

Abstract
Accumulation of connective tissue is a typical feature of chronic liver diseases. Decorin, a small leucine-rich proteoglycan, regulates collagen fibrillogenesis during development, and by directly blocking the bioactivity of transforming growth factor-β1 (TGFβ1), it exerts a protective effect against fibrosis. However no in vivo investigations on the role of decorin in liver have been performed before. In this study we utilized decorin-null (Dcn−/−) mice to establish the role of decorin in experimental liver fibrosis and repair. Not only the extent of experimentally-induced liver fibrosis was more severe in Dcn−/− animals, but also the healing process was significantly delayed vis-à-vis wild-type mice. Collagen I, III, and IV mRNA levels in Dcn−/−livers were higher than those of wild-type livers only in the first two months, but no difference was observed after four months of fibrosis induction, suggesting that the elevation of these proteins reflects a specific impairment of their degradation. Gelatinase assays confirmed this hypothesis as we found decreased MMP-2 and MMP-9 activity and higher expression of TIMP-1 and PAI-1 mRNA in Dcn−/− livers. In contrast, at the end of the recovery phase increased production rather than impaired degradation was found to be responsible for the excessive connective tissue deposition in livers of Dcn−/− mice. Higher expression of TGFβ1-inducible early responsive gene in decorin-null livers indicated enhanced bioactivity of TGFβ1 known to upregulate TIMP-1 and PAI-1, as well. Morever, two main axes of TGFβ1-evoked signaling pathways were affected by decorin deficiency, namely the Erk1/2 and Smad3 were activated in Dcn−/− samples, while no significant difference in phospho-Smad2 was observed between mice with different genotypes. Collectively, our results indicate that the lack of decorin favors the development of hepatic fibrosis and attenuates its subsequent healing process at least in part by affecting the bioactivity of TGFβ1.
Keywords: Decorin, Erk1/2, Liver Fibrosis, MMP-2, MMP-9, TGFβ1
Accumulation of connective tissue is a typical feature of several chronic diseases1–3. The development of fibrosis is the result of imbalance between the synthesis and degradation of extracellular matrix proteins4. Liver is especially prone to fibrotic remodeling5 because this organ is extremely susceptible to chronic and acute viral injury, toxic insults like ethanol, and storage and autoimmune diseases6–8. Without eradication of the etiological agent, liver fibrosis normally progresses to cirrhosis and destroys the normal architecture of the liver culminating with parenchymal and vascular decompensation of the organ. Furthermore, hepatic cirrhosis increases the probability of development of hepatocellular carcinoma9, 10.
In spite of recent progress, human liver cirrhosis is practically still incurable11–13. Our better understanding of pro- and antifibrotic mechanisms failed to uncover curative approaches to combat this debilitating disease. Among factors implicated in the induction and maintenance of excess matrix production, TGFβ1 occupies a central position14–18. This peptide growth factor is capable of activating fibroblast and stellate cells, inhibiting their apoptosis and forcing them to synthesize excess amount of matrix proteins17. This implies that inactivation of TGFβ1 could be a strategic approach in the management of liver fibrosis19.
Decorin, a small leucine-rich proteoglycan (SLRP)20–22 can block the proliferation of cells that are dependent on TGFβ1 for their in vitro growth23. This observation has been exploited in various in vivo models of experimental glomerulonephritis and nephrosclerosis, as well as in scar formation models24, 25. On the other hand, there is no conclusive information on the role of decorin in the fibrotic remodeling of the liver26. Decorin, as a regulator of matrix assembly targets not only TGFβ1, but it is also involved in the maturation of collagen fibrils, as well27–29. Furthermore, it serves as ligand for growth factor receptors modulating signals initiated on EGFR, IGFR and Met receptors30–37. A central question is whether decorin-evoked modulation of signal transduction is directly implicated in fibrogenesis or is acting indirectly via controlling cell proliferation.
The main goal of our current research was to determine whether decorin could play a protective role in liver fibrosis or whether the accumulation of this proteoglycan observed in human cirrhotic liver samples would be just due to a bystander effect. Furthermore, we wanted to determine whether the hypothesized role for decorin could be attributed to modulation of the TGFβ signaling pathway in the liver. In this study, we utilized an experimental animal model of liver fibrosis based on decorin-null and wild type animals sharing the same genetic background. Our results provide evidence that decorin plays a protective role in liver fibrogenesis insofar as the absence of decorin correlates with enhanced hepatic fibrosis and attenuation of its subsequent healing process.
MATERIALS AND METHODS
Decorin-null Mice
All animal experiments were performed under an institutionally approved protocol. Decorin deficient mice were generated as previously described29. In brief, the inactivation of the decorin gene was achieved by targeted disruption of the exon 2 inserting a PGK-Neo cassette. Two male and two female C57Bl/6 mice heterozygous for decorin gene (Dcn+/−), which were backcrossed into C57Bl/6 background for nine generations, were bred until homozygosity. The genotype of the offspring was determined by PCR. Tail DNA was isolated by using high salt method. Subsequently 3 primers were applied, sense and antisense specific for the exon 2, and one corresponding to the PGK-Neo cassette. PCR products were analyzed by 2% agarose gel electrophoresis.
Thioacetamide Treatment
For induction of hepatic fibrosis, we utilized a total of 48 one month-old male mice, all in a C57Bl/6 background. Twenty-four wild-type, and 24 Dcn−/− animals were exposed to thioacetamide (TA) dissolved in drinking water (150 mg/L). To obtain fully-developed hepatic fibrosis, the animals were subjected to TA treatment for four months followed by an additional four months (recovery phase) in which the drug was withdrawn. Three animals were sacrificed at each time point at monthly intervals. Age-matched untreated animals with identical genetic background served as controls. Half of the liver samples was frozen for further processing and the other half was fixed in 10% formaldehyde and embedded in paraffin for histological analysis. Paraffin sections were dewaxed in xylene and stained with hematoxylin and eosin (HE) or picrosirius red (PS), or processed further for immunohistochemistry. Stained sections were used for histological diagnosis and picrosirius red (PS)-stained sections were used for morphometric analysis.
Cell Cultures, Decorin Gene Silencing and TGFβ1 Treatment
We utilized human hepatic stellate LX2 cells (a kind gift from Dr. Scott Friedman) which were spontaneously immortalized by continuous growth in low serum38. The cells were cultured in a humified atmosphere containing 5% CO2 at 37°C in Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml of penicillin and 100 μg/ml of streptomycin (Sigma). Routinely, 2 × 105 cells were seeded in 6-well culture plates and transfected with Silencer Select Pre-Designed siRNA specific for decorin (siRNA ID: s223389, Applied Biosystems) using SiPORT NeoFX Transfection Agent (Applied Biosystems) according to the instructions of the manufacturer. Control cells were transfected with Silencer Select negative control siRNA (siRNA ID: 4390843). After 24 h the medium was replaced with DMEM supplemented with 0.2% FBS alone or in the presence of 2 ng/ml human recombinant TGFβ1 (Cat. No.: PHP143, AbD Serotec, Oxford, UK) for 48 h.
Morphometry, Fluorescence Immunostaining and Quantitative Real-time RT-PCR
PS-stained sections were analyzed by using the Cue-2 program (Olympus, Japan) At least 5 fields were analyzed in each section. Frozen sections of the liver and cells grown on coverslips in a 24-well plate (4 × 104 cells/well) were fixed in ice-cold methanol, washed in phosphate-buffered saline (PBS), blocked with 5 w/v% BSA/PBS containing 10% nonimmune serum of secondary antibody at 37 °C for 30 min. After washing, sections were incubated with the primary antibody diluted in PBS containing 1 w/v% BSA at 37 °C for 1.5 h or at 4 °C for 16 h. Appropriate fluorescent secondary antibodies were applied at room temperature for 30 min. Nuclei were stained with 4′-6′-diamidino-phenylindole (DAPI). Primary and secondary antibodies with their appropriate dilutions are shown in Supplementary Table 1. Pictures were taken by Nikon Eclipse E600 microscope with the help of Lucia Cytogenetics version 1.5.6 program.
For RT-PCR, total RNAs were isolated from frozen livers and LX2 cells. After homogenization in liquid nitrogen Trizol (Invitrogen, Carlsbad, CA) method was used for RNA extraction. cDNAs were generated from 1 μg of total RNA by M-MLV Reverse Transcriptase kit (Invitrogen) following the instructions of the supplier. Real-time PCR was performed by ABI Prism 7000 Sequence Detection System (Applied Biosystems, Welterstadt, Germany), using ABI Taqman Gene Expression Assays for mouse: procollagen α1(I) (assay ID: Mm00801666_g1), procollagen α1(III) (assay ID: Mm00802331_m1), procollagen α1(IV) (assay ID: Mm00802372_m1), TIMP-1 (assay ID: Mm00441818_m1), PAI-1 (assay ID: Mm00435860_m1), TIEG (assay ID: Mm00449812_m1), TGFβ1 (assay ID: Mm01178819_m1) according to the manufacturer’s protocol. Mouse β-actin was used as endogenous control (assay ID: Mm00607939_s1). For human procollagen α1(I) (assay ID: Hs01076780_g1) and α-smooth muscle actin (assay ID: Hs00426835_g1), human GAPDH served as endogenous control (Part no.:4326317E). All samples were run in duplicates in 20 μl with 50 ng of cDNA. Results were obtained as threshold cycle (CT) values. Expression levels were calculated by using the 2−ΔΔCT method.
Dot Blot, Western Blot and Zymography Assays
Total proteins were extracted from frozen liver tissues. After homogenization in liquid nitrogen 1 ml of lysis buffer was added to the samples (20 mM TRIS pH=7.5, 2 mM EDTA, 150 mM NaCl, 1% Triton-X100, 0.5% Protease Inhibitor Cocktail (Sigma, St. Luis, MO) 2 mM Na3VO4, 10 mM NaF. Cultured cells were washed with PBS followed by addition 1 ml of lysis buffer described above. Cells were scraped off with cell scraper and lysates were transferred into microcentrifuge tubes. After incubation for 30 min on ice, samples were centrifuged at 15000 g for 20 min. Supernatants were kept and protein concentrations were measured as described before39.
For dot blot, approximately 2 μg of total protein was applied on PVDF membrane (Millipore, Billerica, MA), endogenous peroxidase was blocked by incubating the membrane in 1% H2O2 for 10 min. Subsequently the membrane was blocked with 3 w/v% BSA in Tris-buffered saline (TBS). Primary antibody was applied at 4 °C for 16 h. Appropriate HRP-conjugated secondary antibody was used for 45 min, dots were detected by SuperSignal West Pico Chemiluminescent Substrate Kit (Pierce/Thermo Scientific, Waltham, MA), and visualized by Kodak Image Station 4000MM Digital Imaging System. The density of the dots was also measured by the same system. A detailed list of the antibodies used in this study is presented in Supplementary Table 1.
For gelatinase (zymography) assays, proteins were isolated from frozen livers after homogenization in liquid nitrogen. The lysis buffer contained 50 mM TRIS pH 7.5, 500 mM NaCl, 5 mM CaCl2. Protein aliquots (15 μg) of each sample were analyzed on a 7.5% sodium dodecyl-sulfate-polyacrylamide gel, containing 300 μg/ml gelatin. After electrophoresis at 200 V for 35 min, the gel was washed in 2.5% Triton X-100 for 30 min, then incubated for 18 hours at 37°C in a solution containing 50 mM Tris, pH 7.5, and 10 mM CaCl2. Gels were fixed in 30% methanol/10% acetic acid for 30 min, then stained with Coomassie Brilliant Blue for another 30 min. Areas of gelatinase activity appeared as transparent bands on the blue background. The intensity of the bands was determined by densitometry using a Kodak IS4000MM Digital Imaging System.
For Western blotting, proteins were isolated as for dot blot, 30 μg of total proteins were mixed with loading buffer containing β-mercaptoethanol and were incubated at 95°C for 5 min. Denatured samples were loaded onto a 10% polyacrylamide gel and were run for 30 min at 200 V on a Mini Protean vertical electrophoresis equipment (Bio-Rad, Hercules, CA). Proteins were transferred to PVDF membrane (Millipore) by blotting for 1.5 h at 100 V. Ponceau staining was applied to determine blotting efficiency. Membranes were blocked with 3 w/v% non-fat dry milk (Bio-Rad) in TBS for 1 h followed by incubation with the primary antibodies at 4 °C for 16 h. Mouse β-actin or human GAPDH served as loading control. Membranes were washed 5 times with TBS containing 0.5 v/v% Tween-20, then were incubated with appropriate HRP-conjugated secondary antibodies for 1 h.
Hydroxyproline Determination
Liver samples were homogenized with 10 volumes of physiological salt solution and precipitated with 1 ml of 10 v/v trichloroacetic acid. Pellets were washed ethanol/diethyl ether in a ratio of 3:1, dried under vacuum and transferred to glass cuvettes. Hydrolysis was performed in 6 N HCl followed by incubation at 120°C for 16 h. Hidroxyproline determination was performed as described before40, 41. In brief, pH of hydrolysates was adjusted to 6–7 by adding 5 N NaOH in a final volume of 12 ml. Two ml aliquots of the samples were transferred into clean tubes and in the presence of 30% of methoxyethanol. 1–1 ml of 50 mM Chloramine T (dissolved in citrate-acetate buffer pH=6) was added. After 20 min of incubation at room temperature the reaction was stopped with 3.15 N perchloric acid followed by of 20% p-dimethylaminobenzaldehyde dissolved in methoxyethanol. The color was developed by incubation for 20 min at 60°C. After cooling, the absorbance was measured at 557 nm. Hydroxyproline content was calculated from standard curves generated with pure hydroxyproline solutions of known concentrations.
Statistical Analysis
All the data were graphed and analyzed using the SigmaPlot and SigmaStat software packages (SPSS, Version 11.0). Statistical analysis among groups was assessed using two-tailed Student’s t-test after data normalization by logarithmic transformation (log10). Normal distribution was further proved by F-test. The values were considered statistically significant with P<0.05.
RESULTS
Hepatic Fibrosis is Accentuated and Healing is Protracted in Decorin-null Mice
Picrosirius red staining for connective tissue clearly showed that Dcn−/− mice accumulated more connective tissue in their livers as compared to the wild-type mice in response to chronic liver injury (Figure 1a). The fibrotic tissue localized mainly to the acino-peripheral regions but its deposition in the sinusoids could also be observed. In Dcn−/− livers, following a four month-exposure to TA, formation of pseudolobules, typically seen in early cirrhosis, could be detected in contrast to the wild-type mice (Figure 1a). Four months after the cessation of TA treatment, the majority of connective tissue fibrils positive for picrosirius red were resolved in the liver of wild-type animals. In contrast, Dcn−/− livers still contained considerable amounts of fibrous tissue (Figure 1a). Morphometric analysis of picrosirius-positive tissue confirmed our visual observations (Figure 1b). The intensity of picrosirius staining was higher already in the liver of untreated Dcn−/− mice. At each time point throughout fibrogenesis and regression higher amounts of connective tissue were detected in mice lacking decorin (Figure 1b), and these differences were statistically significant (P<0.05). Hydroxyproline determination provided further confirmation for the elevated amount of collagens (Figure 1c). In the liver of wild-type mice the production of fibrous tissue stopped after 4 month of TA exposure and no additional connective tissue deposition was observed thereafter. In contrast, in Dcn−/− mice extracellular matrix deposition progressed until the end of the experimental period (8 months). Comparable patterns were observed for changes in hydroxyproline content (Figure 1c).
Figure 1.

Hepatic fibrosis is accentuated and healing is delayed in Dcn−/− mice. (a) Picrosirius-stained sections from the liver of wild-type and Dcn−/− mice without treatment (first column), after 4 months of thioacetamide treatment (second column), and 4 months after the withdrawal of the thioacetamide (third column). Scale bar = 150 μm. (b) Quantification of hepatic fibrosis by morphometric analysis of livers from wild-type and Dcn−/− animals as indicated. Values represent the mean (%) ± SEM of three animals. All the values from 2 months to 4+4 months have P<0.01. (c) Quantification of hydroxyproline in whole liver extracts of wild type and decorin-null livers. Values represent the mean (%) ± SEM of n=3; *P<0.05.
Next, we immunostained the livers of the various groups using antibodies against collagen type I, a major indicator of hepatic fibrosis, and against decorin protein core (Figure 2a). The results showed a significant deposition of collagen type I in the decorin-null mice both at the end of the experimental liver injury and after four months of recovery with TA-free water (Figure 2a). Similar results were obtained with antibodies against collagen type III and IV (Supplementary Figure S1). Quantification of collagen type I showed an increase after four months of TA exposure which was statistically significant (P<0.05); the enhanced deposition of collagen I in the Dcn−/− livers was also statistically significant at the end of the regression period (P<0.05, Figure 2b).
Figure 2.

Changes in collagen type I and decorin in hepatic fibrosis and its healing in Dcn−/− mice. (a) Double immunostaining for collagen I and decorin in representative liver samples from all the various experimentally treated animals as indicated. Collagen is shown as red, decorin as green, nuclei are stained with DAPI. Representative Wt and Dcn−/− controls are shown in Supplementary Figure S2. Scale bar = 200 μm. (b) Quantification of collagen I content in livers from wild-type and decorin-null mice. Results are expressed as mean intensity of dots after densitometry. Wild-type non-treated control was considered as 100%. Values represent the mean ± SD of three animals (*P<0.05). (c) Quantification of decorin levels by immunoblotting of total liver lysates from wild-type untreated mice, and from mice treated for four months with TA or for an additional four months in TA-free conditions (**P<0.01).
Fibrotic Remodeling Enhances Decorin Levels in Wild-type Livers
By Immunostaining, as expected, decorin was detectable only in wild-type animals where it was deposited in periportal connective tissue and around the central veins (Supplementary Figure S2). Notably, during fibrogenesis decorin levels increased in the wild-type animals and accumulated along fibrotic septa (Figure 2a). By quantitative immunoblotting, a significant increase (P<0.01) in the amount of decorin was detected in lysates of fibrotic livers vis-à-vis the untreated control samples (Figure 2c). After 4 months of regression, a statistically relevant (P<0.01) decrease was seen in wild-type mice (Figure 2c). The amount of decorin in control livers and in those taken at the end point of regression was practically identical (P=0.2).
Elevation of Collagens I, III and IV mRNA Levels in the Liver of Dcn−/− Animals Following Hepatic Injury
Next, we determined the mRNA levels of collagens type I, III, and IV using quantitative real-time RT PCR analysis. These mRNA levels were determined in each liver sample at each time-point. Notably, we found no statistically significant changes in mRNA levels of any collagens tested after 4 months of liver injury. However, at the end of the regression period (TA4+4) the steady state levels of all the 3 types of collagens were significantly elevated in Dcn−/− animals vis-à-vis wild-type mice (P<0.05, Figure 3 a–c).
Figure 3.

Elevation of collagen I, III and IV mRNA levels in the liver of Dcn−/− animals following hepatic injury. (a–c) Quantification of mRNA levels for procollagens type I, III and type IV as determined by quantitative real-time RT PCR analysis. The data represent mean ±SD of n=3 *P<0.05. Levels are expressed in folds, normalized to endogenous β-actin amplified in parallel.
MMP-2 and MMP-9 Activity and Expression of their Inhibitors TIMP-1 and PAI-1
Gelatinase activity of liver homogenates was determined by using zymography (Figure 4a). Quantification of the zymography data revealed no difference in either MMP-2 or in MMP-9 activity in the control samples between wild-type and decorin-null mice (Figure 4 b,c). In contrast, in the wild-type fibrotic livers, both MMP-2 and MMP-9 activity significantly increased (P<0.01) and to a greater extent than that of decorin-null animals (P<0.01). These changes regressed by the end of the recovery period displaying only subtle differences between the two genotypes (Figure 4 b,c).
Figure 4.
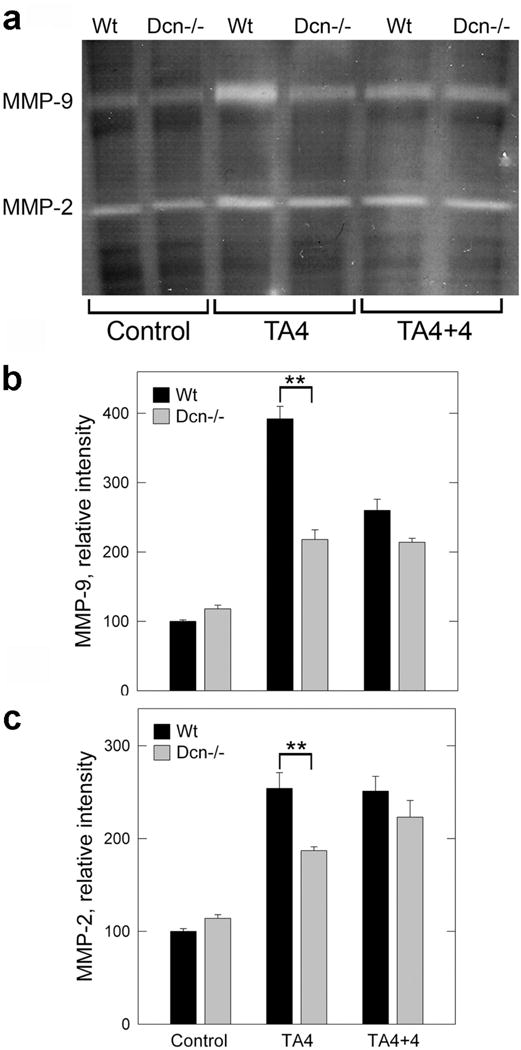
MMP elevation during hepatic fibrosis. (a) Zymogram using liver homogenates of wild-type and Dcn−/− mice without treatment (control), at 4 months of thioacetamide treatment (TA4), and at 4 months after the withdrawal of the chemical (TA4+4). (b,c) Quantification of zymograms as those shown in panel A using scanning densitometry. The values represent the mean intensity (%) ±SD of two experiments run in triplicate; **P<0.01
Fibrogenesis in Dcn−/− livers was characterized not only by low activity of MMP-2 and MMP-9 but also by enhanced expressions of TIMP-1 and PAI-1, two well-known matrix metalloprotease inhibitors, especially at the end of the recovery period (Figure 5).
Figure 5.

Elevation of TIMP-1 and PAI-1 mRNA levels in Dcn−/− liver following experimental hepatic fibrosis. (a,b) Real-time RT-PCR analysis of TIMP-1 andPAI-1 mRNA in whole liver of wild-type and decorin-null animals as indicated. The designations are the same as those of Figure 1.The data represent mean ±SD of n=3; **P<0.01 and *P<0.05. The relative expression levels were calculated by using 2−ΔΔCT method, using β-actin as endogenous control.
Collectively, these results suggest that decorin is directly involved in regulating the fine balance of synthesis and degradation during hepatic fibrosis and healing.
Lack of Decorin in Experimental Hepatic Fibrosis Enhances the Expression of TGFβ1 and TGFβ1-inducible Early Response Gene (TIEG)
To establish whether TGFβ1 would exert a stronger effect in animals lacking functional decorin, we determined the expression of TGFβ1 and TGFβ1-inducible early responsive gene (TIEG) as a marker for the efficacy and activity of the growth factor. Quantitative real-time RT PCR revealed approximately 6-fold increase in TIEG mRNA level in Dcn−/− liver samples (P<0.01) compared to wild-type both during fibrogenesis and regression (Figure 6a), while TGFβ1 mRNA levels increased by 3-fold even in the Dcn−/− control group (P<0.05) (Figure 6b). After 4 months of TA treatment, there was a 20-fold increase of TGFβ1 in Dcn−/− samples in contrast to only a 3-fold increase in wild-type (P<0.01) (Figure 6b). After the regression period, TGFβ1 expression decreased to barely detectable levels, even lower than the wild-type controls (Figure 6b). Thus, the absence of decorin potentiates TGFβ1 bioactivity during hepatic fibrosis and therapeutic approaches to enhance decorin expression in the liver could represent a novel mechanism to counteract this important fibrogenic factor.
Figure 6.

Genetic ablation of decorin gene enhances the expression of the TGFβ1-inducible early responsive gene (TIEG) and TGFβ1 in experimental hepatic fibrosis. Real-time RT-PCR analysis of (a) TGFβ1-inducible early response gene (TIEG) and (b) TGFβ1 expression in whole liver extracts. The designations are the same as those of Figure 1. The data represent mean ±SD of three experiments run in triplicate and are presented as expression relative to endogenous β-actin levels; *P<0.05, **P<0.01.
Increased α-Smooth Muscle Actin and Enhanced Phosphorylation of Erk1/2 and Smad3 in Decorin-null Animals
To investigate what molecular pathways could be operational in our experimental animal model, we determined by immunoblotting the amounts of Erk1/2 and their phosphorylated forms (P-Erk1/2), as well as phospho-Smad2 (P-Smad2) and phospho-Smad3 (P-Smad3), all known downstream effectors of the TGFβ1 signaling pathways. In addition, we determined the levels of α-smooth muscle actin, a known marker of fibrogenic cells such as the hepatic stellate cells (Figure 7). In fibrotic livers (TA4), regardless of their genetic background, the amounts of Erk1/2 and P-Smad2 significantly (P<0.05) increased, as determined by scanning densitometry of several immunoblottings (Supplementary Figure S3). However, at the end of regression period, the amounts of these proteins did not statistically differ from controls. Concurrently, the levels of α−smooth muscle actin were significantly elevated after 4 months of treatment (P<0.01) and after the recovery period in the decorin-null animals compared to wild type (P<0.01, Figure. 7 a,b).
Figure 7.

Amounts of Erk1/2, phospho-Erk1/2 (P-Erk1/2), phospho-Smad2 (P-Smad2), phospho-Smad3 (P-Smad3) and α−smooth muscle actin (αSMA) as detected by Western immunoblotting (a). Pictures are representative of immunoblotting analyses of liver homogenates from two independent experiments run in triplicate. The genotype is indicated at the top and the various categories are listed at the bottom, including animals with no treatment (control), following a 4-month exposure to thioacetamide (TA4), and at the end of the regeneration period (TA4+4). β-actin served as loading control. (b) Densitometric quantification of immunoblottings for αSMA P-Smad3, P-p44 and P-p42. The values were normalized on β-actin levels. Data are expressed as mean ±SD of duplicate experiments run in triplicate; **P<0.01.
In Dcn−/− samples a slight increase was observed in case of Erk1/2 and P-Smad2 compared to wild type. However, while Erk1/2 were not phosphorylated in the wild-type fibrotic liver, the level of phosphorylation increased to 183% in decorin-null samples (P<0.05) (Figure 7b). At the end point of the recovery phase, lower amounts of active Erk1/2 were detected vis-à-vis untreated samples, and Dcn−/− samples contained significantly higher amounts of Erk1/2 as compared to wild type (P<0.05). The level of phospho-Smad3 protein increased during fibrogenesis showing a 2.3-fold increase in wild type and 8.37-fold increase in decorin-null livers after 4 month of TA treatment (P<0.01). At the end of the recovery period (TA4+4) similar amounts of P-Smad3 were detected as in control samples (Figure 7b).
These results suggest that the lack of decorin leads to enhanced accumulation of α-smooth muscle actin positive cells, and to increased phosphorylation of Erk1/2 and Smad3 proteins without significantly affecting the levels of P-Smad2.
Lack or Reduced Decorin Content Results in Enhanced Activation of Hepatic Stellate Cells
To determine whether hepatic stellate cells, known as the main fibrogenic cell type in the liver, could be involved in the action of decorin, we investigated the effects of TGFβ1 in the presence or absence of endogenous decorin. To this end, we utilized human hepatic stellate LX2 cells, which were spontaneously immortalized by continuous growth in low serum38. These cells expresss α-smooth muscle actin, vimentin, glial fibrillary acid protein and display a retinoid phenotype typical of stellate cells. Decorin was silenced by specific siRNA, whereas control cells were transfected with scrambled siRNA. The siRNA-mediated silencing reduced the decorin mRNA level to ~38% (not shown). After TGFβ1 exposure for 48 h, cells with downregulated decorin content showed a 4.42-fold increase in procollagen α1(I) steady state mRNA levels in contrast to cells transfected with scrambled siRNA which exhibited a 2.78-fold increase (P<0.01, Figure 8a). Similar results were obtained with α-smooth muscle actin, insofar as treatment with the growth factor caused a 1.34-fold increase in mRNA level of cells with normal decorin content, while 2.32-fold increase was detected in decorin knock-down samples (P<0.01, Figure 8a).
Figure 8.

Analysis of LX2 cell activation by TGFβ. (a) Quantitative real-time RT-PCR analysis of procollagen type 1, and α-smooth muscle actin mRNA levels normalized on GAPDH. The data represent the mean ±S.D. of duplicate experiments run in triplicates. (b) Determination of collagen 1 protein amount by dot blot in medium of LX2 cells with normal (scr) or silenced decorin content (siDcn) with or without TGFβ1 treatment. (c) Diagram of the relative decorin proteoglycan content from media of cells transfected with either scrambled or decorin-specific siRNAs. The data represent the mean ±SD of duplicate experiments run in triplicates; *P<0.05, **P<0.01.
At the protein level, decorin silencing caused an increase in collagen type 1 in the media of cells exposed to TGFβ1 (P<0.05, Figure 8b), and only a modest increase in siRNA-treated cells without exposure to the growth factor. In agreement with the in vivo data, TGFβ1 caused enhanced decorin production (164%) in LX2 stellate cells (P<0.05, Figure 8c).
In full support of the results presented above, immunofluorescence staining for α-smooth muscle actin of hepatic tissues from various animals showed increased immunopositive cells in the decorin-null livers, especially at 4 months (Figure 9a). Similarly, enhanced α-smooth muscle actin immunostaining was observed when endogenous expression of decorin was decreased in LX2 hepatic stellate cells (Figure 9b), and these effects were more enhanced by TGFβ1 treatment. These findings support and corroborate the in vivo data which showed elevated amounts of α-smooth muscle actin in Dcn−/− liver samples both at 4 months of TA treatment and after the recovery period (Figure 7a.). The correlation between our in vivo and in vitro data was further corroborated by the fact that LX-2 hepatic stellate cells exhibited a significant increase in the α-smooth muscle actin (Figure 10 a,b). In addition, TGFβ1-evoked induction of P-Smad3 was significantly enhanced in cells with reduced expression of endogenous decorin (P<0.05, Figure 10b), see also below.
Figure 9.

Changes in α-smooth muscle actin (αSMA) in liver sections and in LX2 cells. (a) Immunostaining of αSMA on sections of wild type (Wt) and decorin-null (Dcn−/−) livers after 4 months of TA-exposure and at the end of the regression period (TA4+4). (b) Immuncytochemistry of αSMA performed on cells transfected with scrambled (scr) siRNA or with siRNA specific for decorin (siDcn) in the absence or presence of TGFβ1 for 48 h. αSMA is shown as red and nuclei are counterstained with DAPI. Scale bar = 100 μm for each picture.
Figure 10.

Western blot analysis of α-smooth muscle actin (αSMA), phospho-Smad2 (P-Smad2) and phospho-Smad3 (P-Smad3) in LX2 hepatic stellate cells (a). Pictures are representative of immunoblotting analyses of cell lysates from two independent experiments run in triplicate. (b) Quantification of αSMA, P-Smad2 and P-Smad3 content normalized on GAPDH levels. *P<0.05.
Collectively, our findings indicated that endogenous hepatic decorin plays a key role in the regulation of stellate cell activation insofar as genetic ablation of decorin or RNA interference strategies culminate in enhanced activation of stellate cells and a pronounced profibrotic phenotype.
Knockdown of Endogenous Decorin Potentiates TGFβ1-evoked Stimulation of Smad3 Phosphorylation in LX2 Stellate Cells
To investigate the underlying mechanisms regulating the phosphorylation of Smad2 and Smad3 proteins, we performed immunoblotting of cells in which endogenous decorin was either left intact (scrambled siRNA) or reduced by addition of decorin gene-specific siRNA. We have shown above that the in vivo phosphorylated form of both proteins was elevated due to TA-treatment but only P-Smad3 was significantly higher in Dcn−/− samples (Figure 7a). The results of our cell culture model system correlated well with those obtained with the animal experiments because after 48 h of TGFβ1 exposure the extent of increase in P-Smad2 was comparable in cells transfected with either scrambled or decorin-specific siRNAs (Figure 10b). In contrast, after TGFβ1 treatment, P-Smad3 levels were significantly higher in siDcn cells showing a 3.2-fold vs a 1.9-fold elevation in cells trasfected with scrambled siRNA (P<0.05; Figure 10b).
Thus, decorin deficiency potentiates TGFβ1/Smad3 axis suggesting that decorin in vivo might regulate the fine balance of connective tissue remodeling.
DISCUSSION
Several studies have shown that TGFβ1 is a profibrotic cytokine and it has a key role in development of diseases characterized by fibrotic remodeling, such as glomerulosclerosis, pulmonary and hepatic fibrosis17, 42, 43. Since the original discovery that decorin binds to this growth factor23 it has been shown that decorin/TGFβ1 interaction can block TGFβ1 bioactivity both in vitro and in vivo24, 44. These studies reported the efficacy of decorin in inhibiting not only fibroplasia, but also migration of fibroblasts, trophoblasts, and differentiation of myocytes45, 46. Previously, utilizing a cohort of liver pathologies including chronic hepatitis, fibrosis and cirrhosis, we found high amounts of TGFβ1 colocalized with decorin within the fibrotic areas of the liver26. However, these findings could not distinguish between a decorin-mediated block of TGFβ1 activity and a bystander effect. Indeed, in contrast to dermal fibroblasts47, TGFβ1 upregulates decorin production in hepatic stellate cells48. Thus, enhanced deposition of decorin could reflect the stimulatory effect of overproduced TGFβ1, without necessarily implying a protective role of decorin against fibrosis. In the only in vitro liver fibrogenesis model, treatment of hepatic stellate cell cultures with the TGFβ1 resulted in enhanced production of extracellular matrix proteins and decorin could counteract this effect49.
In the present investigation, we utilized a defined experimental animal model of liver fibrosis using mice with identical genetic background (C57Bl/6) except for the absence or presence of decorin. By comparing the fibrogenic response in wild-type and decorin-deficient livers we provide strong genetic evidence for a role of decorin in protecting against fibrogenesis. By further following the animals for an additional four months of recovery period, during which time the animals were not subjected to TA in their diet, we answer an additional question as to whether decorin is also involved in the healing process.
Notably, without any experimental challenge, the amount of connective tissue in the livers from control and Dcn−/− animals did not differ from each other, in agreement with previous studies which have shown that decorin-null animals mature to adulthood without any major disease processes29. However, soon after the start of TA exposure the regular dose given to wild-type mice had to be reduced by 50% as the compound proved to be toxic for the Dcn−/− animals. The degree of TA-induced matrix deposition was higher in the liver from decorin-null mice, and this was observed as early as the first month, lasting throughout the entire experimental period. This response correlated well with an enhancement of collagen I, III and IV proteins in the decorin-deficient mice. However, during the same time interval, the steady state mRNA levels for these three major collagens did not increase in parallel to the increased protein levels. These findings suggest that the clearance rather than the synthesis of collagens was impaired. This hypothesis was further corroborated by relative decrease in two major matrix metalloproteases, MMP-2 and MMP-9, and by a concurrent increase in the hepatic levels of two major MMP inhibitors, namely, TIMP-1 and PAI-1. Nonetheless other possible explanations need to be considered, i.e., changes in transcription/translation efficiency or in mRNA and protein half-lives. Whether the changes observed in our model relate directly to lack of decorin or reflect enhanced TGFβ1 activity needs to be addressed in future studies. Indeed, an adenovirus-mediated direct effect of decorin on metalloproteases has been previously described, although the effect is cell-dependent50, 51. On the other hand, TIMP-1 and PAI-1 levels can also be modulated by TGFβ152, 53.
One of the most interesting findings of the present study is the potential involvement of decorin not only in fibrogenesis, but also in the healing process, during which time numerous enzymes and cells, such as macrophages, would coordinate the clearance of the accumulated connective tissue. Wild-type animals completely recovered, with little or no residual fibrosis, within four months from the cessation of experimental liver injury. In contrast, not only the severity of the hepatic fibrosis was more marked in the decorin-null animals, but also the ability to clear their deposited connective tissue. At the end of the experimental period, the decorin-null livers exhibited elevated collagens I, III and IV, as well as elevated TIMP-1 and PAI-1 mRNA steady state levels. Moreover, the Dcn−/− livers exhibited significantly higher collagen I protein level, while the MMP activities were comparable to the wild-type livers. Thus, we conclude that decorin is a central player not only in the initial matrix deposition following hepatic injury, but also during the subsequent phases of healing and remodeling. This is in agreement with studies indicating that decorin plays a key role during the healing of myocardial infarction54 during cutaneous wound healing55, and in various animal models of renal fibrosis and injury35, 56, 57.
To reveal the underlying mechanism of the cellular processes we measured the state of Erk1/2 activation and P-Smad2/3 proteins, all known downstream effectors of the TGFβ1 signaling pathway. Phosphorylation of Smad2/3 proteins in fibrotic diseases has been described many times previously58–61. In the present study, we did not detect any significant difference in P-Smad2 between wild-type and decorin-deficient animals. On the other hand significant difference was detected in the activation of Smad3 both in Dcn−/− liver after 4 months of TA exposure as well as in decorin deficient LX2 human stellate cells after TGFβ1 exposure, suggesting that the signaling pathway causing the increase in matrix production in Dcn−/− animals might be related to the Smad pathway. The prominent role of Smad3 is well documented as targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice62. Moreover, it is well documented that maximal expression of collagen type I in activated hepatic stellate cells, i.e., the most fibrogenic cell type in liver, requires Smad3 rather than Smad2, both in vivo and in cultured cells63.
Activation of the Erk1/2 was also remarkably higher in Dcn−/− mice both during the fibrotic and regenerative periods. Indeed, several studies have reported the importance of non-Smad pathways of TGFβ signaling60, 64, 65, which culminates in elevated collagen production, in transdifferentiation of hepatic stellate cells into myofibroblasts64, and in epithelial-to-mesenchymal transition65. We do not know whether activation of Erk1/2 occurs through the TGFβ receptor or through crosstalk with different receptor tyrosine kinases. Notably, Erk1/2 is downstream of both EGFR and Met, and decorin is a known negative regulator of both these receptors. Thus, the lack of decorin might cause a protracted activation of one or both of these receptors, especially the Met receptor which is highly expressed in hepatocytes.
In conclusion, although we do not know whether collagen-bound decorin is as effective as soluble decorin, our studies provide the first evidence that decorin, and perhaps other SLRP members, might play a direct role in liver fibrogenesis and healing. It is conceivable that not the absolute amount of decorin, but the ratio of decorin to active TGFβ1 might be important in combating hepatic fibrosis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are very thankful to Dr. Scott Friedman (Mount Sinai School of Medicine, NY) for providing the LX2 stellate cells. This work was supported in part by Hungarian Scientific Research Fund, grants 67925, 042672, 75468, and by the National Institutes of Health grant RO1 CA39481 (to RVI).
Footnotes
SUPPLEMENTARY INFORMATION IS AVAILABLE ON LABORATORY INVESTIGATION’S WEB SITE
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Hope TA, Herfkens RJ, Denianke KS, et al. Nephrogenic Systemic Fibrosis in Patients With Chronic Kidney Disease Who Received Gadopentetate Dimeglumine. Invest Radiol. 2009 doi: 10.1097/RLI.0b013e31819343ba. [DOI] [PubMed] [Google Scholar]
- 2.Zois CD, Baltayiannis GH, Karayiannis P, et al. Systematic review: hepatic fibrosis -regression with therapy. Aliment Pharmacol Ther. 2008;28:1175–1187. doi: 10.1111/j.1365-2036.2008.03840.x. [DOI] [PubMed] [Google Scholar]
- 3.Calvaruso V, Maimone S, Gatt A, et al. Coagulation and fibrosis in chronic liver disease. Gut. 2008;57:1722–1727. doi: 10.1136/gut.2008.150748. [DOI] [PubMed] [Google Scholar]
- 4.Schnaper HW. Balance between matrix synthesis and degradation: a determinant of glomerulosclerosis. Pediatr Nephrol. 1995;9:104–111. doi: 10.1007/BF00858986. [DOI] [PubMed] [Google Scholar]
- 5.Balsano C, Alisi A, Nobili V. Liver fibrosis and therapeutic strategies: the goal for improving metabolism. Curr Drug Targets. 2009;10:505–512. doi: 10.2174/138945009788488459. [DOI] [PubMed] [Google Scholar]
- 6.Epple A, Kuhn HA, Leveringhaus M, et al. Etiology, complications and prognosis of liver cirrhosis in 917 patients. II. Causes of death and prognosis in liver cirrhosis. Lebensversicher Med. 1979;31:117–122. [PubMed] [Google Scholar]
- 7.Fartoux L, Serfaty L. Liver cirrhosis in adults: etiology and specific treatments. Rev Prat. 2005;55:1539–1548. [PubMed] [Google Scholar]
- 8.Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 9.Kato I, Tominaga S, Ikari A. The risk and predictive factors for developing liver cancer among patients with decompensated liver cirrhosis. Jpn J Clin Oncol. 1992;22:278–285. [PubMed] [Google Scholar]
- 10.La Vecchia C, Negri E, Cavalieri d’Oro L, et al. Liver cirrhosis and the risk of primary liver cancer. Eur J Cancer Prev. 1998;7:315–320. doi: 10.1097/00008469-199808000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Albillos Martinez A. What is the treatment of choice in a patient with liver cirrhosis and esophageal varices that have bled to prevent rebleeding: beta-blockers, endoscopic ligation or both? Gastroenterol Hepatol. 2009;32:124–125. doi: 10.1016/j.gastrohep.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Tsao G, Lim J. Management and Treatment of Patients With Cirrhosis and Portal Hypertension: Recommendations From the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol. 2009 doi: 10.1038/ajg.2009.191. [DOI] [PubMed] [Google Scholar]
- 13.Vallet-Pichard A, Mallet V, Costentin CE, et al. Treatment of HBV-related cirrhosis. Expert Rev Anti Infect Ther. 2009;7:527–535. doi: 10.1586/eri.09.28. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt A, Lorkowski S, Seidler D, et al. TGF-beta1 generates a specific multicomponent extracellular matrix in human coronary SMC. Eur J Clin Invest. 2006;36:473–482. doi: 10.1111/j.1365-2362.2006.01658.x. [DOI] [PubMed] [Google Scholar]
- 15.De Bleser PJ, Niki T, Rogiers V, et al. Transforming growth factor-beta gene expression in normal and fibrotic rat liver. J Hepatol. 1997;26:886–893. doi: 10.1016/s0168-8278(97)80257-7. [DOI] [PubMed] [Google Scholar]
- 16.Gauldie J, Bonniaud P, Sime P, et al. TGF-beta, Smad3 and the process of progressive fibrosis. Biochem Soc Trans. 2007;35:661–664. doi: 10.1042/BST0350661. [DOI] [PubMed] [Google Scholar]
- 17.Kanzler S, Lohse AW, Keil A, et al. TGF-beta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol. 1999;276:G1059–1068. doi: 10.1152/ajpgi.1999.276.4.G1059. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Mengsteab S, Tag CG, et al. Transforming growth factor-beta1 gene polymorphisms are associated with progression of liver fibrosis in Caucasians with chronic hepatitis C infection. World J Gastroenterol. 2005;11:1929–1936. doi: 10.3748/wjg.v11.i13.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shek FW, Benyon RC. How can transforming growth factor beta be targeted usefully to combat liver fibrosis? Eur J Gastroenterol Hepatol. 2004;16:123–126. doi: 10.1097/00042737-200402000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Iozzo RV. The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J Biol Chem. 1999;274:18843–18846. doi: 10.1074/jbc.274.27.18843. [DOI] [PubMed] [Google Scholar]
- 21.Schaefer L, Iozzo RV. Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J Biol Chem. 2008;283:21305–21309. doi: 10.1074/jbc.R800020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ameye L, Young MF. Mice deficient in small leucine-rich proteoglycans: novel in vivo models for osteoporosis, osteoarthritis, Ehlers-Danlos syndrome, muscular dystrophy, and corneal diseases. Glycobiology. 2002;12:107R–116R. doi: 10.1093/glycob/cwf065. [DOI] [PubMed] [Google Scholar]
- 23.Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346:281–284. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Li XJ, Liu Y, et al. Recombinant human decorin inhibits cell proliferation and downregulates TGF-beta1 production in hypertrophic scar fibroblasts. Burns. 2007;33:634–641. doi: 10.1016/j.burns.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 25.Isaka Y, Brees DK, Ikegaya K, et al. Gene therapy by skeletal muscle expression of decorin prevents fibrotic disease in rat kidney. Nat Med. 1996;2:418–423. doi: 10.1038/nm0496-418. [DOI] [PubMed] [Google Scholar]
- 26.Dudas J, Kovalszky I, Gallai M, et al. Expression of decorin, transforming growth factor-beta 1, tissue inhibitor metalloproteinase 1 and 2, and type IV collagenases in chronic hepatitis. Am J Clin Pathol. 2001;115:725–735. doi: 10.1309/J8CD-E9C8-X4NG-GTVG. [DOI] [PubMed] [Google Scholar]
- 27.Weber IT, Harrison RW, Iozzo RV. Model structure of decorin and implications for collagen fibrillogenesis. J Biol Chem. 1996;271:31767–31770. doi: 10.1074/jbc.271.50.31767. [DOI] [PubMed] [Google Scholar]
- 28.Zhang G, Ezura Y, Chervoneva I, et al. Decorin regulates assembly of collagen fibrils and acquisition of biomechanical properties during tendon development. J Cell Biochem. 2006;98:1436–1449. doi: 10.1002/jcb.20776. [DOI] [PubMed] [Google Scholar]
- 29.Danielson KG, Baribault H, Holmes DF, et al. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J Cell Biol. 1997;136:729–743. doi: 10.1083/jcb.136.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santra M, Reed CC, Iozzo RV. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J Biol Chem. 2002;277:35671–35681. doi: 10.1074/jbc.M205317200. [DOI] [PubMed] [Google Scholar]
- 31.Reed CC, Gauldie J, Iozzo RV. Suppression of tumorigenicity by adenovirus-mediated gene transfer of decorin. Oncogene. 2002;21:3688–3695. doi: 10.1038/sj.onc.1205470. [DOI] [PubMed] [Google Scholar]
- 32.Csordas G, Santra M, Reed CC, et al. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J Biol Chem. 2000;275:32879–32887. doi: 10.1074/jbc.M005609200. [DOI] [PubMed] [Google Scholar]
- 33.Fiedler LR, Schonherr E, Waddington R, et al. Decorin regulates endothelial cell motility on collagen I through activation of insulin-like growth factor I receptor and modulation of alpha2beta1 integrin activity. J Biol Chem. 2008;283:17406–17415. doi: 10.1074/jbc.M710025200. [DOI] [PubMed] [Google Scholar]
- 34.Goldoni S, Humphries A, Nystrom A, et al. Decorin is a novel antagonistic ligand of the Met receptor. J Cell Biol. 2009;185:743–754. doi: 10.1083/jcb.200901129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaefer L, Tsalastra W, Babelova A, et al. Decorin-mediated regulation of fibrillin-1 in the kidney involves the insulin-like growth factor-I receptor and Mammalian target of rapamycin. Am J Pathol. 2007;170:301–315. doi: 10.2353/ajpath.2007.060497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schönherr E, Sunderkotter C, Iozzo RV, et al. Decorin, a novel player in the insulin-like growth factor system. J Biol Chem. 2005;280:15767–15772. doi: 10.1074/jbc.M500451200. [DOI] [PubMed] [Google Scholar]
- 37.Merline R, Lazaroski S, Babelova A, et al. Decorin deficiency in diabetic mice: aggravation of nephropathy due to overexpression of profibrotic factors, enhanced apoptosis and mononuclear cell infiltration. J Physiol Pharmacol. 2009;60(Suppl 4):5–13. [PMC free article] [PubMed] [Google Scholar]
- 38.Xu L, Hui AY, Albanis E, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54:142–151. doi: 10.1136/gut.2004.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 40.Woessner JF., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 41.Fels IG. Determination of hydroxyproline in liver. Clinical Chemistry. 1958;4:62–65. [PubMed] [Google Scholar]
- 42.Clouthier DE, Comerford SA, Hammer RE. Hepatic fibrosis, glomerulosclerosis, and a lipodystrophy-like syndrome in PEPCK-TGF-beta1 transgenic mice. J Clin Invest. 1997;100:2697–2713. doi: 10.1172/JCI119815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El-Gamel A, Awad MR, Hasleton PS, et al. Transforming growth factor-beta (TGF-beta1) genotype and lung allograft fibrosis. J Heart Lung Transplant. 1999;18:517–523. doi: 10.1016/s1053-2498(98)00024-2. [DOI] [PubMed] [Google Scholar]
- 44.Huijun W, Long C, Zhigang Z, et al. Ex vivo transfer of the decorin gene into rat glomerulus via a mesangial cell vector suppressed extracellular matrix accumulation in experimental glomerulonephritis. Exp Mol Pathol. 2005;78:17–24. doi: 10.1016/j.yexmp.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 45.Droguett R, Cabello-Verrugio C, Riquelme C, et al. Extracellular proteoglycans modify TGF-beta bio-availability attenuating its signaling during skeletal muscle differentiation. Matrix Biol. 2006;25:332–341. doi: 10.1016/j.matbio.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Fischer JW, Kinsella MG, Levkau B, et al. Retroviral overexpression of decorin differentially affects the response of arterial smooth muscle cells to growth factors. Arterioscler Thromb Vasc Biol. 2001;21:777–784. doi: 10.1161/01.atv.21.5.777. [DOI] [PubMed] [Google Scholar]
- 47.Kahari VM, Larjava H, Uitto J. Differential regulation of extracellular matrix proteoglycan (PG) gene expression. Transforming growth factor-beta 1 up-regulates biglycan (PGI), and versican (large fibroblast PG) but down-regulates decorin (PGII) mRNA levels in human fibroblasts in culture. J Biol Chem. 1991;266:10608–10615. [PubMed] [Google Scholar]
- 48.Breitkopf K, Haas S, Wiercinska E, et al. Anti-TGF-beta strategies for the treatment of chronic liver disease. Alcohol Clin Exp Res. 2005;29:121S–131S. doi: 10.1097/01.alc.0000189284.98684.22. [DOI] [PubMed] [Google Scholar]
- 49.Shi YF, Zhang Q, Cheung PY, et al. Effects of rhDecorin on TGF-beta1 induced human hepatic stellate cells LX-2 activation. Biochim Biophys Acta. 2006;1760:1587–1595. doi: 10.1016/j.bbagen.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 50.Dong FQ, Li H, Wu F, et al. Effects of overexpression of decorin on matrix metalloproteinases 2 and 9 in rat mesangial and tubular cells. Zhonghua Yi Xue Za Zhi. 2008;88:3444–3447. [PubMed] [Google Scholar]
- 51.Al Haj Zen A, Lafont A, Durand E, et al. Effect of adenovirus-mediated overexpression of decorin on metalloproteinases, tissue inhibitors of metalloproteinases and cytokines secretion by human gingival fibroblasts. Matrix Biol. 2003;22:251–258. doi: 10.1016/s0945-053x(03)00018-0. [DOI] [PubMed] [Google Scholar]
- 52.Akool el S, Doller A, Muller R, et al. Nitric oxide induces TIMP-1 expression by activating the transforming growth factor beta-Smad signaling pathway. J Biol Chem. 2005;280:39403–39416. doi: 10.1074/jbc.M504140200. [DOI] [PubMed] [Google Scholar]
- 53.Kutz SM, Hordines J, McKeown-Longo PJ, et al. TGF-beta1-induced PAI-1 gene expression requires MEK activity and cell-to-substrate adhesion. J Cell Sci. 2001;114:3905–3914. doi: 10.1242/jcs.114.21.3905. [DOI] [PubMed] [Google Scholar]
- 54.Weis SM, Zimmerman SD, Shah M, et al. A role for decorin in the remodeling of myocardial infarction. Matrix Biol. 2005;24:313–324. doi: 10.1016/j.matbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 55.Jarvelainen H, Puolakkainen P, Pakkanen S, et al. A role for decorin in cutaneous wound healing and angiogenesis. Wound Repair Regen. 2006;14:443–452. doi: 10.1111/j.1743-6109.2006.00150.x. [DOI] [PubMed] [Google Scholar]
- 56.Schaefer L, Macakova K, Raslik I, et al. Absence of decorin adversely influences tubulointerstitial fibrosis of the obstructed kidney by enhanced apoptosis and increased inflammatory reaction. Am J Pathol. 2002;160:1181–1191. doi: 10.1016/S0002-9440(10)64937-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Williams KJ, Qiu G, Usui HK, et al. Decorin deficiency enhances progressive nephropathy in diabetic mice. Am J Pathol. 2007;171:1441–1450. doi: 10.2353/ajpath.2007.070079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 59.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 61.Miyazono K, ten Dijke P, Heldin CH. TGF-beta signaling by Smad proteins. Adv Immunol. 2000;75:115–157. doi: 10.1016/s0065-2776(00)75003-6. [DOI] [PubMed] [Google Scholar]
- 62.Latella G, Vetuschi A, Sferra R, et al. Targeted disruption of Smad3 confers resistance to the development of dimethylnitrosamine-induced hepatic fibrosis in mice. Liver Int. 2009;29:997–1009. doi: 10.1111/j.1478-3231.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- 63.Schnabl B, Kweon YO, Frederick JP, et al. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. doi: 10.1053/jhep.2001.25349. [DOI] [PubMed] [Google Scholar]
- 64.Pannu J, Nakerakanti S, Smith E, et al. Transforming growth factor-beta receptor type I-dependent fibrogenic gene program is mediated via activation of Smad1 and ERK1/2 pathways. J Biol Chem. 2007;282:10405–10413. doi: 10.1074/jbc.M611742200. [DOI] [PubMed] [Google Scholar]
- 65.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.