

Abstract
Nitric oxide (NO) plays a critical role in the regulation of a wide variety of physiological processes. It is a potent inhibitor of platelet adhesion and aggregation, inhibits bacterial adhesion and proliferation, is implicated in mediating the inflammatory response toward implanted devices, plays a role in tumor growth and proliferation, and is a neurotransmitter. Herein, we describe the synthesis and NO-release properties of a modified polydimethylsiloxane that contains S-nitroso-N-acetyl-D-penicillamine covalently attached to the cross-linking agent (SNAP–DMS). Light from a C503B-BAN-CY0C0461 light-emitting diode (470 nm) was used as an external trigger to allow precise control over level and duration of NO release ranging from a surface flux of zero to approximately 3.5×10−10 mol cm-2 min-1. SNAP–PDMS films stored in the dark released NO after 297 days, indicating the long-term stability of SNAP–PDMS.
Keywords: nitric oxide release, polymer, photoinitiated, controlled release
Introduction
Nitric oxide (NO) plays a critical role in the regulation of a wide variety of physiological processes. It is a potent inhibitor of platelet adhesion and aggregation [1, 2], inhibits bacterial adhesion and proliferation [3], is implicated in mediating the inflammatory response toward implanted devices [4], inhibits smooth muscle cell growth and proliferation [5, 6], and is a neurotransmitter. There has been much interest in developing NO-releasing and NO-generating materials in an effort to harness the power of NO to mediate biological responses toward materials used to fabricate or coat biomedical devices.
Reviews by Reynolds, Frost, and Meyerhoff [7, 8] detail approaches that have been used to impart NO-release properties to hydrophobic polymer systems. Keefer [9] reviews the application of NO-release materials in clinical medicine. Diazeniumdiolates have been one of the most widely studied molecules used to release NO from polymers in clinical applications. Decomposition of diazeniumdiolates is driven by protons or heat. There is a huge array of structural variability that can be used to fine-tune NO-release properties of these molecules for a wide variety of applications. The disadvantage of using diazeniumdiolates as an NO donor in polymer systems in particular is that release cannot be changed once the polymer system is synthesized and applied to a medical device, and the surface flux of NO cannot be changed if too much or too little NO is being released. Despite this limitation, very promising results have been obtained that show reduction in thrombus formation on the surface of blood-contacting devices [10], in bacterial adhesion on orthopedic implants [3], and in smooth muscle cell proliferation to potentially lower restenosis rate after balloon angioplasty [11].
Because of the great clinical promise of NO-releasing materials, there is a need for developing materials that will allow dynamically controlling NO release after a biomedical device has been implanted (for example, an intravascular sensor may need a higher level of NO to inhibit thrombus formation on the sensor surface immediately after implantation and require lower levels of NO to maintain the thrombus-free state of the sensor after initial implantation reactions have subsided, or a critically ill patient may require higher levels of NO to maintain sensor function if the patient becomes more hypercoagulable over time). One approach to achieve this dynamic control of NO release is to develop photosensitive NO-releasing agents that release NO in response to intensity and wavelength of light. S-nitrosothiols are a class of NO donors that are capable of photoinitiated NO release. Etchenique et al [12] used photolytic release of NO from monolayers of nitrosated dithiothreitol attached to gold surfaces. Although this work suggests the possibility of photoinitiated temporal and spatial control of NO release, it is limited in application by the small amount of NO available within the monolayer. Frost and Meyerhoff [13] demonstrated that RSNOs (S-nitrosocysteine, S-nitroso-N-acetyl-L-cysteine, and S-nitroso-N-acetylpenicillamine (SNAP)) can be immobilized onto the surface of fumed silica particles that can then be blended in biomedical grade silicone rubber and cast into films. These polymer films were able to generate NO when triggered by white light. This result clearly showed that light can be used to precisely control the level of NO that is released from the polymer.
Herein, we describe the synthesis and NO-release properties of a modified polydimethylsiloxane (PDMS) that contains SNAP covalently attached to the cross-linking agent (SNAP–PDMS) of the polymer. Light from a light-emitting diode (LED) can be used as an external trigger to allow precise temporal and spatial control of the release of NO from this material.
Experimental details
Materials
Silanol-terminated polydimethylsiloxane (viscosity 2,000 cSt) was purchased from Gelest, Inc (Morrisville, PA, USA). (3-Aminopropyl)trimethoxysilane, dibutyltin dilaurate, and concentrated hydrochloric acid were obtained from Aldrich Chemical Co (Milwaukee, WI, USA). N-acetyl-D-penicillamine was purchased from Fluka (St. Louis, MO, USA). Tert-butyl nitrite, 90% technical grade (Acros Organics) and toluene were obtained from Fisher Scientific (Pittsburgh, PA, USA). Alfa Aesar acetic anhydride, magnesium sulfate (MgSO4), and cyclam were purchased from VWR (West Chester, PA, USA). Sterilized reverse-osmosis water (18 MΩ) was produced in the laboratory. Dow Corning RTV-3140 was used as a control (non-modified) PDMS (Ellsworth Adhesives, Germantown, WI, USA).
Synthesis of thiolactone
A self-protected thiolactone was synthesized according to the procedure of Moynihan and Robert [14]. Briefly, 5 g of N-acetyl-D-penicillamine were dissolved in 10 ml pyridine and 10 ml of acetic anhydride were dissolved in 10 ml pyridine. Both solutions were ice cooled for 1 h and then combined. The reaction mixture was allowed to stir for 48 h at room temperature. A dark red-amber color developed. Solvent was rotoevaporated at 50 °C to obtain a thick orange/amber colored oil. This was dissolved in 30 ml chloroform and extracted with 30 ml 1 M HCl three times. The chloroform solution was dried with MgSO4 and suction filtered to remove the drying agent. Chloroform was then removed with the rotoevaporator and resultant crystals were collected and rinsed with hexanes and suction filtration. Light yellow/white colored, small needle-type crystals with a pungent odor were collected and allowed to air dry overnight.
Synthesis of SNAP–PDMS
Figure 1 shows the synthetic scheme of SNAP–PDMS: 1.6 g of silanol-terminated PDMS were dissolved in 4 ml of toluene with the aid of vortex mixing; 0.3 g of (3-aminopropyl)trimethoxysilane were dissolved in 2 ml toluene. The PDMS and (3-aminopropyl)trimethoxysilane were combined and vortex mixed; 1.0 ml of a stock solution containing 20 mg of dibutyltin dilaurate in 20 ml of toluene was added to the PDMS/(3-aminopropyl)trimethoxysilane solution and vortex-mixed immediately. The PDMS was continually stirred and allowed to cross-link at room temperature for 24 h; 50 mg of the thiolactone were dissolved in 1 ml of toluene and 2 ml of the cross-linked PDMS solution were added and stirred for 24 h. The thiol groups were then nitrosated with t-butyl nitrite to form SNAP that is covalently linked to the PDMS cross-links. A dark green color developed after approximately 15 min, and the reaction was allowed to continue for approximately 45 min while protected from light before the SNAP–PDMS was used. The t-butyl nitrite was first cleaned by extraction with 15 mM aqueous cyclam, and 1 ml of t-butyl nitrite was vigorously shaken with 2 ml of 15 mM cyclam. The cyclam was replaced for a total of 3 extraction steps. Freshly cleaned t-butyl nitrite was stored in an amber vial in the refrigerator, and 0.5 ml of cleaned t-butyl nitrite were added to 3 ml SNAP–PDMS. SNAP–PDMS films were cast by pipetting 2 ml of the SNAP–PDMS solution in 2.4 cm diameter polytetrafluoroethylene (PTFE) rings on a PTFE base plate and allowed to cure overnight, in darkness. Dark green films (approximately 200 μm thick) were obtained that released NO upon illumination.
Figure 1.
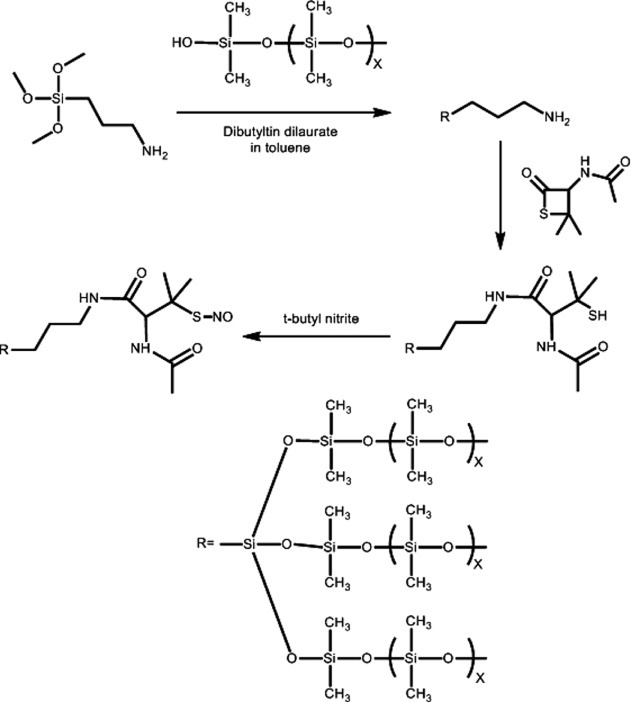
Schematic of the synthesis of S-nitroso-N-acetyl-D-penicillamine covalently linked to polydimethylsiloxane (SNAP–PDMS).
Photoinitiation of NO release
A C503B-BAN-CY0C0461 LED (Mouser Electronics, Mansfield, TX, USA) was used as the light source to release NO from SNAP–PDMS. The LED was connected in series with a resistance of 138 Ω and a voltage source operated at different applied potentials (0, 3, 4.5 and 6 V that corresponds to 0, 3, 12 and 20 mA current, respectively). The LED has a peak of wavelength of 470 nm and its emission spectrum is shown in figure 2.
Figure 2.
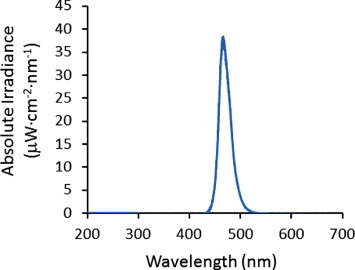
Spectrum of light emitted from the C503B-BAN-CY0C0461 LED (peak wavelength 470 nm) under 6 V applied potential.
Nitric oxide release measurements
Nitric oxide release was measured using a Sievers 280i Nitric Oxide Analyzer (GE Instruments, Boulder, CO, USA) using 200 ml min-1 flow of nitrogen carrier gas. SNAP–PDMS films were cut into 7 mm diameter discs and placed in a cell holder with the LED epoxied on the inner side of the top of the amber sample holder. Amber cells were used to exclude ambient radiation from the SNAP–PDMS test discs. The LED illuminated the broad side of a SNAP–PDMS disc from a distance of 5 cm.
Results and discussion
The great utility for the SNAP–PDMS material is the ability to control NO release, thereby allowing different durations and levels of NO to be delivered on a continually variable basis as needed for a given biological application. A primary amine group was incorporated into the PDMS by using a cross-linking agent that contains an aminopropyl group. Zhang et al [15] first used this cross-linking agent to form diazeniumdiolate-containing silicone rubber. The cross-linked PDMS was reacted with the self-protected thiolactone. The thiolactone then reacted with the primary amine groups to form an amide bond with the N-acetyl-D-penicillamine and result in a free thiol group that is available to form the corresponding S-nitrosothiol. Upon nitrosation, the SNAP–PDMS developed a dark green color that is indicative of the formed S-nitroso-N-acetyl-D-penicillamine. Figure 3 contains a photograph of the SNAP–PDMS after a film has been cast and cured. It is compared to a control PDMS (Dow Corning RTV-3140) film that did not contain the modified cross-linking agent (i.e. no primary amine groups present in the PDMS). PDMS for the control sample was mixed with the thiolactone for 24 h and followed the same procedure for nitrosation that was used with the SNAP–PDMS. No green color developed indicating that the S-nitrosothiol was not formed.
Figure 3.

Photographs of (a) a cast and cured SNAP–PDMS film compared to (b) a control PDMS (Dow Corning RTV-3140) film that did not contain the modified cross-linking agent (i.e., no primary amine groups present in the PDMS). PDMS of the control sample was mixed with the thiolactone for 24 h and nitrosated following the same procedure as that used with the SNAP–PDMS. No color developed, indicating that the S-nitrosothiol was not formed.
Figure 4 shows the surface flux of NO generated from three different 7 mm diameter discs of SNAP–PDMS exposed to light from the 470 nm LED (6 V bias). The average NO surface flux is (3.3 ± 0.5)×10−10 mol cm-2 min-1 (n=3). The variability in the generated surface flux of NO is due in part to variable thicknesses of the films. Figure 5 shows the variable NO generation from a 7 mm diameter disc of SNAP–PDMS at different potentials applied to the LED. In the absence of light, virtually no nitric oxide is generated (trace a). As the light intensity incident on the SNAP–PDMS is increased by stepping through 0, 3, 4.5 and 6 V of applied potential (traces a, b, c and d), the surface flux of NO increases in the sequence (0.20 ± 0.04, 0.90 ± 0.02, 3.10 ± 0.03, and 4.80 ± 0.05)×10−10 mol cm-2 min-1 (averaged over 1 min of release). This is well within the estimated range of physiologically relevant NO produced by endothelial cells ((1–4)×10−10 mol cm-2 min-1) [16]. Figure 6 shows NO generation from the films upon storage in the dark at 4 °C after 13 and 57 days (traces a and b, (5.70 ± 0.06 and 3.20 ± 0.05)×10−10 mol cm-2 min-1, respectively). The films were then kept in the dark at room temperature, and after 297 days (trace c.), were still generating NO at (0.80 ± 0.02)×10−10 mol cm-2 min-1. As a preliminary experiment, this demonstrates that the SNAP–PDMS film is robust enough to continue generating measurable NO for 297 days even under these non-optimal storage conditions.
Figure 4.
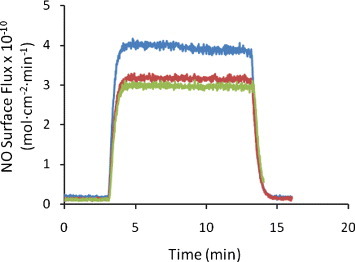
Representative plot of surface flux of NO generated from three different 7 mm diameter discs of SNAP–PDMS exposed to 470 nm LED light (6 V bias). The average NO surface flux is (3.3 ± 0.5)×10−10 mol cm-2 min-1.
Figure 5.
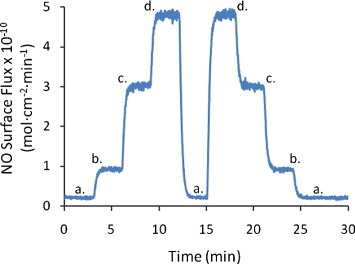
Variable NO generation from a 7 mm diameter disc of SNAP–PDMS when the applied potential is changed to the LED. In the absence of light (a), virtually no nitric oxide is generated, as the light intensity incident on the SNAP–PDMS is increased by stepping through 0, 3, 4.5 and 6 V of applied potential (a, b, c and d), the surface flux of NO increases in the sequence (0.20 ± 0.04, 0.90 ± 0.02, 3.10 ± 0.03 and 4.80 ± 0.05)×10−10 mol cm-2 min-1 (averaged over 1 min of release).
Figure 6.
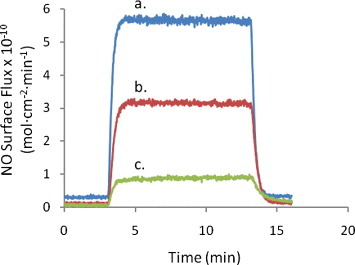
NO generation from the films upon storage in the dark at 4 °C after (a) 13 and (b) 57 days: (5.70 ± 0.06 and 3.20 ± 0.05)×10−10 mol cm-2 min-1, respectively. The films were then kept in the dark at room temperature and after (c) 297 days were still generating NO at (0.80 ± 0.02)×10−10 mol cm-2 min-1, averaged over 10 min of NO release.
SNAP–PDMS shows great promise as a controlled NO-releasing material. Using light as an external trigger to release NO allows precise control of turning NO on and off as well as increasing or decreasing NO flux as desired. This provides an accurate probe of NO levels required to achieve specific physiological responses. We are currently using this SNAP–PDMS material as a substrate upon which cells can be grown to study how the level and duration of NO imposed on the cells affect different cell types. Additionally, we have coated SNAP–PDMS on polymethylmethacrylate (PMMA) optical fibers and generated controlled amounts of NO when the fiber was illuminated with the same 470 nm LED [17]. These coated fibers will be used for probing in vivo responses to intravascular and subcutaneous sensors.
Conclusions
There has been much interest in the development of NO-releasing polymeric materials. Because NO has been identified as a potent molecule to mediate physiological response, NO-releasing and generating materials show great promise for the fabrication and/or coating of medical devices that are safer and more effective when implanted in the body. We have synthesized a silicone rubber material that contains S-nitroso-N-acetyl-D-penicillamine covalently linked to the polymer cross-linking agents. SNAP–PDMS releases NO in response to light generated from a 470 nm LED. Because we have precise control of the level and dose of NO released from SNAP–PDMS by changing the illumination intensity, we can evaluate the levels of NO needed to achieve specific physiological responses. This information will allow tailored design of NO-releasing materials for specific biomedical applications. Complete physical characterization and optimization of storage conditions of SNAP–PDMS is currently underway.
Acknowledgment
The authors are grateful for financial support from the National Science Foundation, Division of Materials Research DMR-0906709-2009.
References
- Mellion B T.et al1981Blood 57946. [PubMed] [Google Scholar]
- Radomski M W, Palmer R M. and Moncada S. Br. J. Pharmacol. 1987;92:639. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nablo B J, Rothrock A R. and Schoenfisch M H. Biomaterials. 2005;26:917. doi: 10.1016/j.biomaterials.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Gifford R.et al2005J. Biomed. Mater. Res. A75A755. 10.1002/(ISSN)1552-4965 [DOI] [PubMed] [Google Scholar]
- Baek S H, Harabie J A, Keefer L A, Hou D M, Finebergm N, Rhoades R. and March K L. Circulation. 2002;105:2779. doi: 10.1161/01.CIR.0000017432.19415.3E. [DOI] [PubMed] [Google Scholar]
- Chaux A, Ruan X M, Fishbein M C, Ouyang Y, Kaul S, Pass J A. and Matloff J M. J. Thorac. Cardiavasc. Surgery. 1998;115:604. doi: 10.1016/S0022-5223(98)70325-3. [DOI] [PubMed] [Google Scholar]
- Frost M C, Reynolds M M. and Meyerhoff M E. Biomaterials. 2005;26:1685. doi: 10.1016/j.biomaterials.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Reynolds M M, Frost M C. and Meyerhoff M E. Free Rad. Biol. Med. 2004;37:926. doi: 10.1016/j.freeradbiomed.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Keefer L K. Annu. Rev. Pharmacol. Toxicol. 2003;43:585. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]
- Frost M C.et al2002Anal. Chem. 745942. 10.1021/ac025944g [DOI] [PubMed] [Google Scholar]
- Bohl K S. and West J L. Biomaterials. 2000;21:2273. doi: 10.1016/S0142-9612(00)00153-8. [DOI] [PubMed] [Google Scholar]
- Etchenique R, Furman M. and Olabe J A. J. Am. Chem. Soc. 2000;122:3967. doi: 10.1021/ja9939845. [DOI] [Google Scholar]
- Frost M C. and Meyerhoff M E. J. Biomed. Mater. Res. 2005;72A(A):409. doi: 10.1002/(ISSN)1552-4965. [DOI] [PubMed] [Google Scholar]
- Moynihan H A. and Robert S M. J. Chem. Soc. Perkins Trans. 1994;1:797. [Google Scholar]
- Zhang H P, Annich G M, Miskulin J, Osterholzer K, Merz S I, Bartlett R H. and Meyerhoff M E. Biomaterials. 2002;23:1485. doi: 10.1016/S0142-9612(01)00274-5. [DOI] [PubMed] [Google Scholar]
- Vaughn M W, Kuo L. and Liao J C. Am. J. Physiol. 1998;274:H2163. doi: 10.1152/ajpheart.1998.274.6.H2163. [DOI] [PubMed] [Google Scholar]
- Starrett M A. Houghton Michigan Technological University; 2010. Electrical and Computer Engineering. [Google Scholar]