

Abstract
The highly regulated pH of cells and the less-regulated pH of the surrounding extracellular matrix (ECM) is the result of a delicate balance between metabolic processes and proton production, proton transportation, chemical buffering, and vascular removal of waste products. Malignant cells show a pronounced increase in metabolic processes where the 10- to 15-fold rise in glucose consumption is only the tip of the iceberg. Aerobic glycolysis (Warburg effect) is one of the hallmarks of cancer metabolism that implies excessive production of protons, which if stayed inside the cells would result in fatal intracellular acidosis (maintaining a strict acid–base balance is essential for the survival of eukaryotic cells). Malignant cells solve this problem by increasing mechanisms of proton transportation which expel the excess acidity. This allows cancer cells to keep a normal intracellular pH, or even overshooting this mechanism permits a slightly alkaline intracellular tendency. The proton excess expelled from malignant cells accumulates in the ECM, where chronic hypoxia and relative lack of enough blood vessels impede adequate proton clearance, thus creating an acidic microenvironment. This microenvironment is quite heterogeneous due to the tumor’s metabolic heterogeneity and variable degrees of hypoxia inside the tumor mass. The acidic environment (plus other necessary cellular modifications) stimulates migration and invasion and finally intravasation of malignant cells which eventually may result in metastasis. Targeting tumor pH may go in two directions: 1) increasing extracellular pH which should result in less migration, invasion, and metastasis; and 2) decreasing intracellular pH which may result in acidic stress and apoptosis. Both objectives seem achievable at the present state of the art with repurposed drugs. This hypothesis analyzes the altered pH of tumors and its implications for progression and metastasis and also possible repurposed drug combinations targeting this vulnerable side of cancer development. It also analyzes the double-edged approach, which consists in pharmacologically increasing intracellular proton production and simultaneously decreasing proton extrusion creating intracellular acidity, acid stress, and eventual apoptosis.
Keywords: metabolic processes, malignant cells, acid, apoptosis, Warburg effect
Introduction
Enhanced growth and proliferation in cancer cells require a nutrient- and oxygen-rich environment in order to fuel anabolic processes that increase metabolism more than 10-fold. But such a rich environment does not exist. As soon as increased proliferation and growth start, high demand depletes oxygen and nutrients from the tumor milieu.
The direct consequence of this hypoxic state is the overexpression of HIF, which may be already increased due to oncogenic mutations.
HIF induces the expression of multiple genes, most of them related to a metabolic switch toward a low (or nil) oxygen consumption/metabolism and stimulation of growth of new vessels (neoangiogenesis). The process of new vessels production achieved through the stimulation of HIF/VEGF in cancer cells and in peritumoral cells like fibroblasts, macrophages, myofibroblasts, and endothelial cells is an imperfect process because the new vessels usually do not reach a fully functional entity. This means that the hypoxic environment becomes a permanent feature in cancer, in spite of these new vessels.
The metabolic shift, which is a consequence of oncogenic mutations and HIF overexpression, is toward a glycolytic phenotype (Warburg effect). Abandoning partially oxidative phosphorylation (OXPHOS) and adopting the glycolytic pathway as the main source of energy has two important advantages for the cancer cells, in spite of this pathway’s low energetic efficiency:
Decreases reactive oxygen species production that is much higher under OXPHOS metabolism.
Generates biological building blocks for other molecules needed in the anabolic process of biomass building.
But at the same time, it has a disadvantage: an excess of protons is produced through different mechanisms; three outstanding are:
Excess lactic acid production; lactic acid dissociates into lactate plus a proton.1
ATP hydrolysis also generates excess of protons.
Hydration of CO2 forming CO3H− through the activity of carbonic anhydrases (CAs), particularly CA IX.
Cancer cells adapt to this excessive intracellular acidity by increasing the number and function of proton-exporting mechanisms. This adaptation keeps intracellular pH at normal or slightly alkaline levels, while the ECM receives the burden of the exported acidity.2 In this way, a pH gradient is quickly established with a normal or slightly alkaline interior and an acidic exterior.3
This picture favors cancer growth and progression through diverse mechanisms:
Intracellular alkaline pH stimulates proliferation.
Extracellular acidity is a necessary feature for the activation of matrix-degrading enzymes like cathepsin and metalloproteases. This matrix degradation is necessary for migration, invasion, and eventual metastasis.
ECM acidity blocks immunologic attacks against malignant cells and decreases tumor access of certain chemotherapeutics.
Hypoxia plus extracellular acidity contribute to tumor progression and the Darwinian selection of resistant cells that may survive in this harsh environment.
The highly regulated pH of cells and the less-regulated pH of the surrounding ECM is the result of a delicate balance between metabolic processes, proton production, proton transportation, chemical buffering, and vascular removal of waste products, supported by sophisticated mechanisms.
Microenvironmental and intracellular pH are major issues that influence processes like proliferation, differentiation, metastasis, and angiogenesis.
Malignant cells show a pronounced increase in metabolic processes where a 10- to 15-fold increase in glucose consumption is only the tip of the iceberg. Aerobic glycolysis (Warburg effect) is one of the hallmarks of cancer metabolism which implies excessive production of protons, which if stayed inside the cell would result in fatal intracellular acidosis (maintaining a strict acid–base balance is essential for the survival of eukaryotic cells). Also, an anaerobic glycolysis is found in hypoxic areas of the tumor. So, the main cause of environmental acidification is the accumulation of protons in the ECM, as byproducts of intracellular metabolism. Cancer cells require a normal or slightly alkaline pH inside the cell which favors protein synthesis and mitosis. Zetterberg and Engstrom4 in 1981 described that a pH between 7.3 and 8.2 produced a linear increase in DNA synthesis in quiescent serum-starved cells. According to Tannock and Rotin,5 the cytoplasmic alkalization may precede proliferation in certain cells. Malignant cells are in a permanent fight against excessive acid load, and solve this chronic problem on a short- and a long-term basis. In the short term, malignant cells use chemical buffering and transfer of acids into organelles.5
In the long term, the solution comes through increasing mechanisms of proton transportation which expel the excess acidity. This allows cancer cells to keep a normal intracellular pH, or even overshooting this mechanism permits a slightly alkaline intracellular tendency.
Not all cancer cells express or overexpress the same combination of proton transporters, and sometimes, there may be important differences among different tumors.6
The proton excess expelled from malignant cells accumulates in the ECM, where chronic hypoxia and relative lack of functional blood vessels hinder adequate proton clearance, thus creating an acidic microenvironment which shows a pH 0.5–1 unit lower than nonmalignant tissues. Cancer actually shows an inverted pH gradient.7 This microenvironment is quite heterogeneous due to the metabolic heterogeneity of the tumor and varying degrees of hypoxia inside the tumor mass, which is partially a consequence of a highly disorganized neovascular structure. Darwinian selection is responsible for the survival and evolution of the best adapted cells to the acidic microenvironment, so almost all malignant tumors show heterogeneous extracellular acidity with well-adapted malignant cells.
The acidic environment (plus other necessary cellular modifications) stimulates migration and invasion and finally intravasation of malignant cells which eventually may result in metastasis. This acidic environment also plays an important role in drug uptake, facilitating the entry of weak acids (cyclophosphamide and cisplatin) into the cells and working the opposite way with weakly alkaline drugs (doxorubicin).
Targeting tumors pH may go in three directions:
increasing extracellular pH which should result in less migration, invasion, and metastasis (“acid-mediated invasion hypothesis”);
decreasing intracellular pH which may result in acidic stress and apoptosis; and
increasing extracellular pH and at the same time decreasing intracellular pH.
These objectives seem achievable at the present state of the art with repurposed drugs.
This review analyzes the altered pH of tumors and its implications for progression and metastasis and also possible repurposed drug combinations to target this vulnerable side of cancer development. Targeting acidic microenvironment should not represent an aggression against normal tissues because they lack this feature, and it may be part of conventional chemoradiotherapy treatments because it does not interfere with them. On the contrary, in certain cases, targeting the acidic microenvironment may be an advantageous companion of conventional treatments.
Many recent studies have emphasized the role of extracellular pH in cancer, while to a certain extent neglecting the importance of intracellular pH in cancer growth and progression. We think that both should be addressed simultaneously in order to achieve better therapeutic results. This is what we have called double-edged pH targeting.
The double-edged pH targeting
Having said this, an interesting question arises: what would happen if proton extrusion is handicapped?
Protons would remain in the cell, decreasing the pH of the intracellular milieu, and the ECM would not receive an acid burden so that acidity of this nano-environment would be reduced. An increase in intracellular acidity would eventually drive the cells toward apoptosis due to acidic stress and migration, and invasion would be decreased.
What would be the result if in addition to handicapping proton extrusion we increase intracellular proton production?
More proton production and less proton extrusion would increase the acidic stress and its consequence: cell death.
Can this be achieved with repurposed nontoxic drugs?
The answer is absolutely “yes”.
Drugs like metformin and doxycycline are inhibitors of mitochondrial complex I decreasing OXPHOS and increasing aerobic glycolysis. Atovaquone is a selective inhibitor of mitochondrial complex III, and it also decreases OXPHOS. The consequence of this process is an increase in lactic acid production. We shall call these drugs “mitochondrial poisons”.
Amiloride, quercetin, proton pump inhibitors (PPIs), and voltage-gated sodium channel (VGSC) blockers like phenytoine decrease the activity of different proton exporters. We shall dub these drugs as “proton extrusion inhibitors”.
The combination of mitochondrial poisons with proton extrusion inhibitors may achieve this double-edged effect we are looking for: increased acidic intracellular environment with decreased extracellular acidity.
But how will this affect noncancer cells?
Cancer cells have a 10- to 17-fold higher consumption of glucose than nonmalignant cells, and glycolysis is the predominant metabolic pathway under normoxia (aerobic glycolysis or Warburg effect) and under hypoxia (anaerobic glycolysis) in cancer cells. Nonmalignant cells on the other hand use the tricarboxylic acid OXPHOS pathway as the predominant metabolic pathway.8 This means that the lactic acid burden is significantly higher in malignant cells and consequently intracellular pH will decrease markedly in relation to nonmalignant cells when treated with mitochondrial poisons.
In addition to the extracellular and the intracellular compartments that are classically considered for pH targeting, there is a third compartment which is frequently forgotten: the intra-organelle compartment with the intra-lysosomal compartment. This compartment is usually acidic in tumor cells and “helps” the cell “put away” part of the excess of acidic load.
The importance of tumor pH targeting is clearly visible in the work of Robey et al,9 where using oral sodium bicarbonate to increase tumor pH, in an in vivo trial of a mouse model of metastatic breast cancer, the number of metastases was decreased.
The review by Leanza et al10 gives an excellent view of ion channel targeting in cancer; nevertheless, it does not discuss the pH modifications that this targeting produces. pH targeting is essentially (though not solely) a targeting of ion channels and ion transporters.
This concept of targeting cancer cells from two different sides was described for the first time in 2011 by McCarty and Whitaker,11 but it had no follow-up in practice.
The double-edged effect of increasing intracellular proton production and inhibiting proton extrusion simultaneously has a proof of concept in the research by Lee et al.12 To achieve this, they used hydrogen sulfide which increases glucose uptake and lactate production through an increased glycolytic rate, and simultaneously reduces the activity of proton extrusion mechanisms by sodium–proton exchanger (NHE) activity reduction. The final result was increased intracellular acidity in cancer cells which selectively killed malignant cells but not the nonmalignant counterparts. The results found in this research showed that at the cellular level at least, it was possible to:
increase intracellular acidity selectively in cancer cells by increasing glycolytic pathways;
use increased intracellular acidity as a lethal weapon against cancer cells; and
preserve normal cells from any major damage due to this procedure.
There are partial aspects of this double-edged approach with sufficient and adequate evidence to support the double-edged hypothesis as described in this review.
Cellular pH sensors
Normal cells keep intracellular pH within a narrow range, which runs between 7.1 and 7.2 by regulating membrane proton pumps and proton transporters that are under the control of intracytoplasmic pH sensors. These sensors recognize pHi and induce adequate cellular responses to keep it in the above-mentioned range. In many cases, this implies acidifying the extracellular pH due to excessive proton extrusion.
Expression of proton-sensing G protein-coupled receptors (GPCRs) regulates certain aspects of tumorigenesis, migration, and invasion. There is no evidence of a relation between intracellular and extracellular pH sensors.
Decreasing proton export produces apoptosis in cancer cells
Six important pH regulators have been identified at the cellular level (there are probably more, but these six account for most of the activity; Figure 1):
Vacuolar ATPase proton pump.
NHE family.
Bicarbonate transporter family.
Monocarboxylate transporter (MCT) family.
VGSCs.
CA isoforms family.
Figure 1.
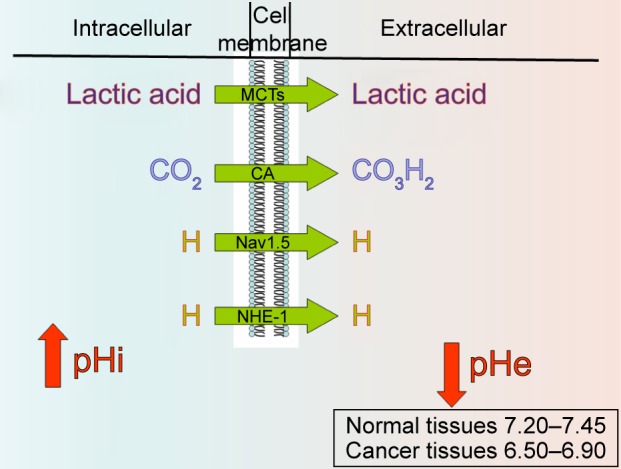
Main pH regulators’ mechanism of action. Nav1.5 represents the voltage-gated sodium channel.
Abbreviations: MCTs, monocarboxylate transporters; CA, carbonic anhydrase; NHE-1, sodium–proton exchanger-1.
Another pH regulator that is showing increased importance in cancer is Na+/CO3H− cotransporter (NBCn1).
Vacuolar ATPase
Vacuolar H+ ATPase is a highly conserved enzyme and a vital component of all eukaryotes, located in the membranes of many organelles and is responsible for low intravacuolar pH, mainly in lysosomes and endosomes. It can also be found in the cell surface.13 V-ATPase pumps H+ from the cytoplasm into vacuoles with energy expenditure (ATP hydrolysis) regulating cytoplasmic pH.
Overexpression of a yeast plasma membrane V-ATPase in normal mouse fibroblasts increased intracellular pH and induced a tumorigenic phenotype.14,15
Inhibition of tumor vacuolar ATPase produces intracellular acidification and induces apoptosis16,17 and increases the cytotoxic activity of chemotherapeutic drugs like paclitaxel.18 Unfortunately, there are no highly effective and specific V-ATPase inhibitors that could reach medical practice, due to their usually high toxicity (like Bafilomycin). Disulfiram is a V-ATPase inhibitor which has been used in the treatment of alcoholism and shows interesting antitumor activity. Probably, most of its activity is not related to V-ATPase inhibition. PPIs usually used for the treatment of gastroduodenal ulcers, gastroesophagic reflux disease, and Zollinger–Ellison syndrome have shown antitumor activity. PPI’s effect is particularly elicited in acidic environment19 like the one usually found in tumors.
In 1993, Martinez-Zaguilan et al20 identified the presence of functional V-ATPases in cell membranes of various human tumors. Bafilomycin A1, an experimental V-ATPase-specific inhibitor, significantly lowered the intracellular pH of those tumors with high V-ATPase expression. A few years later, Sennoune et al21 found that breast cancer cells with high metastatic potential expressed high levels of V-ATPase. The opposite was found in cell lines with low metastatic risk. V-ATPase also regulates Notch signaling in triple-negative breast cancer22 and modulates metalloprotease isoforms in pancreatic cancer cells,23 immunomodulates neutrophils associated with tumors24 and is an essential proton pump for cancer invasion.25 Inhibition of V-ATPase also decreased the population of macrophages associated with the tumor microenvironment which develop a pro-tumor activity,26 decreased tumor growth, and overcame cisplatin resistance in ovarian cancer cells.16 V-ATPases also play an important role as a mediator of cancer-related inflammation.27
These publications represent a proof of concept that V-ATPase is not only present in membranes of intracellular organelles but also located in plasmatic membranes of malignant cells, and its presence has a functional significance: homeostasis of cytoplasmic pH plus other functions related to malignant phenotype. But V-ATPases not only acidify the extracellular compartment through proton extrusion and consequently increase cytoplasmic pH but also acidify intracellular compartments like lysosomes and endoplasmic vesicles.28
It was the work of Yeo et al29 which called attention to the apoptotic effect on gastric cancer of repurposed PPIs already being used to treat gastritis and acid gastroduodenal syndromes.
Selecting the PPI
According to Lugini et al,30 lansoprazole is the best choice for targeting tumor pH because it showed better results when compared to other PPIs in the experimental setting (cell culture). Furthermore, lansoprazole is an inhibitor of fatty acid synthase, which is a necessary enzyme in lipogenic phenotype malignancies. Omeprazole too has been recognized as a fatty acid synthase inhibitor.
Clinical trial NCT02595372 (https://ClinicalTrials.gov) is based on a preliminary retrospective study that showed that PPIs intake in breast cancer patients during chemotherapy significantly improved overall survival. The trial tries to determine the exact role of PPIs in cancer (omeprazole is used). The study is still ongoing at the Indiana University School of Medicine.
Clinical trial NCT01748500 ongoing at the University Health Network in Toronto is an interventional study to determine the benefits of pantoprazole in the treatment of castrate-resistant prostate cancer treated with docetaxel.
Clinical trial NCT01069081 explores whether adding a PPI (esomeprazole at high doses) to docetaxel and cisplatin chemotherapy improves efficacy and does not affect tolerability in metastatic breast cancer.
Clinical trial NCT01163903 is a dose-finding, phase I study to determine the recommended phase II dose for the combination of doxorubicin and pantoprazole in patients with advanced tumors and no standard treatment options.
Additional evidence of PPIs’ effects in cancer is summarized in Table 1.
Table 1.
PPIs in cancer
| Study | PPI effect |
|---|---|
| Mattsson et al120 | In 1991, omeprazole was found to exhibit specific inhibitory activity on the H+/K(+)-ATPase |
| Mizunashi et al121 | In 1993, this research group established that omeprazole decreased bone reabsorption through inhibition of V-ATPase at lysosomal level in osteoclasts in a human clinical setting |
| Luciani et al122 | PPI pretreatment (omeprazole or esomeprazole or pantoprazole) sensitized tumor cell lines (melanoma, adenocarcinoma, and lymphoma) to cisplatin, 5FU, and vinblastine. PPI pretreatment inhibited V-ATPase activity and increased pHe and the pH of lysosomes. Oral pretreatment with omeprazole induced sensitivity of human solid tumors to cisplatin |
| De Milito et al123 | PPIs affected viability of human B cells and increased sensibility to vinblastine. They also induced lysosomal permeabilization which was probably related to apoptosis, which induced cytosolic acidification. PPIs resulted in cytotoxicity for leukemic cells in ALL |
| Capodicasa et al124 | Omeprazole induced apoptosis in polymorphonuclear cells |
| Ferrari et al125 | PPIs chemosensitized human osteosarcoma cells to chemotherapy with cisplatin in cell cultures and xenografts |
| Patel et al126 | Pantoprazole increased the cytotoxicity of doxorubicin in solid tumors (cell culture). Pantoprazole increased endosomal pH |
| Avnet et al127 | The targeting of V-ATPase with siRNA and omeprazole in Ewing sarcoma produced a reduction in cell viability |
| Chen et al128 | Pantoprazole decreased multidrug resistance in gastric adenocarcinoma and decreased cell viability. Pantoprazole decreased pHi and reversed pHi–pHe gradient. Experiments were carried out on cell cultures and xenografts. Also, downregulation of the V-ATPases-mTOR-HIF-1 signaling was found |
| Shen et al129 | Pantoprazole inhibited tumor growth and decreased HIF-1 expression in human gastric adenocarcinoma |
| Patlolla et al130 | Rats fed with omeprazole showed decreases in aberrant crypt formation in a murine model of azoxymethane-induced crypt formation. Omeprazole also increased p21 expression in colon cancer cell lines and decreased antiapoptotic proteins expression |
| Perut et al131 | Sarcomas show increased numbers of acidic lysosomes. Esomeprazole induced dose-dependent cytotoxicity by interfering with proton compartmentalization |
| Azzarito et al132 | Lansoprazole increased sensitization of human melanoma cells to low doses of paclitaxel. This was confirmed in a xenograft model |
| Huang et al133 | Pantoprazole induces apoptosis in gastric cancer cells probably through inhibition of STAT3 |
| Goh et al134 | Esomeprazole increased the antitumor effect of doxorubicin on triple-negative breast cancer cell MDA-MB-468 and showed growth-suppressive activity when used alone |
| Zhang et al135 | Human breast cancer cells treated with lansoprazole showed apoptosis in a dose-dependent way. In xenografts, lansoprazole produced alkalization of lysosomes and increase in ROS |
| Jin et al136 | Omeprazole showed ligand capacity to aryl hydrocarbon receptor, decreasing cell invasion and metastasis in ER-negative breast cancer |
| Salerno et al137 | Rhabdomyosarcoma stem cells showed a very high level of V-ATPase and lysosomal acidity with high invasiveness and reduced cytotoxicity with doxorubicin. Omeprazole increased doxorubicin cytotoxicity, and decreased growth and invasion even at low concentrations of omeprazole |
| Yeo et al29 | Pantoprazole in vivo and in vitro induced apoptosis in gastric cancer cells |
| Udelnow et al138 | Omeprazole inhibited proliferation of pancreatic cancer cells and modulated autophagy |
| Marino et al139 | Esomeprazole induced apoptosis in melanoma cells but also induced autophagic defenses. The administration of an autophagia inhibitor increased malignant cell death due to esomeprazole |
| Vishvakarma and Singh140 | Pantoprazole in a murine model of T cell lymphoma produced an increase in tumoricidal activity of TAMs |
| Yeo et al141 | PPIs induced apoptosis in gastric cancer cells |
Abbreviations: PPIs, proton pump inhibitors; 5FU, 5-fluorouracil; ALL, acute lymphoid leukemia; siRNA, small interfering RNA; ROS, reactive oxygen species; TAMS, tumor associated macrophages.
As can be seen in Table 1, many antitumor effects of PPIs are independent of V-ATPase inhibition.
Table 2 shows other anticancer effects of PPIs independent of V-ATPase inhibition.
Table 2.
Other anticancer effects of PPIs
| Study | Other effects of PPIs related to cancer therapy |
|---|---|
| Shen et al142 | Pantoprazole downregulates pyruvate kinase M2 |
| Zhang et al143 | Pantoprazole decreases Wnt/beta catenin signaling and epithelial–mesenchymal transition in gastric cancer cells, decreasing invasiveness |
| Tan et al144 | Pantoprazole decreases autophagia and increases docetaxel effects increasing growth delay in human breast carcinoma MCF-7 cells, human vulvar epidermoid cells and prostate cancer PC-3 cells |
| Hahm145 | Pantoprazole has anti-inflammatory activity |
| Vishvakarma and Singh146 | Pantoprazole decreases tumor-induced myelosuppression in T cell lymphoma |
| Mishima et al147 | Lansoprazole increases osteoblast genesis through enhancement of nuclear accumulation of Runx2 and stimulating osteoblast differentiation |
| Matsui et al148 | Omeprazole inhibits melanogenesis in rat melanoma cells and in normal human melanocytes by blocking ATP4A (a membrane P-type H+/K+ ATPase) and also increases tyrosinase degradation |
| Shiizaki et al149 | Omeprazole activates aryl hydrocarbon receptor in human hepatoma cells and human hepatocytes |
Abbreviation: PPIs, proton pump inhibitors.
Recently, Canitano et al31 found that lansoprazole, and to a lesser extent omeprazole, showed significant antitumor activity in multiple myeloma cells. Lansoprazole’s cytotoxicity was caspase independent.
NHE-1 and VGSC
The expression of VGSCs appears increased in cancer cells in many cases where it is not expressed in their normal counterparts, and plays a significant role in disease progression. The overexpressed VGSCs are usually Nav1.5, Nav1.6, and Nav1.7 and their splicing variants. The embryonic or neonatal isoform of Nav1.5 was identified in breast cancer. The mechanism of action of VGSCs is depicted in Figure 2.
Figure 2.

V-ATPase using energy and extruding H+.
This overexpression has been identified in breast, cervical, non-small-cell lung, small-cell lung, prostate, ovarian, colon, pancreas, and mesothelial cancer.32 This increased expression induces invasiveness by increasing the proteolytic activity in the ECM without regulating cellular multiplication or migration.33
Activities of NHE-1 and VGSC are summarized in Figure 3.
Figure 3.
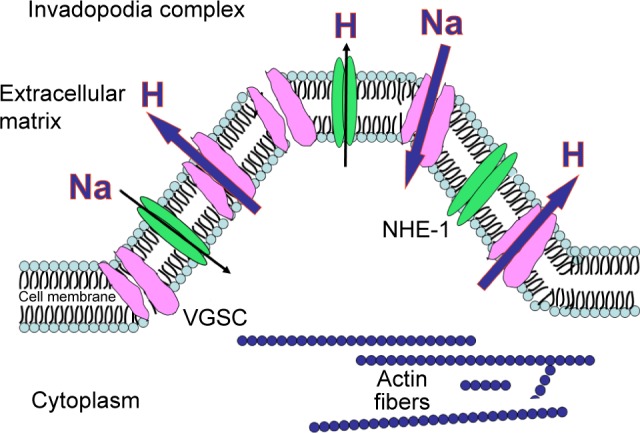
NHE-1 and VGSC working as H+ extruders.150 (Invadopodia complexes are actin-rich protrusions of the cell membrane with active degradation of the extracellular matrix and invasion.)151 The presence of high levels of NHE-1 is fundamental for invadopodia formation.152 There is a relationship between NHE-1 enhanced activity and VGSC, but it has not been fully elucidated yet. Reproduced from Koltai T. Voltage-gated sodium channel as a target for metastatic risk reduction with re-purposed drugs. F1000Res. 2015;4:297.32
Abbreviations: NHE-1, sodium–proton exchanger-1; VGSC, voltage-gated sodium channel.
In 1981, Moolenaar et al34 observed in neuroblastoma cells that there was a Na–proton interchange that increased intracellular pH upon proton extrusion or Na entering the cell. They also described that this mechanism was blocked by amiloride. Three years later, Comoli et al35 described that in a hepatoma model of ascitic cells, pH was higher in proliferating cells than in nonproliferating ones, and this difference was eliminated by amiloride. When glucose was added to the culture, pH decreased in the extracellular medium of proliferating and nonproliferating cells due to lactate production, but intracellular pH only decreased in proliferating cells. Sparks et al36 showed that amiloride had the ability to inhibit H6 hepatoma growth and mammary adenocarcinoma growth in a dose-dependent manner in mice. Not only do the injections of amiloride inhibited tumor growth and proliferation, but there was also a correlation with an important decrease in tumor cell sodium content.
Greco et al37 demonstrated that the acidification of the ECM by proton extrusion through NHE-1 was a fundamental condition for the activation of proteases at the invadopodial extracellular level (Figure 2), and in 1990, Delvaux et al38 found that amiloride and analogs had the capacity to inhibit NHE-1 and malignant cell proliferation.
NHE-1 has three basic functions, all of which are related to cancer evolution:
Extrusion of protons.
Structural anchor for actin filaments.
Assembler of signaling complexes in specialized plasma membrane domains.39
NHE-1 is activated by growth factors, GPCRs, integrins, different tyrosine kinase receptors and p42/p44 MAPK cascade,40,41 HIF-1,42 p38 MAPK,43 cytoplasmic acidification,44 Akt,45 CD44,46 and insulin.47
There is now enough evidence about amiloride’s activities and its analogs to consider this diuretic as an NHE-1 blocker and a pH modifier, including antiproliferative activity. The evidence is summarized in Figure 4.
Figure 4.

Summary of NHE-1 activators on the left side and effects on the right side.
Abbreviation: NHE-1, sodium–proton exchanger-1.
But there are other remarkable effects of amiloride which were described by Davis and Czech:48 it blocks EGFR autophosphorylation, and also the PDGFR autophosphorylation. The authors attribute the antiproliferative activity of amiloride to this anti-tyrosine-kinase activity.
Rich et al49 studying leukemia cells found that pHi was increased not only in leukemia cells but also in peripheral blood cells in relation to normal hematopoietic tissues. Treating leukemia cells with an amiloride analog (5-(N,N-hexamethylene) amiloride) decreased pHi and induced apoptosis.
In addition to its actions on proton extrusion, NHE-1 has other effects that are of capital importance in cell migration: cytoskeletal anchoring. This is particularly important at the invadopodia level where NHE-1 not only creates polarity through proton extrusion that increases pHi but also acts as an anchor of actin filaments to plasma membrane.50 Inhibiting NHE-1 decreases or even eliminates cell migration.
Table 3 provides further evidence of amiloride’s anticancer activity.
Table 3.
Evidences of amiloride’s anticancer activity
| Study | Activity |
|---|---|
| Rowson-Hodel et al153 | The amiloride derivative 5-(N,N-hexamethylene) amiloride has cytotoxic properties against breast cancer cells |
| Sanhueza et al154 | The authors proposed amiloride as a possible treatment of ovarian cancer |
| Acevedo-Olvera et al155 | The authors demonstrated a suppressive effect of 5-(N-ethyl-N-isopropyl) amiloride on the proliferation of leukemia cell line stimulated with SCF, by decreasing the mitochondrial membrane potential and decreasing intracellular alkalization |
| Pieri et al156 | Amiloride blocks the growth of murine leukemia cells |
| Sparks et al36 | Amiloride decreases intranuclear sodium content and inhibits cell proliferation in hepatoma and breast adenocarcinoma cells |
| Kellen et al157 | Amiloride inhibits the urokinase-type activity of plasminogen activator |
| Zheng et al158 | Amiloride increases erlotinib anticancer activity on human pancreatic cancer cells through Akt inhibition |
| Hrgovic et al96 | Ion pump inhibitors reduce tumor growth. Amiloride decreases tumor growth, and synergizes with other ion pump inhibitors |
Abbreviation: SCF, stem cell factor.
Cariporide is a sodium–hydrogen ion exchange inhibitor developed by Aventis Pharma (Mumbai, India) with the purpose of decreasing myocardial ischemic damage during the reperfusion process, but it has shown interesting anticancer activities. Research in the cardiovascular area has slowed down, due to adverse effects when used at high dose. In the field of oncology, cariporide has demonstrated apoptotic effects on cancer cells overexpressing NHE-1.
Cariporide mesylate may be administered by the oral or parenteral route.
Cariporide and amiloride have a guanidine function that is probably responsible for NHE-1 inhibition.51
In cancer, cariporide had been shown to reduce hypoxia-induced invasion in human squamous cell carcinoma of the tongue,52 and cholangiocarcinoma,53 and overcome multidrug resistance.33
Cariporide has not been clinically tested in oncology.
Another mechanism that has been found to partially decrease the activity of NHE-1 is ATP depletion, in spite of the fact that NHE-1 activity is not energy consuming, and therefore, it should not theoretically depend on the ATP level.54 Although it has not been tested experimentally, but based on this finding, we may assume that ATP depletion produced by mitochondrial poisons, as we propose here, may have an inhibitory effect on NHE-1.
Another possible way to decrease NHE-1 activity is through PPARγ agonists in those tumors that overexpress PPARγ, like certain breast cancers. Exposure of these cell lines to natural or synthetic PPARγ agonists like rosiglitazone decreases NHE-1 gene expression.55
Voltage-gated sodium channels
In 1995, Grimes et al56 studied differential electrophysiological characteristics in two different rodent prostate cancer cell lines: Mat-Ly-Lu cell line (highly metastatic) and AT-2 cell line (lower metastatic potential). These two cell lines exhibited different electrophysiological features that maintained direct relationship with in vitro invasiveness. Inward sodium currents were detected only in the Mat-Ly-Lu cell line, and inhibition of VGSC protein with tetrodotoxin (TDX) reduced significantly the capacity for invasion. TDX showed no effect on invasion of AT-2 cell lines. The TDX-induced reduction in invasion kept a direct correlation with the amount of cells expressing VGSC in the culture.
They concluded that ion channels may be involved in malignant cell behavior and VGSCs could play a role in the metastatic process.
Laniado et al57 found similar differences in two human prostate cell lines: one was highly metastatic and the other was not. As in the work by Grimes et al, they found that PC-3, the more malignant cell line, expressed VGSC protein and the inhibition of this channel protein with TDX reduced invasion in a significant way. LNCap cells did not express VGSC.
One conclusion reached by the authors is that cells expressing functional VGSC develop a selective advantage regarding migration and distant metastasis.
The correlation between VGSC protein expression and invasiveness in human and rat prostate cancer cells was confirmed by Smith et al58 by comparing seven lines of rat prostate carcinoma cells with different metastatic ability, and nine human prostate carcinoma cell lines. In general, invading capacity of the basement membrane and metastatic ability kept a positive correlation with the percentage of cells expressing VGSC. But this positive correlation occurred only in a certain range of cells being invasive (27% in rats and 12% in humans). The authors suggest that the discrepancies may be due to the need of other factors besides the presence of VGSC so that this protein may represent a prerequisite for the invasive phenotype but other requirements must also be met for a full-blown invasive phenotype. Fraser et al59 determined the key role played by VGSCs in prostate cancer cells regarding invasion and motility and showed that TDX and phenytoin that are known VGSC blockers decreased motility and invasiveness, while channel openers increased motility.
The increased invasion capacity in VGSC-expressing cancer cells is not limited to prostate. It can be found in breast cancer cell lines.60
Batcioglu et al61 showed the importance of VGSCs inhibition in a rat model of induced breast cancer in order to inhibit antioxidant response. They observed a survival improvement in rats treated with a VGSC blocker.
An important location of VGSCs in cancer cells is in a cellular region directly involved in migration and invasion: the invadopodia.
Invadopodias are actin-rich protrusions of the plasma membrane, which are strongly related to degradation of the ECM.
The Na(+)/HCO3(−) cotransporter SLC4A4
The Na/HCO3 cotransporter SLC4A4 plays an important role in intracellular pH regulation because it intervenes in bicarbonate recapture and helps maintain a slightly alkaline intracellular environment. Parks and Pouyssegur62 found that hypoxia induces overexpression of SLC4A4 cotransporter in a colon adenocarcinoma cell line in a HIF-dependable manner. Inhibition of SLC4A4 reduced proliferation and increased apoptosis during external acidosis. In a breast cancer line overexpressing SLC4A4, the knockdown of this transporter had an important impact on proliferation, migration, and invasion.
Carbonic anhydrase
CAs are a family of hypoxia-inducible enzymes that catalyze the reversible hydration of carbon dioxide to bicarbonate and a proton.
There are 15 CA isoforms expressed in humans; two of them, CA IX and CA XII, were found to be associated with tumors. Both are transmembrane isoforms where the catalytic domain is extracellular. Both may be highly expressed in tumors and almost insignificantly expressed in non- tumor cells.63
CA IX plays a key role in extracellular pH regulation in the tumor environment.
Oncogenic metabolism is characterized by high production of lactic acid and carbon dioxide which are exported to the environment generating an acid extracellular milieu. Bicarbonate generated by CA IX is incorporated into the cell-buffering pHi.
Inhibition of CA IX has shown important antitumor effects and is actually considered a valid target in cancer treatment. Many small molecules with selective ability to inhibit CA IX are in the experimental phase. Also, monoclonal antibodies have been developed and are now under clinical trial.
Acetazolamide is a pan-CA inhibitor with no selectivity for CA IX but has been tested with good results in many tumors like bronchial carcinoid,64 renal carcinoma cells,65 breast cancer cells,66,67 colon cancer cells,68 bladder cancer,69 glioblastoma,70 and gastric carcinoma.71
Acetazolamide improves the efficacy of mTOR inhibitors by increasing its activity in hypoxic areas of the tumor72 and potentiates bevacizumab in cholangiocarcinoma.73
It has been suggested that saccharin may be a potential CA IX and CA XII inhibitor.74
Monocarboxylate transporters
MCT isoforms 1–4 are enzymes that catalyze the proton-linked transport of monocarboxylates such as L-lactate, pyruvate, and ketone bodies across the plasma membrane. MCT4 expression is increased in response to hypoxia by mediation of HIF-1α. It is frequently overexpressed in malignant cells.
MCT1 is present in almost all tissues, and its main role is to catalyze lactic acid influx or efflux from the cell.
When aerobic or anaerobic glycolysis is increased, as in cancer cells, MCT1 reduces intracellular acidity by exporting lactate with a proton. This prevents the toxic accumulation of lactate and acidification in the intracellular milieu. Very aggressive tumors may overexpress MCT4 with similar functions.75
MCTs play a fundamental role in shuttling lactic acid between different cells (Figure 5). This is particularly noticeable in cancer cells. Izumi et al76 found that MCT1 and MCT4 expression in human lung cancer cell lines was significantly correlated with invasiveness.
Figure 5.

Glycolytic cancer cells and “enslaved” mesenchymal cells expel lactic acid through the activity of MCT1. Oxidative cancer cells uptake lactic acid and complete its catabolism through OXPHOS. MCT4 intervenes in this step.
Abbreviations: MCT, monocarboxylate transporter; OXPHOS, oxidative phosphorylation; GLUT, glucose transporter; TCA, tricarboxylic acid.
There are no MCT inhibitors currently used in clinical practice. Astra Zeneca developed an oral molecule (AZD3965) that inhibits MCT1 but not MCT4 which is being tested in clinical trials. It seems to be useful in small-cell lung cancer with overexpression of MCT1 and no overexpression of MCT4.77 The problem with MCT inhibitors is that these drugs are ineffective in hypoxic regions of the tumor because HIF-1α induces MCT4 production.78
The inhibition of MCTs becomes a serious impediment for cancer cell growth if both MCT1 and MCT2 are inhibited.
MCT1, besides its transporter activity, also seems to intervene in cell motility, migration and metastasis. The knockdown of MCT1 decreased HGF-induced and EGF-induced cell motility.79
Flavonoid quercetin seems to inhibit MCTs.80 Simvastatin decreases the activity of MCT4,81 but almost all statins have an inhibitory effect (atorvastatin, fluvastatin, cerivastin, simvastatin, lovastatin).82
Mitochondrial poisons
Atovaquone is an antimalarial drug used for the treatment of pneumocystis pneumonia and toxoplasmosis and is approved by the US Food and Drug Administration. At the molecular level, it is a potent and selective inhibitor of OXPHOS by targeting mitochondrial complex III and inducing aerobic glycolysis and oxidative stress in cancer stem cells.83,84 Atovaquone decreases the pyrimidine synthetic pathway in Plasmodium falciparum dependent on mitochondria.85
Metformin is the most widely prescribed drug for the treatment of diabetes. Its main mechanism of action is inhibition of mitochondrial complex I, increasing the glycolytic pathway through reduction of OXPHOS. Due to lower production of mitochondrial ATP, the AMP/ATP index increases and activates AMPK which further inhibits mTOR.86 Used at high doses, it may produce lactic acidosis due to increased lactic acid production.
Phenformin is metformin’s predecessor with similar effects on lactic acid production but is a more powerful inhibitor of the mitochondrial respiratory chain which entails an increased risk of lactic acidosis. This adverse effect led to the withdrawal of this drug from the market.87–89 As the effect we are looking for is precisely a strong inhibition of lactate oxidation, phenformin may be more appropriate for this purpose than metformin, although it is more toxic. Regarding cancer cytotoxicity, phenformin also seems to be more powerful than metformin.90
Doxycycline is an antibiotic that exerts inhibition of mitochondrial protein synthesis and reduces mitochondrial complex I activity.91–94 (mechanism described in Figure 6).
Figure 6.

Mechanism of action of tetracyclines.
Abbreviation: OXPHOS, oxidative phosphorylation.
All of the three pharmaceuticals described as mitochondrial poisons have in common their inhibitory activity on OXPHOS and increase in lactic acid production through increased aerobic glycolysis.
Metformin, phenformin, and doxycycline are weak inhibitors of mitochondrial complex I with no effect on the rest of the mitochondrial complexes,95 so for a more potent inhibition of the OXPHOS process, it would be convenient to associate atovaquone as an inhibitor of complex III and possible synergistic activity with biguanides. This needs experimental testing.
By reducing OXPHOS activity, mitochondrial poisons decrease ATP production, and NHE activity is reduced at a low intracellular concentration of ATP.52 This means than in theory, at least, mitochondrial poisons may increase inhibition of proton extrusion mechanisms. In spite of this finding, NHE-1 is not energy dependent, and its inhibition is due to modulation of intracellular proton-dependent mechanisms.52
Hypothesis
It has been demonstrated that the different ion pump inhibitors decrease tumor cell proliferation. Using inhibitors that act on different mechanisms show synergy in antiproliferative activity.96 In this review, we propose the use of multiple ion pump inhibitors plus an increase in intracellular acidity by increasing the lactic acid production through mitochondrial poisons like metformin, atovaquone and doxycycline. The excess of intracellular acidity that cannot be extruded due to proton pump inhibition should generate an acidic stress that induces apoptosis.
Precisely, we propose using the association of eight pharmaceuticals (and a possible ninth) to achieve this goal:
Lansoprazole or pantoprazole
Amiloride or an analog of amiloride and cariporide could be another option
Phenytoin
Quercetin
Lipophilic statins like simvastatin, atorvastatin, cerivastatin and lovastatin
Metformin or phenformin
Doxycycline
Atovaquone
If the tumor overexpresses CA, acetazolamide should be added to the combination.
Each of these drugs has low toxicity at therapeutic doses. Except for cariporide, there is adequate experience with all of them, and they are FDA-approved. The drugs are inexpensive and require no phase I clinical trials. They can all be associated with conventional chemotherapy and radiotherapy.
Discussion
The importance of acid–base homeostasis in keeping normal cellular responses has long been known, and the importance of targeting cancer pH has been recognized by the scientific community, to the extent that in 2010, the International Society for Proton Dynamics in Cancer was created97 with the intention of bringing together basic and clinical investigators to stimulate translational research and interdisciplinary work for the development of therapeutic strategies based on proton dynamics in cancer.
The peculiarities of proton dynamics in cancer are a direct consequence of deep metabolic changes in cancer cells. Interfering adequately with the extracellular and intracellular acidity of cancer cells may represent a resource that shows low or no toxicity for normal cells and at the same time decrease proliferation, migration, invasion, and metastasis.
Inhibiting the compensatory mechanisms that tumor cells use to adapt themselves to a high load of toxic metabolites may deprive these cells of a capital resource for detoxification. And what is more important: this inhibition can be achieved with already existing drugs that have no toxic effect on normal cells.
Despite the ease and low cost of interference in proton dynamics as an anticancer strategy, it has been neglected in the clinical setting.
The role of pH in cancer development, evolution, and metastasis has been underlined by many fundamental investigations.98–103 The role of the acidic environment in cancer is a serious drawback for natural and induced immunotherapy,104,105 and neutralizing this acidity improved immunological defenses in an experimental in vivo setting. As proof of concept, we should mention that the combination of bicarbonate with anti-PD-1 drugs or anti-CTLA-4 drugs improved antitumor responses.101
At the same time, an increase in intracellular pH is responsible for increased DNA and protein synthesis, increased metabolic rate, and cell proliferation.106
The simultaneous attack (double-edged) on intracellular pH leading it to acidosis through increased lactic acid production, and extracellular pH pushed toward a higher pH by reduction of the proton export mechanism will have two desired effects:
acidic intracellular stress that may increase apoptosis; and
lower extracellular acidity that decreases migration and invasion by reduced cathepsin and matrix metalloproteinases activity.
The eight drugs (eventually nine) chosen to achieve the double-edged approach were selected on the assumption that they will probably act in synergy to reach the goals outlined. No MCT inhibitors were included in this multidrug compound because no effective drug has been developed yet. But there are evidences that the combination of two flavonoids, phloretin, and luteolin, may show inhibitory activity on MCT178 or phloretin alone.107,108 The interesting issue with phloretin is that it also inhibits MCT4.109 Phloretin has also other anticancer properties and deserves further research.110–112
More than one proton extrusion mechanisms have to be inhibited because tumors show heterogeneous expression of these transporters and there is redundancy in the mechanisms for acid extrusion. This explains why it is necessary to use at least four different compounds to eliminate the main transporters activity.
Targeting extracellular acidity in cancer with a simple and nontoxic resource as PPIs may overcome immune escape that is unleashed by low pHe.113,114 Another simple salt like sodium bicarbonate administered orally elevated peritumoral pHe and inhibited local growth and invasion.115
Tumor pH has a strong influence on therapeutic response:3
Acidity suppresses radiation-induced apoptosis.
Weakly acid drug uptake is enhanced.
Retards the uptake of weakly basic drugs.
The simplicity and low toxicity of pH targeting is so important that there are no sound reasons for neglecting it in cancer treatment. (The fundamentals of the double-edged approach are illustrated through Figures 7–9.)
Figure 7.

Intracellular and extracellular pH in tumor cells. Proton extrusion mechanisms create an extracellular acidic milieu and a slightly alkaline intracellular environment. Low extracellular pH contributes to the activation of enzymes that digest extracellular matrix, while the slightly alkaline cytoplasm is appropriate for increased proliferation. Organelles like lysosomes are highly acidic in cancer cells. Migrating cells exhibit an intracellular pH gradient along the migration axis which is related to NHE-1 activity.159
Abbreviation: NHE-1, sodium–proton exchanger-1.
Figure 8.
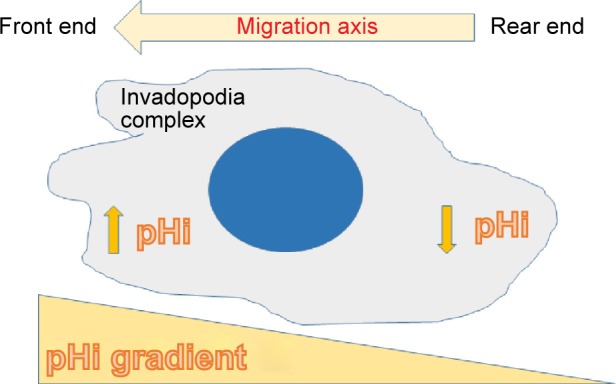
Intracellular pH gradient in migrating cells.112 Malignant cells show a tendency to a higher gradient between the front and rear ends. Inhibition of NHE-1 makes the gradient flatten or disappear. There is also an NHE-1 distribution gradient similar to the pHi gradient112 which cannot be modified by pHe160 when NHE-1 is inhibited.
Abbreviation: NHE-1, sodium–proton exchanger-1.
Figure 9.

The double-edged approach increases intracellular acid burden and decreases extracellular acidity by limiting exportation of protons.
Abbreviation: OXPHOS, oxidative phosphorylation.
Final comment
pHi changes are proportional to the difference between acid extrusion and acid loading.116 The double-edged approach increases acid loading through mitochondrial poisons and reduces acid extrusion by inhibiting acid extruders. The net result is a decrease in pHi which carries cellular acid stress and cytotoxicity. The acid load is strongly enhanced in malignant cells as compared to nonmalignant cells because the production of lactic acid in these tumor cells is much higher than in their normal counterparts. So, we can expect low toxicity in normal cells and a high toxicity in malignant cells.
Inhibition of ion transport alone should have minimal or no cytotoxic effects on malignant cells. Mild mitochondrial poisons alone should have no cytotoxic effects at usual doses, either. But the association of both types of drugs would create a significant acid stress inside the cell that produce cytotoxicity and a decrease of extracellular acidity which may result in decreased migration, invasion, and eventual metastasis. By reducing OXPHOS, mitochondrial poisons produce inhibition of stem cell proliferation, which is an additional feature favoring the use of these kinds of drugs.
Although the acid extrusion mechanisms are highly redundant, a partial inhibition of many of these mechanisms is enough to reduce cancer proliferation and invasion because a complete inhibition of proton extrusion would result in unacceptable toxicity for normal cells.117
The synergistic effect of the association of lansoprazole with an inhibitor of CA IX against human melanoma cells has been recently demonstrated.118
Many of the drugs proposed in the double-edged approach are in clinical use and approved by the FDA and other regulatory authorities. The combination of pharmaceuticals proposed in this review can be associated with most of the chemotherapy protocols currently in use.
This scheme deserves adequate and well-planned clinical trials as an adjunct cancer treatment.
Future perspectives
Based on a mathematical theoretical study, Webb et al119 determined that the transfer of acids from the cytosol into acidic organelles like endoplasmic reticulum, endosomes, Golgi apparatus, and lysosomes had a capital importance in keeping an alkaline pHi. Up to now, no specific drugs have been developed to reduce or abort this sequestration of protons. Probably, the future will present us with advances in this area.
Anticancer vaccines, activated T lymphocytes utilized against tumors, anti-PD-1 and anti-CTLA-4 monoclonal preparations, and anticancer immunotherapy in general will be benefited by modulating tumor acidity that causes a reversible state of anergy.
As new molecules for selective CA IX inhibition, new monocarboxylate inhibitors and new amiloride derivatives are developed and brought into medical practice, the spectrum of pH-targeted therapies will increase and probably become part of the oncological armamentarium. In the meantime, there are excellent drugs that may do the job when adequately combined.
Footnotes
Disclosure
The author reports no conflict of interest in this work.
References
- 1.Walsh M, Fais S, Spugnini EP, et al. Proton pump inhibitors for the treatment of cancer in companion animals. J Exp Clin Cancer Res. 2015;34:93. doi: 10.1186/s13046-015-0204-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffiths JR, McIntyre DJ, Howe FA, Stubbs M. Why are cancers acidic? A carrier-mediated diffusion model for H+ transport in the interstitial fluid. Novartis Found Symp. 2001;240:46–62. doi: 10.1002/0470868716.ch4. [DOI] [PubMed] [Google Scholar]
- 3.Song CW, Griffin R, Park HJ. Cancer Drug Resistance. Humana Press Inc; Totowa, NJ: Springer; 2006. Influence of tumor pH on therapeutic response; pp. 21–42. [Google Scholar]
- 4.Zetterberg A, Engstrom W. Mitogenic effect of alkaline pH on quiescent, serum-starved cells. Proc Natl Acad Sci U S A. 1981;78(7):4334–4338. doi: 10.1073/pnas.78.7.4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49(16):4373–4384. [PubMed] [Google Scholar]
- 6.Lee AH, Tannock IF. Heterogeneity of intracellular pH and of mechanisms that regulate intracellular pH in populations of cultured cells. Cancer Res. 1998;58(9):1901–1908. [PubMed] [Google Scholar]
- 7.Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11(9):671–677. doi: 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- 8.Gatenby R, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 9.Robey IF, Baggett BK, Kirkpatrick ND, et al. Bicarbonate increases tumor pH and inhibits spontaneous metastasis. Cancer Res. 2009;69(6):2260–2268. doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leanza L, Managò A, Zoratti M, Gulbins E, Szabo I. Pharmacological targeting of ion channels for cancer therapy: in vivo evidences. Biochim Biophys Acta. 2016;1863(6 Pt B):1385–1397. doi: 10.1016/j.bbamcr.2015.11.032. [DOI] [PubMed] [Google Scholar]
- 11.McCarty MF, Whitaker J. Manipulating tumor acidification as a cancer treatment strategy. Altern Med Rev. 2010;15(3):264–272. [PubMed] [Google Scholar]
- 12.Lee ZW, Teo XY, Tay EY, et al. Utilizing hydrogen sulphide as a novel anti-cancer agent by targeting glycolysis and pH imbalance. Br J Pharmacol. 2014;171(18):4322–4336. doi: 10.1111/bph.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finbow ME, Harrison MA. The vacuolar H+ ATPase: a universal proton pump of eukaryotes. Biochem J. 1997;324(Pt 3):697–712. doi: 10.1042/bj3240697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perona R, Serrano R. Increased pH and tumorigenicity of fibroblasts expressing a yeast proton pump. Nature. 1988;334(6181):438–440. doi: 10.1038/334438a0. [DOI] [PubMed] [Google Scholar]
- 15.Perona R, Portillo F, Giraldez F, Serrano R. Transformation and pH homeostasis of fibroblasts expressing yeast H(+)-ATPase containing site-directed mutations. Mol Cell Biol. 1990;10(8):4110–4115. doi: 10.1128/mcb.10.8.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulshrestha A, Katara GK, Ginter J, et al. Selective inhibition of tumor cell associated Vacuolar-ATPase ‘a2’ isoform overcomes cisplatin resistance in ovarian cancer cells. Mol Oncol. 2016;10(6):789–805. doi: 10.1016/j.molonc.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fais S, De Milito A, You H, Qin W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 2007;67(22):10627–10630. doi: 10.1158/0008-5472.CAN-07-1805. [DOI] [PubMed] [Google Scholar]
- 18.Whitehurst AW, Bodemann BO, Cardenas J, et al. Synthetic lethal screen identification of chemo sensitizer loci in cancer cells. Nature. 2007;445(7137):815–819. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 19.Larsson H, Mattson H, Sundell G, Carlsson E. Animal pharmacodynamics of omeprazole. A survey of its pharmacological properties in vivo. Scand J Gastroenterol Suppl. 1985;108:23–35. doi: 10.3109/00365528509095817. [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Zaguilan R, Lynch RM, Martinez GM, Gillies RJ. Vacuolar-type H(+) ATPases are functionally expressed in plasma membranes of human tumor cells. Am J Physiol. 1993;265(4 Pt 1):1015–1029. doi: 10.1152/ajpcell.1993.265.4.C1015. [DOI] [PubMed] [Google Scholar]
- 21.Sennoune SR, Bakunts K, Martinez GM, et al. Vacuolar H+ ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am J Physiol Cell Physiol. 2004;286(6):1443–1452. doi: 10.1152/ajpcell.00407.2003. [DOI] [PubMed] [Google Scholar]
- 22.Pamarthy S, Jaiswal MK, Kulshreshtha A, Katara GK, Gilman-Sachs A, Beaman KD. The Vacuolar ATPase a2-subunit regulates Notch signaling in triple-negative breast cancer cells. Oncotarget. 2015;6(33):34206–34220. doi: 10.18632/oncotarget.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung C, Mader CC, Schmitz JC, et al. The vacuolar-ATPase (V-AT-Pase) modulates matrix metalloproteinase (MMP) isoforms in human pancreatic cancer. Lab Invest. 2011;91(5):732–743. doi: 10.1038/labinvest.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ibrahim SA, Katara GK, Kulshrestha A, Jaiswal MK, Amin MA, Beaman KD. Breast cancer associated a2 isoform vacuolar ATPase immunomodulates neutrophils: potential role in tumor progression. Oncotarget. 2015;6(32):33033–33045. doi: 10.18632/oncotarget.5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cotter K, Capecci J, Sennoune S, et al. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J Biol Chem. 2015;290(6):3680–3692. doi: 10.1074/jbc.M114.611210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katara GK, Kulshrestha A, Jaiswal MK, Pamarthy S, Gilman-Sachs A, Beaman KD. Inhibition of vacuolar ATPase subunit in tumor cells delays tumor growth by decreasing the essential macrophage population in the tumor microenvironment. Oncogene. 2015;35(8):1058–1065. doi: 10.1038/onc.2015.159. [DOI] [PubMed] [Google Scholar]
- 27.Kwong C, Gilman Sachs A, Beaman K. Tumor associated a2 vacuolar ATPase acts as a key mediator of cancer-related inflammation by inducing pro-tumorigenic properties in monocytes. J Immunol. 2011;186(3):1781–1789. doi: 10.4049/jimmunol.1002998. [DOI] [PubMed] [Google Scholar]
- 28.Spugnini EP, Citro G, Fais S. Proton pump inhibitors as antivacuolar-ATPases drugs: a novel anticancer strategy. J Exp Clin Cancer Res. 2010;29:44. doi: 10.1186/1756-9966-29-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeo M, Kim DK, Kim YB, et al. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin Cancer Res. 2004;10(24):8687–8696. doi: 10.1158/1078-0432.CCR-04-1065. [DOI] [PubMed] [Google Scholar]
- 30.Lugini L, Federici C, Borghi M, et al. Proton pump inhibitors while belonging to the same family of generic drugs show different anti-tumor effect. J Enzyme Inhib Med Chem. 2015;31(4):538–545. doi: 10.3109/14756366.2015.1046062. [DOI] [PubMed] [Google Scholar]
- 31.Canitano A, Iessi E, Spugnini EP, Federici C, Fais S. Proton pump inhibitors induce a caspase-independent antitumor effect against human multiple myeloma. Cancer Lett. 2016;376(2):278–283. doi: 10.1016/j.canlet.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 32.Koltai T. Voltage-gated sodium channel as a target for metastatic risk reduction with re-purposed drugs. F1000Res. 2015;4:297. doi: 10.12688/f1000research.6789.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gillet L, Roger S, Besson P, et al. Voltage-gated sodium channel activity promotes cysteine cathepsin dependent invasiveness and colony growth of human cancer cells. J Biol Chem. 2009;284(13):8680–8691. doi: 10.1074/jbc.M806891200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moolenaar WH, Boonstra J, van der Saaq PT, de Laat SW. Sodium/proton exchange in mouse neuroblastoma cells. J Biol Chem. 1981;256(24):12883–12887. [PubMed] [Google Scholar]
- 35.Comoli R, Casale A, Mariotti D. Amiloride and glucose effects on the intracellular pH of Yoshida rat ascites hepatoma AH130 cells grown in vivo. Cell Biol Int Rep. 1984;8(4):297–307. doi: 10.1016/0309-1651(84)90156-5. [DOI] [PubMed] [Google Scholar]
- 36.Sparks RL, Pool TB, Smith NK, Cameron IL. Effects of amiloride on tumor growth and intracellular element content of tumor cells in vivo. Cancer Res. 1983;43(1):73–77. [PubMed] [Google Scholar]
- 37.Greco MR, Antelmi E, Busco G, et al. Protease activity at invadopodial focal digestive areas is dependent on NHE1-driven acidic pHe. Oncol Rep. 2014;31(2):940–946. doi: 10.3892/or.2013.2923. [DOI] [PubMed] [Google Scholar]
- 38.Delvaux M, Bastie MJ, Chentoufi J, Cragoe EJ, Jr, Vaysse N, Ribet A. Amiloride and analogues inhibit Na(+)-H+ exchange and cell proliferation in AR42J pancreatic cell line. Am J Physiol. 1990;259(5 Pt 1):G842–G849. doi: 10.1152/ajpgi.1990.259.5.G842. [DOI] [PubMed] [Google Scholar]
- 39.Baumgartner M, Patel H, Barber DL. Na+/H+ exchanger NHE1 as plasma membrane scaffold in the assembly of signaling complexes. Am J Physiol Cell Physiol. 2004;287(4):C844–C850. doi: 10.1152/ajpcell.00094.2004. [DOI] [PubMed] [Google Scholar]
- 40.Wakabayashi S, Bertrand B, Shigekawa M, Fafournoux P, Pouysségur J. Growth factor activation and “H(+)-sensing” of the Na+/H+ exchanger isoform1 (NHE1). Evidence for an additional mechanism not requiring direct phosphorylation. J Biol Chem. 1994;269(8):5583–5588. [PubMed] [Google Scholar]
- 41.Bianchini L, L’Allemain G, Pouyssegur J. The p42/p44 mitogen-activated protein kinase cascade is determinant in mediating activation of the Na+/H+ exchanger (NHE1 isoform) in response to growth factors. J Biol Chem. 1997;272(1):271–279. doi: 10.1074/jbc.272.1.271. [DOI] [PubMed] [Google Scholar]
- 42.Shimoda LA, Fallon M, Pisarcik S, Wang J, Semenza GL. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2006;291(5):L941–L949. doi: 10.1152/ajplung.00528.2005. [DOI] [PubMed] [Google Scholar]
- 43.Khaled AR, Moor AN, Li A, et al. Trophic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinization. Mol Cell Biol. 2001;21(22):7545–7557. doi: 10.1128/MCB.21.22.7545-7557.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lacroix J, Poet M, Maehrel C, Counillon L. A mechanism for the activation of the Na/H exchanger NHE-1 by cytoplasmic acidification and mitogens. EMBO Rep. 2004;5(1):91–96. doi: 10.1038/sj.embor.7400035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snabaitis AK, Cuello F, Avkiran M. Protein kinase B/Akt phosphorylates and inhibits the cardiac Na+/H+ exchanger NHE1. Circ Res. 2008;103(8):881–890. doi: 10.1161/CIRCRESAHA.108.175877. [DOI] [PubMed] [Google Scholar]
- 46.Bourguignon LY, Singleton PA, Diedrich F, Stern R, Gilad E. CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J Biol Chem. 2004;279(26):26991–27007. doi: 10.1074/jbc.M311838200. [DOI] [PubMed] [Google Scholar]
- 47.Sauvage M, Mazière P, Fathallah H, Giraud F. Insulin stimulates NHE1 activity by sequential activation of phosphatidylinositol 3-kinase and protein kinase C ζ in human erythrocytes. Eur J Biochem. 2000;267(4):955–962. doi: 10.1046/j.1432-1327.2000.01084.x. [DOI] [PubMed] [Google Scholar]
- 48.Davis RJ, Czech MP. Amiloride directly inhibits growth factor receptor tyrosine kinase activity. J Biol Chem. 1985;260(4):2543–2551. [PubMed] [Google Scholar]
- 49.Rich IN, Worthington-White D, Garden OA, Musk P. Apoptosis in leukemic cells accompanies reduction in intracellular pH after targeted inhibition of the Na(+)/H(+) exchanger. Blood. 2000;95(4):1427–1434. [PubMed] [Google Scholar]
- 50.Denker SP, Barber DL. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J Cell Biol. 2002;159(6):1087–1096. doi: 10.1083/jcb.200208050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dhein S, Salameh A. Na+/H+-exchange inhibition by cariporide (Hoe 642): a new principle in cardiovascular medicine. Cardiovasc Drug Rev. 1999;17(2):134–146. [Google Scholar]
- 52.Lv C, Yang X, Yu B, Ma Q, Liu B, Liu Y. Blocking the Na+/H+ exchanger 1 with cariporide (HOE642) reduces the hypoxia-induced invasion of human tongue squamous cell carcinoma. Int J Oral Maxillofac Surg. 2012;41(10):1206–1210. doi: 10.1016/j.ijom.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Di Sario A, Bendia E, Omenetti A, et al. Selective inhibition of ion transport mechanisms regulating intracellular pH reduces proliferation and induces apoptosis in cholangiocarcinoma cells. Dig Liver Dis. 2007;39(1):60–69. doi: 10.1016/j.dld.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 54.Cassel D, Katz M, Rotman M. Depletion of cellular ATP inhibits Na+/H+ antiport in cultured human cells. Modulation of the regulatory effect of intracellular protons on the antiporter activity. J Biol Chem. 1986;261(12):5460–5466. [PubMed] [Google Scholar]
- 55.Kumar AP, Quake AL, Chang MK, et al. Repression of NHE1 expression by PPARγ activation is a potential new approach for specific inhibition of the growth of tumor cells in vitro and in vivo. Cancer Res. 2009;69(22):8636–8644. doi: 10.1158/0008-5472.CAN-09-0219. [DOI] [PubMed] [Google Scholar]
- 56.Grimes JA, Fraser SP, Stephens GJ, et al. Differential expression of voltage-activated Na+ currents in two prostatic tumour cell lines: contribution to invasiveness in vitro. FEBS Lett. 1995;369(2–3):290–294. doi: 10.1016/0014-5793(95)00772-2. [DOI] [PubMed] [Google Scholar]
- 57.Laniado ME, Lalani EN, Fraser SP. Expression and functional analysis of voltage-activated Na+ channels in human prostate cancer cell lines and their contribution to invasion in vitro. Am J Pathol. 1997;150(4):1213–1221. [PMC free article] [PubMed] [Google Scholar]
- 58.Smith P, Rhodes NP, Shortland AP, et al. Sodium channel protein expression enhances the invasiveness of rat and human prostate cancer cells. FEBS Lett. 1998;423(1):19–24. doi: 10.1016/s0014-5793(98)00050-7. [DOI] [PubMed] [Google Scholar]
- 59.Fraser SP, Salvador V, Manning EA, et al. Contribution of functional voltage-gated Na+ channel expression to cell behaviors involved in the metastatic cascade in rat prostate cancer: I. Lateral motility. J Cell Physiol. 2003;195(3):479–487. doi: 10.1002/jcp.10312. [DOI] [PubMed] [Google Scholar]
- 60.Roger S, Besson P, Le Guennec JY. Involvement of a novel fast inward sodium current in the invasion capacity of a breast cancer cell line. Biochim Biophys Acta. 2003;1616(2):107–111. doi: 10.1016/j.bbamem.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 61.Batcioglu K, Uyumlu AB, Satilmis B, et al. Oxidative stress in the in vivo DMBA rat model of breast cancer: suppression by a voltage-gated sodium channel inhibitor (RS100642) Basic Clin Pharmacol Toxicol. 2012;111(2):137–141. doi: 10.1111/j.1742-7843.2012.00880.x. [DOI] [PubMed] [Google Scholar]
- 62.Parks SK, Pouyssegur J. The Na(+)/HCO3(−) co-transporter SLC4A4 plays a role in growth and migration of colon and breast cancer cells. J Cell Physiol. 2015;230(8):1954–1963. doi: 10.1002/jcp.24930. [DOI] [PubMed] [Google Scholar]
- 63.Mahon BP, Pinard MA, McKenna R. Targeting carbonic anhydrase IX activity and expression. Molecules. 2015;20(2):2323–2348. doi: 10.3390/molecules20022323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mokhtari RB, Islam SS, Baluch N, et al. The anti-tumor effects of acetazolamide and sulphorane on bronchial carcinoids: preclinical modeling and mechanism. Cancer Res. 2014;74(19):3133. [Google Scholar]
- 65.Parkkila S, Rajaniemi H, Parkkila AK, et al. Carbonic anhydrase inhibitor suppresses invasion of renal cancer cells in vitro. Proc Natl Acad Sci U S A. 2000;97(5):2220–2224. doi: 10.1073/pnas.040554897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohammadpour R, Shahrokh S, Ejeian F, Sheikholya-Lavasani Z, Abdolmohammadi MH, Sheinabi N. Acetazolamide triggers death inducing autophagy in T-47D breast cancer cells. Cell Biol Int. 2014;38(2):228–238. doi: 10.1002/cbin.10197. [DOI] [PubMed] [Google Scholar]
- 67.Belkaid A, Cuperlović-Culf M, Touaibia M, Ouellette RJ, Surette ME. Metabolic effect of estrogen receptor agonists on breast cancer cells in the presence or absence of carbonic anhydrase inhibitors. Metabolites. 2016;6(2) doi: 10.3390/metabo6020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bin K, Shi-Peng Z. Acetazolamide inhibits aquaporin-1 expression and colon cancer xenograft tumor growth. Hepatogastroenterology. 2011;58(110–111):1502–1506. doi: 10.5754/hge11154. [DOI] [PubMed] [Google Scholar]
- 69.Islam SS, Mokhtari RB, Akbari P, Hatina J, Yeger H, Farhat WA. Simultaneous targeting of bladder tumor growth, survival and epithelial-to-mesenchymal transition with a novel therapeutic combination of acetazolamide (AZ) and sulforaphane (SFN) Target Oncol. 2016;11(2):209–227. doi: 10.1007/s11523-015-0386-5. [DOI] [PubMed] [Google Scholar]
- 70.Said HM, Hagemann C, Carta F, et al. Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivates including acetazolamide in human glioblastoma. Bioorg Med Chem. 2013;21(13):3949–3957. doi: 10.1016/j.bmc.2013.03.068. [DOI] [PubMed] [Google Scholar]
- 71.Huang YH, Zhou XY, Wang HM, Xu H, Lv NH. Aquaporin 5 promotes the proliferation and migration of human gastric carcinoma cells. Tumour Biol. 2013;34(3):1743–1751. doi: 10.1007/s13277-013-0712-4. [DOI] [PubMed] [Google Scholar]
- 72.Faes S, Planche A, Uldry E, et al. Targeting carbonic anhydrase IX improves the anti-cancer efficacy of mTOR inhibitors. Oncotarget. 2016 May 2; doi: 10.18632/oncotarget.9134. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaeteewoottacharn K, Kariya R, Dana P, et al. Inhibition of carbonic anhydrase potentiates bevacizumab treatment in chlangiocarcinoma. Tumour Biol. 2016;37(7):9023–9035. doi: 10.1007/s13277-016-4785-8. [DOI] [PubMed] [Google Scholar]
- 74.Mahon BP, Okoh CO, McKenna R. Targeting aggressive cancers with an artificial sweetener: could saccharin be a lead compound in anticancer therapy? Future Oncol. 2015;11(15):2117–2119. doi: 10.2217/fon.15.137. [DOI] [PubMed] [Google Scholar]
- 75.Halestrap AP, Wilson MC. The monocarboxylate transporter family – role and regulation. IUBMB Life. 2012;64(2):109–119. doi: 10.1002/iub.572. [DOI] [PubMed] [Google Scholar]
- 76.Izumi H, Tarigoe T, Ishiguchi T, et al. Cellular pH regulators: potentially promising molecular targets for cancer chemotherapy. Cancer Treatment Rev. 2003;29(6):541–549. doi: 10.1016/s0305-7372(03)00106-3. [DOI] [PubMed] [Google Scholar]
- 77.Blackhall F. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Ann Oncol. 2015;26(Suppl 2):15. doi: 10.1158/1078-0432.CCR-13-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Le Floch R, Chiche J, Marchiq I, et al. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc Natl Acad Sci U S A. 2011;108(40):16663–16668. doi: 10.1073/pnas.1106123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gray AL, Coleman DT, Shi R, Cardelli JA. Monocarboxylate transporter 1 contributes to growth factor induced tumor cell migration independent of transporter activity. Oncotarget. 2016;7(22):32695–32706. doi: 10.18632/oncotarget.9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang W, Morris ME. Flavonoids modulate monocarboxylate transporter 1-mediated transport of gamma-hydroxybutyrate in vitro and in vivo. Drug Metab Dispos. 2007;35(2):201–208. doi: 10.1124/dmd.106.012369. [DOI] [PubMed] [Google Scholar]
- 81.Morris ME, Felmlee MA. Overview of the proton-coupled MCT (SLC16A) family of transporters: characterization, function and role in the transport of the drug of abuse gamma-hydroxybutyric acid. AAPS J. 2008;10(2):311–321. doi: 10.1208/s12248-008-9035-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kobayashi M, Otsuka Y, Itagaki S, Hirano T, Iseki K. Inhibitory effects of statins on human monocarboxylate transporter 4. Int J Pharm. 2006;317(1):19–25. doi: 10.1016/j.ijpharm.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 83.Fiorillo M, Lamb B, Tanowitz HB, et al. Repurposing atovaquone: targeting mitocondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget. 2016 Apr 30; doi: 10.18632/oncotarget.9122. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fry M, Pudney M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80) Biochem Pharmacol. 1992;43(7):1545–1543. doi: 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- 85.Hammond DJ, Burchell JR, Pudney M. Inhibition of pyrimidine biosynthesis de novo in Plasmodium falciparum by 2-(4-t-butylcyclohexyl)-3-hydroxy-1,4-naphthoquinone in vitro. Mol Biochem Parasitol. 1985;14(1):97–109. doi: 10.1016/0166-6851(85)90109-4. [DOI] [PubMed] [Google Scholar]
- 86.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66(21):10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 87.Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333(9):550–554. doi: 10.1056/NEJM199508313330903. [DOI] [PubMed] [Google Scholar]
- 88.Pernicova I, Korbnits M. Metformin mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 89.Janzer A, German NJ, Gonzalez Herrera KN, Asara JM, Haigis MC, Struhl K. Metformin and phenphormin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc Natl Acad Sci U S A. 2014;111(29):10574–10579. doi: 10.1073/pnas.1409844111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung YS, Choi JY. Synergistic anti-cancer effect of phenformin and oxamate. PLoS One. 2014;9(1):e85576. doi: 10.1371/journal.pone.0085576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Du Buy HG, Showacre JL. Selective localization of tetracycline in mitochondria of living cells. Science. 1961;133(3447):196–197. doi: 10.1126/science.133.3447.196. [DOI] [PubMed] [Google Scholar]
- 92.Journey LJ, Goldstein MN. The effect of terramycin on the fine structure of HELa cell mitochondria. Cancer Res. 1963;23(4):551–554. [PubMed] [Google Scholar]
- 93.de Vries H, Kroon AM. On the effect of chloramphenicol and oxytetracycline on the biogenesis of mammalian mitochondria. Biochim Biophys Acta. 1970;204(2):531–541. doi: 10.1016/0005-2787(70)90173-5. [DOI] [PubMed] [Google Scholar]
- 94.de Vries H, Arendzen AJ, Kroon AM. The interference of the macrolide antibiotics with mitochondrial protein synthesis. Biochim Biophys Acta. 1973;331(2):264–275. doi: 10.1016/0005-2787(73)90439-5. [DOI] [PubMed] [Google Scholar]
- 95.Bridges HR, Jones AJY, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. 2014;462(3):475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hrgovic I, Glavic Z, Kovacic Z, Mulic S, Zunic L, Hrgovic Z. Repeated administration of inhibitors for ion pumps reduce markedly tumor growth in vivo. Med Arch. 2014;68(2):76–78. doi: 10.5455/medarh.2014.68.76-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huber V, De Milito A, Harguindey S, et al. Proton dynamics in cancer. J Transl Med. 2010;8:57. doi: 10.1186/1479-5876-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cardone RA, Casavola V, Reshkin SJ. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat Rev Cancer. 2005;5(10):786–795. doi: 10.1038/nrc1713. [DOI] [PubMed] [Google Scholar]
- 99.Martinez-Zaguilan R, Seftor EA, Seftor RE, Chu YW, Gillies RJ, Hendrix MJ. Acidic pH enhances the invasive behavior of human melanoma cells. Clin Exp Metastasis. 1996;14(2):176–186. doi: 10.1007/BF00121214. [DOI] [PubMed] [Google Scholar]
- 100.Parolini I, Federici C, Raggi C, et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem. 2009;284(49):34211–34222. doi: 10.1074/jbc.M109.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Harguindey S, Arranz JL, Wahl ML, Orive G, Reshkin SJ. Proton transport inhibitors as potentially selective anticancer drugs. Anticancer Res. 2009;29(6):2127–2136. [PubMed] [Google Scholar]
- 102.De Milito A, Canese R, Marino ML, et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int J Cancer. 2010;127(1):207–219. doi: 10.1002/ijc.25009. [DOI] [PubMed] [Google Scholar]
- 103.Fais S. Proton pump inhibitor-induced tumour cell death by inhibition of a detoxification mechanism. J Intern Med. 2010;267(5):515–525. doi: 10.1111/j.1365-2796.2010.02225.x. [DOI] [PubMed] [Google Scholar]
- 104.Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016;76(6):1381–1390. doi: 10.1158/0008-5472.CAN-15-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol. 2001;69(4):522–530. [PubMed] [Google Scholar]
- 106.Mahnensmith RL, Aronson PS. The plasma membrane sodium hydrogen exchanger and its role in physiological and pathophysiological processes. Circ Res. 1985;56(6):773–788. doi: 10.1161/01.res.56.6.773. [DOI] [PubMed] [Google Scholar]
- 107.Müller F, Huber K, Pfannkuche H, Aschenbach JR, Breves G, Gäbel G. Transport of ketone bodies and lactate in the sheep ruminal epithelium by monocarboxylate transporter 1. Am J Physiol Gastrointest Liver Physiol. 2002;283(5):G1139–G1146. doi: 10.1152/ajpgi.00268.2001. [DOI] [PubMed] [Google Scholar]
- 108.Meredith D, Bell P, McClure B, Wilkins R. Functional and molecular characterization of lactic acid transport in bovine articular chondrocytes. Cell Physiol Biochem. 2002;12(4):227–234. doi: 10.1159/000066282. [DOI] [PubMed] [Google Scholar]
- 109.Manning Fox JE, Meredith D, Halestrap AP. Characterization of human monocarboxylate transporter 4 substantiates its role in lactic acid efflux from skeletal muscle. J Physiol. 2000;529(Pt 2):285–293. doi: 10.1111/j.1469-7793.2000.00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kobori M, Shinmoto H, Tsushida T, Shinohara K. Phloretin-induced apoptosis in B16 melanoma 4A5 cells by inhibition of glucose trans-membrane transport. Cancer Lett. 1997;119(2):207–212. doi: 10.1016/s0304-3835(97)00271-1. [DOI] [PubMed] [Google Scholar]
- 111.Nelson JA, Falk RE. The efficacy of phloridzin and phloretin on tumor cell growth. Anticancer Res. 1993;13(6A):2287–2292. [PubMed] [Google Scholar]
- 112.Kim MS, Kwon JY, Kang NJ, Lee KW, Lee HJ. Phloretin induces apoptosis in H-Ras MCF10A human breast tumor cells through the activation of p53 via JNK and p38 mitogen-activated protein kinase signaling. Ann N Y Acad Sci. 2009;1171:479–483. doi: 10.1111/j.1749-6632.2009.04692.x. [DOI] [PubMed] [Google Scholar]
- 113.Bellone M, Calcinotto A, Filipazzi P, De Milito A, Fais S, Rivoltini L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. Oncoimmunology. 2013;2(1):e22058. doi: 10.4161/onci.22058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Calcinotto A, Filipazzi P, Grioni M, et al. Modulation of microenvironmental acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012;72(11):2746–2756. doi: 10.1158/0008-5472.CAN-11-1272. [DOI] [PubMed] [Google Scholar]
- 115.Estrella V, Chen T, Lloyd M, et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73(5):1524–1535. doi: 10.1158/0008-5472.CAN-12-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Boron WF. Regulation of intracellular pH. Adv Physiol Educ. 2004;28(1–4):160–179. doi: 10.1152/advan.00045.2004. [DOI] [PubMed] [Google Scholar]
- 117.Oosterwijk E, Gillies RJ. Targeting ion transport in cancer. Philos Trans R Soc Lond B Biol Sci. 2014;369(1638):20130107. doi: 10.1098/rstb.2013.0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Federici C, Lugini L, Marino ML, et al. Lanzoprazle and carbonic anhydrase IX inhibitors synergize against human melanoma cells. J Enzyme Inhib Med Chem. 2016 2016 May 3;:1–7. doi: 10.1080/14756366.2016.1177525. Epub. [DOI] [PubMed] [Google Scholar]
- 119.Webb SD, Sherratt JA, Fish RG. Mathematical modelling of tumor acidity: regulation of intracellular pH. J Theor Biol. 1999;196(2):237–250. doi: 10.1006/jtbi.1998.0836. [DOI] [PubMed] [Google Scholar]
- 120.Mattsson JP, Vaananen K, Wallmark B, Lorentzon P. Omeprazole and bafilomycin, two proton pump inhibitors: differentiation of their effects on gastric, kidney and bone H(+)-translocating ATPases. Biochim Biophys Acta. 1991;1065(2):261–268. doi: 10.1016/0005-2736(91)90238-4. [DOI] [PubMed] [Google Scholar]
- 121.Mizunashi K, Furukawa Y, Katano K, Abe K. Effect of omeprazole, an inhibitor of H+, K(+)-ATPase, on bone resorption in humans. Calcif Tissue Int. 1993;53(1):21–25. doi: 10.1007/BF01352010. [DOI] [PubMed] [Google Scholar]
- 122.Luciani F, Spada M, De Milito A, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst. 2004;96(22):1702–1713. doi: 10.1093/jnci/djh305. [DOI] [PubMed] [Google Scholar]
- 123.De Milito A, Iesi E, Logozzi M, et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 2007;67(11):5408–5417. doi: 10.1158/0008-5472.CAN-06-4095. [DOI] [PubMed] [Google Scholar]
- 124.Capodicasa E, Cornacchione P, Natalini B, et al. Omeprazole induces apoptosis in normal human polymorphonuclear leucocytes. Int J Immunopathol Pharmacol. 2008;21(1):73–85. doi: 10.1177/039463200802100109. [DOI] [PubMed] [Google Scholar]
- 125.Ferrari S, Perut F, Fagioli F, et al. Proton pump inhibitor chemosensitization in human osteosarcoma: from the bench to the patients’ bed. J Transl Med. 2013;11:268. doi: 10.1186/1479-5876-11-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Patel KJ, Lee C, Tan Q, Tannock IF. Use of the proton pump inhibitor pantoprazole to modify the distribution and activity of doxorubicin: a potential strategy to improve the therapy of solid tumors. Clin Cancer Res. 2013;19(24):6766–6776. doi: 10.1158/1078-0432.CCR-13-0128. [DOI] [PubMed] [Google Scholar]
- 127.Avnet S, Di Pompo G, Lemma S, et al. V-ATPase is a candidate therapeutic targer for Ewing sarcoma. Biochim Biophys Acta. 2013;1832(8):1105–1116. doi: 10.1016/j.bbadis.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 128.Chen M, Huang SL, Zhang XQ, et al. Reversal effects of pantoprazole on multidrug resistance in human gastric adenocarcinoma cells by down-regulating the V-ATPases/mTOR/HIF-1α/P-gp and MRP1 signaling pathway in vitro and in vivo. J Cell Biochem. 2012;113(7):2474–2487. doi: 10.1002/jcb.24122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shen Y, Wu Y, Chen M, et al. Effects of pantoprazole as a HIF-1α inhibitor on human gastric adenocarcinoma sgc-7901 cells. Neoplasma. 2012;59(2):142–149. doi: 10.4149/neo_2012_019. [DOI] [PubMed] [Google Scholar]
- 130.Patlolla JM, Zhang Y, Li Q, Steele VE, Rao CV. Anticarcinogenic properies of omeprazole against human colon cancer cells and azoxymethane-induced colonic aberrant crypt foci formation in rats. Int J Oncol. 2012;40(1):170–175. doi: 10.3892/ijo.2011.1214. [DOI] [PubMed] [Google Scholar]
- 131.Perut F, Avnet S, Fotia C, et al. V-ATPase as an effective therapeutic target for sarcomas. Exp Cell Res. 2014;320(1):21–32. [PubMed] [Google Scholar]
- 132.Azzarito T, Venturi G, Cesolini A, Fais S. Lansoprazole induces sensitivity to suboptimal doses of paclitaxel in human melanoma. Cancer Lett. 2015;356(2 Pt B):697–703. doi: 10.1016/j.canlet.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 133.Huang S, Chen M, Ding X, Zhang X, Zou X. Proton pump inhibitor selectively suppresses proliferation and restores the chemosensitivity of gastric cancer cells by inhibiting STAT 3 signaling pathway. Int Immunopharmacol. 2013;17(3):585–592. doi: 10.1016/j.intimp.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 134.Goh W, Sleptsova-Friedrich I, Petrovic N. Use of proton pump inhibitors as adjunct treatment for triple-negative breast cancer. An introductory study. J Pharm Pharm Sci. 2014;17(3):439–446. doi: 10.18433/j34608. [DOI] [PubMed] [Google Scholar]
- 135.Zhang S, Wang Y, Li SJ. Lansoprazole induces apoptosis of breast cancer cells through inhibition of intracellular proton extrusion. Biochem Biophys Res Commun. 2014;448(4):424–429. doi: 10.1016/j.bbrc.2014.04.127. [DOI] [PubMed] [Google Scholar]
- 136.Jin UH, Lee SO, Pfent C, Safe S. The aryl hydrocarbon receptor ligandomeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer. 2014;14:498. doi: 10.1186/1471-2407-14-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Salerno M, Avnet S, Bonuccelli G, Hosogi S, Granchi D, Baldini N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PLos One. 2014;9(10):e110340. doi: 10.1371/journal.pone.0110340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Udelnow A, Kreyes A, Ellinger S, et al. Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells. PLoS One. 2011;6(5):e20143. doi: 10.1371/journal.pone.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Marino ML, Fais S, Djavaheri-Mergny M, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis. 2010;1(10):e87. doi: 10.1038/cddis.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vishvakarma NK, Singh SM. Immunopotentiating effect of proton pump inhibitor pantoprazole in a lymphoma-bearing murine host: implication in antitumor activation of tumor-associated macrophages. Immunol Lett. 2010;134(1):83–92. doi: 10.1016/j.imlet.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 141.Yeo M, Kim DK, Park HJ, Cho SW, Cheong JY, Lee KJ. Blockage of intracellular proton extrusion with proton pump inhibitor induces apoptosis in gastric cancer. Cancer Sci. 2008;99(1):185. doi: 10.1111/j.1349-7006.2007.00642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shen Y, Chen M, Huang S, Zou X. Pantoprazole inhibits human gastric adenocarcinoma SGC-7901 cells by downregulating the expression of pyruvate kinase M2. Oncol Lett. 2016;11(1):717–722. doi: 10.3892/ol.2015.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang B, Yang Y, Shi X, et al. Proton pump inhibitor pantoprazole abrogates adriamycin-resistant gastric cancer cell invasiveness via suppression of Akt/GSK-β/β catenin signaling and epithelial-mesenchymal transition. Cancer Lett. 2015;356(2 Pt B):7044–7712. doi: 10.1016/j.canlet.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 144.Tan Q, Joshua AM, Saggar JK, et al. Effect of pantoprazole to enhance activity of docetaxel against human tumour xenografts by inhibiting autophagy. Br J Cancer. 2015;112(5):832–840. doi: 10.1038/bjc.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 145.Lee HJ, Han YM, Kim EH, Kim YJ, Hahm KB. A possible involvement of Nrf2-mediated heme oxygenase-1 up-regulation in protective effect of the proton pump inhibitor pantoprazole against indomethacin-induced gastric damage in rats. BMC Gastroenterol. 2012;12:143. doi: 10.1186/1471-230X-12-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vishvakarma NK, Singh SM. Augmentation of myelopoiesis in a murine host bearing a T cell lymphoma following in vivo administration of proton pump inhibitor pantoprazole. Biochimie. 2011;93(10):1786–1796. doi: 10.1016/j.biochi.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 147.Mishima K, Kitoh H, Ohkawara B, et al. Lansoprazole upregulates polyubiquitination of the TNF receptor-associated factor 6 and facilitates Runx2-mediated osteoblastogenesis. EBioMedicine. 2015;2(12):2046–2061. doi: 10.1016/j.ebiom.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Matsui MS, Petris MJ, Niki Y, et al. Omeprazole, a gastric proton pump inhibitor, inhibits melanogenesis by blocking ATP7A trafficking. J Invest Dermatol. 2015;135(3):834–841. doi: 10.1038/jid.2014.461. [DOI] [PubMed] [Google Scholar]
- 149.Shiizaki K, Kawanishi M, Yagi T. Microbial metabolites of omeprazole activate murine aryl hydrocarbon receptor in vitro and in vivo. Drug Metab Dispos. 2014;42(10):1960–1967. doi: 10.1124/dmd.114.058966. [DOI] [PubMed] [Google Scholar]
- 150.Harguindey S, Arranz JL, Polo Orozco JD, et al. Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs – an integral molecular/biochemical/metabolic/clinical approach after one hundred years of cancer research. J Transl Med. 2013;11:282. doi: 10.1186/1479-5876-11-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tolde O, Ròsel D, Vesely P, Folk P, Brábek J. The structure of invadopodia in a complex 3D environment. Eur J Cell Biol. 2010;89(9):674–680. doi: 10.1016/j.ejcb.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 152.Yamaguchi H. Pathological roles of invadopodia in cancer invasion and metastasis. Eur J Cell Biol. 2012;91(11–12):902–907. doi: 10.1016/j.ejcb.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 153.Rowson-Hodel AR, Berg AL, Wald JH, et al. Hexamethylene amiloride engages a novel reactive oxygen species- and lysosome-dependent programmed necrotic mechanism to selectively target breast cancer cells. Cancer Lett. 2016;375(1):62–72. doi: 10.1016/j.canlet.2016.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sanhueza C, Araos J, Naranjo L, et al. Modulation of intracellular pH in human ovarian cancer. Curr Mol Med. 2016;16(1):23–32. doi: 10.2174/1566524016666151222143437. [DOI] [PubMed] [Google Scholar]
- 155.Acevedo Olvera LF, Diaz Garcia H, Parra Barrera A, et al. Inhibition of the Na+/H+ antiporter induces cell death in TF-1 erythroleukemia cells stimulated by rhe stem cell factor. Cytokine. 2015;75(1):142–150. doi: 10.1016/j.cyto.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 156.Pieri C, Giunta S, Giuli C, Bertoni-Freddari C, Muzzioli M. In vitro block of murine L 1210 leukemia cell growth by amiloride, an inhibitor of passive Na+ influx. Life Sci. 1983;32(15):1779–1784. doi: 10.1016/0024-3205(83)90842-1. [DOI] [PubMed] [Google Scholar]
- 157.Kellen JA, Mirakian A, Kolin A. Antimetastatic effect of amiloride in an animal tumour model. Anticancer Res. 1988;8(6):1373–1376. [PubMed] [Google Scholar]
- 158.Zheng YT, Yang HY, Li T, et al. Amiloride sensitizes human pancreatic cancer cells to erlotinib in vitro through inhibition of the PI3K/AKT signaling pathway. Acta Pharmacol Sin. 2015;36(5):614–626. doi: 10.1038/aps.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Martin C, Pedersen SF, Schwab A, Stock C. Intracellular pH gradients in migrating cells. Am J Physiol Cell Physiol. 2011;300(3):C490–C495. doi: 10.1152/ajpcell.00280.2010. [DOI] [PubMed] [Google Scholar]
- 160.Stüwe L, Müller M, Fabian A, et al. pH dependence of melanoma cell migration: protons extruded by NHE1 dominate protons of the bulk solution. J Physiol. 2007;585(Pt 2):351–360. doi: 10.1113/jphysiol.2007.145185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Whitaker-Menezes D, Martinez Outschoorn UE, Lin Z, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is marker of oxidative stress in cancer associated fibroblasts. Cell cycle. 2011;10(11):172–183. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sonveaux P, Copetti T, De Saedeleer CJ, et al. Targeting the lactate transporter MCT-1 in endothelial cells inhibits lactate induced HIF-1 activation and tumor angiogenesis. Plos One. 2012;7(3):e33418. doi: 10.1371/journal.pone.0033418. [DOI] [PMC free article] [PubMed] [Google Scholar]