

Abstract
Macromolecular interactions are used to form supramolecular assemblies, including through the interaction of guest-host chemical pairs. Microstructural heterogeneity has been observed within such physical hydrogels; yet, systematic investigation of the microstructure and its determining inputs are lacking. Herein, we investigated the hierarchical self-assembly of hyaluronic acid (HA) modified by the guest-host pair adamantane (Ad-HA, guest) and β-cyclodextrin (CD-HA, host), as well as with methacrylate groups to both tether fluorescent agents and to covalently stabilize the material structure. We observed microporous materials in the hydrated state, which temporally arose from initially homogenous hydrogels composed of the two polymers. Independent fluorescent labeling of Ad-HA and CD-HA demonstrated spatiotemporal co-localization, indicative of guest-host polymer condensation on the microscale. The hydrogel void fractions and pore diameters were independently tuned through incubation time (0-7 days), polymer concentration (1.25-10 wt%), and polymer modification (25-50% Ad-HA modification). Void fractions as great as 93.3±2.4% were achieved and pore diameters ranged from 2.1±0.5 to 1025.4±209.4 μm. The segregation of discrete solid and solute phases was measured with both atomic force microscopy and diffusive microparticle tracking analysis, where the solute phase contained only dilute polymer. The study represents a systematic investigation of hierarchical self-assembly in binary associating hydrogels, and provides insights on mechanisms that control microstructure within supramolecular hydrogels.
Keywords: Supramolecular assembly, hydrogel, hyaluronic acid, cyclodextrin, porous
INTRODUCTION
Material heterogeneity arises naturally from supramolecular self-assembly processes, leading to complex structures that are adaptive and evolving.1 Indeed, natural materials such as the extracellular matrix are formed via supramolecular self-assembly (e.g., the fibrillar structure of collagen). The formation of such structures has been mimicked with synthetic analogues, including with peptide-based assemblies2-6 that recapitulate many aspects of the native extracellular matrix.7-9 Similar principles have been exploited to form highly ordered and even porous materials through block copolymer assembly,10, 11 hydrogen bonding,12, 13 and with DNA-based materials.14 Though, the utility of such methods is limited largely to the fabrication of structures at the nanoscale.15
Alternatively, supramolecular assemblies have been formed through the directed interaction of modified polymers.16-18 Prominent among these physical hydrogels is the utilization of cyclodextrins, cucurbit[n]urils, and other host macrocycles that form dynamic physical associations with an array of corresponding guest molecules.18, 19 Through polymer conjugation, these macrocycles have bridged the gap between simple supramolecular bonding and complex macromolecular self-assembly processes, enabling formation of precise nano- and micro-particulates, as well as bulk hydrogels.
Within bulk hydrogel supramolecular assemblies, the polymer network has been generally regarded as a homogenous distribution with amorphous structure. However, theoretical explorations have predicted well-ordered states, such as polymer alignment in “railway” complexes, that are thermodynamically preferred under dilute conditions.20 Such states are proposed to arise from the dynamic rearrangement of supramolecular bonds, which enables the polymers to self-sort into their lowest energy state — the configuration in which the number of guest-host complexes is maximized. Monte Carlo simulations of polyelectrolytes have also demonstrated such phenomena in concentrated conditions, including polyelectrolyte collapse into dense heterogeneous structures through the introduction of divalent counterions.21 Experimentally, recent reports have indicated that bulk physical hydrogels can form heterogeneous structures, including those formed through macrocyclic assembly.22-26 Despite this work, there remains a lack of experimental investigation to bridge the gap between directed polymeric self-assembly and supramolecular microstructures.
Towards the application of these physical hydrogels, a better understanding of these underlying phenomena is needed. Particularly, hydrogels from supramolecular assemblies are finding widespread use in biomedical applications, including as injectable hydrogels for the delivery of therapeutics and cells.27-31 In these cases, heterogeneity may influence outcomes such as drug release profiles and cellular interactions, where the scale at which biochemical and biophysical signals are presented to cells may be important.32, 33 The control of microstructural evolution in supramolecular assemblies may also add to techniques that are currently used to introduce structural heterogeneity, such as particle leaching, gas foaming, freeze-drying, and electrospinning.34
Herein, we investigated the evolution of heterogeneity in supramolecular hydrogels formed through the guest-host association of β-cyclodextrin (CD) and adamantane (Ad) when separately conjugated to hyaluronic acid (HA). Investigation was performed via confocal microscopy in the hydrated state to elucidate hierarchical assembly via guest-host induced polymer condensation. The temporal evolution and influence of hydrogel composition on porosity were explored via quantitative image analysis. Further, multiple modalities of micromechanical analysis were used to explore the extent of polymer segregation. The work represents a systematic study of hierarchical microporous assembly in supramolecular hydrogels, offering insights into mechanisms of formation and control over the hydrogel structure.
EXPERIMENTAL
Materials synthesis
Reagents were purchased from Sigma-Aldrich, unless otherwise indicated. Hyaluronic acid (HA, 90kDa; Lifecore Biomedical) was methacrylated (MeHA) via esterification with methacrylic anhydride (pH 8.25-8.5, 3 hrs, 30% modification)35 and subsequently converted to the tetrabutylammonium salt (MeHA-TBA) by ion exchange against Dowex 50Wx8 hydrogen form and neutralization with aqueous tetrabutylammonium hydroxide (0.4M, Fisher Scientific).36 MeHA-TBA was modified by 1-adamantane acetic acid (Acros Organics) to form Ad-MeHA or 6-O-monotosyl-6-deoxy-β-cyclodextrin to form CD-MeHA by anhydrous reaction in DMSO according to our previously published methods.37 Briefly, coupling of cyclodextrin (0.6 eq) and HA (1.0 eq disaccharides) was accomplished by reaction in the presence of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP, 0.6 eq), yielding 25% of disaccharides modified. Esterification of adamantane (3.0 eq) and HA (1.0 eq disaccharides) proceeded by reaction with di-tertbutyl dicarbonate (Boc2O) and 4-dimethylaminopyridine (DMAP, 1.5 eq) where the amount of Boc2O was varied to alter HA modification: 25% (0.47 eq) and 50% (1.13 eq). For all HA derivatives, purification was performed by exhaustive dialysis, filtration to remove insoluble byproducts where necessary, and lyophilization. 1H NMR spectra were acquired in deuterated water (360 MHz, Bruker) and determination of HA modifications performed as previously described.37, 38
In order to fluorescently label the hydrogels, the peptide GCKKG was prepared by solid phase peptide synthesis (PS3, Protein Technologies) from FMOC protected amino acids and glycinol 2-chlorotrityl resin (Novabiochem) and terminated with either 5(6)-carboxyfluorescein (GCKKG-Fluor) or rhodamine B (GCKKG-Rho). Molecular weights were confirmed by MALDI-TOF mass spectrometry (Applied Biosystems Voyager 6030; GCKKG-Fluor: m/z = 893.3, expected 893.5 Da and GCKKG-Rho: m/z = 959.5, expected 959.5 Da).
Ad-MeHA and CD-MeHA were labeled by the fluorescent peptides to form Ad-MeHA-Fluor and CD-MeHA-Rho, respectively. The desired peptides were dissolved in deionized water (0.015 eq) and added dropwise to a solution of the methacrylated polymer (220 mmol disaccharides, 1.0 eq) dissolved in triethanolamine buffer (0.2 M TEOA in PBS, 10 mL, pH 10). Following reaction at room temperature for two hours, purification was performed by exhaustive dialysis and the products were recovered by lyophilization.
Hydrogel formation
Ad-MeHA of 25% modification was used for all samples, except where indicated. The concentration (1.25-10.0 wt%) denotes the combined weight percent of both guest and host polymers. Adamantane and β-cyclodextrin were maintained in a one-to-one ratio (mass ratio of Ad-HA/CD-HA = 0.615 and 0.339 for 25% and 50% modified Ad-HA, respectively). Hydrogels were prepared from separate solutions of the guest and host polymers in PBS containing 5.0 mM lithium phenyl-2,4,6-trimethylbenxoylphosphonate (LAP, photoinitiator). The two component solutions were combined, manually mixed, and centrifuged to remove entrapped air.
Microscopic characterization
For analysis of polymer co-localization, separately labeled guest (Ad-MeHA-Fluor) and host (CD-MeHA-Rho) polymers were utilized. Hydrogels were formed as described, cast in cylindrical molds (8 mm diameter, 75 μL hydrogel), covered in PBS, and incubated for the prescribed period. Prior to confocal imaging (Leica TCS SP5, 20 μm z-spacing, 20x immersion lens), buffer was changed to PBS supplemented with 5.0 mM LAP (2 hr incubation) and photocrosslinked (EXFO OmniCure Series 1000, 320-390 nm filter, 10 mW/cm2, 3 min) to stabilize the structures.
Examination of hydrogel porosity was similarly performed, with hydrogels consisting of Ad-MeHA and CD-MeHA. For imaging and quantification, GCKKG-Fluor was included (5.0 mM) in the buffer at the time hydrogels were dissolved as well as during LAP incubation prior to photocrosslinking. Samples were repeatedly washed by PBS prior to image acquisition, which was performed as described. For quantification (ImageJ) of hydrogel void fraction (Figure S1), images were thresholded and converted to binary (n = 4 hydrogels/group, 10 slices/hydrogel) with the void fraction taken as the ratio of non-zero pixels to total pixel count. The pore diameter (n = 4 hydrogels/group, > 3 pores/hydrogel) is reported as the maximum diameter for the observed pore. Data is reported as mean ± standard deviation and a comparison between 50% and 25% modified Ad-HA samples was performed with a two-tailed Student’s t-test with significance determined at P < 0.05.
Atomic force microscopy
Hydrogels were formed as described and cast into ~ 200 μm thick layers between two glass slides. To aid in sample recovery, one slide was hydrophobically treated (Rain-X). Following incubation for 3 days, 5.0 mM LAP was added (2 hr incubation) and the samples were photocrosslinked (10 mW/cm2, 3 min). The top surface was exposed by removal of the treated slide and then subjected to atomic force microscopy (AFM)-based nanoindentation. Nanoindentation was performed on the top surface and at pore centers (n ≥ 2 locations/sample, 3 repeats/location) at a 10 μm/s indentation depth rate using a borosilicate microspherical tip (R = 12.5 μm, nominal spring constant k= 0.03 N/m) and a Dimension Icon AFM (BrukerNano). The effective indentation modulus, Eind, was calculated by fitting the loading portion of each indentation force-depth (F-D) curve to the elastic Hertz model:
| eq. (1) |
where R is the tip radius and v is Poisson’s ratio (0.49 for highly swollen hydrogels).39 Data are reported as mean ± standard error and comparison was performed by a two-tailed Student’s t-test with significance determined at P < 0.05.
Diffusive microparticle tracking
Hydrogels were cast in thin layers, as described for AFM characterization, with hydrogels containing 0.2 μm fluorescent carboxylate-modified polystyrene beads (Fisher, 6.7×10-3 wt%). Following swelling at 37°C for 7 days, a concentrated solution of microbeads (< 5 μL, 0.04 wt%) was introduced into the aqueous phase via a microinjector and beads were allowed to passively diffuse throughout the construct. Controls included suspension of microbeads (6.7×10-3 wt%) in PBS containing soluble unmodified HA (0, 2.5, 5.0, and 10.0 wt%) as well as beads adhered to a glass surface (fixed, non-diffusive control). Fluorescent beads were imaged for 30 seconds at 62.5 fps (Basler acA640-90uc) at 40x magnification (Olympus BX51).
Images were corrected via background subtraction and sharpening (ImageJ) prior to analysis (Trackpy) to determine the trajectory and mean squared displacement (MSD) of individual particles; drift correction was used as necessary to account for non-diffusive motion. Data was analyzed (MATLAB) by assessment of MSD during the first second of recorded motion, where data for each particle was approximated by a power law:
| eq. (2) |
where D represents the diffusion coefficient and the power, α, describes the diffusivity law of the sample (i.e., superdiffusive, diffusive, or subdiffusive). Data for MSD (n = 3 sample/group, >25 particles/sample) and fit parameters are reported as mean ± standard error. Fit parameters were determined by least-squares regression in MATLAB (Mathworks) and comparisons performed by ANOVA with post hoc Tukey’s HSD; significance was determined at P < 0.05.
RESULTS AND DISCUSSION
Material Preparation and Polymer Co-localization
Macrocyclic guest-host assembly has been used to form hydrogels with diverse polymeric structures and guest-host pairs.18 Here, we utilized hyaluronic acid (HA) modified (Fig. 1A) by 1-adamantane acetic acid (Ad-HA) and aminated β-cyclodextrin (CD-HA) as previously described.37 Upon aqueous dissolution and combination of Ad-HA and CD-HA polymers, assembly (Fig. 1B) is initiated at the molecular scale through guest-host (GH) complexation. At the macromolecular level, it has been proposed that binary associations between polymers with such structure (i.e., linear and pendantly modified) thermodynamically favor the formation of specifically structured polymer pairs, including lateral assembly in a “railway” type complex to minimize free energy,20 resulting in GH polymer condensation. Extrapolating this concept to more dense combinations of guest and host modified polymers, we hypothesized that the initially homogenous mixture would not represent the thermodynamically favored state. Rather, stochastic rearrangement of bonding pairs was expected to result in polymer condensation, leading to microscale organization of dense polymer regions and concurrent formation of polymer void pores throughout the hydrogel.
Fig. 1.
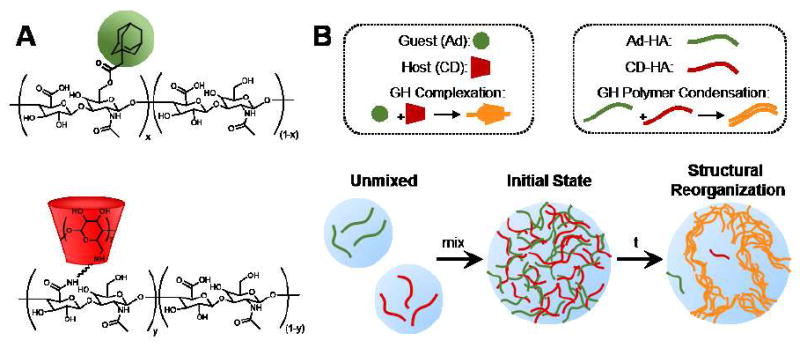
Polymer structure and self-assembly. (A) Chemical structures of the component polymers, including hyaluronic acid (HA, black) modified by 1-adamantane acetic acid (Ad, green) or β-cyclodextrin (CD, red). (B) Schematic of guest-host (GH) hydrogel hierarchical organization at the molecular, polymeric, and microscales.
Indeed, macromolecular self-assembly has resulted in ordered structures, including in porous network assembly by crown ethers26 as well as in thin film and bulk hydrogel assemblies in cucurbit[8]uril systems.24, 25 Though, such observations have primarily been made by scanning electron microscopy (SEM) of dried samples, where freeze-drying may induce porosity via ice crystal formation as an artifact of the methodology. Such artifacts have been observed in cucurbit[8]uril hydrogels by examination via small-angle neutron scattering (SANS) which revealed no change in nanostructure with increased crosslink density, despite observation of a porous structure by SEM.40
Given these prior observations, direct visualization of hydrogels in the hydrated state was deemed essential. To enable microscale imaging in aqueous conditions, guest and host polymers were methacrylated to allow fluorescent labeling via Michael addition reactions—where Ad-MeHA and CD-MeHA were labeled by thiolated fluorescein (Fluor) and rhodamine (Rho), yielding Ad-MeHA-Fluor and CD-MeHA-Rho, respectively. Additionally, methacrylate functionalization was utilized to stabilize the supramolecular hydrogels via photopolymerization41 in the presence of lithium phenyl-2,4,6-trimethylbenxoylphosphonate (LAP) to prevent disruption of the structures, which may occur due to physical agitation or thermally induced GH complex dissociation.
Solutions of Ad-MeHA-Fluor (0.95 wt%), CD-MeHA-Rho (1.55 wt%), and their corresponding GH hydrogel (2.5 wt%: 0.95 wt% Ad-MeHA-Fluor + 1.55 wt% CD-MeHA-Rho) were incubated for three days prior to photopolymerization and examination. Control solutions for Ad-MeHA-Fluor and CD-MeHA-Rho remained translucent at the macroscale, both before and following photopolymerization (Fig. 2A); whereas, GH hydrogels were translucent upon formation but then turbid by day 3, indicative of light diffraction by microstructural heterogeneity. When these samples were examined with confocal microscopy and quantified with intensity profiles (Fig. 2B), the CD-MeHA-Rho and Ad-MeHA-Fluor hydrogels were homogeneous at day 3, whereas the GH hydrogel assumed a heterogeneous and highly porous architecture. This microscale polymer condensation has not been previously observed for GH hydrogels in the hydrated state. Since the controls did not develop this condensation, the observed GH hydrogel structure is therefore the direct result of hierarchical assembly and not an artifact of hydrophobic polymer association or polymerization-induced aggregation.
Fig. 2.
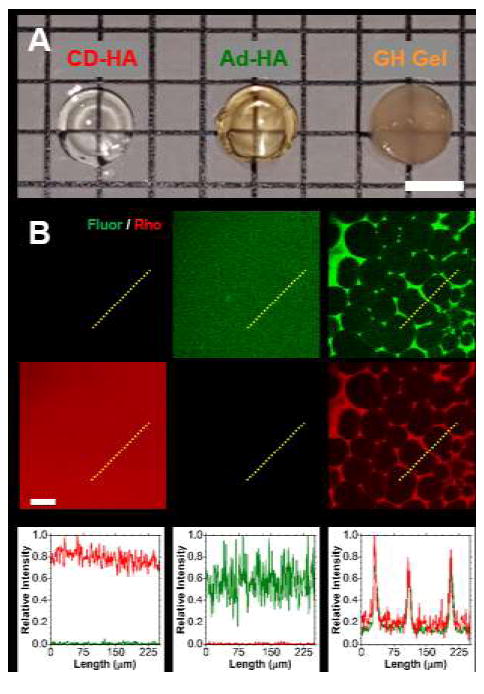
Binary associations and heterogeneity in guest-host hydrogels. (A) Micrograph of CD-MeHA-Rho (left, 1.55 wt%), Ad-MeHA-Fluor (middle, 0.95 wt%), and corresponding GH hydrogel (2.5 wt%: 0.95 wt% Ad-MeHA-Fluor + 1.55 wt% CD-MeHA-Rho) polymerized at day 3 following incubation. Scale bar: 5.0 mm. (B) Fluorescent confocal images (top, red and green channels separated) and normalized intensity profiles (bottom, paths indicated by yellow dotted lines) of separate guest-host hydrogel components and the corresponding GH hydrogel. Scale bar: 50 μm.
In order to probe the initial state and evolution of the porous structure, GH hydrogels were formed (2.5 wt%) and imaged over time to evaluate temporal changes (Fig. 3). Initially (day 0), the guest and host polymers were uniformly distributed, as confirmed by their corresponding intensity profiles. Over time, porous structures developed throughout the hydrogels (day 1 and day 3). Intensity profiles further demonstrated a high degree of spatial co-localization between the fluorescein and rhodamine signals, indicating co-localization of the modified polymers. Likewise, examination of three-dimensional (3D) reconstructions (Fig. 4) demonstrated co-localization of the separate polymers throughout space with relative heterogeneity of pore size throughout the depth (400 μm) examined. Such co-localization is requisite for the hierarchical assembly mechanism proposed, as both guest and host polymers are required to achieve GH polymer condensation.
Fig. 3.
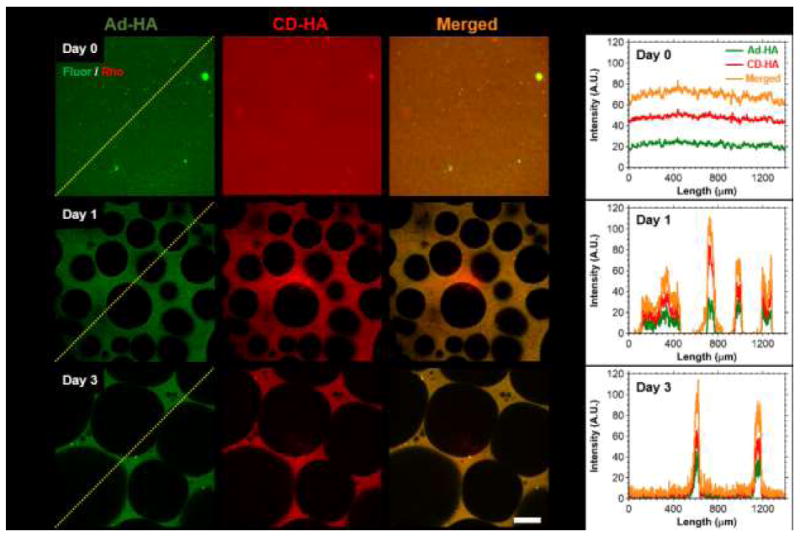
Temporal evolution of hydrogel porosity and component co-localization. (A) Representative fluorescent confocal microscopy images of GH hydrogels (2.5 wt%) at day 0 (top), day 1 (middle) and day 3 (bottom), where components were separately labeled for imaging: Ad-MeHA-Fluor (green) and CD-MeHA-Rho (red). Scale bar: 100μm. (B) Corresponding normalized intensity profiles along the paths indicated (yellow dotted line), illustrating spatiotemporal co-localization of guest and host polymers.
Fig. 4.
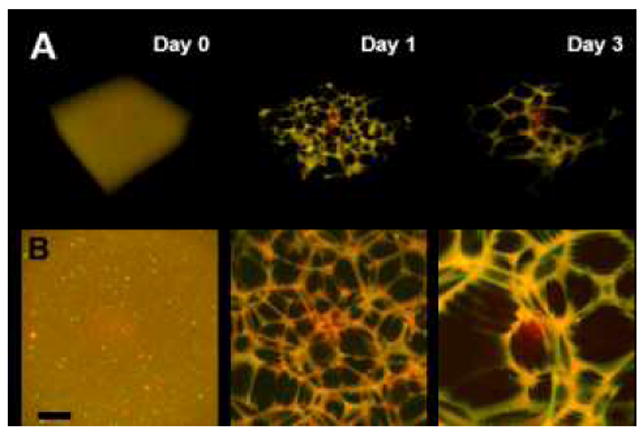
Orthogonal view of three-dimensional (3D) reconstructions (A) and maximum projection (B) of the guest-host hydrogels (Day 0-3; 2.5 wt%). Images are merged channels from confocal Z-stacks at day 0 (left), day 1 (middle) and day 3 (right). Scale bar: 100μm.
Control of Heterogeneity
In various self-assembled systems, the resulting superstructure is dependent upon the material composition, including component topology and concentration.2, 12, 13, 42 These factors govern the configurations in which macromolecules may assemble and the influence of secondary interactions (i.e., entanglement, additional hydrogen bonding, and electrostatic repulsion) on the assembly structure. Additionally, the formation of the supramolecular structure is temporally dependent, since macromolecular re-arrangement is required.12 Accordingly, both temporal dependence and hydrogel composition were systematically investigated by microscopy methods, where hydrogels were allowed to evolve to the determined time point prior to photocrosslinking in the presence of thiolated fluorescein (5 mM). Inclusion of the fluorophore during the crosslinking process enabled improved contrast for quantification of pore features (Fig. S1), such as void fraction and pore diameter.
Heterogeneity rapidly developed within GH hydrogels (2.5wt%, Fig. 5A), with void fractions of 67.8±4.2% observed within 6 hours that slowly increased to 93.3±2.4% by day 7. This indicated relatively rapid GH polymer condensation to achieve thermodynamic equilibrium. During this timeframe, the pore size steadily increased from 58.3±3.5 μm at 6 hr to 1025.4±209.4 μm at day 7. The drastic growth in pore size despite reduced relative changes in void fraction indicated that pore growth may have resulted from microscopic restructuring, such as the merging of adjacent pores due to wall thinning (Fig 5A). Wall thinning may be the result of forces within the wall due to bulk swelling, as well as surface erosion that is known to occur in these dynamic hydrogel systems over the course of months.37, 43 Minimal changes in void fraction occurred beyond the first day, concurrent with drastic growth in pore diameters, suggesting that wall tension and not polymer erosion may be the more dominant process.
Fig. 5.
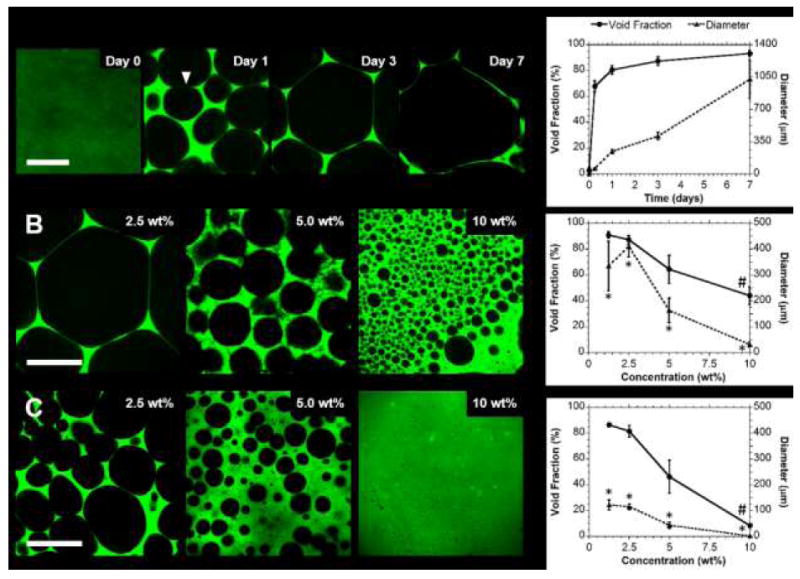
Control over hydrogel porosity. (A) Representative confocal images (left) and corresponding quantification (right) of hydrogel void fractions and pore diameters over time (Day 0-7; 2.5 wt%; 25% modified Ad-HA). Wall thinning and pore merging, proposed to contribute to restructuring, is indicated (▼). Pore features were dependent on polymer concentration (B; Day 3; 1.25-10.0 wt%; 25% modified Ad-HA), including with change in Ad-HA modification (C; Day 3; 1.25-10 wt%; 50% modified Ad-HA). (mean ± SD; n ≥ 4; p < 0.05 for 25% vs 50% modified Ad-HA for pore diameter (*) and void fraction (#))
To evaluate how hydrogel composition influenced porosity, samples were evaluated at day 3 (Fig. 5B,C). Both void fraction (90.9±2.6% to 44.1±6.8%) and pore diameter (418.8±1.5 μm to 31.5±9.8 μm) were inversely proportional to polymer concentration (1.25-10 wt%). With increased modification of the guest polymer (50% modified Ad-HA), pore size was controlled independent of polymer concentration. Void fractions (86.6±1.7% to 8.5±0.9%) were similar to those of 25% modified Ad-HA, with a significant reduction observed only at high (10 wt%) concentrations. Drastic reduction in pore size (122.4±19.4 μm to 2.1 ± 0.5 μm, approximately fourfold) was observed, which was significant relative to the 25% modified Ad-HA across all concentrations. Taken together, these results indicate that polymer concentration is the dominant governing factor in controlling the void fraction. Secondary polymeric interactions, including polymer solvation and electrostatic repulsion of HA are therefore implicated in suppression of void formation as they counter guest-host polymer condensation. Independent of these mechanisms, increased multivalence (i.e., increased Ad modification) is known to drastically reduce dynamic rearrangement of guest-host hydrogels as a result of increased polymer avidity and formation of multifold junctions, as examined through prior characterization of bulk relaxation behaviors.37, 44 Here, reduction in macromolecular dynamics due to increased avidity translated to reduced microstructural rearrangement and hence a reduced pore size. Multivalence also influenced void fraction, likely due to suppressed dynamics that inhibited pore segregation at high polymer concentrations. Due to the influence of macromolecular dynamics on the observed porosity, it is notable that the feature size and evolution timescale may also be drastically altered through selection of the supramolecular binding pairs, as the timescale for the transient associations is related to the equilibrium association constant (Keq) which varies by greater than ten orders of magnitude.18, 45
Micromechanical Characterization
Imaging in the hydrated state allowed discrete quantification of hydrogel heterogeneity. However, the presence and concentration of polymer within the porous phase of the hydrogel could not be determined by optical methods due to light diffraction which produced a non-zero intensity throughout the sample. In order to confirm segregation of a solvent phase, devoid of an elastic network and containing only dilute polymer in solution, both atomic force microscopy (AFM) and diffusive microparticle tracking analysis were performed.
To probe heterogeneous structures via AFM, hydrogels (2.5 wt%) were cast in thin layers (~200 μm thick) between coverslips, as described, and allowed to evolve for three days prior to photocrosslinking. The top surface was exposed and subject to indentation testing (Fig. 6) at the hydrogel surface (i), where an indentation modulus of 0.41±0.07 kPa was observed. The probe was re-located to the pore center (ii) at a height identical to (i), where indentation yielded no measurable modulus. Assisted by the z-step motor of AFM (resolution = 0.1 μm), the indenter tip was lowered for ≈50 μm until the pore surface (iii) was located. Throughout this process, nanoindentation measurements were repeated (approximately every 10 μm) with no measureable modulus observed. At location (iii), nanoindentation yielded a modulus of 0.47±0.13 kPa. Measurements at location (ii) and throughout the depth of the pore demonstrated a lack of crosslinked solid phase material, as there was no resistance to indentation. Moreover, the moduli of the gel phase were similar (p = 0.77) at both the top surface (i) and the bottom of the pore region (iii), indicating homogeneity throughout the gel phase. It should be noted that these moduli are representative of the GH hydrogel only after the introduction of covalent crosslinks with photocrosslinking and are used only towards assessing structural heterogeneity.
Fig. 6.
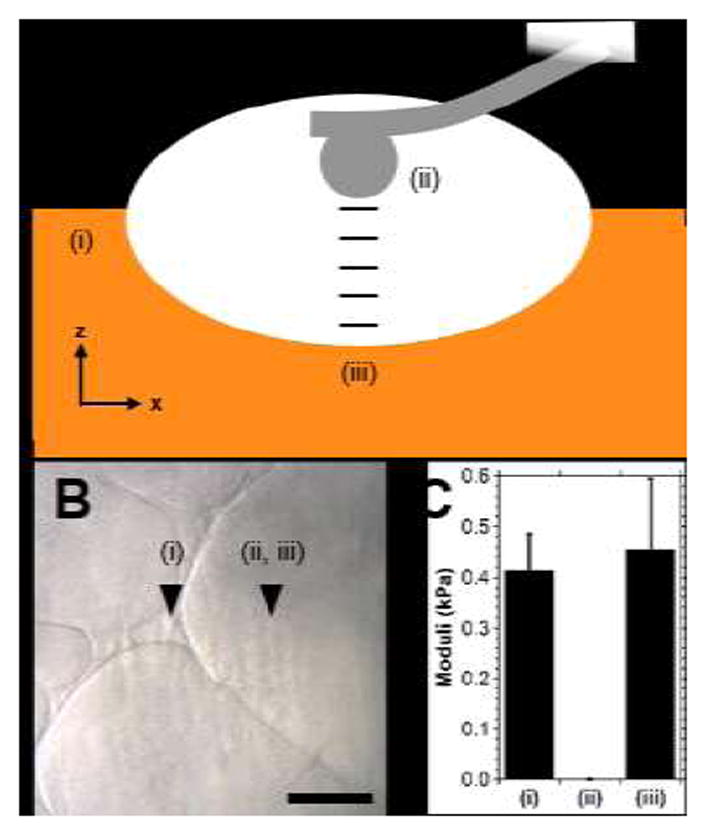
Examination of guest-host hydrogel heterogeneity via atomic force microscopy (AFM). (A) Schematic illustration of testing (side view), where moduli of the hydrogel (Day 3; 2.5 wt%) were determined at the top surface (i), in the pore center (ii, same z position as (i)) and at increments of approximately 10 μm until the bottom surface of the pore region (iii) was reached. (B) Bright field image, approximate location of testing indicated. Scale bar: 100 μm. (C) Indentation moduli at the locations tested. (mean ± SE; n ≥6; p = 0.77).
Microparticle tracking was employed to independently validate observations of discrete phase segregation and to further characterize regional composition and viscoelastic properties in the native physically crosslinked state. While AFM nanoindentation is useful to illustrate varied hydrogel moduli in space, it is unable to account for changes in solvent viscosity which may occur due to un-crosslinked polymer in solution. Microparticle tracking has become a standard methodology, including in its active (i.e., traction force microscopy and driven microbead rheology) and passive (i.e., diffusive microbead rheology) forms. Moreover, these techniques have recently enabled investigation of micro-heterogeneous environments, including in the intracellular and pericellular space.46, 47 Thus, diffusive microbead rheology was explored herein, as it enables facile investigation and precision suitable to discern changes in viscosity resulting from small changes in HA concentrations.48, 49
To assess properties via microrheology, samples were prepared in thin layers with inclusion of 0.2 μm fluorescent beads without photocrosslinking. As included microbeads were entrapped within the polymer mesh during structural evolution, diffusive beads were not initially observed within the pores and were thus introduced by microinjection of concentrated suspensions and allowed to diffuse throughout the constructs prior to image acquisition. Particle motion and trajectories within the hydrogels (Fig. 7A,B, Movie S1) indicated two discrete populations—a population of diffusive particles grossly similar to those in buffer (PBS) and a separate population with constrained trajectories. Examination of mean-squared distance (MSD, Fig. 7C,D) and fitting of diffusion parameters (eq. 2) confirmed diffusive, unconstrained motion (α = 1.04 ± 0.013) and subdiffusive motion (α = 0.76 ± 0.049) of particles within the pore and gel phases, respectively. Between the two populations, diffusion coefficients differed significantly (Fig. 7E, Tukey p < 0.0001). Significant differences were also noted in the pore phase relative to measurement in buffer alone (PBS) and in the gel phase relative to particles fixed to a glass surface, indicating some viscosity of the pore phase and some motion in the gel phase.
Fig. 7.
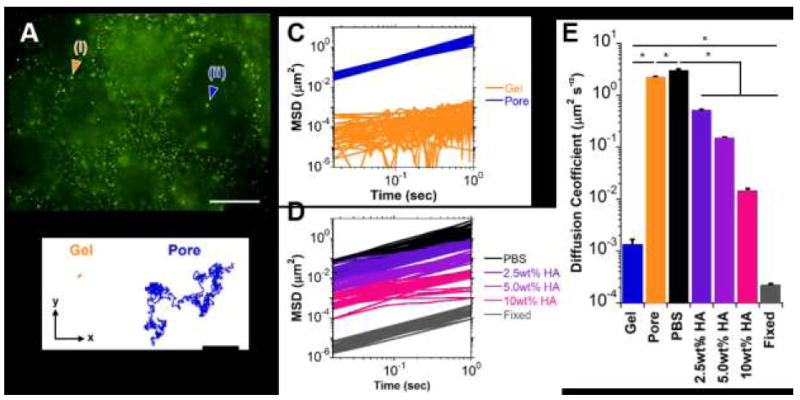
Microrheological examination of guest-host hydrogels (Day 7; 2.5 wt%). (A) Representative epifluorescent image of microbeads (0.2 μm diameter) entrapped within the hydrogel (i) or diffusing throughout the pores (ii). Scale bar: 50μm. (B) Trajectories of particle motion within the x-y plane over a 30 second period for a microbead engrafted within the hydrogel (Gel, orange) or diffusing throughout a pore (Pore, blue). (C-D) Mean squared distance (MSD) traces of particle motion as a function of time for representative subpopulations (n=20 particles/group shown) within the hydrogel (C) and for solutions consisting of PBS or soluble hyaluronic acid (2.5, 5.0, 10.0 wt% HA in PBS) and fixed (non-diffusive) controls (D). (E) Diffusion coefficients, determined by fit to eq. (2) (mean ± SE; n > 25; *p < 0.05).
To further validate sensitivity of the method toward viscosity changes, particle motion was examined in HA solutions of varying concentration (Fig. 7D, Movie S2). Drastic changes in gross particle motion and diffusion coefficients (Tukey p < 0.001) were observed with varied concentration. Exponential fits were performed to diffusion coefficients (D) as a function of HA concentration ([HA] wt%), (D = 3.72e−0 58[HA]; R2 > 0.97), from which the HA concentration within the pores was approximated to be 0.032 wt%. Low concentrations of unbound polymer within the pore were likely a result of erosion of guest or host polymers from the gel surface, a reversible process known to occur in guest-host type hydrogels due to stochastic association and dissociation of the binding pairs.43
Overall, the formation of such porous structures is of potential interest towards development of scaffolds for cellular investigation or towards biomedical therapies, as the hollow pores readily allow diffusion that is needed for cell seeding and viability, as well as the presentation of mechanical signals in pseudo-three dimensional culture settings.32, 33 Moreover, the polymer condensation into a solid phase as shown here may have implications in the release of entrapped molecules. In prior studies utilizing guest-host hydrogels, prolonged release of molecules has been demonstrated, including for bovine serum albumin (>60 days), peptides, and small molecules (>3 weeks).28, 29, 37 These results rival or surpass timeframes of many conventional covalently crosslinked hydrogels, and this unanticipated behavior may depend largely upon reductions in mesh size, induced by phase separation of the polymer into a more condensed state.
CONCLUSIONS
Supramolecular interactions were utilized to produce physical hydrogels that evolved through hierarchical assembly to produce highly porous microstructures over time. Assembly resulted from macromolecular condensation, driven by dynamic molecular guest-host complexation. Micromechanical analyses indicated that the pores were indeed devoid of solid hydrogel—containing only low concentrations of dissociated polymer from stochastic erosion. Furthermore, the porosity evolved temporally to increase both void fraction and pore diameter. Owing to polymer solvation and electrostatic repulsion which counter polymer condensation, polymer concentration was primarily responsible for reduction in void fraction. Reduction in network dynamics (e.g., through increased polymer modification) influenced the timescale of macromolecular rearrangement, altering pore diameter. Through these mechanisms, pore diameters spanning three orders of magnitude and void fractions as great as 93.3±2.4% were achieved. These studies begin to close the gap in our knowledge of directed polymeric assembly and resultant microstructure within supramolecular hydrogels, furthering the development of physically assembled hydrogels systems with structural complexity.
Supplementary Material
Acknowledgments
This work was financially supported by the National Institutes of Health (R01 HL107938, T32 AR007132), Drexel Faculty Start-up Grant (LH) as well as a Predoctoral Fellowship (CBR) and an Established Investigator Award (JAB) from the American Heart Association.
References
- 1.Service RF. Science. 2005;309(5731):95–95. doi: 10.1126/science.309.5731.95. [DOI] [PubMed] [Google Scholar]
- 2.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294(5547):1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 3.Capito RM, Azevedo HS, Velichko YS, Mata A, Stupp SI. Science. 2008;319(5871):1812–1816. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- 4.Murnen HK, Rosales AM, Jaworski JN, Segalman RA, Zuckermann RN. J Am Chem Soc. 2010;132(45):16112–16119. doi: 10.1021/ja106340f. [DOI] [PubMed] [Google Scholar]
- 5.Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417(6887):424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 6.Percec V, Dulcey AE, Balagurusamy VS, Miura Y, Smidrkal J, Peterca M, Nummelin S, Edlund U, Hudson SD, Heiney PA. Nature. 2004;430(7001):764–768. doi: 10.1038/nature02770. [DOI] [PubMed] [Google Scholar]
- 7.Krishna OD, Kiick KL. Biomacromolecules. 2009;10(9):2626–2631. doi: 10.1021/bm900551c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Leary LER, Fallas JA, Bakota EL, Kang MK, Hartgerink JD. Nat Chem. 2011;3(10):821–828. doi: 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- 9.Reyes CD, García AJ. J Biomed Mater Res, Part A. 2003;65(4):511–523. doi: 10.1002/jbm.a.10550. [DOI] [PubMed] [Google Scholar]
- 10.Cui H, Chen Z, Zhong S, Wooley KL, Pochan DJ. Science. 2007;317(5838):647–650. doi: 10.1126/science.1141768. [DOI] [PubMed] [Google Scholar]
- 11.Jenekhe SA, Chen XL. Science. 1999;283(5400):372–375. doi: 10.1126/science.283.5400.372. [DOI] [PubMed] [Google Scholar]
- 12.Dankers PYW, Hermans TM, Baughman TW, Kamikawa Y, Kieltyka RE, Bastings MMC, Janssen HM, Sommerdijk N, Larsen A, van Luyn MJA, Bosman AW, Popa ER, Fytas G, Meijer EW. Adv Mater. 2012;24(20):2703–2709. doi: 10.1002/adma.201104072. [DOI] [PubMed] [Google Scholar]
- 13.Cheng C-C, Chang F-C, Wang J-H, Chu Y-L, Wang Y-S, Lee D-J, Chuang W-T, Xin Z. RSC Advances. 2015;5(93):76451–76457. [Google Scholar]
- 14.Vyborna Y, Vybornyi M, Häner R. J Am Chem Soc. 2015;137(44):14051–14054. doi: 10.1021/jacs.5b09889. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S. Nat Biotechnol. 2003;21(10):1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 16.Webber MJ, Appel EA, Meijer E, Langer R. Nat Mater. 2016;15(1):13–26. doi: 10.1038/nmat4474. [DOI] [PubMed] [Google Scholar]
- 17.Wang HY, Heilshorn SC. Adv Mater. 2015;27(25):3717–3736. doi: 10.1002/adma.201501558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodell CB, Mealy JE, Burdick JA. Bioconjugate Chem. 2015;26(12):2279–2289. doi: 10.1021/acs.bioconjchem.5b00483. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Jiang M. Chem Soc Rev. 2011;40(5):2254–2266. doi: 10.1039/c0cs00153h. [DOI] [PubMed] [Google Scholar]
- 20.Semenov A, Chariot A, Auzely-Velty R, Rinaudo M. Rheol Acta. 2007;46(5):541–568. [Google Scholar]
- 21.Yin D-W, de la Cruz MO, de Pablo JJ. Journal Chemical Phys. 2009;131(19):194907. doi: 10.1063/1.3264950. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Ma H, Chieng YY, Hou S, Li X, Tan Y. RSC Advances. 2015;5(27):20684–20690. [Google Scholar]
- 23.McGann CL, Akins RE, Kiick KL. Biomacromolecules. 2015;17(1):128–140. doi: 10.1021/acs.biomac.5b01255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Nie C-B, Zhou T-Y, Qi Q-Y, Fu J, Wang X-Z, Dai L, Chen Y, Zhao X. Polym Chem. 2015;6(11):1923–1927. [Google Scholar]
- 25.Appel EA, Biedermann F, Rauwald U, Jones ST, Zayed JM, Scherman OA. J Am Chem Soc. 2010;132(40):14251–14260. doi: 10.1021/ja106362w. [DOI] [PubMed] [Google Scholar]
- 26.Yan X, Xu D, Chi X, Chen J, Dong S, Ding X, Yu Y, Huang F. Adv Mater. 2012;24(3):362–369. doi: 10.1002/adma.201103220. [DOI] [PubMed] [Google Scholar]
- 27.Rodell CB, Rai R, Faubel S, Burdick JA, Soranno DE. J Controlled Release. 2015;206(0):131–139. doi: 10.1016/j.jconrel.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 28.Mealy JE, Rodell CB, Burdick JA. J Mat Chem B. 2015;3(40):8010–8019. doi: 10.1039/C5TB00981B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appel EA, Loh XJ, Jones ST, Dreiss CA, Scherman OA. Biomaterials. 2012;33(18):4646–4652. doi: 10.1016/j.biomaterials.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 30.Gaffey AC, Chen MH, Venkataraman CM, Trubelja A, Rodell CB, Dinh PV, Hung G, MacArthur JW, Soopan RV, Burdick JA, Atluri P. J Thorac Cardiovasc Surg. 2015;150(5):1268–1277. doi: 10.1016/j.jtcvs.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeom J, Kim SJ, Jung H, Namkoong H, Yang J, Hwang BW, Oh K, Kim K, Sung YC, Hahn SK. Adv Healthcare Mater. 2015;4(2):237–244. doi: 10.1002/adhm.201400304. [DOI] [PubMed] [Google Scholar]
- 32.Marklein RA, Soranno DE, Burdick JA. Soft Matter. 2012;8(31):8113–8120. [Google Scholar]
- 33.Huebsch N, Lippens E, Lee K, Mehta M, Koshy ST, Darnell MC, Desai RM, Madl CM, Xu M, Zhao X. Nat Mater. 2015;14(12):1269–1277. doi: 10.1038/nmat4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Annabi N, Nichol JW, Zhong X, Ji CD, Koshy S, Khademhosseini A, Dehghani F. Tissue Eng Part B Rev. 2010;16(4):371–383. doi: 10.1089/ten.teb.2009.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodell CB, MacArthur JW, Dorsey SM, Wade RJ, Wang LL, Woo YJ, Burdick JA. Adv Fund Mater. 2015;25(4):636–644. doi: 10.1002/adfm.201403550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smeds KA, Grinstaff MW. J Biomed Mater Res. 2001;54(1):115–121. doi: 10.1002/1097-4636(200101)54:1<115::aid-jbm14>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 37.Rodell CB, Kaminski AL, Burdick JA. Biomacromolecules. 2013;14(11):4125–4134. doi: 10.1021/bm401280z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Highley CB, Rodell CB, Burdick JA. Adv Mater. 2015;27:5075–5079. doi: 10.1002/adma.201501234. [DOI] [PubMed] [Google Scholar]
- 39.Ahearne M, Yang Y, El Haj AJ, Then KY, Liu K-K. J R Soc Interface. 2005;2(5):455–463. doi: 10.1098/rsif.2005.0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Appel EA, Loh XJ, Jones ST, Biedermann F, Dreiss CA, Scherman OA. J Am Chem Soc. 2012;134(28):11767–11773. doi: 10.1021/ja3044568. [DOI] [PubMed] [Google Scholar]
- 41.Lu HD, Soranno DE, Rodell CB, Kim IL, Burdick JA. Adv Healthcare Mater. 2013;2(7):1028–1036. doi: 10.1002/adhm.201200343. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Wang X, Yang F, Shen H, You Y, Wu D. Langmuir. 2015;31(51):13834–13841. doi: 10.1021/acs.langmuir.5b03823. [DOI] [PubMed] [Google Scholar]
- 43.Rodell CB, Wade RJ, Purcell BP, Dusaj NN, Burdick JA. ACS Biomater Sci Eng. 2015;1(4):277–286. doi: 10.1021/ab5001673. [DOI] [PubMed] [Google Scholar]
- 44.Charlot A, Auzely-Velty R. Macromolecules. 2007;40(26):9555–9563. [Google Scholar]
- 45.Appel EA, del Barrio J, Loh XJ, Scherman OA. Chem Soc Rev. 2012;41(18):6195–6214. doi: 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- 46.Selvaggi L, Salemme M, Vaccaro C, Pesce G, Rusciano G, Sasso A, Campanella C, Carotenuto R. Methods. 2010;51(1):20–26. doi: 10.1016/j.ymeth.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 47.Schultz KM, Kyburz KA, Anseth KS. Proc Natl Acad Sci U S A. 2015;112(29):E3757–E3764. doi: 10.1073/pnas.1511304112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mellnik J, Vasquez PA, McKinley SA, Witten J, Hill DB, Forest MG. Soft Matter. 2014;10(39):7781–7796. doi: 10.1039/c4sm00676c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill DB, Vasquez PA, Mellnik J, McKinley SA, Vose A, Mu F, Henderson AG, Donaldson SH, Alexis NE, Boucher RC, Forest MG. PLoS One. 2014;9(2):e87681. doi: 10.1371/journal.pone.0087681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.