

Abstract
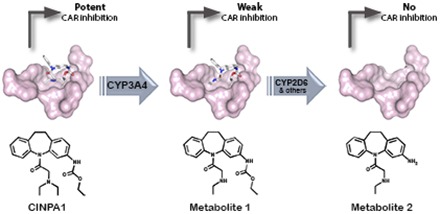
The constitutive androstane receptor (CAR) regulates the expression of genes involved in drug metabolism and other processes. A specific inhibitor of CAR is critical for modulating constitutive CAR activity. We recently described a specific small-molecule inhibitor of CAR, CINPA1 (ethyl (5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate), which is capable of reducing CAR-mediated transcription by changing the coregulator recruitment pattern and reducing CAR occupancy at the promoter regions of its target genes. In this study, we showed that CINPA1 is converted to two main metabolites in human liver microsomes. By using cell-based reporter gene and biochemical coregulator recruitment assays, we showed that although metabolite 1 was very weak in inhibiting CAR function and disrupting CAR-coactivator interaction, metabolite 2 was inactive in this regard. Docking studies using the CAR ligand-binding domain structure showed that although CINPA1 and metabolite 1 can bind in the CAR ligand-binding pocket, metabolite 2 may be incapable of the molecular interactions required for binding. These results indicate that the metabolites of CINPA1 may not interfere with the action of CINPA1. We also used in vitro enzyme assays to identify the cytochrome P450 enzymes responsible for metabolizing CINPA1 in human liver microsomes and showed that CINPA1 was first converted to metabolite 1 by CYP3A4 and then further metabolized by CYP2D6 to metabolite 2. Identification and characterization of the metabolites of CINPA1 enabled structure-activity relationship studies of this family of small molecules and provided information to guide in vivo pharmacological studies.
Introduction
The constitutive androstane receptor (CAR) and pregnane X receptor (PXR) can be activated by structurally diverse compounds (this is referred to as ligand promiscuity), and these receptors are key regulators of expression of metabolic enzymes such as the cytochrome P450s (P450s), transferases, and membrane transporters (Willson and Kliewer, 2002; Timsit and Negishi, 2007). CINPA1 [ethyl (5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate] is a potent inhibitor of human constitutive androstane receptor (hCAR) that was recently discovered by using a directed chemical screening approach; unlike other reported inhibitors of CAR, CINPA1 does not activate PXR (Cherian et al., 2015b). Although the most studied hCAR isoform, hCAR1, is constitutively active when exogenously expressed in cultured cells, it can still be modulated by both activators and inhibitors (Honkakoski et al., 2003; Cherian et al., 2015a). CAR transcriptionally regulates the expression of CYP2B6 and CYP3A4 by binding to the xenobiotic response elements in the promoter regions of these genes (Sueyoshi et al., 1999; Goodwin et al., 2002; Li et al., 2012). CINPA1 acts as an inverse agonist in experimental contexts involving the constitutively active receptor but is a true antagonist in scenarios where hCAR activity is inducible in the presence of an agonist like CITCO [(E)-1-[6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazol-5-yl]-N-[(3,4-dichlorophenyl)methoxy]methanimine]. CINPA1 binds to CAR and reduces its interactions with coactivators and DNA elements, essentially abrogating the transactivation potential of CAR. CINPA1 does not activate PXR and is thus a unique pharmacologic tool for delineating CAR’s functions. In our structure-activity relationship studies, a series of CINPA1 analogs were tested for their CAR-binding function and more potent analogs were identified (Lin et al., 2016). Because of CAR’s ligand promiscuity, it is critical to identify the metabolites of CINPA1 and examine their effects on CAR. This is particularly important when studying the role of CAR in regulating xenobiotic and endobiotic metabolism in animal models or human liver.
Phase I and phase II drug-metabolizing enzymes and membrane transporters play pivotal roles in detoxifying, eliminating, and biotransforming drugs. Identifying the enzyme(s) responsible for metabolizing a chemical and characterizing the resulting metabolites represents the first stage in learning about the in vivo pharmacokinetic properties of a chemical. The clearance of any chemical in humans is markedly dependent on the relative expression and activity of the various P450s (Evans and Relling, 1999). These phase I enzymes can perform an assortment of modifications, including hydroxylation, aromatic oxidation, heteroatom oxidation, N- and O-dealkylation, aldehyde oxidation, dehydrogenation, and aromatase activity (Meunier et al., 2004; Ortiz de Montellano and De Voss, 2005; Zanger and Schwab, 2013). Although there are more than 50 P450 enzymes, six of them metabolize 90% of drugs, with the two most significant enzymes in this regard being CYP3A4 and CYP2D6. CYP3A4 is the principal and most versatile member of the P450 family and is involved in metabolizing the majority of xenobiotics and endobiotics (Guengerich et al., 1994). CYP3A4 is generally thought to be a low-affinity, high-capacity enzyme capable of hydroxylation and N-deethylation reactions (Fabre et al., 1993; Wang et al., 2000; Hodgson, 2001). CYP2D6 is another key phase I enzyme; it has a complex genotype and a trimodal phenotype with slow, fast, or ultrafast metabolizers. The genetic variability of CYP2D6 greatly influences the clinical pharmacokinetics and therapeutic and/or adverse effects of certain drugs (Bertilsson et al., 2002). In addition to P450s, non-P450 enzymes, such as esterases, aldehyde oxidases, and flavin-containing monooxygenases, are also involved in phase I metabolism (Oda et al., 2015). Esterases, for example, help metabolize 10% of clinical drugs to either detoxify or activate them and are commonly expressed in the liver (Fukami and Yokoi, 2012).
The effective use of CINPA1 in preclinical animal studies can be guided by identifying all of the metabolites of CINPA1, their effect on CAR function, and the P450 isoforms involved in forming these metabolites. By using human liver microsomes (HLMs) and recombinant P450s, we determined the primary and secondary metabolism of CINPA1 and the specific P450s involved. The results were confirmed by measuring the percent inhibition of P450-dependent metabolism of CINPA1 in HLMs that is caused by known specific chemical inhibitors of P450 isoforms (Walsky and Obach, 2004). We also tested the effect of the identified metabolites on CAR function and made informed estimations by using protein docking studies to interpret the apparent differences in metabolite function, despite the structural similarities. This in vitro study will enable the in vivo activity of CINPA1 to be predicted and interpreted.
Materials and Methods
Chemicals.
Substrates, metabolite standards, internal standards, inhibitors, and other materials were obtained from the following sources: all anhydrous solvents, dimethylsulfoxide (DMSO), ketoconazole, N,N′,N′′-triethylenethiophosphoramide (thioTEPA), ticlopidine, quinidine, efavirenz, tolbutamide, phenacetin, bis(p-nitrophenyl) phosphate (BNPP), and acetaminophen (Sigma-Aldrich, St. Louis, MO); tranylcypromine (PCPA), quercetin, dextromethorphan, 1′-hydroxymidazolam, dextrorphan, hydroxytolbutamide, and (±)4-hydroxymephenytoin (Cayman Chemical, Ann Arbor, MI); sulfaphenazole, midazolam, and the NADPH regenerating system (Corning, Tewksbury, MA); S-mephenytoin (Santa Cruz Biotechnology, Dallas, TX); and 8-hydroxyefavirenz (Toronto Research Chemicals, Toronto, Canada). All reagent-grade solvents used for chromatography were of high-performance liquid chromatography (LC) grade (Fisher Scientific, Suwanee, GA). CINPA1 was prepared by WuXi AppTec (Hong Kong, China). The first metabolite (metabolite 1 or Met1) of CINPA1, ethyl [5-(N-ethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl]carbamate (catalog no. 5214995), was obtained from ChemBridge Corp. (San Diego, CA).
Synthesis of Metabolite 2.
The general synthetic chemistry procedures and instrumentation used were described previously (Lin et al., 2016). Organic reagents were purchased from commercial suppliers, unless otherwise noted, and were used without further purification. All solvents were analytical grade or reagent grade. All reactions were carried out in flame-dried glassware under argon or nitrogen. Flash column chromatography was performed with Sigma-Aldrich silica gel 60 (200–400 mesh) and was carried out under moderate pressure, with columns of an appropriate size being packed and eluted with appropriate eluents. All reactions were monitored by performing thin-layer chromatography (TLC) on precoated plates (silica gel HLF; Analtech/iChromatography, Newark, DE). TLC spots were visualized either by exposure to iodine vapor or by UV irradiation. Organic solvents were removed under vacuum with a rotary evaporator. The reactions, purities, and identities of the final compounds were monitored or determined by performing TLC or by using a Waters Acquity ultra-performance liquid chromatography (UPLC) mass spectrometry (MS) system (Waters, Milford, MA) with a C18 column in a 2-minute gradient (H2O + 0.1% formic acid → acetonitrile + 0.1% formic acid) and a photodiode array detector (215–400 nm), evaporative light scattering detector, and Acquity SQD electrospray ionization (ESI)–positive MS. Preparative TLC separation was performed by using self-casted preparative TLC plates consisting of Sigma-Aldrich silica gel 60 (200–400 mesh) on 20 cm × 20 cm glass plates. The reaction products were purified by using a Dionex APS 3000 dual purification/analytical LC/photodiode array detector/MS system (Dionex, Sunnyvale, CA) with a C18 column in a 15-minute gradient (H2O with 0.05% NH3·H2O → acetonitrile) and ESI-positive MS. The high-resolution mass spectrum was obtained by using a Waters Acquity UPLC system with a C18 column (H2O + 0.1% formic acid → acetonitrile + 0.1% formic acid gradient over 2.5 minutes) under Xevo G2 (Waters) quadrupole time-of-flight ESI in positive resolution mode. Compounds were internally normalized to leucine-enkephalin lock solution, with a calculated error of less than 3 ppm. The 1H nuclear magnetic resonance spectrum was recorded on a Bruker UltraShield 400 Plus system (Bruker, Billerica, MA). The chemical shift values are expressed in parts per million relative to tetramethylsilane as the internal standard.
Metabolite 2 (Met2, 1-(3-amino-10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-2-(ethylamino)ethan-1-one) was synthesized as follows (Scheme 1). First, 1-(3-amino-10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-2-(ethylamino)ethan-1-one, compound 78 (Lin et al., 2016) (250.00 mg, 591.16 µmol), K2CO3 (81.70 mg, 591.16 µmol), and ethylamine (799.48 mg, 17.73 mmol) in dimethylformamide (2.00 ml) were stirred at room temperature for 2 hours. TLC and LC/MS showed that the starting material, compound 78, was depleted. The reaction mixture was quenched with water (10 ml) and then extracted with ethyl acetate (10 ml × 2). The combined ethyl acetate layer was washed with brine, dried with anhydrous Na2SO4, and concentrated with a Rotavapor (Buchi, New Castle, DE) to obtain intermediate 1 [benzyl (5-(ethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate, 200.00 mg, 463.47 µmol, 78.4% yield] as a crude oil, which was used directly for the next step. Second, intermediate 1 (200.00 mg, 463.47 µmol) and 10% Pd/C (20.00 mg, 463.47 µmol) in MeOH (3.00 ml) were stirred under a balloon of H2 at room temperature for 2 hours. TLC showed that the starting material, intermediate 1, was depleted. The reaction mixture was filtered, and the filtrate was concentrated with a Rotavapor. The residue was then purified by preparative TLC (methylene chloride/MeOH = 10/1) to yield Met2 (103.00 mg, 346.35 µmol, 74.7% yield, 95.1% purity) as a yellow oil. 1H nuclear magnetic resonance (400 MHz, MeOD4) was as follows: δ (in parts per million) 7.2–7.44 (m, 4H), 6.90–7.07 (m, 1H), 6.58–6.77 (m, 2H), 3.43–3.67 (m, 1H), 2.93–3.29 (m, 3H), 2.46–2.89 (m, 4H), and 0.86–1.19 (m, 3H). The ESI time-of-flight high-resolution mass spectrum had mass-to-charge ratios of 296.1778 (C18H21N3O + H+ requires 296.1765) and 318.1585 (C18H21N3O + Na+ requires 318.1593) (Supplemental Figs. 1 and 2).
Scheme 1.

Synthesis of Met2. Reagents and conditions were as follows: (A) K2CO3, ethylamine, dimethylformamide, room temperature, 2 hours; and (B) 10% Pd/C, MeOH, H2, room temperature, 2 hours.
Reagents and Instruments.
LC/MS was performed using a Waters Acquity UPLC/MS/UV system. HLMs represented a mixed-sex pool of 50 individual donors (Sekisui XenoTech LLC, Kansas City, KS). Mouse liver microsomes (MLMs) represented a mixture of male and female CD-1 mice (Gentest; Corning). All of the recombinant human P450 enzymes used—CYP1A2, CYP2B6, CYP2C9 (Arg144), CYP2C19, CYP2D6 (Val374), and CYP3A4 (Supersomes; Corning)—were purified from baculovirus-insect cell lines. The microsomal preparations were stored at −80°C until use. The total P450 content, protein concentrations, and specific activity of each P450 isoform were as supplied by the manufacturer.
HepG2 and Caco-2 cells were obtained from American Type Culture Collection (Manassas, VA); their mycoplasma contamination status was tested periodically and found to be negative. Fetal bovine serum (FBS) was obtained from HyClone (Logan, UT). The CAR expression vector (FLAG-hCAR1 in a pcDNA3.1 vector) and CYP3A4-luciferase reporter (CYP3A4-luciferase in a pGL3 vector) were as described previously (Li et al., 2012; Cherian et al., 2015b). CYP2B6-luc, consisting of a luciferase reporter gene under the control of the CYP2B6 promoter region (the phenobarbital-responsive enhancer module/xenobiotic responsive enhancer module) was kindly provided by Dr. Hongbing Wang (University of Maryland, Baltimore, MD; Wang et al., 2003).
Microsomal Stability and Identification of CINPA1 Metabolites.
Microsomal stability of CINPA1 in HLMs and MLMs was analyzed as described previously (Rakesh et al., 2012). Briefly, a 10 mM CINPA1 stock solution was prepared in DMSO and diluted 1000-fold in 0.5 mg/ml HLMs or MLMs in triplicate wells for six time points. NADPH regenerating agent was added to all plates to initiate the reaction. The plates were incubated at 37°C for the indicated length of time, and then the reactions were quenched by adding cold acetonitrile with the internal standard (4 μg/ml warfarin). The plates were sealed, mixed, and centrifuged at 4000 rpm for 20 minutes. The supernatants were transferred to analytical plates for analysis by LC/MS using a Waters Acquity UPLC/MS/UV system. Metabolic stability was evaluated via the half-life from a least-squares fit of the multiple time points based on first-order kinetics. We used HLMs to follow the metabolism of CINPA1 by using LC/MS analysis as described (Ward et al., 2003). LC/MS analysis was performed on the extracted samples to determine the MS fragmental pattern by using a Waters Acquity UPLC/MS/UV system and an Acquity SQD ESI-positive mass spectrometer. The MS fragmentation patterns were matched with synthetic standards for CINPA1 and its metabolites. Retention times of the CINPA1 metabolite peaks after injecting microsomal incubates of CINPA1 into the UPLC system were also compared with those of the reference metabolite standards (Met1 and Met2).
Aqueous Solubility.
Thirty microliters of 10 mM CINPA1 in DMSO was applied to a well in a stock plate and diluted 100-fold in system solution buffer (pH 7.4; pION Inc., Woburn, MA). The solution was incubated at room temperature for 12–18 hours to allow it to fully stabilize and was then filtered through a 96-well filter plate (pION Inc.). This assay was performed as described previously (Rakesh et al., 2012) on a Biomek FX ADMETox workstation (Beckman Coulter Inc., Fullerton, CA). Fractions were collected from the filtered sample plate and diluted with isopropanol [1:1 (v/v)], and the concentration was determined by UV spectrometry (230–500 nm). The calculations were performed with μSOL Evolution software (pION Inc., Woburn, MA), and the compounds were each tested in triplicate.
Caco-2 Permeability.
Caco-2 permeability determinations were performed in the 96-well Transwell system (Corning, NY) using the method of Uchida et al. (2009) with modifications as described in Rakesh et al. (2012). Caco-2 cells were maintained at 37°C in a humidified incubator with a 5% CO2 atmosphere. The cells were cultured in Eagle’s minimum essential medium containing 10% FBS with 100 U/ml penicillin and 100 μg/ml streptomycin. Caco-2 cells were seeded onto inserts of a 96-well plate at a density of 2 × 104 cells/insert and cultured in minimum essential medium containing 10% FBS for 7 days. Each cultured monolayer on the 96-well plate was then washed twice with Hanks’ balanced salt solution/HEPES (10 mM, pH 7.4). The permeability assay was initiated by adding CINPA1 (50 μM) to the inserts (apical side, A) or receivers (basolateral side, B). The Caco-2 cell monolayers were then incubated for 2 hours at 37°C. Fractions were collected from the receivers (for apical-to-basal permeability) or inserts (for basal-to-apical permeability), and concentrations were assessed by UPLC/MS (Waters). All assays were performed in triplicate. The A → B (or B → A) apparent permeability coefficients (in centimeters per second) were calculated using eq. 1:
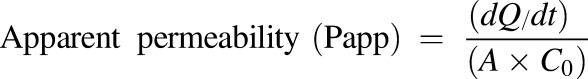 |
(1) |
where dQ/dt (in micromoles per second) represents the flux of the drug across the monolayer, C0 (in micromoles per liter) is the initial drug concentration on the apical side, and A (in square centimeters) is the surface area of the monolayer.
Plasma Protein Binding.
Mouse and human plasma protein binding of CINPA1 was tested as previously described by Rakesh et al. (2012) using the single-use rapid equilibrium dialysis devices from Thermo Scientific (Rockford, IL). Mouse and human plasma was obtained from Lampire Biologic Laboratories (Pipersville, PA) and centrifuged at 1000 rpm for 2 minutes to remove particulates before use. The 10 μM final concentration of CINPA1 in mouse and human plasma was prepared from 10 mM CINPA1 stock solution in DMSO (0.1% final concentration of DMSO).
Plasma solutions with CINPA1 were placed in the sample chamber while phosphate-buffered saline was placed in the adjacent (buffer) chamber. After incubation at 37°C for 4 hours on an orbital shaker (100 rpm), the CINPA1 concentrations in both the buffer and plasma chambers were quantified by measuring the peak areas relative to the internal standard (4 μg/ml warfarin) by UPLC/MS. The percentage of CINPA1 bound to plasma was calculated using eq. 2:
 |
(2) |
Kinetic Analyses in HLMs and Supersomes.
Kinetic studies of CINPA1 metabolism were conducted in an HLM preparation representing a mixed-sex pool of 50 individual donors, as well as in recombinant human P450 enzymes (Ward et al., 2003; Kazui et al., 2010). CINPA1 or Met1 (1.6–200 µM) was incubated for 30 minutes at 37°C with HLMs (0.5 mg protein/ml) and an NADPH regenerating system (1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 3.3 mM magnesium chloride, and 0.4 U/ml glucose-6-phosphate dehydrogenase) in potassium phosphate buffer (pH 7.4) containing 1 mM EDTA in 100-µl reaction volumes. For the recombinant human P450 assays, the incubation mixtures were in either phosphate or Tris buffer at pH 7.4 containing 41 pmol/ml CYP1A2, CYP3A4, CYP2C9, CYP2C19, CYP2D6, or CYP2B6 enzymes and an NADPH regenerating agent. Various concentrations of CINPA1 or Met1 were added in potassium phosphate buffer (pH 7.4, 100 mM) for the CYP1A2, CYP2B6, CYP2C19, CYP2D6, and CYP3A4 reactions or in Tris-HCl buffer (pH 7.4, 50 mM) for the CYP2C9 reactions. After incubation for 30 minutes at 37°C, the reactions were terminated by adding 200 μl acetonitrile containing the internal standard (4 μg/ml warfarin) to each well. The plates were then sealed and centrifuged at 4000 rpm for 10 minutes. The supernatants (200 μl each) were transferred to analytical plates for analysis by LC/MS with a Waters Acquity UPLC/MS/UV system. The formation rates of metabolites versus CINPA1 substrate concentrations were fitted to sigmoidal enzyme kinetic models to estimate the apparent kinetic parameters (e.g., Km and Vmax; see the kinetic data analysis section below). To enable kinetic analysis or observe the effect of substrate concentration on enzymatic activity, various concentrations of CINPA1 or Met1 were used in these assays.
Enzyme Inhibition Assays.
The rates of CINPA1, Met1, and Met2 metabolism in HLMs were evaluated in the absence (control) and presence of the following known isoform-specific inhibitors (Ward et al., 2003; Walsky and Obach, 2004): PCPA (5 μM) for CYP1A2, sulfaphenazole (5 μM) for CYP2C9, ticlopidine (25 μM) for CYP2C19, quinidine (10 μM) for CYP2D6, ketoconazole (5 μM) for CYP3A4, quercetin (25 μM) for CYP2C8, thioTEPA (25 μM) for CYP2B6, and BNPP (250 µM) for esterases. The specific conditions for using these inhibitors were described in detail in earlier publications (Desta et al., 1998; Rae et al., 2002; Walsky and Obach, 2004). All inhibitors were preincubated with the NADPH regenerating system and HLMs (0.5 mg/ml) for 15 minutes at 37°C, before initiating the reaction by adding the substrate and incubating for an additional 30 minutes. CINPA1 or Met1 (5 μM) were incubated in HLMs with or without P450 isoform–specific inhibitor and with an NADPH regenerating system for 30 minutes. The percent inhibition of the metabolite formation rate by each P450 isoform–specific inhibitor was calculated by comparing the activity in samples containing the inhibitor with that in the vehicle control (DMSO with no inhibitor).
Kinetic Data Analysis.
Estimates of the kinetic constants were obtained by nonlinear regression analysis using GraphPad Prism software (version 6; GraphPad Software Inc., La Jolla, CA). The Hill equation [V = Vmax · C^h/ (Km^h + C^h)] was fitted to the formation rates (V) of metabolite versus substrate concentrations (C), where h is the Hill coefficient and a measure of the cooperativity of substrate binding to the enzyme. In cases in which h was equal to 1, the simple single-site Michaelis–Menten equation [V = Vmax · C/ (Km + C)] was used. The models that fit best were selected based on the dispersion of residuals and the standard errors of the parameter estimates. The calculated kinetic constants (Km and Vmax) are presented as means ± S.D.
The rates of formation of metabolites (V) in human recombinant P450 systems were calculated using eq. 3:
 |
(3) |
A similar equation was used to determine the rate of metabolite formation in HLMs, V (picomoles per minute per milligram HLM protein).
The enzyme abundance (picomoles of P450 per milligram protein) of the various P450s in HLMs was obtained from previously reported data (Achour et al., 2014), wherein the authors performed correlation analysis of the abundance of P450 enzymes in data collated from 50 different laboratories representing donors worldwide. In vitro intrinsic clearance (CLint) values were calculated as in eq. 4:
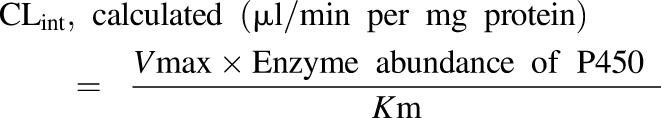 |
(4) |
An estimation of the microsomal CLint contribution for each P450 enzyme (percent enzyme contribution) was calculated using eq. 5:
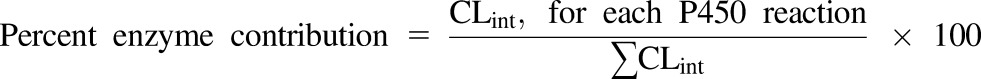 |
(5) |
The percentage of enzymatic activity remaining in the presence of chemical inhibitors was calculated according to eq. 6:
 |
(6) |
Luciferase Assays.
HepG2 cells grown in flasks were transfected with FLAG-hCAR1 and CYP2B6-luciferase reporter or CYP3A4-luciferase reporter as described previously (Cherian et al., 2015b) and incubated for 24 hours. The cells were then trypsinized, plated in 96-well plates, and treated with chemicals for 24 hour before measuring the luciferase reporter activity with SteadyLite firefly luciferase reagent and an EnVision plate reader (PerkinElmer, Waltham, MA). The percentage of CAR inhibition was calculated by setting 10 µM CINPA1 (positive control) as 100% inhibition and DMSO (negative control) as 0%. The final DMSO concentrations in all assays were maintained at 0.2%. To investigate the dose-response manner, the compounds were tested in a dose-response format (40 µM to 0.02 µM, 1:2 serial dilutions for 12 concentrations for CINPA1; 40 µM to 0.3125 µM, 1:2 serial dilutions for eight concentrations for Met1 and Met2).
Time-Resolved Fluorescence Resonance Energy Transfer Coactivator Recruitment Assay.
The effect of CINPA1 and the metabolites on the recruitment or repression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) binding to hCAR was evaluated by using a LanthaScreen time-resolved fluorescence resonance energy transfer (TR-FRET) assay (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions and as described previously (Cherian et al., 2015b; Lin et al., 2016). The final chemical concentrations ranged from 70 µM to 1.19 nM (1:3 serial dilutions for 11 concentration levels). DMSO and clotrimazole (42 µM) were used as negative (0% inhibition) and positive (100% inhibition) controls, respectively. The final DMSO concentration was 0.7% in all assay wells. Emission signals at 490 nm and 520 nm were collected on a PHERAstar plate reader (BMG Labtech, Durham, NC) and used to calculate the TR-FRET ratio normalized to positive and negative controls.
Docking Studies.
The Protein Data Bank (PDB) file for the hCAR–ligand-binding domain (LBD) protein crystal structure (PDB code 1XVP) was obtained from the RCSB Protein Data Bank (http://www.rcsb.org). The PyMOL molecular graphics system (http://www.pymol.org; Schrödinger, New York, NY) was used to remove all of the water molecules and ligand from the crystal structure. The ligand structures were generated in ChemBio3D (CambridgeSoft Corp., Waltham, MA) with energy minimization. The protein and ligand PDBQT files needed for docking were subsequently created in AutoDockTools (http://mgltools.scripps.edu/, version 1.5.6; Scripps Research Institute, La Jolla, CA). Docking of the ligands to hCAR-LBD was performed in the AutoDock Vina program (version 1.1.1) (Trott and Olson, 2010). The exhaustiveness value was set to 300, and the protein search space at the XYZ dimensions was set to 22 Å × 22 Å × 22 Å with the coordinates 34.231 (x), 59.568 (y), and 78.066 (z). Analysis and visualization of the docking results was performed in PyMOL with the assistance of LigPlot+ (European Bioinformatics Institute, Cambridge, UK) (Laskowski and Swindells, 2011).
Results
Identification of CINPA1 Metabolites in HLMs.
To identify the metabolites of CINPA1, we incubated CINPA1 (200 μM) with HLMs and analyzed the terminated reaction by using LC/MS. Figure 1A provides a representative UPLC trace of CINPA1 and its metabolites in an HLM incubate. Two major metabolite peaks, designated as Met1 and Met2, were separated at retention times of 1.55 and 1.24 minutes, respectively. Warfarin (the internal standard) and CINPA1 appeared at retention times of 1.68 and 1.56 minutes, respectively.
Fig. 1.

Identification of CINPA1 metabolites in HLMs and their structures. (A) LC/MS chromatograms obtained after in vitro incubation of CINPA1 with HLMs. CINPA1 (200 µM) was incubated with HLMs (0.5 mg/ml protein) and an NADPH regenerating system for 30 minutes at 37°C. The peaks corresponding to CINPA1, Met1, Met2, and the internal standard (warfarin) were monitored by UPLC with single ion reaction channels. The y-axis represents the percent relative abundance of each chemical in a single reaction well. (B) Structure of CINPA1 and two of its metabolites, Met1 and Met2. I.S., internal standard; MW, molecular weight; SIR, selected ion recording.
We confirmed the identity of the metabolite peaks in Fig. 1A by LC/MS analysis and by comparing LC retention times with those for synthetic reference compounds (Supplemental Fig. 3). When we compared the UPLC retention times of the CINPA1 metabolite peaks in microsomal incubates with those of ethyl [5-(N-ethylglycyl)-10,11-dihydro-5H-dibenzazepin-3-yl]carbamate, the retention time of Met1 was the same as that of ethyl [5-(N-ethylglycyl)-10,11-dihydro-5H-dibenzazepin-3-yl]carbamate (1.55 minutes). Similarly, the retention time of Met2 matched that of 1-(3-amino-10,11-dihydro-5H-dibenzazepin-5-yl)-2-(ethylamino)ethan-1-one (1.24 minutes). Figure 1B illustrates the structures of CINPA1 and its major metabolites, Met1 and Met2.
CINPA1 is rapidly converted to Met1 by microsomal enzymes and has a half-life of 0.43 ± 0.01 hours in HLMs and 0.40 ± 0.02 hours in MLMs (Table 1). Table 1 also shows the solubility of CINPA1 at pH 7.4, the Caco-2 permeability, and in vitro protein binding data for CINPA1. CAR protein shows remarkable species specificity in the ligands it binds to and we previously showed that mouse CAR does not seem to be inhibited by CINPA1 (Cherian et al., 2015b). Figure 2 depicts the time course for the formation of CINPA1 metabolites. The formation of Met2 showed a lag time relative to the formation of Met1, suggesting that Met2 is a secondary metabolite of CINPA1. When we incubated Met1 in HLMs and monitored the formation of Met2, Met1 was efficiently converted to Met2 (V = 65.4 ± 4.3 pmol/min per mg protein; Table 2). Our data provide evidence that Met2 is formed from CINPA1 by a stepwise reaction (i.e., first Met1 is formed, and then further ethyl carbamic acid is removed to form Met2). This appears to be major route of CINPA1 metabolism.
TABLE 1.
In vitro pharmacokinetic profiling of CINPA1
Data are presented as means ± S.D.
| Solubility, pH 7.4 | Caco-2 Permeability |
Microsomal Stability t1/2 |
Plasma Protein Binding |
||||
|---|---|---|---|---|---|---|---|
| Papp A/B | Papp B/A | Efflux Ratio | MLMs | HLMs | Mouse | Human | |
| mg/l | nm/s | h | % | ||||
| 17.40 ± 0.95 | 408.7 ± 17.7 | 323.2 ± 12.4 | 0.790 | 0.40 ± 0.02 | 0.43 ± 0.01 | 85.32 ± 0.50 | 92.37 ± 0.27 |
Pharmacokinetic parameters were obtained from aqueous solubility, Caco-2 permeability, microsomal stability, and plasma protein binding assays as described in the Materials and Methods in triplicated reaction wells. A/B, apical-to-basal permeability; B/A, basal-to-apical permeability; Papp, apparent permeability coefficient.
Fig. 2.

Time course for CINPA1 metabolite formation in HLMs. The microsomal stability of CINPA1 (10 µM) incubated in HLMs (0.5 mg/ml) and an NADPH regenerating system (final volume, 100 μl) was evaluated at 37°C. The reactions were terminated at time zero and after 15, 30, 60, 120, and 240 minutes of incubation and were analyzed by LC/MS, as detailed in the Materials and Methods. Each point is an average of triplicate reactions ± S.D. Error bars are present for all data points (note: some error bars are so small that they maybe masked by the squares or triangles).
TABLE 2.
Kinetic parameters obtained for metabolite formation in HLMs
Data are presented as apparent kinetic parameters ± S.D.
| Kinetic Parameter | Met1 Formation | Met2 Formation |
|---|---|---|
| Vmax (pmol/min per mg protein) | 464.2 ± 70.2 | 65.4 ± 4.3 |
| Km (µM) | 65.5 ± 19.5 | 10.3 ± 2.6 |
| Vmax/Km (µl/min per mg protein) | 7.09 | 6.36 |
Kinetic parameters for Met1 formation were estimated by fitting the velocity versus CINPA1 concentration curve to the Hill equation (represented in Fig. 3A). For Met2 formation from Met1, the single-site Michaelis–Menten equation was used (represented in Fig. 3B). In each case, the apparent kinetic parameter is presented as obtained from triplicated reaction wells.
Kinetic Analysis of Metabolite Formation in HLMs.
We first performed the kinetic analysis of CINPA1 metabolite formation in HLMs. The kinetic profiles of CINPA1 and Met1 metabolism are shown in Fig. 3. The formation rate of Met1 in HLMs revealed a sigmoidal saturation curve (Fig. 3A, left) that was fitted to a Hill equation (h = 1.319). The Eadie–Hofstee plot of this metabolite showed a curvilinear relationship (Fig. 3A, right) indicating positive cooperativity. Met2 formation using Met1 as a substrate in HLMs was measured, and the data were fitted into the Michaelis–Menten equation, as shown in Fig. 3B (left; essentially a Hill equation with the Hill slope constrained to 1). The Eadie–Hofstee plot of this metabolite mostly shows a linear relationship (Fig. 3B, right), suggesting the involvement of a single enzyme or more than one enzyme with similar affinities. The kinetic parameters obtained from these plots are listed in Table 2. A comparison of the goodness-of-fit values generated from these data does not implicate a substrate-inhibition enzyme model for these reactions. Met2 is formed from CINPA1 by a two-step reaction, but the kinetic parameters generated for the formation of this metabolite with CINPA1 as a substrate are likely to be compromised by the faster rate of the first reaction (N-deethylation) and the slower second reaction (the removal of ethyl carbamate).
Fig. 3.

Kinetics of metabolite formation in HLMs. (A and B) An increasing concentration of CINPA1 (0–200 µM) (A) or Met1 (0–200 µM) (B) was incubated with HLMs (0.5 mg/ml) and an NADPH regenerating system at 37°C for 30 minutes. The velocity of Met1 or Met2 formation (V, in picomoles per minute per milligram of HLM) versus the substrate concentration was fitted to a Hill equation (see the Materials and Methods) to obtain the kinetic parameters. Corresponding Eadie–Hofstee plots (velocity versus velocity/[substrate]) are shown in the insets. Each point represents the average of triplicate incubations ± S.D.
Identification of P450 Enzymes Involved in Metabolite Formation.
In an effort to pinpoint the major enzymes involved in the breakdown of CINPA1, we performed enzyme phenotyping assays using recombinant forms (Supersomes) of the major human P450 enzymes: CYP3A4, CYP2D6, CYP2C19, CYP2C9, CYP2B6, and CYP1A2. The ability of each enzyme to form Met1 from CINPA1 was measured by using 5 μM or 50 μM CINPA1 as the substrate. As shown in Fig. 4, A and B, CINPA1 was metabolized to Met1 most efficiently by CYP3A4, and to a lesser extent, by CYP2C19, in a concentration-dependent manner. Other isoforms (CYP2D6, CYP2C9, CYP2B6, and CYP1A2) had no detectable activity with respect to Met1 formation. The formation of Met2 from Met1 was also analyzed for each enzyme at two different Met1 concentrations. CYP2D6 appears to be the major enzyme involved in converting Met1 to Met2; CYP2C19 also exhibits slight activity (Fig. 4, C and D). The other P450s investigated (CYP3A4, CYP2C9, CYP2B6, and CYP1A2) failed to catalyze this metabolic conversion. CINPA1 or Met1 metabolism in these recombinant enzyme assays was compared with that of the control substrates specifically recommended for each enzyme (phenacetin O-deethylation for CYP1A2, tolbutamide hydroxylation for CYP2C9, S-mephenytoin hydroxylation for CYP2C19, efavirenz hydroxylation for CYP2B6, dextromethorphan O-demethylation for CYP2D6, and midazolam hydroxylation for CYP3A4).
Fig. 4.

Metabolism of CINPA1 and Met1 by a panel of recombinant human P450s. (A and B) CINPA1 (5 or 50 µM) was incubated with a panel of P450 isoforms (24–41 pmol/ml) and an NADPH regenerating system at 37°C for 30 minutes. The rate of formation of Met1 (in picomoles of Met1 per minute per picomoles of P450) ± S.D. for duplicates measurements is represented. (C and D) Met1 (5 or 50 µM) was incubated with a panel of P450 isoforms and an NADPH regenerating system at 37°C for 30 minutes. Data are presented as the mean rate of formation of Met2 (in picomoles of Met2 per minute per picomoles of P450) ± S.D. for triplicate wells.
Kinetic Analysis of Metabolite Formation in Recombinant Human P450s.
After we identified CYP3A4 and CYP2D6 as the major P450s responsible for converting CINPA1 to Met1 and Met1 to Met2, respectively (Fig. 4), we performed a full kinetic analysis of metabolite formation by using the same recombinant human P450s (Fig. 5). As shown in Fig. 5A for CYP3A4, the formation rates of Met1 versus CINPA1 concentration fitted best to a single-site Michaelis–Menten equation. The Vmax/Km values obtained for recombinant CYP3A4 and CYP2C19 (6.53 and 0.62 μl/min per pmol P450, respectively; Table 3) suggest that although CYP3A4 is the main enzyme involved in Met1 formation, other enzymes may catalyze CINPA1 metabolism in HLMs. Because CYP2C19 showed some activity with regard to Met1 formation (Fig. 4, A and B), we examined the kinetics for the formation rate of Met1 in five other human P450 isoforms and compared the data with the formation rate with CYP3A4. The respective kinetic parameters derived from fitting the data to a Hill equation are shown in Table 3. Accordingly, the in vitro efficiency (Vmax/Km value) for Met1 formation by CYP3A4 was manyfold (10- to 82-fold) higher than that of the other isoforms. Most CINPA1 (96%; Table 3) was metabolized by CYP3A4, rather than by the other enzymes examined here.
Fig. 5.

Kinetics of metabolite formation in recombinant human P450s. (A) An increasing concentration of CINPA1 (0–200 µM) was incubated with recombinant human CYP3A4 (41 pmol/ml) and an NADPH regenerating system at 37°C for 30 minutes. (B) Met1 (0–200 µM) was incubated with recombinant human CYP2D6 (41 pmol/ml) and an NADPH regenerating system at 37°C for 30 minutes. The velocity of metabolite formation (V, in picomoles per minute per picomoles of enzyme) versus the substrate concentration was fitted to a Michaelis–Menten equation (see the Materials and Methods) to obtain the kinetic parameters. Corresponding Eadie–Hofstee plots (velocity versus velocity/[substrate]) are shown in the insets. Each point represents the average of triplicate incubations ± S.D.
TABLE 3.
Kinetic parameters obtained for metabolite formation in P450s
| P450 Enzyme | Relative Enzyme Abundancea | Vmax | Km | Vmax/Km | CLint | Enzyme Contribution |
|---|---|---|---|---|---|---|
| pmol P450/mg protein | pmol/min per pmol P450 | µM | µl/mg protein per min | % | ||
| Formation of Met1 from CINPA1 | ||||||
| CYP3A4 | 93 | 83.22 | 12.74 | 6.53 | 607.49 | 96.28 |
| CYP2D6 | 12.6 | 20.43 | 261.40 | 0.08 | 0.98 | 0.16 |
| CYP2C19 | 11 | 13.09 | 21.06 | 0.62 | 6.84 | 1.08 |
| CYP2C9 | 61 | 3.92 | 32.63 | 0.12 | 7.33 | 1.16 |
| CYP2B6 | 16 | 22.50 | 80.58 | 0.28 | 4.47 | 0.71 |
| CYP1A2 | 39 | 4.17 | 41.89 | 0.10 | 3.88 | 0.61 |
| Formation of Met2 from Met1 | ||||||
| CYP3A4 | 93 | — | — | — | — | — |
| CYP2D6 | 12.6 | 89.67 | 119.20 | 0.752 | 9.48 | 94.60 |
| CYP2C19 | 11 | 11.98 | 745.90 | 0.016 | 0.18 | 1.76 |
| CYP2C9 | 61 | 1.41 | 303.60 | 0.005 | 0.28 | 2.84 |
| CYP2B6 | 16 | — | — | — | — | — |
| CYP1A2 | 39 | 0.52 | 250.90 | 0.002 | 0.08 | 0.80 |
Kinetic parameters calculated for formation of Met1 from CINPA1 were estimated by fitting the velocity versus CINPA1 concentration curve for each enzyme to the Hill equation (curves represented in Supplemental Fig. 4A, as obtained from triplicate reaction wells). Kinetic parameters calculated for formation of Met2 from Met1 were estimated by fitting the velocity versus concentration curve in each enzyme, with Met1 as substrate, to the Hill equation (curves represented in Supplemental Fig. 4B, as obtained from triplicate reaction wells). Dashes indicate that the data for Met1 metabolism in CYP3A4 and CYP2B6 did not fit to the Hill equation and hence Vmax and Km could not be obtained (flagged as ambiguous by GraphPad Prism software).
The enzyme abundance of various P450s in HLMs was obtained from data reported by Achour et al. (2014) and is a meta-analysis based combination of multiple studies using numerous microsomal donors.
The analysis of Met1 breakdown to Met2 was performed in recombinant CYP2D6 enzyme assays, and the formation rate of Met2 was fitted into a single-site Michaelis–Menten equation (Fig. 5B). The rate of Met2 formation from Met1 was slower than that of Met1 formation from CINPA1, which is consistent with the observation shown in Fig. 2. This apparent difference in rates might reflect a better metabolic stability of Met1 in the microsomes and/or a lower binding efficiency of CYP2D6 for Met1. The catalytic efficiency (Vmax/Km) of Met1 in the CYP2D6 enzyme assay (0.75 μl/min per pmol P450; Table 3) was much lower than expected, suggesting that other enzymes not examined here might have a role in Met1 metabolism in HLMs. In addition, CYP2D6 is considered a low-capacity, high-affinity enzyme that can be easily saturated by substrate (Bertilsson et al., 2002). It should be noted that the CYP2D6 enzyme we employed is the Val374 isoform (catalog no. 456217; Corning). Again, we measured Met2 formation in other human P450 isoforms and compared the results with those for CYP2D6 (Table 3). Although CYP2C19 makes some contribution to Met2 formation, CYP2D6 appears to be the major isoform capable of metabolizing Met1 to Met2, accounting for 94.6% of Met1 metabolism (compared with the other enzymes examined) (Table 3). The kinetic parameters obtained from the six P450s tested are presented in Table 3, and the actual data graphs are illustrated in Supplemental Fig. 4.
The data obtained from the recombinant P450 assays were corroborated by incubating substrates with isoform-specific inhibitors of the major P450 enzymes in HLMs and are illustrated in Fig. 6. We used 5 μM ketoconazole to inhibit CYP3A4 enzyme activity, 10 μM quinidine for CYP2D6, 25 μM ticlopidine for CYP2C19, 5 μM sulfaphenazole for CYP2C9, 25 μM quercetin for CYP2C8, and 25 μM thioTEPA for CYP2B6 and 5 μM PCPA for CYP1A2 inhibition. All inhibitors were preincubated with the NADPH regenerating system and HLMs for 15 minutes at 37°C before the reaction was initiated by adding CINPA1 or Met1. The incubation then continued for an additional 30 minutes. Met1 formation from CINPA1 was greatly hindered in the presence of the CYP3A4 inhibitor ketoconazole (Fig. 6A), whereas the conversion of Met1 to Met2 was most suppressed in the presence of the CYP2D6 inhibitor quinidine (Fig. 6B). Neither CINPA1 nor its metabolites exhibited any detectable inhibition of the recombinant enzymes tested using their respective isoform-diagnostic substrates. For example, CINPA1 did not inhibit the activity of the recombinant CYP3A4 enzyme as observed in reactions using midazolam as the substrate (Supplemental Fig. 5A). Preincubation of CYP3A4 with ketoconazole (isoform-specific inhibitor of CYP3A4) completely inhibited midazolam metabolism but CINPA1, Met1, or Met2 (5 µM) did not alter CYP3A4-mediated midazolam hydroxylation. In HLMs, preincubation with an even higher concentration (200 µM) of CINPA1, Met1, or Met2 only marginally affected midazolam hydroxylation, whereas ketoconazole strongly inhibited midazolam metabolism (Supplemental Fig. 5B).
Fig. 6.

Metabolite formation in HLMs in the presence of specific P450 enzyme inhibitors. Specific inhibitors of various P450 isoforms (or DMSO control) were preincubated at the indicated concentrations with HLMs (0.5 mg/ml) and an NADPH regenerating system at 37°C for 15 minutes. (A and B) 5 µM CINPA1 (A) or Met1 (B) was added to the reaction mixtures, which were then incubated for an additional 30 minutes at 37°C. The reactions were terminated with acetonitrile and analyzed by LC/MS. The enzyme activity was calculated by measuring the amount of metabolite (Met1 or Met2) formed. (C) An esterase inhibitor, BNPP (250 µM), or DMSO (control) was preincubated with HLMs and an NADPH regenerating system at 37°C for 15 minutes as described earlier. Met1 (5 µM) was added to this mixture as the substrate, and the reaction was terminated after an additional 30 minutes of incubation at 37°C. The percent enzyme activity was calculated by normalizing the amount of metabolite formed in the absence of the inhibitor (DMSO) to 100%. Each bar represents the average of triplicate reaction wells ± S.D. One-way analysis of variance was used to compare the percent enzyme activity in the presence of each inhibitor to DMSO control. *P < 0.05. Unmarked bars indicate no significant change observed. The inhibitors used were ketoconazole (KTZ; CYP3A4), quinidine (Quin; CYP2D6), ticlopidine (Ticlo; CYP2C19), sulfaphenazole (SFZ; CYP2C9), quercetin (Querc; CYP2C8), thioTEPA (TT; CYP2B6), PCPA (CYP1A2), and BNPP (esterase). ns, not significant.
We considered the possibility that an esterase catalyzes the conversion of Met1 to an intermediate compound before Met2 formation. To test this hypothesis, we used the esterase inhibitor BNPP in HLM assays with 5 μM Met1 as substrate. The microsome-NADPH system was preincubated with BNPP (or DMSO control) for 15 minutes at 37°C before the reaction was initiated with Met1. We found no change in Met2 formation in the presence of the BNPP (Fig. 6C), indicating that esterases are probably not involved in the breakdown of Met1.
Effect of CINPA1 Metabolites on hCAR-Mediated Transcriptional Activity.
CINPA1 is a potent inhibitor of hCAR. Because of the ligand promiscuity of CAR, we investigated the effect of the metabolites of CINPA1 on CAR activity, first in HepG2 cells transiently expressing hCAR, along with a CYP2B6-luciferase or CYP3A4-luciferase reporter (the ectopically expressed hCAR activates the CYP2B6 and CYP3A4 promoter, respectively). Whereas CINPA1 is capable of inhibiting hCAR function, as revealed by its inhibiting the promoter activity of CYP2B6 and CYP3A4, Met1 was only a weak inhibitor and Met2 was inactive (Fig. 7, A and B). The reduced activity of the metabolites was confirmed by a TR-FRET assay using CAR-LBD and a coactivator peptide (PGC-1α), interaction between which is enhanced by an activating ligand but reduced by an inhibiting ligand. CINPA1 efficiently inhibits CAR-LBD interaction with the coactivator peptide with an IC50 of 0.6 μM, suggesting that CINPA1 interacts with the LBD of CAR (Cherian et al., 2015b). In comparison, Met1 is a much weaker inhibitor of CAR-coactivator interaction, with an approximate IC50 of 28 μM, which is about 45-fold higher than that of CINPA1 in the same assay (Fig. 7C). Since the dose-response curve for Met1 does not plateau at its highest concentrations, the IC50 value obtained is approximate at best. Met2 has little or no effect on CAR interaction with this coactivator peptide, confirming the observation made in the transcriptional activation assay (Fig. 7, A and B).
Fig. 7.

Effect of CINPA1 metabolites on hCAR-mediated transactivation. (A and B) HepG2 cells were transfected with plasmids expressing hCAR1 and CYP2B6-luciferase reporter (A) or CYP3A4-luc reporter (B). After 24-hour incubation, the cells were treated with increasing concentrations of CINPA1 (0.02–40 µM) or Met1 or Met2 (0.3125–40 µM). Luciferase reporter activity was measured 24 hours after treatment. The activity in DMSO-treated (negative control) samples was set as 0% inhibition; the activity in samples treated with 40 µM CINPA1 (positive control) was set as 100% hCAR inhibition. (C) CAR binding to PGC-1α coactivator peptide was determined by using the LanthaScreen TR-FRET coactivator recruitment assay. Fluorescein-labeled PGC-1α peptide (125 nM) was complexed with GST-hCAR-LBD (5 nM) and Tb–anti-GST antibody (5 nM), as described in the Materials and Methods. CINPA1, Met1, or Met2 at concentrations ranging from 70 µM to 1.19 nM (1:3 dilutions for 11 concentration levels) were added to the peptide mixture, and TR-FRET emissions at 490 and 520 nm were measured. The percentage inhibition of binding was calculated for each treatment, and the data were normalized to the positive control (42 µM clotrimazole, 100% inhibition) and negative control (DMSO, 0% inhibition). Each data point represents the average of at least three replicates ± S.D. GST, glutathione S-transferase; luc, luciferase.
Docking of the Ligands to hCAR-LBD.
Studies determining the docking of CINPA1 and Met1 to the hCAR-LBD provide a rationale for the differences in potency between both of these ligands and the lack of activity observed for Met2, in spite of their structural similarities. According to the models, the ligands reside in the binding pocket of the protein (Fig. 8), with a potentially important hydrogen bond interaction between the ethyl carbamate of CINPA1 or Met1 with either Asn165 or His203 (Fig. 8A). In addition, the ethyl component of the ethyl carbamate moiety in CINPA1 (Fig. 8B) and Met1 extends to a hydrophobic pocket, which would enhance ligand binding.
Fig. 8.

Docking of CINPA1 and Met1 to hCAR-LBD. (A) Potential hydrogen bonding between the ethyl carbamate of CINPA1 and Asn165 or His203. Met1, but not Met2, would also display similar interactions because of its ethyl carbamate moiety. The carbon atoms of Asn165 and His203 are represented in pink, and potential hydrogen bonding is indicated with dash lines. (B) Interaction of the ethyl component of the ethyl carbamate moiety in CINPA1 (represented as white spheres) with relevant protein residues, which would also represent a similar hydrophobic contact by Met1. (C) Interaction of the diethyl amino group of CINPA1 (white spheres) with relevant protein residues. (D) Interaction of the ethyl amino group of Met1 (white spheres) with one of the hydrophobic pockets. The ligands are shown as sticks and the relevant protein residues are shown as surface representations. Ligand and protein (surface representation) carbon, oxygen, nitrogen, and sulfur atoms are shown in white, red, blue, and yellow, respectively.
The diethyl amino group of CINPA1 interacts with two highly hydrophobic cavities (Fig. 8C), whereas the ethyl amino group of Met1 interacts with only one of them (Fig. 8D). This single difference between CINPA1 and Met1 is sufficient to explain the weaker potency of Met1. Because the ethyl carbamate group is absent and the diethyl amino moiety is simplified to a monoethyl counterpart in Met2, noticeable binding of the ligand is not observed because of a lack of hydrogen bonding and the above-described hydrophobic contacts.
Discussion
CINPA1 is a recently identified potent small-molecule inhibitor of hCAR (Cherian et al., 2015b), which is unique in its ability to not activate PXR. In stability assays, CINPA1 has a half-life of approximately 26 minutes in HLMs. Here, we detailed the identification of CINPA1 metabolites and the enzymes involved in this catalytic process. The two main metabolites of CINPA1 were denoted Met1 and Met2. Compared with CINPA1 in terms of their action on CAR function, Met1 is a weak inverse agonist of hCAR in transactivation assays, whereas Met2 has almost no effect on CAR. CINPA1 strongly disrupts the interaction between CAR-LBD and a coactivator peptide in biochemical assays (IC50 = 0.6 μM), where it behaves as a strong inverse agonist. Consistent with its weak inhibition of CAR function, Met1 has an IC50 of 28 µM, whereas Met2 shows very little activity even at the highest concentrations tested. We studied the docking of CINPA1 and the metabolites to the hCAR-LBD structure to help us further understand the basis for this apparent structure-activity relationship. CINPA1 makes several prominent contacts with the protein, including potential hydrogen bonding with either Asn165 or His203 and hydrophobic interactions. Met1, which has an ethyl amino group instead of a diethyl amino group found in CINPA1 (Fig. 8D) but can possibly still interact with the LBD through the ethyl carbamate moiety, displays lower potency in functional assays. Met2, which lacks the ethyl carbamate group and also has the diethyl amino moiety simplified to a monoethyl counterpart, is incapable of binding to CAR because of a lack of hydrogen bonding and hydrophobic contacts with CAR. CAR mutants involving the putative amino acids that form hydrogen bonds with these compounds will be used to validate our docking studies in our future studies.
We present here a detailed characterization of the P450 enzymes involved in the in vitro human metabolism of CINPA1. We demonstrated that: 1) CINPA1 undergoes primary N-deethylation to Met1, predominantly catalyzed by CYP3A4; 2) secondary metabolism of Met1 to Met2 is primarily catalyzed by CYP2D6; and 3) Met1 is a weak inverse agonist of CAR, whereas Met2 is mostly inactive as interaction with the CAR-LBD is curtailed. Our findings provide direct evidence that Met2 is a secondary metabolite of CINPA1, in that its formation proceeds stepwise from CINPA1, with Met1 as an intermediate.
Our data provide strong evidence that CYP3A4 is the major P450 isoform involved in the rapid conversion of CINPA1 to Met1. First, the formation rates of Met1 from CINPA1 were potently inhibited by ketoconazole (Fig. 6A), a specific inhibitor of CYP3A4 (Walsky and Obach, 2004). Second, CYP3A4 formed Met1 from CINPA1 (Figs. 4A and 5A) with the highest specific activity. CYP3A4 is reportedly involved in N-deethylation reactions (Fabre et al., 1993; Wang et al., 2000), and Met1 is formed from CINPA1 by an N-deethylation reaction. We also noted that recombinant human CYP2C19 could convert CINPA1 to Met1, but the contributions of this P450 isoform to CINPA1 metabolism appear to be minor, because a CYP2C19-specific inhibitor (ticlopidine) only marginally inhibited the rate of formation of Met1 in HLMs (Fig. 6A). The estimated CLint of CINPA1 metabolism by CYP2C19 is only 12.04 μl/min per mg protein, which is more than 80-fold lower than the contribution of CYP3A4 (Table 3). Because of the high efficiency and abundance of CYP3A4 in the liver, almost 96% of CINPA1 appears to be metabolized by CYP3A4 as opposed to one of the other five isoforms examined (i.e., CYP1A2, CYP2B6, CYP2C9, CYP2C19, and CYP2D6). Pre-incubation with CINPA1, Met1, or Met2 at equimolar or higher concentrations as midazolam did not inhibit CYP3A4-mediated midazolam hydroxylation, unlike ketoconazole. This suggests that as a CYP3A4 substrate, CINPA1 does not compete strongly with midazolam which is a diagnostic substrate of CYP3A4 enzymatic activity (Supplemental Fig. 5).
Our data also indicate that CYP2D6 is one of the isoforms involved in converting Met1 to Met2 (Figs. 4B and 5B). This is validated by the complete inhibition of Met2 formation in the presence of the CYP2D6-specific inhibitor quinidine (Fig. 6B). However, the lower in vitro catalytic efficiency rate of Met1 metabolism in CYP2D6 enzyme assays (Table 3) suggests that there may be other phase I enzymes involved in Met2 formation.
Almost every enzyme involved in drug metabolism is subject to common genetic polymorphisms that may contribute to interindividual variability in drug response. CYP2D6, in particular, is highly polymorphic, which can translate into clinically relevant variations in the enzyme activity of CYP2D6. The vagaries of CYP2D6 polymorphisms have been extensively observed in the case of opioid-mediated pain management and in outcomes among women treated with tamoxifen for early stage breast cancer (Paar et al., 1997; Schroth et al., 2009). We used a single version of CYP2D6, the commercially available CYP2D6 (Val374) Supersome, to study the conversion of Met1 to Met2; hence, we cannot gauge the variation in Met2 formation in the presence of other CYP2D6 polymorphic forms. The Km for Met1 binding to CYP2D6 was quite high at 119 μM, suggesting that Met1 is not a high-affinity substrate of CYP2D6. Coupled with the fact that CYP2D6 is a low-capacity P450, this explains the lower rate of Met1 conversion to Met2. We think it is highly possible that Met2 formation in HLMs is also driven by a different phase I enzyme or an array of enzymes not examined here.
The small but flexible LBD of CAR allows it to bind and respond to xenobiotics with a variety of chemical structures (Cherian et al., 2015a). The structurally similar CINPA1, Met1, and Met2 display potent, weak, and no activity, respectively, in inhibiting the constitutive hCAR. Although they share the same chemical core structurally, CINPA1, Met1, and Met2 have vastly different effects on CAR binding and inhibition. Thus, these chemicals represent the first set of structurally related but functionally different compounds useful as tools for studying structure-activity relationships in CAR, which is known for its ligand promiscuity.
These data provide a scientific basis upon which to design focused preclinical studies that will help in understanding the pharmacokinetic and pharmacogenetic factors influencing CINPA1 efficacy and safety. Identifying the main metabolites of CINPA1 and analyzing their functional effect on hCAR provide useful information for guiding and interpreting data from in vivo studies of CINPA1. Furthermore, these studies enable structure-activity studies of hCAR, which might lead to the development of hCAR modulators with specific properties.
Acknowledgments
The authors thank Dr. Hongbing Wang for kindly providing the CYP2B6-luciferase construct, Dr. David Moore for kindly providing the hCAR1 construct, WuXi AppTec for technical assistance in chemical synthesis, other members of the Chen research laboratory for valuable discussions, Dr. Philip M. Potter for discussions regarding esterases, and Dr. Keith A. Laycock for editing the manuscript.
Abbreviations
- BNPP
bis(p-nitrophenyl) phosphate
- CAR
constitutive androstane receptor
- CINPA1
ethyl (5-(diethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl)carbamate
- CITCO
(E)-1-[6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazol-5-yl]-N-[(3,4-dichlorophenyl)methoxy]methanimine
- CLint
intrinsic clearance
- DMSO
dimethylsulfoxide
- ESI
electrospray ionization
- FBS
fetal bovine serum
- hCAR
human constitutive androstane receptor
- HLM
human liver microsome
- LBD
ligand-binding domain
- LC
liquid chromatography
- Met1
metabolite 1 (ethyl [5-(N-ethylglycyl)-10,11-dihydro-5H-dibenzo[b,f]azepin-3-yl]carbamate)
- Met2
metabolite 2 1-(3-amino-10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-2-(ethylamino)ethan-1-one
- MLM
mouse liver microsome
- MS
mass spectrometry
- P450
cytochrome P450
- PCPA
tranylcypromine
- PDB
Protein Data Bank
- PGC-1α
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PXR
pregnane X receptor
- thioTEPA
N,N′,N′′-triethylenethiophosphoramide
- TLC
thin-layer chromatography
- TR-FRET
time-resolved fluorescence resonance energy transfer
- UPLC
ultra-performance liquid chromatography
Authorship Contributions
Participated in research design: Cherian, Chen.
Conducted experiments: Cherian, Yang, Chai, Lin.
Performed data analysis: Cherian, Yang, Chai, Lin.
Wrote or contributed to the writing of the manuscript: Cherian, Chai, Lin, Chen.
Footnotes
This research was supported by ALSAC, St. Jude Children’s Research Hospital, the National Institutes of Health National Institute of General Medical Sciences [Grants R01GM086415, R01GM110034, and R35GM118041], and the National Institutes of Health National Cancer Institute [Grant P30CA21765].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Achour B, Barber J, Rostami-Hodjegan A. (2014) Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: a meta-analysis. Drug Metab Dispos 42:1349–1356. [DOI] [PubMed] [Google Scholar]
- Bertilsson L, Dahl ML, Dalén P, Al-Shurbaji A. (2002) Molecular genetics of CYP2D6: clinical relevance with focus on psychotropic drugs. Br J Clin Pharmacol 53:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian MT, Chai SC, Chen T. (2015a) Small-molecule modulators of the constitutive androstane receptor. Expert Opin Drug Metab Toxicol 11:1099–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian MT, Lin W, Wu J, Chen T. (2015b) CINPA1 is an inhibitor of constitutive androstane receptor that does not activate pregnane X receptor. Mol Pharmacol 87:878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desta Z, Kerbusch T, Soukhova N, Richard E, Ko JW, Flockhart DA. (1998) Identification and characterization of human cytochrome P450 isoforms interacting with pimozide. J Pharmacol Exp Ther 285:428–437. [PubMed] [Google Scholar]
- Evans WE, Relling MV. (1999) Pharmacogenomics: translating functional genomics into rational therapeutics. Science 286:487–491. [DOI] [PubMed] [Google Scholar]
- Fabre G, Julian B, Saint-Aubert B, Joyeux H, Berger Y. (1993) Evidence for CYP3A-mediated N-deethylation of amiodarone in human liver microsomal fractions. Drug Metab Dispos 21:978–985. [PubMed] [Google Scholar]
- Fukami T, Yokoi T. (2012) The emerging role of human esterases. Drug Metab Pharmacokinet 27:466–477. [DOI] [PubMed] [Google Scholar]
- Goodwin B, Hodgson E, D’Costa DJ, Robertson GR, Liddle C. (2002) Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol Pharmacol 62:359–365. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, Gillam EMJ, Martin MV, Baba T, Kim BR, Shimada T, Raney KD, Yun CH. (1994) The importance of cytochrome P450 3A enzymes in drug metabolism, in Assessment of the Use of Single Cytochrome P450 Enzymes in Drug Research (Waterman MR, Hildebrand M. eds) pp 161–186, Springer-Verlag, Berlin. [Google Scholar]
- Hodgson E. (2001) In vitro human phase I metabolism of xenobiotics I: pesticides and related chemicals used in agriculture and public health, September 2001. J Biochem Mol Toxicol 15:296–299. [DOI] [PubMed] [Google Scholar]
- Honkakoski P, Sueyoshi T, Negishi M. (2003) Drug-activated nuclear receptors CAR and PXR. Ann Med 35:172–182. [DOI] [PubMed] [Google Scholar]
- Kazui M, Nishiya Y, Ishizuka T, Hagihara K, Farid NA, Okazaki O, Ikeda T, Kurihara A. (2010) Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos 38:92–99. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51:2778–2786. [DOI] [PubMed] [Google Scholar]
- Li L, Sinz MW, Zimmermann K, Wang H. (2012) An insulin-like growth factor 1 receptor inhibitor induces CYP3A4 expression through a pregnane X receptor-independent, noncanonical constitutive androstane receptor-related mechanism. J Pharmacol Exp Ther 340:688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Yang L, Chai SC, Lu Y, Chen T. (2016) Development of CINPA1 analogs as novel and potent inverse agonists of constitutive androstane receptor. Eur J Med Chem 108:505–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier B, de Visser SP, Shaik S. (2004) Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem Rev 104:3947–3980. [DOI] [PubMed] [Google Scholar]
- Oda S, Fukami T, Yokoi T, Nakajima M. (2015) A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug Metab Pharmacokinet 30:30–51. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P, De Voss J. (2005) Substrate oxidation by cytochrome P450 enzymes, in Cytochrome P450 (Ortiz de Montellano P. ed) pp 183–245, Springer, New York. [Google Scholar]
- Paar WD, Poche S, Gerloff J, Dengler HJ. (1997) Polymorphic CYP2D6 mediates O-demethylation of the opioid analgesic tramadol. Eur J Clin Pharmacol 53:235–239. [DOI] [PubMed] [Google Scholar]
- Rae JM, Soukhova NV, Flockhart DA, Desta Z. (2002) Triethylenethiophosphoramide is a specific inhibitor of cytochrome P450 2B6: implications for cyclophosphamide metabolism. Drug Metab Dispos 30:525–530. [DOI] [PubMed] [Google Scholar]
- Rakesh BD, Bruhn D, Madhura DB, Maddox M, Lee RB, Trivedi A, Yang L, Scherman MS, Gilliland JC, Gruppo V, et al. (2012) Antitubercular nitrofuran isoxazolines with improved pharmacokinetic properties. Bioorg Med Chem 20:6063–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroth W, Goetz MP, Hamann U, Fasching PA, Schmidt M, Winter S, Fritz P, Simon W, Suman VJ, Ames MM, et al. (2009) Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA 302:1429–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueyoshi T, Kawamoto T, Zelko I, Honkakoski P, Negishi M. (1999) The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J Biol Chem 274:6043–6046. [DOI] [PubMed] [Google Scholar]
- Timsit YE, Negishi M. (2007) CAR and PXR: the xenobiotic-sensing receptors. Steroids 72:231–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida M, Fukazawa T, Yamazaki Y, Hashimoto H, Miyamoto Y. (2009) A modified fast (4 day) 96-well plate Caco-2 permeability assay. J Pharmacol Toxicol Methods 59:39–43. [DOI] [PubMed] [Google Scholar]
- Walsky RL, Obach RS. (2004) Validated assays for human cytochrome P450 activities. Drug Metab Dispos 32:647–660. [DOI] [PubMed] [Google Scholar]
- Wang H, Faucette S, Sueyoshi T, Moore R, Ferguson S, Negishi M, LeCluyse EL. (2003) A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. J Biol Chem 278:14146–14152. [DOI] [PubMed] [Google Scholar]
- Wang JS, Backman JT, Taavitsainen P, Neuvonen PJ, Kivistö KT. (2000) Involvement of CYP1A2 and CYP3A4 in lidocaine N-deethylation and 3-hydroxylation in humans. Drug Metab Dispos 28:959–965. [PubMed] [Google Scholar]
- Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. (2003) The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther 306:287–300. [DOI] [PubMed] [Google Scholar]
- Willson TM, Kliewer SA. (2002) PXR, CAR and drug metabolism. Nat Rev Drug Discov 1:259–266. [DOI] [PubMed] [Google Scholar]
- Zanger UM, Schwab M. (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138:103–141. [DOI] [PubMed] [Google Scholar]