

Abstract
The advent of next generation sequencing (NGS) in 2010 has transformed medicine and particularly the growing field of monogenic inborn errors of immunity, including primary immunodeficiencies (PID). NGS has facilitated the discovery of novel disease-causing genes and the genetic diagnosis of patients with PID. Whole-exome sequencing (WES) is presently the most cost-effective approach for PID research and diagnostics, though whole genome sequencing (WGS) offers several advantages. The scientific or diagnostic challenge consists in selecting one or two candidate variants among thousands of NGS calls. Variant- and gene-level computational methods as well as immunological hypotheses can help to narrow down this genome-wide search. The key to success is a well-informed genetic hypothesis on three key aspects: mode of inheritance, clinical penetrance, and genetic heterogeneity of the condition. This determines the search strategy and the frequency cut-offs for candidate alleles. Subsequent functional validation of the disease-causing effect of the candidate variant is critical. Even the most up-to-date dry lab cannot clinch this validation without a seasoned wet lab. The multifariousness of variations entails an experimental rigor even greater than traditional Sanger sequencing-based approaches, in order not to assign PID to false positives. Finding the needle in the haystack takes patience, prudence, and discernment.
Keywords: Next generation sequencing, Whole exome sequencing, Whole genome sequencing, Targeted sequencing, Primary immunodeficiency
Introduction
Primary immunodeficiencies (PID) are monogenic inborn errors of immunity that underlie a growing variety of phenotypes in at least one of five categories: infection, auto-immunity, auto-inflammation, allergy, and tumor (1, 2). The penetrance of the first PIDs to be described displayed complete penetrance, accounting for these conditions to be referred to as Mendelian traits. It has now become clear that most PIDs are monogenic but with variable expressivity and, often, incomplete penetrance. From 2010 onward, next-generation sequencing (NGS) has boosted the discovery of novel genetic etiologies of known and novel PID phenotypes (3, 4). Sanger sequencing of a small set of candidate genes remains a useful diagnostic or research process if the patient’s phenotype is typical of specific genotypes. However, this is seldom the case. There can be a large number of genotypes underlying the best-known phenotypes. Sanger sequencing as a first approach has become especially obsolete for research purposes. NGS-based gene panel sequencing, whole-exome sequencing (WES), and whole-genome sequencing (WGS) are ideal in the field of PID, for research or diagnostic goals, as a variety of clinical and immunological phenotypes may result from mutations in a single gene (genetic pleiotropy), and mutations in multiple genes can underlie the same phenotype (genetic heterogeneity). In addition, many genotypes of known PID phenotypes are yet to be unraveled and many novel phenotypes have yet to be ascribed to the field of PID (5, 6). This review aims to provide clinical immunologists involved in the care for PID patients with basic information about the technical aspects of NGS and the advantages and shortcomings of PID gene panels, WES, and WGS. NGS quality parameters will be explained and strategies for selecting and validating candidate mutations will be clarified. We will not discuss RNA sequencing, which uses NGS technology for the analysis of the transcriptome and can also be useful in the search for novel PID genes, as it is currently not a standard approach in the discovery or diagnosis of genetic etiologies of PID. Finally, we will briefly review the novel PID genes identified by NGS.
I. Generating next generation sequencing data
NGS – also known as deep sequencing, massive parallel sequencing, or second generation sequencing – is a sequencing method in which hundreds of millions of small DNA fragments are sequenced in parallel (Figure 1). NGS can be used to sequence entire genomes (WGS), or a targeted panel of genes, ranging from a small number of genes (e.g. all genes known to cause PID, hereafter referred to as a gene panel) to the whole exome (WES)(7–10). The technical process of NGS is summarized in Figure 1 and the related vocabulary is explained in Table I. The human genome contains 3.2 billion base pairs. The exome is being defined as all of the exons for the 20,000 protein coding genes in the human genome, and all the exons pertaining to microRNA, small nucleolar RNA, and large intergenic noncoding RNA genes as defined in Ensembl (http://mar2016.archive.ensembl.org/Homo_sapiens/Info/Annotation) (11). WES thus requires the DNA template to be enriched in exons. This “capture” step is (i) not needed in WGS, and (ii) the Achilles’ heel of gene panels and WES, because the capture or enrichment process is not perfectly homogeneous across all exons introducing some bias. Moreover, gene panels, WES, and some WGS protocols rely on DNA amplification by Polymerase Chain Reaction (PCR) for sequencing library preparation. This is associated with guanine-cytosine (GC) bias errors, stochastic errors, template switch errors, and polymerase errors. PCR-free library preparation is available for WGS. NGS allows for rapid and efficient sequencing of targeted panels and even whole genomes, but is associated with biases and disparities based on the different regions of the genome that must be taken into account during data analysis.
Figure 1. Next generation sequencing: WES and WGS.
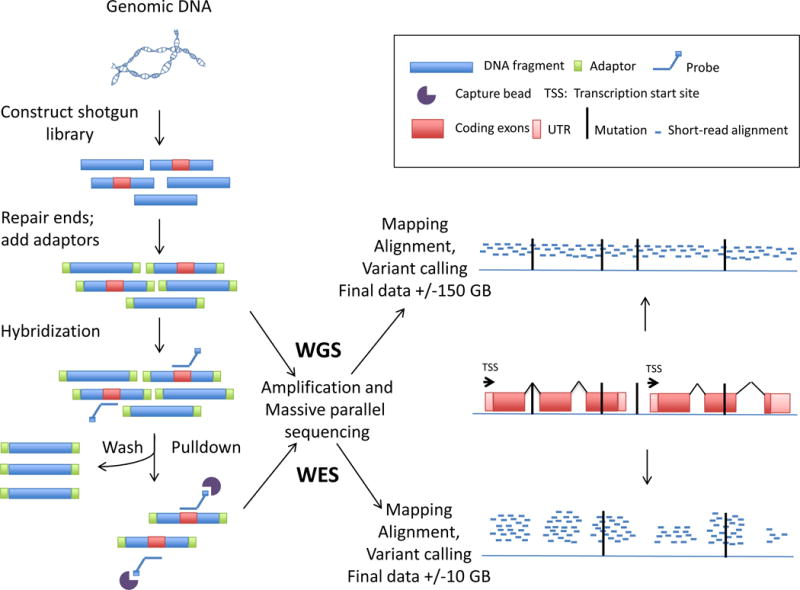
Starting from the patient’s genomic DNA, short fragments are created by either sonication or by restriction enzymes. A shotgun library is made in which fragments of DNA are fused with adaptors. In WES the coding exome (or another genomic region of interest in targeted capture) is enriched by a “capture” step before sequencing. Ideally, each base or each coding region is then read at least 20 times to discriminate sequencing errors from true variants. After the sequencing cycles the reads are computationally mapped to a reference genome. Differences between the patient’s DNA sequence are compared to this reference and “called” as variants. Alterations in the patient’s DNA sequence and the reference genome are identified and “called” as variants. Various “calling” pipelines are available to call variants, e.g. the Genome Analysis Toolkit.
Table I.
Vocabulary in NGS
| Allele frequency | Frequency at which the allele occurs in the population |
| Capture | The enrichment of the DNA of interest for instance by hybridization |
| Capture bait library kit | RNA baits that are complementary to target DNA sequences and which can be used to capture the target by hybridization |
| Copy number variation | Structural variation in the genome either through deletion or duplication of pieces of DNA / chromosomes |
| Coverage | May refer to the percentage of the target genome that is sequenced to the pre-set depth |
| Dominance | Interaction between two alleles at the same locus with the phenotype resulting from heterozygosity differing from the average of the homozygotes |
| Gene damage index | The accumulated mutational damage of each human gene in the healthy human population, based on the 1000 Genomes Project and the CADD score for predicting impact (34) |
| Genotyping quality | A combined score of NGS outcomes (from 0 to 99) quantifying the reliability of a genotype result. A genotyping quality > 20 is generally considered as the threshold for retaining genotypes for further analyses. |
| Haploinsufficiency | A single defective allele results in the disease phenotype |
| Linkage analysis | Familial studies searching for chromosomal regions shared in excess by affected relatives. A specific method is homozygosity mapping searching for chromosomal regions that are homozygous in an affected subject born to consanguineous parents. Linkage studies are generally performed over the whole genome with genome-wide SNP arrays. These data are analyzed by an appropriate statistical linkage method (e.g. the LOD score method). |
| Mutation significance cutoff | Represents for a given gene the threshold for a given variant-level score (such as CADD/PolyPhen-2/SIFT score) above which there is likely to be a significant impact. For each gene, a high phenotypic impact (i.e. possibly damaging) is defined as any CADD/PolyPhen-2/SIFT score equal or above the MSC generated by the specific method, and a low phenotypic impact (i.e. benign) is defined as any score below the MSC (28) |
| Non-synonymous variant | A change in nucleotide in an exonic region of a gene altering the amino acid of the encoded protein |
| Penetrance | Probability that an individual carrying a given genotype manifests a given phenotype |
| Pseudogene | Pseudogenes are genomic DNA sequences similar to normal genes but non-functional |
| Purifying selection | Selection against a deleterious allele in a given population. Purging of the deleterious variants occurs when they cause early death or affect reproductive fitness |
| Sequencing depth | The average number of times a particular nucleotide is represented or “read” in a collection of sequences |
| Single nucleotide variant | A variation of a single nucleotide in the genome |
| Variant annotation | Ascribing meta-information to a variant e.g. the exact position in the genome or the predicted functional impact |
| Variant calling | The identification of differences between the sequence results and the reference genome |
The raw sequence output format for NGS is the FASTQ format, which contains the nucleotide sequences of the short reads and quality information for every nucleotide sequenced. These short reads are then mapped to the human genome reference, and the aligned reads are stored in a BAM file. The final step is to “call” the variants, defined as all the alleles identified in the NGS sample that differ from the reference sequence. This list of variants is stored in variant call format (VCF) files. VCF files are smaller in size than FASTQ or BAM files and are the most practical files for researchers or clinicians trying to identify the genetic etiology of the sample, i.e. what allele(s) cause the PID. VCF files provide the physical position of the variant, the alleles identified in the sample, as well as other annotations including a genotype quality (GQ) score, read depth, and the frequency of the allele in different population(s). GQ score is a statistical measure of the accuracy of a genotype call at a given position. Detailed and reference methods for each of these steps have been shared by the Exome Aggregation Consortium who have analyzed over 60,000 human exomes (12). Researchers or clinicians cannot directly change GQ scores but they can influence the read depth. Read depth or coverage refers to the number of reads per nucleotide. Increasing the read depth makes it easier to distinguish technical errors from genuine variants. Read depth can be increased for a fixed cost by restricting sequencing to a defined set of genes, such as all known PID genes, as opposed to the whole exome.
Coverage can also refer to the percentage of the targeted region or of the whole genome sequenced to a pre-set depth (13). This is a key factor in the choice and the design of a specific panel: the best diagnostic panel should have 100% of the targeted nucleotides covered at a minimum read depth, usually > 20 reads, to allow for a good GQ score. However the overall mean read depth does not have to be very high (>200 for example). This is preferable to a panel with an elevated overall mean read depth (e.g. 500) but with only 95% of the targeted bases covered at a read depth of > 20. The importance of a minimum coverage across all nucleotides targeted favors well-designed panels and WGS. A recent study compared WES and WGS for DNA from six individual patients (11) and showed that more high-quality variants were called by WGS, despite mean read depth for the WES (73×) being almost twice the mean read depth for the WGS (39×) across all nucleotides targeted by the WES kit. This difference in performance can be attributed to much more homogeneous coverage, with a read depth of at least 20× for a higher percentage of the exome in the WGS samples. Read depth is also the main parameter used by algorithms to call copy number variants (CNVs) for gene panels and WES because most CNVs do not start or end within exons (14). WGS remains far superior to any gene panel or WES for CNV identification.
The targeting and coverage of coding regions by the exome capture kit is another important issue. Exon capture for WES is subject to reference bias: as the oligonucleotide baits are designed from known coding sequences, they fail to capture unknown exons. WES cannot target and capture deep intronic regions, which can contain disease-causing mutations (15–18). Most commercial exome kits cover a core set of exons, and target between 50 and 75Mb, but it is important to know which exons are targeted by the kit as there are differences between the various kits. For example, Belkadi et al. found that 1.5% of exons belonging to the consensus coding sequence transcript were excluded from the target enrichment of the popular Agilent Human All-Exon Kit (v5+utr;75 Mb), in other words these exons were entirely outside the capture bait library (11). These exons belonged to 588 genes, including 50 genes relevant to human monogenic disorders, such as BRCA1. Moreover, not all targeted regions are sequenced equally well and not all sequences can be aligned to the reference sequence to allow for variant calling. In the study by Belkadi et al., the coding regions with the lowest WES coverage common to all six individuals, included 47 genes underlying Mendelian diseases (including IFNGR2 and IL12B). Differences in hybridization efficiency during capture, due to GC content variation in the genome, account for the poor coverage of some target exon regions. DNA regions with nucleotide repeats may also result in coverage problems (19, 20). Pseudogenes, as for IKBKG and NCF1, also hamper correct data generation. The optimization of exon capture kits has resulted in increased uniformity of coverage, but an average read depth of 90× is required to cover at least 80% of the target with a read depth of 10× (13, 21).
II. Filtering and selecting appropriate variants
NGS identifies between 20,000 and 50,000 high-quality variants per exome depending on the kit used and some of the criteria for data processing (11, 12, 22). The variants/calls are analyzed and selected according to criteria both at the variant level (allele frequency (AF), variant annotation, potential functional impact) and at the gene level (gene expression, gene function, gene population genetics). An allele with an AF >1% is regarded as common; the remainder are rare or private to the individual or the kindred studied. Public databases of variants (e.g. dbSNP, 1000 Genomes, Exome Aggregation Consortium (ExAC)) and disease causing variants (Human Gene Mutation Database, HGMD) contain data from thousands of individuals of various ethnicities (23). It is nevertheless useful to have an in-house database of variants from >500 individuals of the same ethnicity obtained with the same sequencing technology and analysis pipeline (10) (24).
Many variant annotation approaches aim to predict the functional impact of a variant. This is challenging for variants in intronic regions, regulatory regions, non-coding exons, RNA genes, and for synonymous variants. Nonsense variants, variants affecting splice sites, in-frame and out-of-frame deletions/insertions, mutations of the stop or start codon are likely to be deleterious. Missense variants are the most abundant non-synonymous coding variants. They have received the most attention in variant-level approaches, as their impact is less easily predictable. Common variant-level algorithms for missense variants include “sorting intolerant from tolerant” (SIFT) and polymorphism phenotyping v2 (PolyPhen2) (25, 26). The combined annotation-dependent depletion (CADD) score combines information from these and other annotations including conservation, regulatory information, and transcript information to score both protein-altering and regulatory variants. CADD scores range from 1 to 99 with increasing deleteriousness, and 15 has been proposed as a standard cutoff for all human genes (27). CADD outperforms PolyPhen2 and SIFT for predicting deleteriousness, pathogenicity and molecular functionality, but it has some limitations. First CADD-estimated deleteriousness is not necessarily biologically correct. For instance a stop mutation can have a high CADD score but, if it occurs sufficiently downstream, the truncated protein’s function may be preserved. If sufficiently upstream, the stop codon may be overruled by re-initiation of translation. At any position, alternative splicing can occasionally bypass the mutant exon and generate an alternative, functional isoform. Second, reliable data input is lacking for intronic variants. Finally, CADD score has a high false negative rate when using a fixed cutoff. The mutation significance cut-off (MSC) can be used to overcome this problem (23, 28).
Variant selection based on gene-level criteria takes information about the individual genes into account. There are three reasons for selecting a mutated gene as a candidate disease-causing gene. First it may encode a protein, which acts in a pathway related to the phenotype. The Human Gene Connectome describes the biological distance and route (genes located between two other genes) between all human genes based on protein-protein interaction prediction. It can be used to rank novel candidate genes by their biological distance to core genes (i.e. genes already known to cause the specific disease or the disease group) (29, 30). Second, knowledge about the expression of a gene in an array of human cell lines, cell types, tissues, and organs is essential. For instance, in the prioritization of 144 WES derived variants with AF <1% in a previously healthy child with severe influenza the homozygous, de novo, and two of the three compound heterozygous variants affected genes unrelated to immunity, lungs, or leukocytes. The third gene harboring two compound heterozygous variants was IRF7 which is expressed in the lung and leukocytes, particularly dendritic cells, and was therefore selected as a strong candidate gene (31). However, a mutation in a ubiquitously expressed gene can result in a phenotype that is highly tissue-specific. Third, the relevance of a gene to human disease depends on knowledge from population genetics. It is important to know the degree of purifying selection acting on the gene of interest: autosomal dominant (AD) inherited disease genes involved in essential host defense mechanisms, particularly those encoding components of the innate immune system, are under strong purifying selection, whereas genes with deleterious mutations in the general population are unlikely to be cause a rare reproduction-threatening phenotype with complete penetrance (32, 33). The gene damage index (GDI) is a genome-wide, gene-specific metric which correlates with evolutionary pressure, protein complexity, coding sequence length, and number of paralogs. It is an efficient method for filtering out false positive variants (34). Combined impact prediction provides the most information: a benign variant in a gene with a high GDI is expected to have the lowest phenotypic impact whereas a variant that is predicted to be damaging in a gene displaying low GDI would be expected to have the greatest phenotypic impact (35). The importance of a combined approach is also reflected in the mutation significance cutoff (MSC), which can be used in a quantitative approach with gene-level and gene-specific cutoff values to improve the use of existing variant-level methods, CADD in particular (28, 35) (Table II).
Table II.
Criteria for prioritizing a variant in a gene as a candidate disease-causing variant
| Genetic hypothesis on the mode of inheritance, clinical penetrance and genetic heterogeneity | |
|---|---|
| Variant-Level Criteria | In practice… |
| Allele-frequency is low | AF >1% is a common allele |
| Predicted functional impact is “damaging” | e.g. CADD > 15 |
| Gene-Level Criteria | In practice |
| The gene encodes a protein that is involved in a pathway relevant to the phenotype | The gene is ranked closely to a known disease causing gene in the HGC |
| The gene is expressed in a cell-type or tissue relevant to the phenotype | Knowledge on expression of the gene in an array of cells and tissues is needed |
| The gene is under strong purifying selection | The GDI of the gene is low; CADD > MSC |
| Experimental Validation of the causal relationship between genotype and phenotype | |
AF: allele frequency; CADD: combined annotation-dependent depletion; GDI: gene damage index; HGC: human gene connectome; MSC: mutation significance cutoff
III. Testing a genetic hypothesis
The experimenter who does not know what he is looking for will not understand what he sees. This is true in human genetics and particularly for unbiased, genome-wide, so-called “hypothesis-generating” approaches based on NGS. These approaches are unbiased from a physiological point of view and may generate immunological hypotheses, but in the first place they must be designed and interpreted in the light of a genetic hypothesis. A thorough knowledge of the clinical and cellular phenotype, its prevalence in the general population, its ethnic distribution, associated with the familial segregation and degree of consanguinity is essential in the listing and prioritization of genetic hypotheses. This will underlie the three key hypotheses, concerning mode of inheritance, clinical penetrance, and genetic heterogeneity (36–38). In an autosomal recessive (AR) setting, homozygous and compound heterozygous variants are selected. In AD and in X-linked recessive (XR) models, a single heterozygous or hemizygous variant is selected. Knowledge of the prevalence of the disease and estimates of clinical penetrance and genetic heterogeneity are decisive to determine the cutoffs for the frequency of the candidate mutant allele. For example, for an AR disease that has an estimated prevalence of 1/106, the cutoff AF for a candidate variant assuming complete penetrance should not exceed 10−3 (10−3 × 10−3= 1/106).
Fundamentally, there are two types of studies: those based on a single kindred and those based on multiple kindreds. The ideal situation for single-kindred studies arises when the proband descends from consanguineous parents in a pedigree with presumed AR inheritance. In this context filtering can prioritize homozygous variants with low AF. The larger the number of healthy or diseased siblings, the easier the selection becomes. Compound heterozygous variants are also strong candidates in the AR inheritance model. However, consanguineous families are not protected from AD or XR inheritance, or de novo mutations (even in an AR model). In a proband born to non-consanguineous parents, three genetic hypotheses can be pursued and should be prioritized: XR, AR, and AD. In a male proband displaying a sporadic phenotype, it is difficult to prioritize a specific genetic hypothesis. A single kindred with multiple affected relatives is also instructive, as linkage analysis can help to narrow down the search by mapping the potentially disease-causing variant to a specific locus. A recent study showed that WES can be used for powerful linkage analyses in coding regions (41). A third situation arises when a single patient presents with an early onset, distinct and highly deleterious phenotype. The possibility of a heterozygous or hemizygous de novo mutation can then be considered. Trio design (sequencing of patient and both parents) is an excellent strategy in this setting. The variant should be found in the proband but not in the parents. De novo mutations are often private and do not appear in public databases (allele frequency virtually 0). In each generation, about 30 to 100 de novo mutations arise at the genome level so at most one or two, on average, are likely to be detected in each exome (39, 40).
When studying multiple unrelated kindreds, genetic and phenotypic homogeneity is assumed for at least a subset of patients (several unrelated affected individuals display the same clinical phenotype based upon a mutant allele in the same gene). This is the basis of tests aggregating variants within genes (sometimes called gene burden tests), and searching for an enrichment of variants in a given gene in patients compared to controls (41). Healthy individuals, ideally of the same ethnic origin as the patients, must be used as controls, and exome or genome data must ideally be obtained with the same NGS method. In case of complete penetrance, the hypothetic disease-causing variants found in patients cannot be present in unaffected individuals. In case of incomplete penetrance, the situation is more complex as these hypothetic disease-causing variants can also be present in asymptomatic subjects, including unaffected subjects of the same pedigree. The candidate variants obtained after filtering using the criteria based on the genetic hypothesis must be analyzed further at the variant and gene levels and validated by experimental studies. A flow chart of the process, from NGS to experimental validation, is crystallized in Table II and shown in Figure 2 for the example of human FADD deficiency, the first PID gene identified by WES (42). The initial discovery and the approach used are validated by the description of a novel case of FADD deficiency (43).
Figure 2.
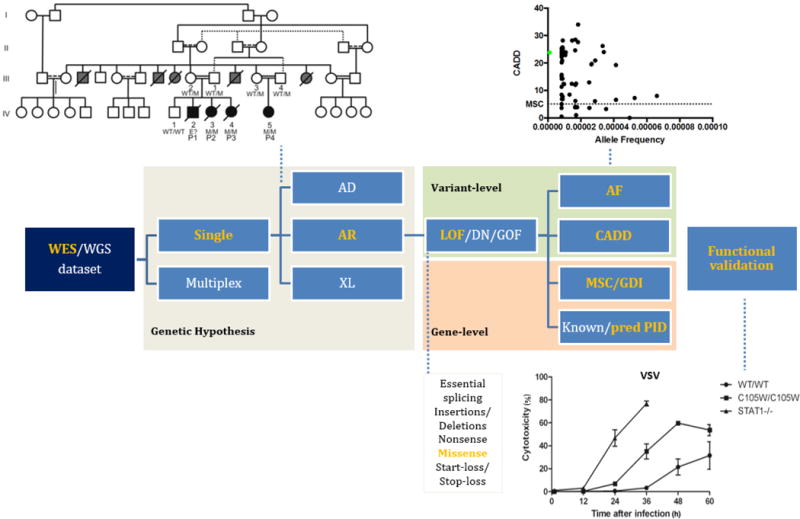
Flow diagram from the raw NGS dataset to validation of the mutation for FADD deficiency. Bolze et al. (42) investigated the WES dataset for members of a large, consanguineous, multiplex kindred in which biological features of auto-immune lymphoproliferative syndrome were found to be associated with severe bacterial and viral infections, hepatic encephalopathy and cardiac malformations. The AR genetic hypothesis set the choice of variant analysis algorithm and allowed the identification of a homozygous missense candidate variant in FADD, encoding the Fas-associated death domain protein (FADD). The variant was identified in all patients. The FADD variant was not found in ExAC, 1000 genomes or the in-house HGID database (MAF=0) and had a high CADD score (23.8) exceeding the FADD MSC (5.07). Connectome analysis predicted FADD to be a candidate novel PID gene as it directly interacts with FAS (p=0.0006). Functional analysis revealed an impaired type I IFN antiviral response in assessments of the antiviral effect of IFN-alpha in vesicular stomatitis virus infected cells. This accounted for the phenotype in vivo and validated the variant as the disease-causing mutation. For clarity, this flow shows only the steps performed to unravel FADD deficiency (in yellow). This flow diagram is applicable to all other genetic hypotheses (white).
IV. Validating genetic findings experimentally
Experimental validation of the causal relationship between genotype and phenotype is crucial. NGS can easily lead to a false-positive being associated with disease if the genetic hypothesis is flawed, population genetics is neglected, or experimental validation is insufficient (e.g. the recent report of a presumed novel disease-causing mutation in seven patients with multiple sclerosis from two multiplex families) (44, 45). Only in rare cases of phenotypic and genetic homogeneity can a strong suspicion of causality be established by genetic means alone (e.g. haploinsufficiency at the RPSA locus and isolated congenital asplenia (46). Over 20% of the PID-causing genes identified to date derive from single-patient studies. Guidelines for experimental validation have been proposed and should be respected, especially in single-patient genetic studies (47). A causal relationship between a candidate genotype and a relevant phenotype requires an experimentally proven thread of causes and consequences. This step may be straightforward (the exception) or may necessitate substantial investigation (the rule), accounting for the lion’s share of time from sequencing to final validation (48). Solid knowledge of physiology and pathology is essential for the design of optimal ways to test candidate variants experimentally (38). The ultimate challenge is validating the causal relationship when the encoded protein cannot easily be linked to known signaling pathways or when the variant is in an inter-genic region or in an RNA gene: for variants in RNA genes there are technical issues (difficulties studying expression by gene transfer techniques) and because of insufficient information on the function of RNA genes.
Experimental studies must show that the variant (or two genetic variants in cases of compound heterozygosity) destroys, impairs, or alters expression or function of the gene product. The first step consists in testing the impact of the putative disease-causing variant on protein expression. Impaired protein expression provides a strong experimental hint that the variant may be disease-causing (but it is not necessary or sufficient to establish causality). The subcellular distribution and trafficking of the mutant protein can also be informative. For synonymous variants, impact on mRNA structure or quantity must be assessed. However assessing the impact of the variant on protein expression is not sufficient, as the loss of some proteins can be harmless (38, 49, 50). Subsequently, the functional impact of the variant should be demonstrated in a cell type and an assay that is relevant to the clinical phenotype such as a relevant leukocyte subset or a primary or inducible pluripotent stem cell (iPSC) derived non-hematopoietic cell (e.g. iPSC derived pulmonary epithelial cells to study the phenotype in IRF7 deficiency) (31). For de novo mutations, it is important to test several cell types to prove that the mutation is germline and not somatic, in which case it may however remain disease-causing (51). In all cases, the mechanism by which a disease-causing variant alters the gene function must be demonstrated to be loss or gain of function for at least one biological function related to the PID.
The cellular phenotype must be rescued by transduction with a wild-type allele for LOF variants. New molecular biology tools, such as the iPSC technology and CRISPR/CAS9 editing, have revolutionized our capacity to prove the causality of a given variant (47, 52). Nevertheless, experimental validation can be challenging. For instance, negative dominance models are difficult to test. In this model, the variant allele encodes an altered protein that impairs the function of the normal protein encoded by the other, wild-type allele. This may even occur for loss-of-expression alleles, when the altered and normal protein requires oligomeric complexes for stability, as demonstrated for TRAF3 (53). Co-transfecting the mutant and the wild-type allele in a cell line deficient for the gene product should prove negative dominance. Rescue must be by knockdown, knockout, or correction of the mutant (47). If experiments at the cellular level do not deliver robust evidence, or in addition to these in vitro studies, animal models can be used to demonstrate causality. When experimenting with mice, scientists have to remember that laboratory mice are not necessarily good disease models, as they are inbred and kept under highly controlled experimental conditions (54). In spite of all available technologies, there can be a dichotomy between the validation of the clinical phenotype and the validation of the cellular phenotype. For instance in AD hyper-IgE syndrome, the validation of the dominant negative STAT3 mutations underlying the clinical phenotype is clear. However many clinical phenotypes remain unexplained at the cellular and tissue levels (55).
V. Discoveries of PID-causing mutations by NGS
The introduction of NGS in research settings has resulted in an exponential increase in the number of disease-causing genotypes identified for PID. AR disorders remain 4 times more common than AD disorders because they are easier to identify by classic molecular methods and NGS (Table III, Figure 3). Since 2010, the number of AD conditions identified, has been steadily increasing as NGS has made it possible to decipher disease-causing mutations in small pedigrees and in multiple unrelated individuals with the same phenotype (56). NGS has also indirectly provided insight into the vast phenotypic heterogeneity of PIDs affecting a given locus (57–60). For instance, AR complete LOF mutations in STAT1 result in life-threatening viral diseases and severe infections with weakly virulent mycobacteria and other intracellular bacteria (61). GOF mutations in STAT1 underlie most cases of chronic mucocutaneous candidiasis (62). A similar phenomenon has been observed for STAT3. LOF mutations in STAT3 cause AD hyper-IgE syndrome whereas GOF mutations, sometimes at the same position, lead to lymphoproliferation and auto-immunity (63–66). Mutations in DOCK8 typically result in AR combined immunodeficiency (CID), with severe eczema and elevated serum IgE levels, but they can also produce a CID phenotype without eczema or hyper-IgE (67, 68). Perhaps the most beautiful example of phenotypic diversity is that provided by RAG1 and RAG2 mutations, ranging from severe combined immunodeficiency to CID with autoimmunity. Moreover, knowledge of the crystal structure of the RAG complex and assays for assessing recombination activity have provided a molecular and cellular basis for this continuum of disease manifestations (69–71). Beside hypomorphic mutations, somatic mosaicism (in which different cell populations display different genotypes) adds to the challenge of genetic diagnosis in PID. True somatic mosaicism has been identified in auto-immune lymphoproliferative syndrome, multisystem inflammatory disease and severe combined immunodeficiency (51, 72, 73). Other reports on somatic mosaicism have described somatic reversions from the mutated to the wild-type form, variably attenuating the phenotype (74). Although cases of “true” somatic mosaicism and somatic revertants have been identified by classic sequencing methods, NGS with high read depth will boost the study of this genetic mechanism in PID.
Table III. PID genes categorized by year and method of discovery, with indication of the mode of inheritance.
If inheritance can be either AR or AD for a single gene, the gene is counted in both categories. The initial description of the genes indicated in gray involved NGS. Many genes have both LOF and hypomorphic mutations – this is not considered separately. A growing number of genes harbor both GOF and LOF mutations: STAT1, STAT3, ZAP70.
| YEAR | AD | AR | XL |
|---|---|---|---|
| 1980 – ‘85 | ADA, IGKC | ||
| 1986 – ‘90 | SERPING1 | C1QB, C1R, C3, CYBA, ITGB2, PNP | CYBB, G6PD |
| 1991 | NCF1 | ||
| 1992 | C2, CD3G, TAP1, TAP2 | ||
| 1993 | C8B, CD3E, CIITA | BTK, CD40LG, IL2RG | |
| 1994 | LIPA, MPO, TCN2, ZAP70 | WAS | |
| 1995 | FAS | ATM, BLM, C1QC, C5, C6, FCGR1, JAK3, MBL2, NFC2, RFXANK | CFP |
| 1996 | IFNGR1, FASLG | C1QC, C7, C8G, CFI, IGHM, IFNGR1, LYST, RAG1, RAG2 | TAZ |
| 1997 | MEFV | AIRE, C9, CFH, IL2RA, MEFV, RFX5, RFXAP, SLC37A4 | |
| 1998 | C1S, C8A, IGLL1, IL7R, IL12B, IL12RB1, IFNGR2, NBN | DKC1, SH2D1A | |
| 1999 | CASP10, ELANE, TNFRSF1A | AP3B1, BLNK, CD79A, CEBPE, CTSC, DNMT3B, FOXN1, GINS1, MRE11A, MVK, PRF1 | |
| 2000 | RAC2 | AICDA, MOGS, PTPRC, RAB27A, SPINK5 | FOXP3, IKBKG |
| 2001 | NLRP3, NOD2, SH3BP2, SLC35C1, STAT1, TERC | CD8A, CD40, CFD, DCLRE1C, LIG4, RMRP, SLC35C1 | |
| 2002 | CASP8, PSTPIP1 | SMARCAL1, TAPBP, TMC6, TMC8 | |
| 2003 | CD46, CXCR4, GFI1, LRRC8A, NFKBIA, TBX1 | CD3D, ICOS, IRAK4, MASP2, SBDS, STAT1, STAT5B, UNC13D, UNG, VPS13B | |
| 2004 | |||
| 2005 | SLC11A1, TERT, TNFRSF13B | LPIN2, TNFRSF13B | |
| 2006 | APOL1, B2M, CD19, CD247, CD46, NHEJ1, ORAI1, RNASEH2A/2B/2C, SP110, TREX1, TYK2, UNC93B1 | XIAP | |
| 2007 | CFH, NRAS, STAT3, TLR3, TREX1 | CD79B, CFHR1, HAX1, LAMTOR2, NOP10 | |
| 2008 | AIRE, NLRP12, TINF2 | CORO1A, MYD88, NHP2, PMS2, SLC29A3, TERT | CSF2RA |
| 2009 | THBD | AK2, CARD9, CCBE1, CLEC7A, DOCK8, FCN3, FERMT3, G6PC3, IL10RA/B, IL1RN, ITK, NCF4, PRKDC, RNF168, SAMHD1, STIM1, STXBP2, TNFRSF13C | |
| 2010 | PSMB8, TRAF3 | CD81, FADD, ITCH, MASP1, MS4A1, USB1 | |
| 2011 | GATA2, IL17F, IRF8, KRAS, TICAM1 | ACP5, ADAM17, BLOC1S6p, COLEC11, CSF2RB, IL17RA, IL36RN,IRF8, TRAC, ZBTB24, TICAN1 | MAGT1 |
| 2012 | ACTB, CARD11/14, IKZF1, PIK3CD/R1, PLCG2, TBK1, UNC119 | ADAR, CD27, CR2, ISG15, LCK, LRBA, MCM4,PIK3R1, POLE, RBCK1, RHOH, STK4, WIPF1 | |
| 2013 | NFKB2, RPSA, RTEL1, TCF3, TNFSF12 | CARD11, CD59, CFB, IKBKB, IL21R, MALT1, PRKCD, RTEL1, STAT2, TNFRSF4, TRAF3IP2, TTC7A, VPS45 | |
| 2014 | CTLA4, IFIH1, NLRC4, TGFBR1/2, TMEM173 | ACD, BCL10, CECR1, CSF3R, CTPS1, IL21, INO80, JAGN1, MAP3K14, PCNA, PGM3, STAT4, TRNT1, XRCC4 | |
| 2015 | COPA, IRF3, NFAT5, NFKB1 | CDCA7, CLPB, DOCK2, HELLS, IL17RC, IRF7, MYSM1, PARN, RNU4ATAC, RNF31, RORC, TTC37 | |
| 2016 | IRF2BP2 | AP3D1, ERCC6L2, LAT, NSMCE3, POLE2, STX11, TFRC | POLA1 |
Figure 3.
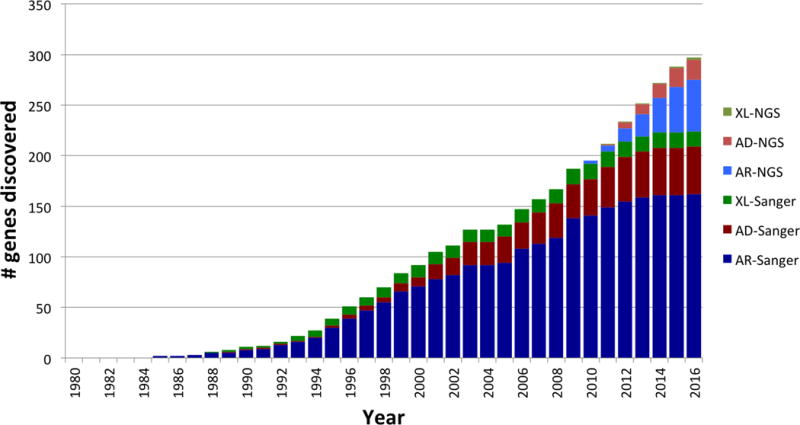
Cumulative number of single-gene defects underlying PIDs described since 1980 until now with either conventional molecular techniques or NGS and their mode of inheritance. If inheritance can be either AR or AD for a given gene, the gene is counted in both categories. The initial description of genes indicated in red involved NGS. Many genes have both LOF and hypomorphic mutations – this is not considered separately. A number of genes harbor both GOF and LOF mutations: STAT1, STAT3, ZAP70. They are not considered separately except for STAT1 for which mutations with both AR and AD inheritance have been described.
Blue: AR inheritance, Red: AD inheritance, Green: XR inheritance. Pale colors indicate genes discovered by NGS.
The NGS-guided discovery of new PID genotypes and phenotypes has revealed a spectrum of penetrance of the phenotype, which remains poorly explained (36, 75, 76). One possibility is a phenotype of genetic susceptibility to infection with a particular microbe that is not expressed until exposure to the microbe occurs. In addition, incomplete penetrance after exposure has been observed in several severe conditions due to viruses as HSV-1 or influenza virus (31). Hypomorphic mutations are also a potential explanation. NGS has also boosted new insights into disease mechanisms leading to new therapies, e.g. magnesium treatment in MAGT1 deficiency (48). The genetic dissection of PIDs identified by NGS has also demonstrated new functions for known proteins. For instance, the catalytic subunit of polymerase alpha plays a crucial role in the synthesis of cytoplasmic RNA-DNA hybrids which avoid excessive type I interferon responses to other nucleic acids (77). Likewise ISG15, an anti-IFN-γ-inducing molecule was shown to be a strong negative regulator of IFN-α/β immunity through USP18 (78–80). An overview of PID disease-causing genes, their mode of inheritance and phenotypes unraveled by NGS or classical methods is provided in Table III.
What about the choice for gene panels, WES, or WGS? Gene panels are not feasible for research purposes, as will be explained below. WES is sufficient in the vast majority of cases. There are two independent lines of evidence in favor of WES. First, the last 30 years of human genetics have shown that mutations underlying monogenic disorders are overwhelmingly found in the exome, and more specifically in the coding exome. Second, the exome is the most purified and conserved region of the whole genome, implying that variations elsewhere, even in enhancers and promoters, are unlikely to disrupt gene expression and function (35). However, within the total group of single-gene defects related to PIDs, singular diseases like cartilage hair hypoplasia and Roifman syndrome are caused by mutations in non-coding RNA genes, respectively RMRP and snRNAU4atac. RNA genes are typically less well covered by WES than by WGS (11). In addition, WGS has resulted in the elucidation of the gene defect underlying X-linked reticulate pigmentary disorder after two decades of research had failed to identify the causative gene, the reason being that the mutation was located deeply into an intron (77). We can only speculate on the maximal proportion of disease-causing mutations not located in the exome – this may range from 5 up to 50%, depending on the PID category (81, 82).
VI. The thin line between diagnostic and research settings
NGS is increasingly being used for the molecular diagnosis of PID (83). Limited resources have invited some to question the benefits of providing a molecular diagnosis to PID patients (84). However, a molecular diagnosis is a definitive diagnosis. Second, in case of a strong genotype-phenotype correlation, molecular diagnosis offers prognostic information. Third, genetic analysis allows for identifying potentially fatal PIDs prior to onset of symptoms enabling timely intervention (e.g. prevention of infections). Fourth, genetic counseling of the relatives is only relevant if a diagnosis is at hand. Finally, molecular diagnosis promotes new treatment modalities either based on modification of the signaling pathway in which the mutated gene product is involved or by gene therapy or gene editing (85, 86). Nevertheless, a molecular diagnosis comes at a cost. First, there is the financial cost related to the generation, the analysis, and the storage of NGS data. The incidental finding of known pathogenic mutations in disease-causing genes (e.g. BRCA1) is also an issue in NGS for both research as well as diagnostic purposes. In 2013 the American College of Medical Genetics and Genomics working group released a panel of 56 genes harboring mutations, which when identified could lead to prevention of a severe outcome. Recommendations were made to report known pathogenic mutations in these genes (87).
PID gene panels constitute another potential approach in diagnostics. PID panels reduce the risks and benefits of incidental findings. A gene panel is less expensive than WES and could be seen as a rapid first-line test. However, gene panels are not a good diagnostic approach for the following reasons. First, they cannot keep pace with the discovery of new PID-causing genes. Since 2010, 10 new PID-causing genes, on average, have been identified per year. Second, many PID-causing genes remain unknown and the use of panels limits the discovery of new genes. Third, they may lead the clinician to focus on a red herring (e.g. a heterozygous missense), while the true disease-causing mutation is not captured in the panel (e.g. a homozygous nonsense). Finally, as sequencing costs go down, the relative cost of a gene panel will increase, without providing the opportunity to search outside the targeted region in case it does not contain a disease-causing mutation. Although success rates may be higher in selected patient populations and as panels evolve, reports of PID panels in large cohorts delivered a genetic diagnosis for only 15% of patients (88, 89).
WGS has clear potential technical benefits over WES as it can detect intronic and intergenic mutations, as well as CNVs, and it provides uniform coverage. However, sequencing, data storage and analysis remain expensive and troublesome. More than 90% of WGS costs relate to sequencing, whereas most of the cost for WES is accounted for by the cost of the capture kit. With sequencing costs tumbling more rapidly than the cost of capture kits, WGS costs will soon approach those for WES. Moreover, specific analysis pipelines can restrict the initial “diagnostic” analysis to virtual “panel-like analysis” or to an “exome-like analysis”. The analysis can then be extended to the genome in the second tier. Despite its disadvantages, WGS has entered diagnostic strategies for newborns with suspected PID (83). However, for the time being, WES is the most reasonable approach. The criteria for WES-based diagnosis are very simple. If the mono- or bi-allelic genotype is known to cause the patient’s phenotype, based on previous studies, a diagnosis can be made. Otherwise, the diagnostic approach to establish causality between genotype and phenotype becomes a research problem, which should be tackled as such as described above. It is popular but wrong to rely on in silico predictors of pathogenicity, even for a known PID-causing gene. Additionally, it is essential to be aware of known PID genes harboring disease-causing mutations not well covered by WES. The importance of the phenotype description by the clinician is therefore invaluable, in both diagnostic and in research settings. The major pros and cons of panel sequencing, WES and WGS are summarized in Table IV.
Table IV.
Gene Panels, Whole Exome Sequencing and Whole Genome Sequencing in a nutshell
| Targeted sequencing | |||
|---|---|---|---|
| Gene Panel | Whole Exome Sequencing | Whole Genome Sequencing | |
| Pro |
|
|
|
| Con |
|
|
|
| |||
| Detection of large structural variations (inversions, translocations, nucleotide repeat expansions) inaccurate with some platforms | |||
| Gained information | Variants in known disease-causing genes | Variants in exome as targeted by the kit | Variants in the entire genome |
| Possible information | Novel or known disease-causing mutation in a known disease-causing gene (including intronic mutation depending on panel design) | Novel or known disease-causing mutation in known disease-causing genes/Novel disease-causing gene within the exome as targeted by the kit | Novel or known disease-causing mutation in known disease-causing genes/Novel disease-causing gene/Mutations in deep intronic regions – intergenic regions – regulatory domains |
Conclusion
The advent of NGS has revolutionized gene detection for both research and diagnostic purposes. NGS-based gene panel sequencing, WES, and WGS are most useful in the field of PID. WES is presently the most cost-effective approach for PID research and diagnosis but WGS provides more uniform coverage. The scientific or diagnostic challenge is the selection of one or two candidate variants among thousands of NGS calls, a task truly resembling ‘finding a needle in a haystack’. We stress the importance of combining variant-level (CADD score) and gene-level approaches (MSC, GDI) with a well-informed genetic hypothesis for the prioritization of potential disease-causing variants. The crucial step is the meticulous experimental validation of the presumed disease-causing mutation. Rigor in establishing the method of genetic investigation is essential to prevent false-positive NGS results. The ultimate goal of NGS approaches in PID is not only to identify novel or known PID genotypes underlying novel or known PID phenotypes, thereby offering diagnostic, prognostic, and therapeutic insights, but also to continue enhancing our knowledge on the various facets of immunity, ranging from host defense to self-tolerance.
Key concepts.
Next generation sequencing (NGS) is a high throughput sequencing method that permits the targeted sequencing of gene panels including the whole exome (WES), and the sequencing of the whole genome (WGS)
Variant-level and gene-level computational and statistical methods can be used to select candidate mutations out among the thousands of variants generated by targeted sequencing, WES, or WGS
A clear genetic hypothesis is essential for the successful use of NGS research or diagnosis in inborn errors of immunity/primary immunodeficiency (PID)
The pathogenic role of any candidate variant should be rigorously validated by in-depth experimental studies
Clinical implications.
The use of NGS facilitates research and diagnosis for inborn errors of immunity/primary immunodeficiency (PID)
WES is the current method of choice.
The discovery of novel genetic etiologies of PIDs is growing in two directions, searching for causes of known and unknown PID phenotypes
The implementation of a rigorous genetic diagnosis is key to the optimal management of patients with PID
Acknowledgments
Funding
IM is funded by a KOF mandate of the KU Leuven and by the Jeffrey Modell Foundation, B Bosch is funded by a Research Mandate of the FWO Vlaanderen, XB is funded by a research grant of the Research Council of the Catholic University of Leuven. The Laboratory of Human Genetics of Infectious Diseases (JLC, AB, B Boisson, YI) was supported, in part, by grants from the Institut National de la Santé et de la Recherche Médicale (INSERM), University Paris Descartes, the Rockefeller University, the St. Giles Foundation, the European Research Council (grant n°ERC-2010-AdG-268777) (LA), the French National Research Agency (ANR) under the “Investments for the future” program(grant n°ANR-10-IAHU-01), and the National Institute of Allergy and Infectious Diseases (NIAID) (grant n° R37AI095983).
List of abbreviations
- AD
Autosomal Dominant
- AF
Allele frequency
- AR
Autosomal Recessive
- CADD
Combined Annotation-Dependent Depletion
- CD
Coverage depth
- CID
Combined Immunodeficiency
- CNV
Copy number variation
- DN
Dominant negative
- ExAC
Exome Aggregation Consortium
- GDI
Gene damage index
- GC
Guanine-Cytosine
- GQ
Genotyping quality
- GOF
Gain-of-function
- HGMD
Human Gene Mutation Database
- LOF
Loss-of-function
- Mb
Megabases
- mRNA
Messenger RNA
- MSC
Mutation Significance Cutoff
- ncRNA
Non-coding RNA
- NGS
Next-generation sequencing
- PCR
Polymerase chain reaction
- PID
Primary immunodeficiency
- PolyPhen2
Polymorphism Phenotyping v2
- SIFT
Sorting intolerant from tolerant
- SNV
Single-nucleotide variant
- UTR
Untranslated Regions
- WES
Whole-exomesequencing
- WGS
Whole-genome sequencing
- XR
X-linked recessive
Contributor Information
Isabelle Meyts, Department of Immunology and Microbiology, Childhood Immunology, Department of Pediatrics, University Hospitals Leuven and KU Leuven, Leuven, Belgium.
Barbara Bosch, Department of Pediatrics, University Hospitals Leuven and KU Leuven Belgium; St. Giles Laboratory of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, New York, USA.
Alexandre Bolze, St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY 10065; Helix, 1 Circle Star Way, San Carlos, CA 94070.
Bertrand Boisson, St. Giles Laboratory of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, New York, USA; The Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Hospital for Sick Children, Paris, France; Paris Descartes University, Imagine Institute, Paris, France.
Yuval Itan, St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, Rockefeller University, New York, NY 10065.
Aziz Belkadi, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Hospital for Sick Children, 75015 Paris, FranceFrance; Imagine Institute, Paris Descartes University, 75015 Paris, France.
Vincent Pedergnana, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Hospital for Sick Children, 75015 Paris, France; Imagine Institute, Paris Descartes University, 75015 Paris, France; Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford OX3 7BN, United Kingdom.
Leen Moens, Laboratory Medicine, Experimental Laboratory Immunology, Department of Laboratory Medicine, University Hospitals Leuven and KU Leuven, Leuven, Belgium.
Capucine Picard, Paris Descartes University – Sorbonne Paris Cité, Paris, FranceStudy Center for Immunodeficiency, Necker-Enfants Malades Hospital, Assistance Publique-Hôpitaux de Paris (AP-HP; Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM UMR 1163, Institut IMAGINE, Paris, France.
Aurélie Cobat, Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Hospital for Sick Children, 75015 Paris, France; Imagine Institute, Paris Descartes University, Paris, France.
Xavier Bossuyt, Laboratory Medicine, Experimental Laboratory Immunology, Department of Laboratory Medicine, University Hospitals Leuven and KU Leuven, Leuven, Belgium.
Laurent Abel, St. Giles Laboratory of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, New York, USA; The Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Hospital for Sick Children, Paris Descartes University, Imagine Institute, Paris, France.
Jean-Laurent Casanova, St. Giles Laboratory of Infectious Diseases, Rockefeller Branch, The Rockefeller University; the Howard Hughes Medical Institute, New York, New York, USA; The Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM U1163, Necker Hospital for Sick Children; the Imagine Institute, Paris Descartes University; and the Pediatric Hematology and Immunology Unit, Assistance Publique-Hôpitaux de Paris, Necker Hospital for Sick Children, Paris, France.
References
- 1.Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova JL, Chatila T, et al. The 2015 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2015;35(8):727–38. doi: 10.1007/s10875-015-0198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bousfiha AA, Jeddane L, Ailal F, Al Herz W, Conley ME, Cunningham-Rundles C, et al. A phenotypic approach for IUIS PID classification and diagnosis: guidelines for clinicians at the bedside. J Clin Immunol. 2013;33(6):1078–87. doi: 10.1007/s10875-013-9901-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conley ME, Casanova JL. Discovery of single-gene inborn errors of immunity by next generation sequencing. Curr Opin Immunol. 2014;30:17–23. doi: 10.1016/j.coi.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang M, Abolhassani H, Lim CK, Zhang J, Hammarstrom L. Next Generation Sequencing Data Analysis in Primary Immunodeficiency Disorders – Future Directions. J Clin Immunol. 2016;36(Suppl 1):68–75. doi: 10.1007/s10875-016-0260-y. [DOI] [PubMed] [Google Scholar]
- 5.Picard C, Fischer A. Contribution of high-throughput DNA sequencing to the study of primary immunodeficiencies. Eur J Immunol. 2014;44(10):2854–61. doi: 10.1002/eji.201444669. [DOI] [PubMed] [Google Scholar]
- 6.Itan Y, Casanova JL. Novel primary immunodeficiency candidate genes predicted by the human gene connectome. Front Immunol. 2015;6:142. doi: 10.3389/fimmu.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shendure J, Fields S. Massively Parallel Genetics. Genetics. 2016;203(2):617–9. doi: 10.1534/genetics.115.180562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilad Y, Pritchard JK, Thornton K. Characterizing natural variation using next-generation sequencing technologies. Trends Genet. 2009;25(10):463–71. doi: 10.1016/j.tig.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, Petrovski S, et al. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 2013;14(7):460–70. doi: 10.1038/nrg3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–55. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 11.Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A. 2015;112(17):5473–8. doi: 10.1073/pnas.1418631112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Exome Aggregation Consortium ML. Karczewski Konrad, Minikel Eric, Samocha Kaitlin, Banks Eric, Fennell Timothy, O’Donnell-Luria Anne, Ware James, Hill Andrew, Cummings Beryl, Tukiainen Taru, Birnbaum Daniel, Kosmicki Jack, Duncan Laramie, Estrada Karol, Zhao Fengmei, Zou James, Pierce-Hoffman Emma, Berghout Joanne, Cooper David, Deflaux Nicole, DePristo Mark, Do Ron, Flannick Jason, Fromer Menachem, Gauthier Laura, Goldstein Jackie, Gupta Namrata, Howrigan Daniel, Kiezun Adam, Kurki Mitja, Moonshine Ami Levy, Natarajan Pradeep, Orozco Lorena, Peloso Gina, Poplin Ryan, Rivas Manuel, Ruano-Rubio Valentin, Rose Samuel, Ruderfer Douglas, Shakir Khalid, Stenson Peter, Stevens Christine, Thomas Brett, Tiao Grace, Tusie-Luna Maria, Weisburd Ben, Won Hong-Hee, Yu Dongmei, Altshuler David, Ardissino Diego, Boehnke Michael, Danesh John, Donnelly Stacey, Roberto Elosua, Florez Jose, Gabriel Stacey, Getz Gad, Glatt Stephen, Hultman Christina, Kathiresan Sekar, Laakso Markku, McCarroll Steven, McCarthy Mark, McGovern Dermot, McPherson Ruth, Neale Benjamin, Palotie Aarno, Purcell Shaun, Saleheen Danish, Scharf Jeremiah, Sklar Pamela, Sullivan Patrick, Tuomilehto Jaakko, Tsuang Ming, Watkins Hugh, Wilson James, Daly Mark, MacArthur Daniel. Analysis of protein-coding genetic variation in 60,706 humans. BioRXiv. 2016 doi: 10.1038/nature19057. http://dx.doi.org/10.1101/030338. [DOI] [PMC free article] [PubMed]
- 13.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15(2):121–32. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- 14.Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, Coe BP, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22(8):1525–32. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, et al. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39(12):1522–7. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 16.Tassara C, Pepper AE, Puck JM. Intronic point mutation in the IL2RG gene causing X-linked severe combined immunodeficiency. Hum Mol Genet. 1995;4(9):1693–5. doi: 10.1093/hmg/4.9.1693. [DOI] [PubMed] [Google Scholar]
- 17.Picard C, Dogniaux S, Chemin K, Maciorowski Z, Lim A, Mazerolles F, et al. Hypomorphic mutation of ZAP70 in human results in a late onset immunodeficiency and no autoimmunity. Eur J Immunol. 2009;39(7):1966–76. doi: 10.1002/eji.200939385. [DOI] [PubMed] [Google Scholar]
- 18.Butte MJ, Haines C, Bonilla FA, Puck J. IL-7 receptor deficient SCID with a unique intronic mutation and post-transplant autoimmunity due to chronic GVHD. Clin Immunol. 2007;125(2):159–64. doi: 10.1016/j.clim.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meienberg J, Zerjavic K, Keller I, Okoniewski M, Patrignani A, Ludin K, et al. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res. 2015;43(11):e76. doi: 10.1093/nar/gkv216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kebschull JM, Zador AM. Sources of PCR-induced distortions in high-throughput sequencing data sets. Nucleic Acids Res. 2015;43(21):e143. doi: 10.1093/nar/gkv717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29(10):908–14. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adam R, Spier I, Zhao B, Kloth M, Marquez J, Hinrichsen I, et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am J Hum Genet. 2016;99(2):337–51. doi: 10.1016/j.ajhg.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016 doi: 10.1038/ng.3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 26.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Itan Y, Shang L, Boisson B, Scott E, Ciancanelli M, Martinez-Barricarte R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nature Methods. 2016 doi: 10.1038/nmeth.3739. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itan Y, Zhang SY, Vogt G, Abhyankar A, Herman M, Nitschke P, et al. The human gene connectome as a map of short cuts for morbid allele discovery. Proc Natl Acad Sci U S A. 2013;110(14):5558–63. doi: 10.1073/pnas.1218167110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itan Y, Mazel M, Mazel B, Abhyankar A, Nitschke P, Quintana-Murci L, et al. HGCS: an online tool for prioritizing disease-causing gene variants by biological distance. BMC Genomics. 2014;15:256. doi: 10.1186/1471-2164-15-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348(6233):448–53. doi: 10.1126/science.aaa1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alcais A, Quintana-Murci L, Thaler DS, Schurr E, Abel L, Casanova JL. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Annals of the New York Academy of Sciences. 2010;1214:18–33. doi: 10.1111/j.1749-6632.2010.05834.x. [DOI] [PubMed] [Google Scholar]
- 33.Deschamps M, Laval G, Fagny M, Itan Y, Abel L, Casanova JL, et al. Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. Am J Hum Genet. 2016;98(1):5–21. doi: 10.1016/j.ajhg.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itan Y, Shang L, Boisson B, Patin E, Bolze A, Moncada-Velez M, et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci U S A. 2015;112(44):13615–20. doi: 10.1073/pnas.1518646112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quintana-Murci L, Clark AG. Population genetic tools for dissecting innate immunity in humans. Nat Rev Immunol. 2013;13(4):280–93. doi: 10.1038/nri3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Su HC, Lenardo MJ. Genomics is rapidly advancing precision medicine for immunological disorders. Nat Immunol. 2015;16(10):1001–4. doi: 10.1038/ni.3275. [DOI] [PubMed] [Google Scholar]
- 37.Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet. 2011;12(9):628–40. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- 38.Itan Y, Casanova JL. Can the impact of human genetic variations be predicted? Proc Natl Acad Sci U S A. 2015;112:11426–7. doi: 10.1073/pnas.1515057112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–41. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13(8):565–75. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 41.Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95(1):5–23. doi: 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolze A, Byun M, McDonald D, Morgan NV, Abhyankar A, Premkumar L, et al. Whole-exome-sequencing-based discovery of human FADD deficiency. Am J Hum Genet. 2010;87(6):873–81. doi: 10.1016/j.ajhg.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savic S, Parry D, Carter C, Johnson C, Logan C, Gutierrez BM, et al. A new case of Fas-associated death domain protein deficiency and update on treatment outcomes. J Allergy Clin Immunol. 2015;136(2):502–5 e4. doi: 10.1016/j.jaci.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Sadovnick AD, Traboulsee AL, Ross JP, Bernales CQ, Encarnacion M, et al. Nuclear Receptor NR1H3 in Familial Multiple Sclerosis. Neuron. 2016;90(5):948–54. doi: 10.1016/j.neuron.2016.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.International MS Genetics Consortium CC. NR1H3 p.Arg415Gln is not associated to multiple sclerosis risk. BioRXiv. 2016 [Google Scholar]
- 46.Bolze A, Mahlaoui N, Byun M, Turner B, Trede N, Ellis SR, et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 2013;340(6135):976–8. doi: 10.1126/science.1234864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Casanova JL, Conley ME, Seligman SJ, Abel L, Notarangelo LD. Guidelines for genetic studies in single patients: lessons from primary immunodeficiencies. J Exp Med. 2014;211(11):2137–49. doi: 10.1084/jem.20140520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lenardo M, Lo B, Lucas CL. Genomics of Immune Diseases and New Therapies. Annu Rev Immunol. 2016;34:121–49. doi: 10.1146/annurev-immunol-041015-055620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352(6284):474–7. doi: 10.1126/science.aac8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miosge LA, Field MA, Sontani Y, Cho V, Johnson S, Palkova A, et al. Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci U S A. 2015;112(37):E5189–98. doi: 10.1073/pnas.1511585112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351(14):1409–18. doi: 10.1056/NEJMoa040036. [DOI] [PubMed] [Google Scholar]
- 52.Pessach IM, Ordovas-Montanes J, Zhang SY, Casanova JL, Giliani S, Gennery AR, et al. Induced pluripotent stem cells: a novel frontier in the study of human primary immunodeficiencies. J Allergy Clin Immunol. 2011;127(6):1400–7 e4. doi: 10.1016/j.jaci.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33(3):400–11. doi: 10.1016/j.immuni.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Casanova JL, Abel L. The human model: a genetic dissection of immunity to infection in natural conditions. Nat Rev Immunol. 2004;4(1):55–66. doi: 10.1038/nri1264. [DOI] [PubMed] [Google Scholar]
- 55.Kane A, Lau A, Brink R, Tangye SG, Deenick EK. B-cell-specific STAT3 deficiency: Insight into the molecular basis of autosomal-dominant hyper-IgE syndrome. J Allergy Clin Immunol. 2016 doi: 10.1016/j.jaci.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 56.Boisson B, Quartier P, Casanova JL. Immunological loss-of-function due to genetic gain-of-function in humans: autosomal dominance of the third kind. Curr Opin Immunol. 2015;32:90–105. doi: 10.1016/j.coi.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woutsas S, Aytekin C, Salzer E, Conde CD, Apaydin S, Pichler H, et al. Hypomorphic mutation in TTC7A causes combined immunodeficiency with mild structural intestinal defects. Blood. 2015;125(10):1674–6. doi: 10.1182/blood-2014-08-595397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanna S, Beziat V, Jouanguy E, Casanova JL, Etzioni A. A homozygous mutation of RTEL1 in a child presenting with an apparently isolated natural killer cell deficiency. J Allergy Clin Immunol. 2015;136(4):1113–4. doi: 10.1016/j.jaci.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 59.Aguilar C, Lenoir C, Lambert N, Begue B, Brousse N, Canioni D, et al. Characterization of Crohn disease in X-linked inhibitor of apoptosis-deficient male patients and female symptomatic carriers. J Allergy Clin Immunol. 2014;134(5):1131–41 e9. doi: 10.1016/j.jaci.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 60.Netter P, Chan SK, Banerjee PP, Monaco-Shawver L, Noroski LM, Hanson IC, et al. A novel Rab27a mutation binds melanophilin, but not Munc13-4, causing immunodeficiency without albinism. J Allergy Clin Immunol. 2016 doi: 10.1016/j.jaci.2015.12.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol. 2012;24(4):364–78. doi: 10.1016/j.coi.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208(8):1635–48. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357(16):1608–19. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 64.Chandrasekaran P, Zimmerman O, Paulson M, Sampaio EP, Freeman AF, Sowerwine KJ, et al. Distinct mutations at the same positions of STAT3 cause either loss or gain of function. J Allergy Clin Immunol. 2016 doi: 10.1016/j.jaci.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125(4):591–9. doi: 10.1182/blood-2014-09-602763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vogel TP, Milner JD, Cooper MA. The Ying and Yang of STAT3 in Human Disease. J Clin Immunol. 2015;35(7):615–23. doi: 10.1007/s10875-015-0187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Khan S, Kuruvilla M, Hagin D, Wakeland B, Liang C, Vishwanathan K, et al. RNA sequencing reveals the consequences of a novel insertion in dedicator of cytokinesis-8. J Allergy Clin Immunol. 2016;138(1):289–92 e6. doi: 10.1016/j.jaci.2015.11.033. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361(21):2046–55. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016;16(4):234–46. doi: 10.1038/nri.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. 2014;133(4):1099–108. doi: 10.1016/j.jaci.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. 2015;125(11):4135–48. doi: 10.1172/JCI80477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Koning HD, van Gijn ME, Stoffels M, Jongekrijg J, Zeeuwen PL, Elferink MG, et al. Myeloid lineage-restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome. J Allergy Clin Immunol. 2015;135(2):561–4. doi: 10.1016/j.jaci.2014.07.050. [DOI] [PubMed] [Google Scholar]
- 73.Alsina L, Gonzalez-Roca E, Giner MT, Piquer M, Puga I, Pascal M, et al. Massively parallel sequencing reveals maternal somatic IL2RG mosaicism in an X-linked severe combined immunodeficiency family. J Allergy Clin Immunol. 2013;132(3):741–3 e2. doi: 10.1016/j.jaci.2013.03.038. [DOI] [PubMed] [Google Scholar]
- 74.Ban SA, Salzer E, Eibl MM, Linder A, Geier CB, Santos-Valente E, et al. Combined immunodeficiency evolving into predominant CD4+ lymphopenia caused by somatic chimerism in JAK3. J Clin Immunol. 2014;34(8):941–53. doi: 10.1007/s10875-014-0088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet. 2013;132(10):1077–130. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. The Journal of experimental medicine. 2011;208(10):2083–98. doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, et al. DNA polymerase-alpha regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol. 2016;17(5):495–504. doi: 10.1038/ni.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2015;517(7532):89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med. 2016;213(7):1163–74. doi: 10.1084/jem.20151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15(7):429–40. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 81.Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic Diagnosis Using Whole Exome Sequencing in Common Variable Immunodeficiency. Front Immunol. 2016;7:220. doi: 10.3389/fimmu.2016.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Buckley RH. The multiple causes of human SCID. J Clin Invest. 2004;114(10):1409–11. doi: 10.1172/JCI23571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raje N, Soden S, Swanson D, Ciaccio CE, Kingsmore SF, Dinwiddie DL. Utility of next generation sequencing in clinical primary immunodeficiencies. Curr Allergy Asthma Rep. 2014;14(10):468. doi: 10.1007/s11882-014-0468-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Notarangelo LD, Sorensen R. Is it necessary to identify molecular defects in primary immunodeficiency disease? J Allergy Clin Immunol. 2008;122(6):1069–73. doi: 10.1016/j.jaci.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 85.Hubbard N, Hagin D, Sommer K, Song Y, Khan I, Clough C, et al. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood. 2016;127(21):2513–22. doi: 10.1182/blood-2015-11-683235. [DOI] [PubMed] [Google Scholar]
- 86.Fischer A. Gene therapy: Myth or reality? C R Biol. 2016;339(7–8):314–8. doi: 10.1016/j.crvi.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 87.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–74. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nijman IJ, van Montfrans JM, Hoogstraat M, Boes ML, van de Corput L, Renner ED, et al. Targeted next-generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol. 2014;133(2):529–34. doi: 10.1016/j.jaci.2013.08.032. [DOI] [PubMed] [Google Scholar]
- 89.Stoddard JL, Niemela JE, Fleisher TA, Rosenzweig SD. Targeted NGS: A Cost-Effective Approach to Molecular Diagnosis of PIDs. Front Immunol. 2014;5:531. doi: 10.3389/fimmu.2014.00531. [DOI] [PMC free article] [PubMed] [Google Scholar]