

Abstract
The urea-based homogeneous precipitation method was introduced in the preparation of monodisperse colloidal spheres for (Y0.95Eu0.05)2O3 red-emitting phosphors, and the processing window was defined. Particle size and shape are significantly affected by the ion concentration and the urea/RE3+ molar ratio R (RE3+=Y3++Eu3+). A low ion concentration is beneficial in forming monodisperse spheres and extending their formation domain. Increasing R results in a gradual change in the composition of spherical particles from the core-shell Eu(OH)CO3@Y(OH)CO3 structure to a homogeneous solid solution, thereby significantly lowering the calcination temperature at which precursors convert to oxides. Upon UV excitation into the charge-transfer band at 254 nm, the uniform phosphor spheres of (Y0.95Eu0.05)2O3 exhibit typical red emissions at 613 nm; the emission is stronger from larger particles mainly because of their smaller surface area. Both the luminescence intensity and quantum efficiency of the oxide phosphors increase with elevated calcination temperatures. The spherical shape and excellent dispersion of the precursor particles (∼450 nm in diameter) have been well retained after calcination at 1000 ○C for 4 h, and the resultant oxide phosphors exhibit external and internal quantum efficiencies of 50 and 82%, respectively.
Keywords: optical materials, powder processing, optical spectroscopy, colloidal monodisperse spheres
Introduction
Yttrium oxide doped with Eu3+ ions (Y2O3:Eu3+) is a well-known red-emitting phosphor that is widely used in fluorescent lamps, white-light-emitting diodes, plasma display panels, flat-panel displays, field emission displays, cathode-ray tubes and so on [1]. For successful application, phosphor particles must be non-aggregated and have fine size, narrow size distribution and spherical morphology. The spherical shape of the particles facilitates their dense packing and reduces light scattering from the surface. Spray pyrolysis is known to produce dispersed and spherical phosphor particles. However, the hollow and porous morphology and the wide size distribution may degrade the luminescence performance [2, 3]. Recently, spherical phosphor particles have been synthesized by the hydrothermal method, but with the drawbacks of aggregation and low yield [4].
Urea-based homogeneous precipitation (UBHP) is a promising technique that can produce monodisperse basic carbonate colloid spheres of rare-earth elements. It relies on the slow decomposition of urea at elevated temperatures (≥83 ○C), which slowly and homogeneously releases precipitating ligands (mainly OH− and CO32−) into the reaction system [5–12]. Systems containing one rare-earth element (RE) were systematically studied by Matijević and Hsu [5], Hsu et al [6], Aiken et al [7], Sordelet and Akinc [8] and Sordelet and Akinc [9] through UBHP processing. More recently, Li et al [10–12] have extended their compositions to Y/Gd, Y/Eu, Gd/Eu and even Y/Gd/Eu mixed systems. They also proposed using the monodisperse basic carbonate colloid spheres processed via UBHP as the precursors for luminescent oxides, which definitely widens the practical applications of these phosphors. This approach resulted not only in monodispersity of the phosphor spheres but also in markedly enhanced luminescence [11]. While the previous studies largely focused on the effect of RE doping and solvent on the particle morphology and formation mechanism under a fixed processing condition, the range of processing parameters was not well defined, although it is important for applications. This issue was addressed in the present work by widely varying the urea/RE3+ molar ratio R and the ion concentration. A low ion concentration was found to be beneficial to forming monodisperse spheres and extending their formation domain, thus allowing a highly reliable size-selective synthesis. Furthermore, an increase in the R can induce a gradual conversion of spherical particles from the core-shell Eu(OH)CO3@Y(OH)CO3 structure to a homogeneous solid solution, thereby significantly lowering the conversion temperature of precursors to oxides. The size and temperature dependences of luminescence from the resultant Y2O3:Eu3+ phosphors were also characterized. We consider that the outcomes of this work may widen the applications of Y2O3:Eu3+ red-emitting phosphors and are relevant to other phosphors.
Experimental details
Synthesis
We used Y(NO3)3·6H2O (99.99% pure, Kanto Chemical Co., Inc., Tokyo, Japan) and Eu(NO3)3·6H2O (99.95% pure, Kanto Chemical Co., Inc.) as yttrium and europium sources, respectively. The luminescence quenching concentration of Eu3+ in Y2O3 is known to be ∼5 at.%, and thus, in this work, the Y:Eu molar ratio was kept at 19:1. In a typical synthesis, an appropriate of urea (CO(NH2)2, >99% pure, Kanto Chemical Co., Inc.) was dissolved in a mixed nitrate solution to make a total volume of 2 litres, which was then homogenized under magnetic stirring at room temperature for 1 h. The mixed solution, contained in a beaker wrapped with aluminum foil, was heated on a hot plate to 90 ± 1 ○C within 60 min. After reacting at 90 ± 1 ○C for 2 h, the suspension was cooled naturally to ∼50 ○C and the resultant colloidal particles were collected via centrifugation. The wet precipitate was washed with distilled water three times to remove the byproducts, rinsed with absolute ethanol, and dried in air at 70 ○C for 24 h to yield a white powder for further characterization. (Y0.95Eu0.05)2O3 was obtained by calcining the precursors under flowing O2 gas (200 ml min−1) for 4 h at selected temperatures, which were reached using a heating rate of 5 ○C min−1. To monitor the composition evolution of the precursor particles, 30 ml of the colloidal suspension was regularly sampled after visible turbidity appeared in the originally clear solution. The sampled suspension was quenched with water and centrifuged to achieve solid/liquid separation. The clear solution obtained by the above method was used for further analysis. The synthesis parameters adopted in this work are summarized in table 1.
Table 1.
UBHP conditions for precursor synthesis.
| Sample | Initial urea | Initial Y3++Eu3+ | Ratio | Sum | Particle |
|---|---|---|---|---|---|
| ID | concentration | concentration | R | (urea+ Y3++Eu3+) | morphology |
| (mol l−1) | (mol l−1) | (mol l−1) | |||
| S1 | 0.03 | 0.015 | 2 | 0.045 | Spheres |
| S2 | 0.05 | 0.015 | 3.3 | 0.065 | Spheres |
| S3 | 0.10 | 0.015 | 6.7 | 0.115 | Spheres |
| S4 | 0.25 | 0.015 | 16.7 | 0.265 | Spheres |
| S5 | 0.5 | 0.015 | 33.3 | 0.515 | Spheres |
| S6 | 1 | 0.015 | 66.7 | 1.015 | Spheres |
| S7 | 2 | 0.015 | 133.3 | 2.015 | Spheres and plates |
| S8 | 0.5 | 0.030 | 16.7 | 0.530 | Aggregates |
| S9 | 0.5 | 0.020 | 25 | 0.520 | Spheres |
| S10 | 0.5 | 0.005 | 100 | 0.505 | Spheres |
| S11 | 0.5 | 0.002 | 250 | 0.502 | Spheres |
Characterization techniques
Phase identification was performed by x-ray diffraction (XRD) using a RINT 2200 V/PC diffractometer (Rigaku, Tokyo) operated at 40 kV/40 mA, nickel-filtered Cu Kα radiation and a scan speed of 1.0○ (2θ) per minute. Thermogravimetry (TG, Model Thermo Plus TG8120, Rigaku) analysis of the dried precursor was carried out in stagnant air at a heating rate of 10 ○C min−1. Morphologies of the products were observed by field emission scanning electron microscopy (FE-SEM, Model S-5000, Hitachi, Tokyo) at an acceleration voltage of 10 kV. The chemical composition of the precursor compound and reaction solution was determined by elemental analysis, with the Y3+ and Eu3+ contents measured by the inductively coupled plasma method (Model IRIS Advantage, Nippon Jarrell-Ash Co. Ltd, Kyoto, Japan). Photoluminescence/photoluminescence excitation (PL/PLE) and quantum efficiency were performed at room temperature using an FP-6500 fluorescence spectrophotometer (JASCO, Tokyo) equipped with a 150 W Xe lamp as the excitation source. The slit widths were set to 5 nm both in excitation and emission. The spectral response of the spectrophotometer was corrected in the range of 220–850 nm using a Rhodamine-B solution (5.5 g l−1 in ethylene glycol, for 220–600 nm range) and a standard light source unit (ECS-333, JASCO, for 350–850 nm) as the references. The external quantum efficiency (εex, total number of emitted photons divided by total number of excitation photons) and internal quantum efficiency (εin, total number of emitted photons divided by number of photons absorbed by the sample) of the phosphors were derived from the following equations [13] using the built-in analysis software:
Here, E(λ)/hv, R(λ)/hv and P(λ)/hv are the number of photons in the excitation, reflectance and emission spectra of the samples, respectively. The reflection spectrum of a Spectralon diffusive white standard was used for calibration.
Results and discussion
Monodisperse colloidal precursor spheres: processing window and formation mechanism
Monodisperse colloidal precursor spheres with a general chemical composition of RE(OH)CO3·nH2O (RE=Y and Eu) were produced by the UBHP method, which agrees with our previous results [10–12]. In this work, we defined the processing window and found that the urea/RE3+ molar ratio R and the ion concentration in the reaction system significantly affect the precursor morphologies.
Figure 1 shows FE-SEM particle morphologies of samples S1–S11. The particles obtained at the R between 2 and 67 are monodisperse spheres as shown in figures 1(a)–(f). The average particle size is proportional to R for R smaller than 7 (figures 1(a)–(c) and 2) and decreases for R between 7 and 67 (figures 1(c)–(f) and 2). At an even higher R of 133.3, the products were mixtures of spheres and plates (figure 1(g)). At the relatively low R (R<7), the precipitation process is quite slow and yields small particles after 2 h of reaction. The precipitation reaction is incomplete after 2 h for R<7, as determined by the elemental analysis of the solution (shown further in figure 5(a)). A higher R (still R<7) results in a faster precipitation at the already formed nuclei, and thus, larger particles, as shown in figures 1(a)–(c). However, the R also affects the number of the original nuclei, and the increased nuclei concentration at a higher R (R>7) contributes to smaller particles (figures 1(c)–(f) and 2). At the R of 133.3, the decomposition of excess urea generates more OH− ions, which change the medium from acidic to alkaline; thus, carbonate plates appear instead of carbonate spheres [7], as shown in figures 1(g) and 3. In our previous work, the sequential precipitation of Eu3+ and Y3+ was observed in the UBHP method, resulting in homogeneous nucleation of Eu(OH)CO3 in priority [10, 11]. When all the Eu3+ ions were contained in monodisperse carbonate spheres at the initial precipitation stage, a small amount of Y3+ ions still existed in the reaction solution. Thus, together with the change in the solution from acidic to alkaline due to the decomposition of excess urea, plates of Y2(CO3)3·2H2O (figure 3) appeared and coexisted with the already formed monodisperse spheres as shown in figure 1(g).
Figure 1.

FE-SEM images showing morphologies of samples S1–S11 (a–k), respectively.
Figure 2.
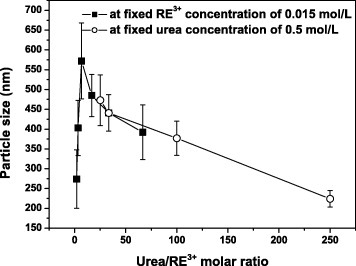
Average diameters of the colloidal precursor spheres plotted versus the urea/RE3+ molar ratio R.
Figure 5.
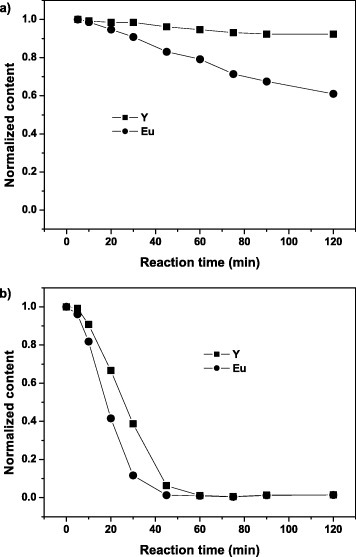
Normalized concentrations of Y and Eu versus reaction time for the reaction solution of samples S3 (a) and S6 (b).
Figure 3.
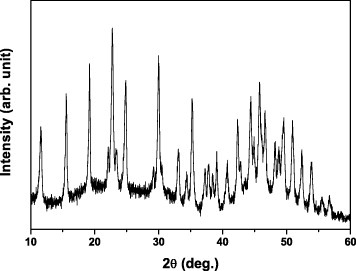
XRD pattern of sample S7. The diffraction peaks correspond to Y2(CO3)3·2H2O (JCPDS file No. 24-1419).
Systematic observations revealed that the particle morphology was affected not only by the R but also the ion concentration. Since the concentrations of urea and OH− and CO32− ions are closely related, it is plausible to assume that the combined concentrations of urea, Y3+ and Eu3+ ions stands for the total ion concentration (table 1). Comparison of samples S4 and S8 reveals that although they have the same R of 16.7, their morphologies differ owing to the dissimilar ion concentrations—monodisperse spheres for S4 and aggregates for S8 (table 1 and figures 1(d) and (h)). Higher ion concentrations result in aggregation, whereas at low concentrations, the products are monodisperse spheres even at a high R of 250. These results extend the processing window of monodisperse spheres, which are beneficial to applications; by adjusting the R, the particle size can be tuned from ∼220 to ∼500 nm (table 1, figures 1(i)–(k) and 2).
The spheres have a Eu content of 8.3 at.% at the R of 2, which is higher than the nominal value of 5 at.%; the Eu content steadily decreases with increasing R and reaches the nominal value at R=33.3. The basic carbonate colloidal spheres are formed via nucleation growth processes [5–12]. Previously, we found that the homogeneous nucleation of Eu(OH)CO3 occurs preferentially, owing to the smaller solubility product of Eu(OH)CO3, and then the precipitation of Y(OH)CO3 proceeds via heterogeneous nucleation on the already formed Eu(OH)CO3 nuclei [11]. Thus, the concentration of Eu increases and that of Y decreases from the particle surface to the core. At lower urea concentrations, more Eu and less Y are incorporated to the particle, as shown in figure 4.
Figure 4.
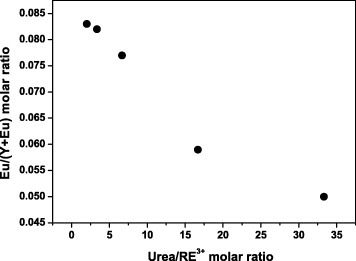
Dependence of the Eu/(Y+Eu) molar ratio on the urea/RE3+ molar ratio R.
To investigate the precipitation mechanism, regular sampling and analysis of the reaction solution at different reaction stages were performed for samples S3 and S6 (figure 5). After a 2 h reaction, there is still much Y3+ and Eu3+ in the reaction solution for S3 (figure 5(a)), indicating incomplete precipitation. However, complete precipitation was observed for S6 after 60 min, indicating that a higher R accelerates the precipitation. Since nucleation starts only when supersaturation S reaches the critical supersaturation S∗ [14], a range of S values reaching S∗ of Y3+ and Eu3+ exists in the precipitation process owing to the different solubility products of these ions. At a lower R, the release of carbonate ions arising from urea decomposition is slower, and thus, the precipitation rates of Y3+ and Eu3+ differ significantly as shown in figure 5(a). This might further result in core-shell Eu(OH)CO3@Y(OH)CO3 particles rather than homogeneous solid solutions. In contrast, a higher R is beneficial to the formation of a homogeneous solid solution owing to the fast and similar precipitation rates of Y3+ and Eu3+ ions (figure 5(b)). Since non-spherical particles, such as plates and aggregates (samples S7 and S8, figures 1(g) and (h)), form at high ion concentrations, a low ion concentration and a high R are both necessary for the formation of monodisperse spheres of homogeneous composition.
Characterization and size-dependent luminescence of oxide spheres
Figure 6 shows thermal decomposition behaviors of the basic carbonate particles for two typical samples. Sample S5 completely converts to oxide by 700 ○C, and the observed weight loss (39.7%) is close to that (40.3%) calculated from the chemical formula (Y0.95Eu0.05)(OH)CO3·1.3H2O. Although the decomposition behavior of sample S3 (chemical formula (Y0.923Eu0.077)(OH)CO3·1.0H2O) is similar to that of S5 up to 650 ○C, the conversion to oxide finalizes only by 850 ○C, resulting in a weight loss of 37.8% (calculated: 37.6%). Since Eu(OH)CO3 converts to oxide at a similar temperature, we suggest that Eu(OH)CO3 existed in sample S3, and this assumption agrees well with the core-shell structure discussed above.
Figure 6.
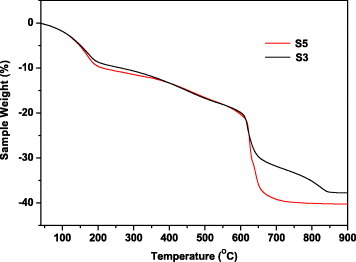
Thermogravimetry curves for samples S3 and S5.
Phase evolution of the precursors upon annealing was studied by XRD for samples S3 and S5 (figure 7). The amorphous precursor S5 did not transform to oxide until the calcination temperature of 600 ○C, and almost all the diffraction peaks corresponding to the Y2O3-based solid solution of cubic structure (JCPDS file No. 43-4036) have appeared at this temperature (figure 7(a)). Owing to Eu3+ homogenization in the Y2O3 lattice, a steady shift of 0.08○ toward lower angles with the temperature increasing from 600 to 1000 ○C was observed by examining the 222 reflection. The shift saturated above 1000 ○C, indicating the formation of a homogeneous solid solution around this temperature [11]. However, the amorphous precursor S3 did not completely transform to oxide at 600 ○C (figure 7(b)), and another phase, Eu2O2CO3, coexisted with the cubic Y2O3 oxide. Eu2O2CO3 completely transformed to oxide at 850 ○C, and no peak shift was observed for Y2O3. A 0.24○ shift toward lower angles, which is larger than that of S5, was detected with the temperature increasing from 850 to 1000 ○C by monitoring the 222 reflection of Y2O3. Since sample S3 had a core-shell Eu(OH)CO3@Y(OH)CO3 structure, the precursor converted to oxide in two steps, that is, Eu2O2CO3@Y2O3 at 600 ○C and Eu2O3@Y2O3 at 850 ○C. However, the transformation from a core-shell structure to a homogeneous solid solution requires a further temperature increase, which results in a significant peak shift due to Eu3+ doping and homogenization in the Y2O3 lattice. As mentioned above, a higher R is beneficial to forming a homogeneous solid solution. In this case, the homogenization of Eu3+ in the Y2O3 lattice with increasing temperature was less required in sample S5 than in S3 that resulted in a smaller peak shift for S5 (figure 7).
Figure 7.
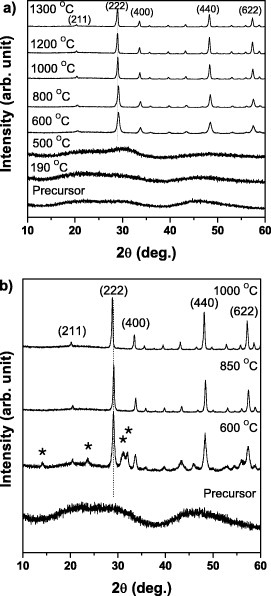
XRD patterns of samples S5 (a) and S3 (b) annealed at various temperatures. The peaks marked by ∗ in (b) correspond to the diffraction of Eu2O2CO3 (JCPDS file No. 23-0245).
Figures 8(a)–(d) show FE-SEM morphologies of sample S5 annealed at various temperatures. The spherical shape and excellent dispersion of the precursor particles have largely been retained to the oxides with the annealing temperatures up to 1000 ○C (figures 8(a) and (b)). At higher temperatures, the resultant particles are still well dispersed, but the shape changed from spherical to cubic (figures 8(c) and (d)). After calcination at 1300 ○C for 4 h, small particles merged in sample S11 (figure 8(e)), but larger cubic particles were dispersed in sample S5 (figure 8(d)).
Figure 8.

FE-SEM images showing morphologies of sample S5 annealed at 800 ○C (a), 1000 ○C (b), 1200 ○C (c) and 1300 ○C (d) for 4 h. Also shown is sample S11 annealed at 1300 ○C for 4 h (e).
The effects of calcination temperature on PL and PLE were studied for sample S5 (figure 9). The strong excitation peak at 254 nm is a charge-transfer band induced by the electronic transitions from the 2p orbital of O2− to the 4f orbital of Eu3+ activators. The three groups of excitation peaks at longer wavelengths are the intra-4f electronic transitions of Eu3+. Upon UV excitation at 254 nm, the oxide solid solutions exhibit sharp PL lines ranging from 500 to 750 nm, which are associated with the transitions from the 5D0 to 7FJ (J=0, 1, 2, 3, 4) states of Eu3+ [4], [10–12], [15–17]. The red emission at 613 nm originates from the hypersensitive 5D0→7F2 forced electric dipole transition. The PLE/PL bands do not shift but strengthen at higher annealing temperatures. Both the external and internal quantum efficiencies show similar temperature dependences as shown in figure 10. The decomposition temperature of the precursors affects the luminosity and quantum efficiency of the resultant phosphor particles, particularly when it is below 1000 ○C. This is mainly due to the Eu3+ homogenization in the Y2O3 lattice, which alleviates the local concentration quenching of PL, although improved crystallinity of the host lattice also contributes to the stronger emission and higher quantum efficiency. After annealing at 1000 ○C, the oxide phosphors had the external and internal quantum efficiencies of 50 and 82%, respectively (figure 10). A 300 ○C temperature increase from 1000 to 1300 ○C only increased the external and internal quantum efficiencies by 14 and 5%, respectively, mainly because of a further crystallinity improvement.
Figure 9.
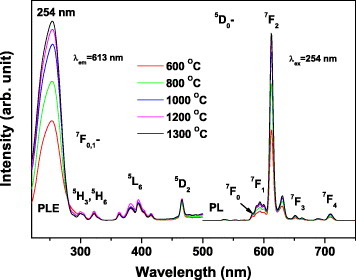
PL and PLE spectra of sample S5 annealed at various temperatures.
Figure 10.
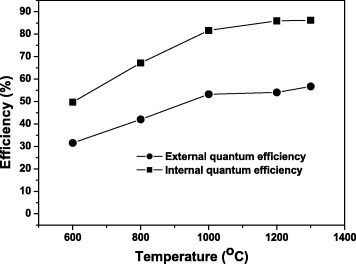
External and internal quantum efficiencies of PL (254 nm excitation) from sample S5 annealed at various temperatures.
Figure 11 shows the size-dependent luminescence of (Y0.95Eu0.05)2O3 annealed at 1300 ○C. Although the particle size does not significantly affect the band positions, a 2.27-fold intensity increase is observed for the 613 nm emission when the particle size increases from ∼220 nm (sample S11) to ∼450 nm (sample S5). The stronger luminescence of larger particles mainly results from their smaller surface area and thus lower concentration of surface defects.
Figure 11.
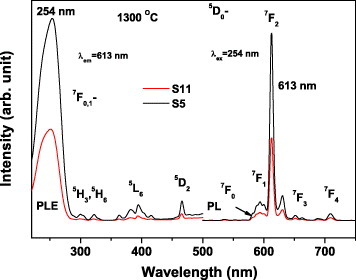
PL and PLE spectra of samples S5 and S11 annealed at 1300 ○C for 4 h.
Conclusions
Monodisperse colloidal spheres (∼220–500 nm in diameter) of (Y0.95Eu0.05)2O3 red-emitting phosphor have been synthesized by the UBHP method and the processing window has been systematically investigated. The particle size and shape were significantly affected by the ion concentration and the urea/RE3+ molar ratio R. Low ion concentrations are beneficial to the formation of monodisperse spheres, and a higher R results in a gradual change from the core-shell Eu(OH)CO3@Y(OH)CO3 structure to a homogeneous solid solution, thereby significantly lowering the conversion temperature of precursors to oxides. Uniform spheres of (Y0.95Eu0.05)2O3 exhibit the characteristic red emission at 613 nm upon UV excitation into the charge-transfer band at 254 nm. The PLE/PL bands did not shift but strengthened with increasing particle size, mainly due to the reduction of the surface area. The luminescence intensity and quantum efficiency of oxide phosphors increased with the elevated calcination temperatures. After annealing at 1000 ○C, the spherical shape and excellent dispersion of the precursor particles (∼450 nm in diameter) were retained and the resultant oxide phosphor exhibited the external and internal PL quantum efficiencies of 50 and 82%, respectively.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (Grants 50972025 and 50990303), the Special Fund for Basic Research in Central Universities (Grant N090502001), the Program for Changjiang Scholars and Innovative Research Teams in University (PCSIRT, IRT0713) and Grants-in-Aid for Scientific Research (KAKENHI, No. 22550135). The authors thank Mr Y Yajima of the Materials Analysis Station, National Institute for Materials Science, for performing elemental analysis. Q Zhu also acknowledges the financial support from the China Scholarship Council.
References
- Blasse G. and Grabmair B C. Berlin Springer; 1994. Luminescent Materials. [Google Scholar]
- Kang Y C, Roh H S. and Park S B. Adv. Mater. 2000;12:451. doi: 10.1002/(ISSN)1521-4095. [DOI] [Google Scholar]
- Kang Y C, Roh H S. and Park S B. J. Am. Ceram. Soc. 2001;84:447. doi: 10.1111/j.1151-2916.2001.tb00675.x. [DOI] [Google Scholar]
- Zhu Q, Li J-G, Li X D. and Sun X D. Acta Mater. 2009;57:5975. doi: 10.1016/j.actamat.2009.08.026. [DOI] [Google Scholar]
- Matijević E. and Hsu W P. J. Colloid Interface Sci. 1987;118:506. doi: 10.1016/0021-9797(87)90486-3. [DOI] [Google Scholar]
- Hsu W P, Rőnnquist L. and Matijević E. Langmuir. 1988;4:31. doi: 10.1021/la00079a005. [DOI] [Google Scholar]
- Aiken B, Hsu W P. and Matijević E. J. Am. Ceram. Soc. 1988;71:845. doi: 10.1111/jace.1988.71.issue-10. [DOI] [Google Scholar]
- Akinc M. and Sordelet D. Adv. Ceram. Mater. 1987;2:232. [Google Scholar]
- Sordelet D. and Akinc M. J. Colloid Interface Sci. 1988;122:47. doi: 10.1016/0021-9797(88)90286-X. [DOI] [Google Scholar]
- Li J-G, Li X D, Sun X D, Ikegami T. and Ishigaki T. Chem. Mater. 2008;20:2274. doi: 10.1021/cm7033257. [DOI] [Google Scholar]
- Li J-G, Li X D, Sun X D. and Ishigaki T. J. Phys. Chem. C. 2008;112:11707. doi: 10.1021/jp802383a. [DOI] [Google Scholar]
- Li J-G, Zhu Q, Li X D, Sun X D. and Sakka Y. Acta Mater. 2011;59:3688. doi: 10.1016/j.actamat.2011.03.004. [DOI] [Google Scholar]
- Okubo K. and Shigeta T. J. Illum. Eng. Inst. Japan. 1999;83:87. [Google Scholar]
- Cushing B L, Kolesnichenko V L. and O'Connor C J. Chem. Rev. 2004;104:3893. doi: 10.1021/cr030027b. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Li J-G, Zhi C Y, Li X D, Sun X D, Sakka Y, Golberg D. and Bando Y. Chem. Mater. 2010;22:4204. doi: 10.1021/cm1011586. [DOI] [Google Scholar]
- Zhu Q, Li J-G, Zhi C Y, Ma R, Sasaki T, Xu J X, Liu C H, Li X D, Sun X D. and Sakka Y. J. Mater. Chem. 2011;21:6903. doi: 10.1039/c1jm00048a. [DOI] [Google Scholar]
- Zhu Q, Li J-G, Li X D. and Sun X D. Curr. Nanosci. 2010;6:496. [Google Scholar]