample individuals over time or whether to use those resources to sample more individuals only once. An argument can be made for both approaches depending on the goal of the study. For instance, specific strains of gut bacteria have been demonstrated to exist for many years within their hosts (Faith et al., 2013). Many studies report that the adult human fecal microbiota are “stable” over time, based on measures of within-subject (alpha) versus between-subject (beta) diversity (see below for further discussion) (HMPC, 2012a; Costello et al., 2009; Ley et al., 2006; Turnbaugh et al., 2009; Wu et al., 2011). Furthermore, overall patterns of diversity across the human body have been reproducible at different time points (Consortium, 2012a, b; Costello et al., 2009). Thus, the stability of the adult gut microbiome can be used to argue that single time points are sufficient to describe an individual’s microbiome. However, within-subject diversity in one individual over time can approach the between-subject diversity of many individuals at one time (Caporaso et al., 2011b). Therefore, repeated sampling over time will provide a more comprehensive view of the diversity.
Besides gaining a more comprehensive view of diversity for an individual, time series have added benefits. Aspects of the microbiota that can be examined more effectively with time-series data include changes in the presence/absence, in the membership in the microbial community, and in the relative abundances of microbes. Time-series data can also reveal interesting characteristics of the microbiome that are not apparent from single time points, such as the volatility of the microbiota (degree of change between time points) and its resilience (bounce back after disturbance), both of which may be altered in disease or preclinical states (Carvalho et al., 2012; Costello et al., 2012; Dethlefsen et al., 2008; Lozupone et al., 2012). Recently developed analytical approaches (discussed below) can test whether an early configuration of the microbiota predicts later configurations, including predictions of the development of pathological states such as colitis (Carvalho et al., 2012).
Obtaining and Storing Samples
Storage and transit conditions can be important variables in microbiome study outcomes because they impact the DNA yields and qualities. Therefore, whatever the conditions used, it is most important to be consistent across samples and to keep conditions constant (e.g., avoid inconsistent freeze thaws). The most widely accepted protocols include immediate freezing either on dry ice or in liquid nitrogen, followed by storage at −80° C (Consortium, 2012a). However, this approach is not always practical, particularly for stool samples, which may be collected at home and then stored for an indeterminate time in home freezers. Whether samples must be immediately frozen (and at what temperature) or whether they can withstand a period at room temperature remains controversial. However, several studies have assessed the effects of storage conditions on the composition of the microbiota by 16S rRNA gene sequencing, and their results can help to guide a storage plan based on which aspects of the microbiota are impacted (for instance, for fecal samples see Carroll et al., 2012, Lauber et al., 2010, and Wu et al., 2010; for vaginal samples, see Bai et al., 2012). Generally, these studies report that the effects of short-term storage conditions on diversity and structure of the communities are surprisingly small. Lauber and colleagues recently showed that storage for 2 weeks at temperatures ranging from −80° C to 20° C did not significantly affect patterns of between-sample diversity or the abundance of major taxa (Lauber et al., 2010). Note, however, that the number of freeze-thaw cycles has been reported to have an effect on the composition of the microbial community (Sergeant et al., 2012) and should be minimized. Studies incorporating metabolomics or metatranscriptomics likely have more stringent requirements, and the use of RNA stabilizers is still being evaluated. If samples do experience big environmental fluctuations, it is important to record these events so that the effect on the data can be assessed using statistical modeling.
Long-term storage at −80° C is currently the norm, but as studies grow in size and number, repeated acquisition of ultra-low freezers is becoming a burden. Another long-term storage approach involves the lyophilization of samples and storage at room temperature (Koren et al., 2012). Again, although storage conditions may not affect the outcome of DNA-based studies (16S rRNA PCR amplicon and metagenomic sequencing), RNA-based studies may require different handling.
DNA Protocols
DNA Extraction and Amplification
Different DNA extraction protocols can result in different diversity profiles because some cell types resist common mechanical or chemical lysis methods (Salonen et al., 2010; Smith et al., 2011). However, most studies (for example, the HMP) use widely employed protocols and/or commercially available kits specific to the sample type (e.g., blood versus stool). Extraction protocols should be employed consistently within a study, and the wide use of commercial kits increases the possibility for consistency between independent studies. For samples from which very little DNA can be extracted, DNA amplification may be necessary. The whole-genome amplification methods MDA (multiple displacement amplification; Dean et al., 2002) and MALBAC (multiple annealing and looping-based amplification cycles; Zong et al., 2012) are generally preferred, as they are best able to maintain the diversity of DNA molecules present in the original sample. Moreover, both of these methods are suitable for amplification from single cells (Lasken, 2007; Zong et al., 2012).
Introduced Contaminants
Contamination issues are particularly important for samples with low microbial DNA concentrations. This concern was noted early in microbial ecology studies: extreme environments with low biomass were thought to contain similar microbes until the realization was made that they were common lab contaminants (Tanner et al., 1998). Testing commercial kits for contamination is particularly important for studies using 16S rRNA gene amplification, which is highly sensitive to low levels of bacterial DNA contamination. Testing involves running a blank extraction control through DNA extraction and PCR prior to working with samples. We have found that all commercial reagents, from extraction kits to primers and polymerases, may be contaminated with microbial DNA, and levels can differ between batches from the same vendor. Within a lab, PCR amplicons can become contaminants, and many labs spatially separate pre- and post-PCR steps.
If the negative extraction controls result in PCR products, albeit less product than the samples, it may seem tempting to “sequence the negative control” and subtract those sequences computationally. This process may seem like the sole recourse if the extraction blank yields PCR product and the sample has been used up. We encountered this issue in a study of the microbial diversity of atheromas obtained from patients. One surprise was that the extraction control replicates had very different diversity profiles (O.K., R.K., and R.E.L., unpublished data). Removal of sequences matching those in the negative controls had a major impact on the study results and interpretation (O.K., R.K., and R.E.L., unpublished data), undermining our confidence in the data. We started the study over completely with new clinical samples and clean blanks (Koren et al., 2011).
PCR
Selection of 16S Primers
In studies using 16S rRNA gene sequencing, the choice of primer set depends on a number of factors, including compatibility with previous studies and the specificities of the primers. For instance, the bacterial 27F/338R primer set is biased against the amplification of bifidobacterial 16S rRNA genes; the 515F/806R primer set amplifies sequences from both Bacteria and Archaea (Kuczynski et al., 2012). Because the phylogenetic information varies along the length of the 16S rRNA gene (i.e., highly conserved areas are the least informative), the choice of region is more important than the length of the amplicon (Soergel et al., 2012). The Earth Microbiome Project has PCR protocol information for use on Illumina instruments and lists barcoded primer sequences (http://www.earthmicrobiome.org). In some cases, sequence lengths obtained from paired-end reads (particularly Illumina MiSeq) may be long enough for overlap and can be joined into a single longer sequence (a “contig”). If there is little or no overlap between sequence reads, both can be used in the downstream analyses, with at best modest performance enhancements over using just one of the two sets (Soergel et al., 2012; Werner et al., 2012b). In other words, if 16S rRNA gene diversity data consist of paired-end Illumina data that cannot be joined, for many applications it is acceptable to use one of the reads (often the first read is of higher quality than the second).
Vetted Barcoded Primers Sets Are Available
Barcoded primers have been designed and optimized for multiplexing samples on the Roche 454 (http://www.hmpdacc.org/tools_protocols/tools_protocols.php) and on Illumina platforms (http://www.earthmicrobiome.org/emp-standard-protocols/16s/). Error-correcting barcodes have the added advantage of reducing the possibility that a sequence will be assigned to the wrong sample (Hamady et al., 2008). We have found through repeated use of barcode sets that the specific barcode sequence used does not influence amplicon yield or diversity.
Effect of PCR Conditions on Results
Low DNA template concentration and high PCR cycle number are known to introduce PCR bias. To minimize bias, it is common practice to perform and pool multiple (i.e., triplicate) PCRs for each sample, to minimize PCR cycle number, and to use a standard but relatively high DNA template concentration across samples (Acinas et al., 2005; Aird et al., 2011; Sipos et al., 2007). Use of error-correcting polymerases may reduce PCR error, and longer annealing times can also assist in reducing chimera formation (Haas et al., 2011) (see below). Use of PCR enhancers such as betaine or BSA can improve yields, and pre-incubations with RNases can facilitate otherwise difficult reactions. In systems with large amounts of host contamination, such as plant endophytic microbiomes, approaches such as PCR clamps can help to reduce unwanted plant plastid contamination (Lundberg et al., 2013). The effects of these protocol variations on the inferred microbial community composition have not been systematically evaluated. Best practice is, of course, to choose a protocol and apply it consistently across all samples in a study and even between studies to facilitate later comparisons.
Software for Detecting Sequence Chimeras
PCR errors are also common and are difficult to detect. Chimeras, which are caused by incomplete template extension and give the appearance of recombination among disparate sequences (Haas et al., 2011), can cause inflated diversity. There are several software options for chimera filtering, including ChimeraSlayer (Haas et al., 2011), UCHIME (Edgar et al., 2011), DECIPHER (Wright et al., 2012), and Perseus (Quince et al., 2011). These different methods often disagree with one another. Noise introduced by PCR and sequencing errors has the greatest impact on alpha diversity but generally has very little effect on beta diversity (Ley et al., 2008).
16S rRNA Gene Sequence Data Analysis
Bacterial and archaeal 16S rRNA gene sequence data from complex microbial communities present bioinformatical, statistical, and computational challenges. Box 2 provides a list of software options and computational requirements for 16S rRNA gene-sequencing data. The most widely used software packages are QIIME (Caporaso et al., 2010b) (http://www.qiime.org) and mothur (Schloss et al., 2009) (http://www.mothur.org). Both packages are open source and have online tutorials and forums. We focus our discussion on the QIIME package (as several of the authors of this Primer are developers of QIIME), but many of the themes discussed here are common to both software packages.
Box 2. Software for Analysis of 16S rRNA Amplicon Data.
Several software packages are available for the analysis of sequenced amplicons. The most commonly used are QIIME (Caporaso et al., 2010b), RDP (Cole et al., 2009), mothur (Schloss et al., 2009), and VAMPS (http://vamps.mbl.edu/). RDP and VAMPS are both web-based tools, whereas QIIME and mothur are primarily accessed through command-line interfaces. Galaxy (Blankenberg et al., 2010; Giardine et al., 2005; Goecks et al., 2010) and CloVR (Angiuoli et al., 2011) wrappers have been developed to enable access to these tools via the web, and QIIME is also web accessible through the IPython Notebook (Caporaso et al., 2012). We focus our discussion of computational resources on QIIME because several of the authors of this Primer have development and leadership roles in the QIIME project. QIIME is implemented as a collection of command-line scripts designed to take users from raw sequence data and sample metadata through publication-quality graphics and statistics. Some of these scripts are primarily wrappers of one or more other software packages, such as uclust (Edgar, 2010), RDP classifier (Cole et al., 2009), muscle (Edgar, 2004), and PyNAST (Caporaso et al., 2010a). Other QIIME scripts directly implement statistical tests, diversity estimators, and data visualization tools. QIIME can be run on systems ranging from personal computers to computing clusters and clouds. Native installation is supported on Linux and Mac OS X; installation via Oracle Virtual-Box is supported on Linux, Mac OS X, and Windows; and installation via MacQIIME is supported on Mac OS X. Additionally, we officially support use of QIIME on the commercial Amazon Web Services (AWS) Elastic Compute Cloud (EC2) and developers of academic computing clouds including iPlant (Goff et al., 2011) and Magellan (https://www.nersc.gov/research-and-development/cloud-computing/). AWS is an excellent resource for users without their own computing hardware, and tools like StarCluster (http://star.mit.edu/cluster/) facilitate the usage of cluster computing environments in the cloud.
Data Filtering and Normalization
Quality Filtering
Marker gene (i.e., 16S rRNA gene) analysis generally begins with demultiplexing of sequence reads, whereby each sequence is assigned to its sample of origin based on the barcode. For computational efficiency, quality filtering is also typically applied at this stage. Reads not matching any barcode are discarded, as are sequences that fail to meet minimum quality thresholds. These quality thresholds are sequencing-platform specific and may be based on features, including quality scores provided by the sequencing instrument, read length, and the presence of ambiguous base calls. Denoising was developed for reducing sequencing errors on the 454 platform and does not apply to Illumina data (Reeder and Knight, 2010). We recently investigated Illumina quality-filtering parameters in detail, and the recommendations from this study (Bokulich et al., 2013a) have been implemented in QIIME. Additional quality filtering can occur downstream in data analysis; for instance, sequences observed only one time or only in a single sample may be considered artifacts and are often discarded. It is rare, though not impossible, for the same errors to be independently generated in different samples (Lahr and Katz, 2009).
Uneven Sequence Counts across Samples
The number of sequences obtained in a sequencing run can vary across samples for technical rather than biological reasons, and these sequencing depth artifacts can affect diversity estimates. One approach to account for variable sequencing depth is to use frequencies of OTUs (operational taxonomic units, described below) within samples (i.e., to normalize by total sample sequence count). We recommend against this approach, as we have found that it is subject to statistical pitfalls and can lead to samples clustering by sequencing depth (Friedman and Alm, 2012; C. Lozupone, J.G.C., and R.K., unpublished data). In a second approach termed rarefaction, equal numbers of sequences are randomly selected from each sample. The number of sequences drawn is usually the sequence count of the sample with the smallest acceptable number of sequences. This sequence count number should reflect a balance between retaining as many sequences as possible without excluding too many low-sequence samples. A major disadvantage of rarefaction is that valuable data from high-sequence count samples are discarded. Thus, rarefaction can lead to a more conservative view of the abundances of rarer taxa across samples. Additionally, rarefaction has recently been shown to introduce errors in analyses, and alternatives to rarefaction have been proposed (McMurdie and Holmes, 2013). Rarefaction is not necessary when OTU abundances are modeled as response variables in statistical models: total sequence counts can be retained and used as a factor in multivariate models (see below).
Identifying the Microbial Groups: OTU Analysis
Approaches to “Picking OTUs.”
After quality filtering, sequences are clustered into operational taxonomic units (OTUs; sometimes referred to as phylotypes), which provide a working name for groups of related bacteria. OTUs are based on sequence identity (%ID), and various thresholds of sequence identity are used to represent different taxonomic levels (e.g., 97% ID for species, 95% for genera). These taxonomic thresholds are known to be very rough estimates: the degree of sequence variability depends on the region of the 16S rRNA gene sequenced, the length of the amplicon, and the specific taxa in question. A sequence identity of 97% is most often used to denote bacterial “species” despite the fact that there is no rigorous species concept for bacteria. Nonetheless, these OTU naming conventions are useful because they have become the shared vocabulary used to discuss sequence-based observations.
Types of OTU-Picking Algorithms
The specific OTU-picking algorithm used can have a major impact on downstream findings and interpretations of the data. OTU clustering algorithms fall into three categories: de novo, closed reference, and open reference. In de novo OTU picking, sequences are clustered into OTUs, without any external reference sequences (Schloss and Handelsman, 2005). In contrast, closed-OTU picking uses a reference sequence database, and sample sequences that fail to match the reference sequence database are discarded. Open-reference OTU picking is a two-step process consisting of first closed-reference OTU picking followed by de novo clustering of sequences that fail to match to the reference database. The pros and cons of these three methods are detailed here (http://qiime.org/tutorials/otu_picking.html). We recommend open-reference OTU picking because it retains all sequence data, though there are circumstances for which this method is not applicable. For example, when combining sequence data from different regions of the 16S rRNA gene, a closed-reference OTU-picking approach must be used because sequences from different regions of the same 16S rRNA gene would otherwise cluster into different de novo OTUs.
In reference-based OTU picking, sequences are clustered against a reference database such as Greengenes (McDonald et al., 2012), Ribosomal Database Project (RDP) (Cole et al., 2009), or SILVA (Quast et al., 2013). Note that these databases are periodically updated, and the taxonomic information can change between versions. The human gut microbiota are well represented in the databases compared to other sample types. We have found that ~5% of the human gut sequences and up to 50% of soil microbial community sequences might be discarded in closed-reference OTU picking due to failure to match to a database sequence (Werner et al., 2012a). Additionally, the mouse gut microbiota are less well represented in databases compared to human gut microbiota. As more sequences are added to the databases, matching efficiencies are improving across sample types.
Binning Sequences by Taxonomy versus %ID
OTU-picking algorithms followed by taxonomic assignment of representative OTU sequences yield lists of OTUs with taxonomic labels. Many OTUs will lack a complete taxonomy label; for example, the classification might include a family level categorization but might lack genus or species categorization. Incomplete taxonomy can result from either a lack of confidence in where the OTU fits in the phylogeny (i.e., several matches are equally likely and there is no consensus) or from matching to a branch in the phylogeny that lacks taxonomic information. There is a temptation to emphasize results for taxa that have associated genus/species names. However, OTUs without genus/species information are frequently both more abundant and more representative of total diversity than are OTUs with genus/species names (Werner et al., 2012a). OTUs with genus/species information are more likely to include a reference strain that is cultured, and these organisms are not randomly distributed across the phylogeny. Some types of Bacteria, such as Proteobacteria, lend themselves willingly to culturing, whereas the majority of phyla do not.
There are two approaches to assessing the abundances of higher-level taxonomic groups, such as orders or families: summing the sequences for all OTUs belonging to the group of interest (collapsing taxonomies) or picking OTUs at a lower %ID. As %ID thresholds do not necessarily map onto the taxonomy well, these can, in principle, yield different results. A word of caution about the genus level: members of the same bacterial genus are not necessarily each other’s closest relatives. For example, the genus Clostridium is found in different bacterial families. Thus, summing sequence counts for OTUs assigned to the genus “Clostridium” will have the effect of combining sequences for potentially very distantly related organisms and will yield a meaningless category. Clostridium species are in the process of being reclassified into new genera (e.g., Blautia). However, until the tree is fully curated, the phylogenetics of organisms assigned common genus names (another example is Eubacterium) should be carefully checked before combining sequence sets. In addition, all OTUs that have no genus designation will be collapsed into a “no-name” genus, which could be an arbitrary mix of OTUs from across their family. Because of these issues, diversity calculations should be performed before collapsing data by taxonomy. We strongly recommend against collapsing OTUs at the genus level unless the study addresses specific known monophyletic genera, i.e., genera in which all species are each other’s close relatives.
Microbiota Diversity Analysis
Microbiome diversity is typically described in terms of within (i.e., alpha) and between samples (i.e., beta) diversities. Methods for analyzing alpha and beta diversity (see Box 3) have been discussed at length in various reviews (Kuczynski et al., 2010; Lozupone and Knight, 2008). Here, we focus instead on the visualization, clustering, and modeling of the diversity in microbiota data.
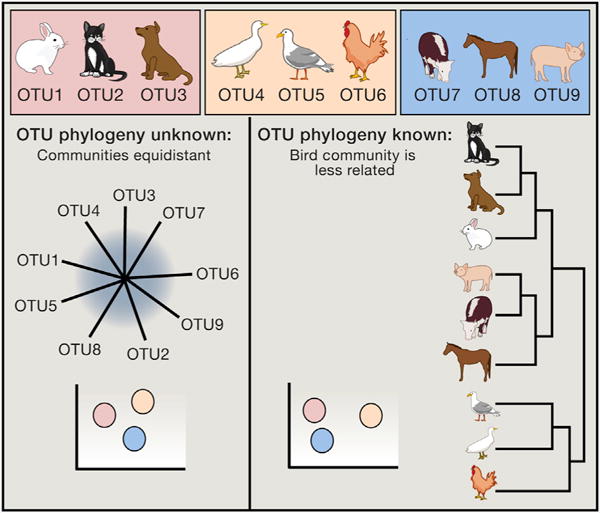
Box 3. Comparing Species Composition between Samples.
Beta diversity metrics provide a measure of the degree to which samples differ from one another and can reveal aspects of microbial ecology that are not apparent from looking at the composition of individual samples. Generally, beta diversity metrics are remarkably robust to issues such as low sequence counts and noise. Beta diversity metrics can be grouped in a couple of different ways. First, they can be quantitative (using sequence abundance, e.g., Bray-Curtis or weighted UniFrac) or qualitative (considering only presence-absence of sequences, e.g., binary Jaccard or unweighted UniFrac). Second, they can be phylogeny based (the UniFrac metrics) or not (Bray-Curtis, etc.). The figure shows an explanation of why phylogeny-based metrics such as UniFrac can outperform other metrics in community comparisons. The top three boxes represent communities, each with three taxa, shown as OTUs and as pictures of animals. When the phylogenetic relationship of the OTUs is not known (see star phylogeny in lower-left), the three communities appear equally unrelated (lower-left PCoA plot). When their pylogeny is taken into account (i.e., mammals are more related to each other than birds in the phylogeny on the right), then the orange (bird) community is shown to be more distant in the in PCoA plot (lower-right).
Data Visualization: Principal Coordinates Analysis
Ordination techniques, such as principal coordinates analysis (PCoA), reduce the dimensionality of microbiome data sets so that a summary of the beta diversity relationships can be visualized in two- or three-dimensional scatterplots. The principal coordinates (PCs), each of which explains a certain fraction of the variability (formally called inertia), observed in the data set are plotted to create a visual representation of the microbial community compositional differences among samples (Figure 3). Observations based on PCoA plots can be substantiated with statistical analyses that assess the clusters (see Figure 3 legend).
Figure 3. Principal Coordinates Analysis and Classification Methods.
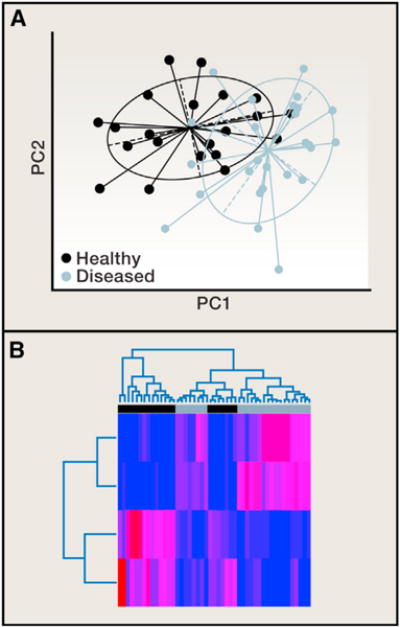
(A) Principal coordinates (PCs) from a principal coordinates analysis (PCoA) are plotted against each other to summarize the microbial community compositional differences between samples. Each point represents a single sample, and the distance between points represents how compositionally different the samples are from one another. The points are colored by health state, showing a clear difference in the microbial community composition between diseased (green) and healthy (purple).
(B) Classification methods can be used to determine which OTUs discriminate between the healthy and diseased groups, and a heatmap can be used to visualize over/under representation of these OTUs in the groups. In this example, the abundances of the four discriminatory OTUs (rows) are colored from low abundance (blue) to high abundance (red) in the 47 samples (columns). Both the PCoA plot and the sample dendrogram in the heatmap show that the separation between disease and health states is not perfect. There is some overlap in the composition of these samples, though the placement of points in the PCoA plot is far from random. This observation should be supported with statistical analysis. For example, a Monte Carlo two-sample t test, comparing the distribution of within-group distances to the distribution of between-group distances applied to these data tells us that this clustering pattern is statistically significant.
PCs Explaining a Low Percentage of Variation Can Yield Biological Insights
When the individual PCs explain small fractions of the total variation, it may nevertheless be possible to infer the factors driving the separation of samples along the PC. For example, Figure 4 illustrates an example of PC1 relating to the OTU abundances across samples (see Koren et al., 2012 for a real example). In many cases, despite having a low percentage of the variance explained, biological patterns are still revealed (Kuczynski et al., 2010).
Figure 4. Use Caution when Applying Unsupervised Classification to Data Gradients.
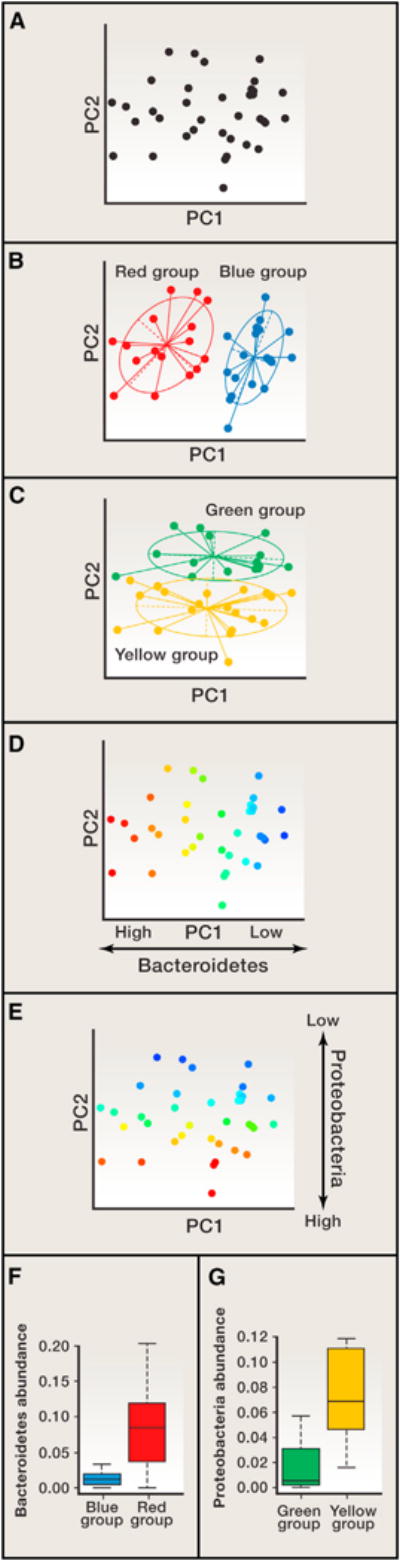
(A–C) In this simulated microbiome data set, a principal coordinates analysis (PCoA) was performed, and the first two principal coordinates, PC1 and PC2, are plotted. The exact same set of points is shown in panels (A–E) but is colored differently. In (A), samples are all colored black to show that they form gradients along PCs 1 and 2. In (B) and (C), two sets of clusters were designated by bisecting the spread of samples. In (B), half of the samples form the red cluster, and the second half form the Blue cluster along PC1. In (C), half of the samples are in the Green cluster, and the second half form the Yellow cluster along PC2. In (B) and (C), starplots display inferred clusters; this display can give the misleading impression of distinct clusters (see A; the data structure consists of gradients, not distinct clusters).
(D–G) In (D) and (E), the samples are colored according to the abundances of the taxa that drive their separation along PCs 1 and 2. (D) The abundance of sequences belonging to the Bacteroidetes phylum drives the spread of samples along PC1; (E) abundances of Proteobacteria in the samples drive their spread along PC2. When the relative abundances for these phyla in samples are averaged (F and G), it is apparent that the Blue samples, which are at the “low end” of the Bacteroidetes gradient, have lower means than the Red samples, which are at the high end (F). Similarly, because the Yellow/Green samples are spread along PC2 according to their abundance of Proteobacteria, these two groups will also exhibit different mean abundances (G). Therefore, plotting mean values of the abundances of taxa that drive the gradients in the PCoA plots does not constitute a validation of the PCoA patterns.
Classification and Clustering
Classification methods can be supervised or unsupervised depending on whether categorical metadata (i.e., discrete sample information like diet, genotype, diseased, etc.) is used. Supervised classification methods require knowledge of which samples belong to which group, whereas unsupervised methods do not. Both methods are useful, and the choice of method depends on the information available about the samples and the question being asked.
Supervised classification methods can be used to determine which taxa differ between predefined groups of samples (e.g., diseased versus healthy) and to build models that use these discriminatory taxa to predict the classification of a new sample. Examples of commonly employed supervised classification methods are described in Knights et al. (2011).
Unsupervised classification (clustering), on the other hand, does not make use of any prior knowledge about the samples. Samples are categorized into clusters based on the abundances of specific taxa. A between-sample distance metric, such as UniFrac or Bray-Curtis, is used to generate these clusters. Clustering approaches can differ based on whether the number of clusters is preset or optimized to produce maximally distinct clusters. The specific methods used in clustering (both distance metrics and clustering algorithms) can affect the outcome and the interpretation of clustering analyses (Koren et al., 2013). For this reason, it is important to perform clustering in several different ways to ensure that the existence of clusters is not dependent on just one set of parameters.
Koren et al. (2013) explored the sensitivities of some of these methods using data sets from the HMP and MetaHIT. As an example, we illustrate in Figure 4 how some of these approaches assume that clusters exist even if the structure of the data set is a gradient. These approaches will delineate distinct clusters regardless of the fact that none actually exist. We have recommended that several approaches to clustering be used in parallel (Koren et al., 2013). If distinct clusters are identified as robust using several methods, the enrichment of predefined sample classes within the clusters can be explored, as well as the biological basis of the clustering. If the samples do appear to form distinct clusters, these can then be labeled as classes, and one can search for OTUs that discriminate these classes.
Clustering of human gut microbiome samples led to the idea of enterotypes, or distinct compositions of the gut microbial communities in different individuals (Arumugam et al., 2011). As data sets have expanded, these discrete categories have emerged as the extremes of continuous gradients, with some configurations being more prevalent than others in certain populations (Koren et al., 2013). Within any given study, it may be possible to group samples into discrete types based on the community composition, but care should be taken to determine whether a discrete clustering pattern is present or whether more data would reveal a gradient or other alternative structure in the data.
Modeling
The data that can be extracted from a microbiome analysis take many forms (e.g., alpha diversity measures, PCs of the beta diversity PCoA, and the abundances of OTUs) that can be used as response variables in statistical models. Abundances of OTUs, in particular, are seldom normally distributed because many samples will have zero counts for rare OTUs. Such zero-inflated, sparse data sets trigger the need for either transformations or nonparametric statistics. Note that a single transformation approach may not work equally well for all OTUs. In addition, care must be taken to avoid multiple comparison issues, as there are generally thousands of OTUs being tested for association with a few states (e.g., healthy or diseased), so many spurious associations are expected. Consultation with a statistician is highly recommended at both the experimental design phase and during the analysis phase.
Recent large-scale analyses provide examples of how to apply multivariate statistics to 16S rRNA sequence data (Bokulich et al., 2013b; Lundberg et al., 2012; McMurdie and Holmes, 2013; Peiffer et al., 2013). The study of Benson et al. (2010) provides an example of how variation induced by the maternal effect can be controlled for statistically. In human studies, covariates such as gender, age, and body mass index can be included in models. Technical sources of variation in microbiome data are also very important to control for statistically. These include, for example, the specific run of a sequencing instrument in studies that include multiple runs. We have found that the Roche 454 instrument in particular can introduce variation from run to run, which can be controlled for statistically by including the run information for each sample in the models (and by randomizing samples across runs). Note that, within the same analysis, we strongly recommend against incorporating data from different instruments (e.g., Illumina and 454 platforms). Other sources of variation include factors such as the date (or season) at which samples were collected, the time maintained in freezers, and the identity of the person handling the samples. When OTU abundances are the response variable in the models, sequence count per sample can be included as a covariate. This alternative approach to rarefying sequence data increases power by retaining the full data set.
Concluding a Study: Standardized Databases
As the microbiome field matures and technology further develops, improvements and standardization of microbiome research are expected. Each new microbiome study pushes forward the guidelines and requirements to conduct and publish a microbiome study. It is therefore imperative that investigators stay abreast of the current practices in the microbiome field. For all projects (16S rRNA amplicon, metagenomics, or metatranscriptomics), an important step is the submission of both the sequence files and the metadata (covariates) associated with each sample to public databases. Several database initiatives exist for this purpose, including QIIME, MG-RAST, and NCBI’s and EBI’s respective short-read archives (SRA). In general, journals and funding agencies require deposition of data into INSDC (the International Nucleotide Sequence Database Collaboration, which encompasses NCBI, EBI and DDBJ). Unstructured archival resources such as Dryad allow public hosting of large data sets but do not require standardization of format; this makes data deposition easy but data reuse essentially impossible. The QIIME database is not intended as an archival repository of data but does provide a number of tools for rapidly comparing data from different microbial communities. Given the growing number of independent microbiome studies, there is great interest in being able to combine data from multiple studies to increase power through meta-analysis (Koren et al., 2013). In addition to standardizing protocols for sample processing and sequencing, meta-analysis will only be possible if metadata collection and reporting is also standardized. To achieve this goal, the Genomic Standard Consortium developed the minimum information about a marker gene sequence (MIMARKS) (Yilmaz et al., 2011), part of the MIxS family of standards that allows description of a wide range of omics data sets.
Final Remarks
The analysis of microbial community diversity is rapidly becoming a component of a vast array of different research programs, ranging from neurobiology to nutrition. We focused this Primer on 16S rRNA gene diversity studies because it is an entry point into the field. However, increasingly powerful algorithms coupled with more accessible sequencing strategies make whole microbial genome reconstructions from metagenomic samples a growing reality and hint at the future of research in the field. For example, metagenomic analysis was recently used to observe characteristics of temporal succession of the gut microbiome in preterm infants and proved capable of identifying relevant strains and functions of putative bacterial pathogens (Morowitz et al., 2011). Similarly, applying these tools allowed reconstruction of genomes from a new uncultivated phylum, Melainabacteria, that is common in the human gut (Di Rienzi et al., 2013).
Regardless of the methodologies employed to study the microbiome, many fundamentals remain the same. These recommended practices distill down to the following: (1) carefully design the study to reduce confounding factors, (2) apply consistent experimental and analytic methods throughout, (3) keep good records so that all possible metadata can be used in statistical models, (4) match the software and the statistical toolkits to the data sets generated and keep detailed records of the bioinformatics steps of the analysis (including versions of software and lists of commands), and (5) deposit all data in public databases using standard formats. This last recommendation incorporates each individual researcher and study into the larger community and allows a second generation of analyses that mine the databases for larger trends.
Acknowledgments
We thank the organizers of the 2012 Keystone Conference on the Microbiome for inviting us to conduct a Workshop on the microbiome, which served as the basis for this Primer.
References
- Abarenkov K, Henrik Nilsson R, Larsson KH, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, et al. The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol. 2010;186:281–285. doi: 10.1111/j.1469-8137.2009.03160.x. [DOI] [PubMed] [Google Scholar]
- Acinas SG, Sarma-Rupavtarm R, Klepac-Ceraj V, Polz MF. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl Environ Microbiol. 2005;71:8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird D, Ross MG, Chen WS, Danielsson M, Fennell T, Russ C, Jaffe DB, Nusbaum C, Gnirke A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angiuoli SV, Matalka M, Gussman A, Galens K, Vangala M, Riley DR, Arze C, White JR, White O, Fricke WF. CloVR: a virtual machine for automated and portable sequence analysis from the desktop using cloud computing. BMC Bioinformatics. 2011;12:356. doi: 10.1186/1471-2105-12-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Gajer P, Nandy M, Ma B, Yang H, Sakamoto J, Blanchard MH, Ravel J, Brotman RM. Comparison of storage conditions for human vaginal microbiome studies. PLoS ONE. 2012;7:e36934. doi: 10.1371/journal.pone.0036934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;19:11–21. doi: 10.1002/0471142727.mb1910s89. Unit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. 2013a;10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich NA, Thorngate JH, Richardson PM, Mills DA. Microbial biogeography of wine grapes is conditioned by cultivar, vintage and climate. Proc Natl Acad Sci USA. 2013b doi: 10.1073/pnas.1317377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010a;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010b;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Knight R, Kelley ST. Host-associated and free-living phage communities differ profoundly in phylogenetic composition. PLoS ONE. 2011a;6:e16900. doi: 10.1371/journal.pone.0016900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Costello EK, Berg-Lyons D, Gonzalez A, Stombaugh J, Knights D, Gajer P, Ravel J, Fierer N, et al. Moving pictures of the human microbiome. Genome Biol. 2011b;12:R50. doi: 10.1186/gb-2011-12-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. Ultrahigh-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll IM, Ringel-Kulka T, Siddle JP, Klaenhammer TR, Ringel Y. Characterization of the fecal microbiota using high-throughput sequencing reveals a stable microbial community during storage. PLoS ONE. 2012;7:e46953. doi: 10.1371/journal.pone.0046953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, González A, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12:139–152. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Aitken JD, Gewirtz AT, Vijay-Kumar M. Gut microbiota drives metabolic disease in immunologically altered mice. Adv Immunol. 2012;116:93–112. doi: 10.1016/B978-0-12-394300-2.00003-X. [DOI] [PubMed] [Google Scholar]
- Cho I, Yamanishi S, Cox L, Methé BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488:621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol. 2012;10:735–742. doi: 10.1038/nrmicro2876. [DOI] [PubMed] [Google Scholar]
- HMPC (Human Microbiome Project Consortium) A framework for human microbiome research. Nature. 2012a;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, H.M.P.; Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012b;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336:1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P, Sun Z, Zong Q, Du Y, Du J, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzi SC, Sharon I, Wrighton KC, Koren O, Hug LA, Thomas BC, Goodrich JK, Bell JT, Spector TD, Banfield JF, Ley RE. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. Elife. 2013;2:e01102. doi: 10.7554/eLife.01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8:e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friswell MK, Gika H, Stratford IJ, Theodoridis G, Telfer B, Wilson ID, McBain AJ. Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice. PLoS ONE. 2010;5:e8584. doi: 10.1371/journal.pone.0008584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J, Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SA, Vaughn M, McKay S, Lyons E, Stapleton AE, Gessler D, Matasci N, Wang L, Hanlon M, Lenards A, et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Front Plant Sci. 2011;2:34. doi: 10.3389/fpls.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley SA, Thackray LB, Zhao G, Presti R, Miller AD, Droit L, Abbink P, Maxfield LF, Kambal A, Duan E, et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell. 2012;151:253–266. doi: 10.1016/j.cell.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz BL, Deng L, Poulos BT, Sullivan MB. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ Microbiol. 2013;15:1428–1440. doi: 10.1111/j.1462-2920.2012.02836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. 2012;336:1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack PJ, Amos-Ritchie RN, Reverter A, Palacios G, Quan PL, Jabado O, Briese T, Lipkin WI, Boyle DB. Microarray-based detection of viruses causing vesicular or vesicular-like lesions in livestock animals. Vet Microbiol. 2009;133:145–153. doi: 10.1016/j.vetmic.2008.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson F, Tremaroli V, Nielsen J, Bäckhed F. Assessing the human gut microbiota in metabolic diseases. Diabetes. 2013;62:3341–3349. doi: 10.2337/db13-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, Hugenholtz P, van der Lelie D, Meyer F, Stevens R, et al. Unlocking the potential of metagenomics through replicated experimental design. Nat Biotechnol. 2012;30:513–520. doi: 10.1038/nbt.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights D, Costello EK, Knight R. Supervised classification of human microbiota. FEMS Microbiol Rev. 2011;35:343–359. doi: 10.1111/j.1574-6976.2010.00251.x. [DOI] [PubMed] [Google Scholar]
- Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62:1505–1510. doi: 10.1136/gutjnl-2012-303954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, Bäckhed F. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4592–4598. doi: 10.1073/pnas.1011383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Bäckhed HK, Gonzalez A, Werner JJ, Angenent LT, Knight R, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150:470–480. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE. A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput Biol. 2013;9:e1002863. doi: 10.1371/journal.pcbi.1002863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J, Liu Z, Lozupone C, McDonald D, Fierer N, Knight R. Microbial community resemblance methods differ in their ability to detect biologically relevant patterns. Nat Methods. 2010;7:813–819. doi: 10.1038/nmeth.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski J, Lauber CL, Walters WA, Parfrey LW, Clemente JC, Gevers D, Knight R. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2012;13:47–58. doi: 10.1038/nrg3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahr DJ, Katz LA. Reducing the impact of PCR-mediated recombination in molecular evolution and environmental studies using a new-generation high-fidelity DNA polymerase. Biotechniques. 2009;47:857–866. doi: 10.2144/000113219. [DOI] [PubMed] [Google Scholar]
- Lasken RS. Single-cell genomic sequencing using Multiple Displacement Amplification. Curr Opin Microbiol. 2007;10:510–516. doi: 10.1016/j.mib.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Lauber CL, Zhou N, Gordon JI, Knight R, Fierer N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol Lett. 2010;307:80–86. doi: 10.1111/j.1574-6968.2010.01965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Clare S, Walker AW, Stares MD, Connor TR, Raisen C, Goulding D, Rad R, Schreiber F, Brandt C, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012;8:e1002995. doi: 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI. Evolution of mammals and their gut microbes. Science. 2008;320:1647–1651. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Knight R. Species divergence and the measurement of microbial diversity. FEMS Microbiol Rev. 2008;32:557–578. doi: 10.1111/j.1574-6976.2008.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, del Rio TG, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488:86–90. doi: 10.1038/nature11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, Dangl JL. Practical innovations for high-throughput amplicon sequencing. Nat Methods. 2013;10:999–1002. doi: 10.1038/nmeth.2634. [DOI] [PubMed] [Google Scholar]
- Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, Holmes S. Waste Not, Want Not: Why Rarefying Microbiome Data is Inadmissible. PLoS Comput Biol. 2013;10:e1003531. doi: 10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morowitz MJ, Denef VJ, Costello EK, Thomas BC, Poroyko V, Relman DA, Banfield JF. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc Natl Acad Sci USA. 2011;108:1128–1133. doi: 10.1073/pnas.1010992108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios G, Quan PL, Jabado OJ, Conlan S, Hirschberg DL, Liu Y, Zhai J, Renwick N, Hui J, Hegyi H, et al. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg Infect Dis. 2007;13:73–81. doi: 10.3201/eid1301.060837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer JA, Spor A, Koren O, Jin Z, Tringe SG, Dangl JL, Buckler ES, Ley RE. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc Natl Acad Sci USA. 2013;110:6548–6553. doi: 10.1073/pnas.1302837110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596. doi: 10.1093/nar/gks1219. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127:423–433. doi: 10.1016/j.cell.2006.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods. 2010;7:668–669. doi: 10.1038/nmeth0910-668b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen A, Nikkilä J, Jalanka-Tuovinen J, Immonen O, Rajilić-Stojanović M, Kekkonen RA, Palva A, de Vos WM. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods. 2010;81:127–134. doi: 10.1016/j.mimet.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol. 2005;71:1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant MJ, Constantinidou C, Cogan T, Penn CW, Pallen MJ. High-throughput sequencing of 16S rRNA gene amplicons: effects of extraction procedure, primer length and annealing temperature. PLoS ONE. 2012;7:e38094. doi: 10.1371/journal.pone.0038094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos R, Székely AJ, Palatinszky M, Révész S, Márialigeti K, Nikolausz M. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol Ecol. 2007;60:341–350. doi: 10.1111/j.1574-6941.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- Smith B, Li N, Andersen AS, Slotved HC, Krogfelt KA. Optimising bacterial DNA extraction from faecal samples: comparison of three methods. Open Microbiol J. 2011;5:14–17. doi: 10.2174/1874285801105010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MI, Yatsunenko T, Manary MJ, Trehan I, Mkakosya R, Cheng J, Kau AL, Rich SS, Concannon P, Mychaleckyj JC, et al. Gut microbiomes of Malawian twin pairs discordant for kwashiorkor. Science. 2013;339:548–554. doi: 10.1126/science.1229000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soergel DA, Dey N, Knight R, Brenner SE. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012;6:1440–1444. doi: 10.1038/ismej.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner MA, Goebel BM, Dojka MA, Pace NR. Specific ribosomal DNA sequences from diverse environmental settings correlate with experimental contaminants. Appl Environ Microbiol. 1998;64:3110–3113. doi: 10.1128/aem.64.8.3110-3113.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MR, Kamboj M, Pamer EG. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest. 2010;120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science. 2011;334:255–258. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, Angenent LT, Knight R, Ley RE. Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J. 2012a;6:94–103. doi: 10.1038/ismej.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner JJ, Zhou D, Caporaso JG, Knight R, Angenent LT. Comparison of Illumina paired-end and single-direction sequencing for microbial 16S rRNA gene amplicon surveys. ISME J. 2012b;6:1273–1276. doi: 10.1038/ismej.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright ES, Yilmaz LS, Noguera DR. DECIPHER, a search-based approach to chimera identification for 16S rRNA sequences. Appl Environ Microbiol. 2012;78:717–725. doi: 10.1128/AEM.06516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Lewis JD, Hoffmann C, Chen YY, Knight R, Bittinger K, Hwang J, Chen J, Berkowsky R, Nessel L, et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 2010;10:206. doi: 10.1186/1471-2180-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz P, Kottmann R, Field D, Knight R, Cole JR, Amaral-Zettler L, Gilbert JA, Karsch-Mizrachi I, Johnston A, Cochrane G, et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol. 2011;29:415–420. doi: 10.1038/nbt.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]