

Abstract
Innate immune signals help break self-tolerance to initiate autoimmune diseases such as type 1 diabetes, but innate contributions to subsequent regulation of disease progression are less clear. Most studies have measured in vitro innate responses of GM-CSF dendritic cells (DCs) that are functionally distinct from conventional DCs (cDCs) and do not reflect in vivo DC subsets. To determine whether autoimmune NOD mice have alterations in type 1 IFN innate responsiveness, we compared cDCs from prediabetic NOD and control C57BL/6 (B6) mice stimulated in vivo with the TLR9 ligand CpG, a strong type 1 IFN inducer. In response to CpG, NOD mice produce more type 1 IFN and express higher levels of CD40, and NOD monocyte DCs make more TNF. However, the overall CpG-induced transcriptional response is muted in NOD cDCs. Of relevance the costimulatory proteins CD80/CD86, signals needed for regulatory T cell homeostasis, are upregulated less on NOD cDCs. Interestingly, NOD Rag1−/− mice also display a defect in CpG-induced CD86 upregulation compared with B6 Rag1−/−, indicating this particular innate alteration precedes adaptive autoimmunity. The impaired response in NOD DCs is likely downstream of the IFN-α/β receptor because DCs from NOD and B6 mice show similar CpG-induced CD86 levels when anti–IFN-α/β receptor Ab is added. IFN-α–induced nuclear localization of activated STAT1 is markedly reduced in NOD CD11c+ cells, consistent with lower type 1 IFN responsiveness. In conclusion, NOD DCs display altered innate responses characterized by enhanced type 1 IFN and activation of monocyte-derived DCs but diminished cDC type 1 IFN response.
In autoimmune diseases such as type 1 diabetes (T1D), autoreactive T cells lend specificity to the killing of target tissues, but innate immune cells also contribute significantly to autoimmune pathogenesis (1). NOD mice model the spontaneous β cell–targeted autoimmunity found in T1D (2). Stages of diabetes pathogenesis have been defined by adaptive immune changes: first, autoreactive T cells are activated (initiation of insulitis), then these cells infiltrate the pancreas (prediabetic phase), and later these pathogenic cells overwhelm immune regulation and destroy insulin-producing cells (overt diabetes) (3–5), but innate immune contributions at each stage are less clear. Deficiencies in innate signaling lowers diabetes incidence in NOD mice, likely via early effects on initiation of insulitis (6). Dendritic cells (DCs) contribute to diabetes pathogenesis at this early stage (7–9), but because of their role in maintaining regulatory T cells (Tregs), increasing DCs at later stages of T1D can block disease (10). Autoimmunity disrupts DC-mediated steady-state tolerance either through intrinsic genetic alterations or because of endogenous innate signals that induce some level of maturation (11, 12). We now ask, how is DC innate responsiveness altered in prediabetic NOD mice?
To date, studies on innate stimulation of NOD DCs have focused on in vitro bone marrow (BM) GM-CSF cultures and have found hyporesponsiveness to LPS stimulation such as lower IL-12 but increased NF-κB activation (13, 14). Importantly, a recent study shows that BM GM-CSF cultures are a complex mix of macrophages, monocytes and related cells, and conventional DCs (cDCs) that varies depending on exact culture conditions, making these studies hard to interpret or align with any DC population in vivo (15, 16). Therefore, to understand how innate responses contribute to diabetes pathogenesis, in vivo DC subsets need to be studied.
Several subsets of DCs have been described that can affect autoimmune pathogenesis (17, 18). cDCs are the migratory and lymphoid resident cells that often control T cell fate. Two main subsets of cDCs are found in both mouse and human (19). CD8+ cDC1s normally contribute to tolerance via induction of Tregs or deletion of autoreactive cells but have tolerance defects in NOD mice (20). CD11b+ cDC2s can induce strong CD4+ T cell stimulation and retain more tolerogenic activity in NOD mice. Distinct from cDCs, MHC class II+ monocyte-derived DCs (moDCs) primarily develop in response to inflammatory signals and may contribute to both Treg homeostasis and activation of pathogenic responses (14, 21, 22). Plasmacytoid DCs (pDCs) are the major producer of type 1 IFN, which then acts on cDCs, and is required for upregulation of genes that are associated with DC maturation and Ag presentation (23, 24).
Type 1 IFN production is critical for induction of protective innate and adaptive immunity against pathogens (25), yet dysregulated type 1 IFN responses can also induce inflammatory tissue damage (26). However, IFN responses can induce regulatory responses, such as production of IL-10 and IDO (27, 28). In fact, IFN-β is successfully used to treat multiple sclerosis, another tissue-specific autoimmune disease. Type 1 IFN contributes to initiation of insulitis in NOD mice, and an IFN-inducible transcriptional signature has been detected in diabetic-prone children that later develop T1D, suggesting that type 1 IFN is critical for the initial break in tolerance for T1D (5, 7, 29, 30), Yet, it is not clear what role innate responses and in particular type 1 IFN play at later stages of disease in setting the balance of pathogenesis and regulation that can ultimately lead to hyperglycemia. Therefore, we set out to determine how cDCs respond to an IFN-inducing stimulus in the context of chronic autoimmunity, focusing on the prediabetic NOD mice.
In this study, we show that, CpG-induced type 1 IFN is higher in prediabetic NOD mice serum and DCs, but the overall transcriptional CpG response is muted in NOD cDCs compared with C57BL/6 (B6) cDCs. In contrast to this overall reduced response, CD40 is higher in NOD cDCs after CpG stimulation, and moDCs were more activated in NOD mice as evidenced by higher inflammatory cytokine production. We hypothesized that NOD cDCs have impaired signaling downstream of the IFN-α/β receptor (IFNAR). Supporting this, the DC response to CpG-treatment is similar in NOD and B6 mice in the presence of IFNAR blocking Abs, and gene targets of the IFN-induced transcriptional complex IFN-stimulated gene factor 3 (ISGF3) are enriched more in B6 DCs after CpG stimulation. Indeed, we find that IFN-induced nuclear localization of STAT1 is lower in NOD. Therefore, NOD cDCs likely have impaired IFNAR responses because of impaired nuclear localization of STAT1. These alterations in innate immunity impact pathways that may have effects on Tregs and diabetes pathogenesis.
Materials and Methods
Mice
B6, B6.H2g7 (B6g7), NOD, and RAG1−/− mice in both B6 and NOD background were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) animal facility. Animals were housed in specific pathogen-free conditions and handled according to the Animal Care and Use Committee of the National Institutes of Health. The B6g7 congenic strain is on the B6 background but carries the NOD-derived H2g7 haplotype. B6g7 mice were used in the experiments where indicated. NOD.MyD88−/− mice were received from D. Mathis (Harvard University, Cambridge, MA) and bred at NIDDK. Age-matched female 8- to 10-wk-old mice with 3–4 mice/group were used in all experiments.
Generation of Flt3 ligand BM-derived DCs in vitro and CpG stimulation
Flt3 ligand (Flt3L) BMDCs were cultured as described previously (31). Cells were stimulated on day 6 for 12 h with CpG-A (InvivoGen, San Diego, CA) + a liposomal transfection reagent, DOTAP (Roche, Mannheim, Germany) (1 μg CpG/well with 2.5 μl DOTAP/well). Where indicated, wells were pretreated with anti-IFNAR Abs (clone MAR1-5A3, IgG1, 20 μg/ml) (BioLegend, San Diego, CA) for 24 h before CpG was added. Following CpG stimulation, cells were collected and analyzed by flow cytometry.
In vivo CpG injection and DC isolation from lymphoid organs
Where indicated, 200 mg anti-IFNAR or isotype (human IFN-γR1 mAb clone GIR208, mouse IgG1) was injected 24 h before CpG injection. Five micrograms of CpG formulated with 30 ml DOTAP (1 μg/μl) was administered i.v., and after 12 h, spleens, pancreatic lymph nodes (pLNs), and skin LNs (sLNs) were harvested separately as indicated for DC enrichment. Untreated (no CpG or anti-IFNAR Ab) mice received 200 μl PBS. DCs were isolated from individual spleens and LNs with collagenase III digestion (Worthington, Lakewood, NJ) as described by Guerrero et al. (31). Cells were then strained using 60-μm filters, stained, and run by flow cytometry.
Flow cytometry
Cells were collected, blocked with anti-CD16/32 (Fcg R III/II; BioLegend), and stained with appropriate Abs against surface proteins in PBS with 2% FCS as described previously (31).
In some experiments, intracellular proteins were stained after fixation and permeabilization using fixation and permeabilization buffers from eBioscience (San Diego, CA). Phosphoflow experiments were performed as previously described but with addition of blocking with anti-mouse 16.2 Abs (FCgRIV; BioLegend) to reduce nonspecific binding (32, 33). For intracellular TNF-α staining, cells were fixed, permeabilized, and stained with appropriate Abs.
The following Abs were used: anti-CD3 (145-2C11), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD49b (DX5), anti-Ly6C (HK1.4), anti-Ly6G (1A8), anti–Siglec-H (551), anti-CD24 (M1/69), anti-CD8α (53-6.7), anti-CD86 (GL-1), anti-CD80 (16-10A1), anti-CD40 (3/23), anti–TNF-α (MP6-XT22), anti-IFNAR (MAR1-5A3), and IL-12p40 (C15.6) from BioLegend; anti-phospho (p)ERK1/2 (T202/Y204), anti-STAT1 (pY701), and anti-STAT4 (pY693) from BD Biosciences (San Jose, CA); and anti-STAT1 (D1K9Y) and anti-STAT4 (C46B10) from Cell Signaling Technology (Danvers, MA). Brilliant Violet 510–conjugated streptavidin (BioLegend) and Pacific Orange–conjugated streptavidin (Life Technologies, Grand Island, NY) were used to detect biotinylated Abs, and Aqua Dead Cell Stain (Life Technologies) was used to gate on viable cells. Fluorescence minus one controls include all fluorophores, except the color being analyzed. Samples were collected on a BD LSRII flow cytometer with four lasers and analyzed using FlowJo software 9.8.2 (Tree Star, Ashland, OR).
Affymetrix microarray analysis
CD8+ cDCs and CD11b+ cDCs from NOD and B6 mice were sorted, and RNA was purified for GeneChip Mouse Genome 430 array analysis. Generation of principal component analysis (PCA) plots and heat maps were performed using Partek Genomic Suite 6.5 (Partek, St. Louis, MO). Differentially expressed (DE) genes that belong to each mouse strain (NOD or B6) following CpG treatment were determined using the samr package with 0.1 false-discovery rate (FDR) and were represented using a Venn diagram. Comparison of genes that were DE before or after treatment with CpG was further analyzed by Ingenuity Pathway Analysis software (National Institutes of Health library). Normalized intensity data were log base 2 transformed for all the analysis unless otherwise stated for some analysis where fold changes were calculated from the untransformed values. Gene lists for gene enrichment analysis (Table I) were obtained through the Qiagen Web site. The array data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE75883 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75883).
Table I.
Genes increased by CpG stimulation are enriched for genes containing binding sites of key IFN pathway regulators in B6 but not NOD DCs
| Gene Groups | IRF-7A | STAT1 | ISGF3 | STAT3 | STAT4 | CREB | NF-κB |
|---|---|---|---|---|---|---|---|
| Induced more with CpG in NOD CD8+ DCs | 0.597 | 0.791 | 0.724 | 0.889 | 0.918 | 0.75 | 0.549 |
| Induced more with CpG in B6 CD8+ DCs | 0.038* | 0.001** | 0.001** | 0.001** | 0.001** | 0.001** | 0.003* |
Kolmogorov–Smirnov test was used to evaluate the difference between the p value profiles of genes in the seven binding site gene lists against that of DE genes in comparing different mouse strains.
p < 0.05,
p < 0.01.
Cytokine measurements
Supernatant from either Flt3L-supplemented BMDC cultures or in vitro enriched DC stimulation was collected 12 h poststimulation with CpG at day 6; serum samples were collected 6 h after in vivo CpG treatment of mice and stored at −70°C for cytokine analysis. TNF-α and IL-12p70 were measured using eBioscience Mouse Cytokine ELISA kits, and IFN-α and IFN-β were measured using the ELISA kit from PBL assay science. In all the ELISA kits, 3,3′,5,5′-Tetramethylbenzidine Liquid Substrate was common for colorimetric reaction as supplied by the manufacturer, and the OD was measured in a multiscan ELISA reader (Synergy H1, Winooski, VT) at 450 nm. The concentrations were calculated from the standard curves established with corresponding purified recombinant mouse cytokines.
Western blot analysis
Equal numbers of spleen lymphocytes were taken to separate cytoplasmic and nuclear protein fractions using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific, Waltham, MA). Fractionated protein samples were run on a NuPAGE 4–12% or 12% Bis-Tris gel (Invitrogen) and blotted onto a nitrocellulose membrane (Invitrogen). Western blots were developed using an ECL system, according to the manufacturer’s instructions (Amersham Biosciences, Pittsburgh, PA). The results were visualized by ChemiDoc Touch Gel and Western blot Imaging System (Bio-Rad, Hercules, CA). Data were subjected to densitometric analysis using Image Lab software included with the ChemiDoc. Abs used for Western blotting include the following: anti-STAT1 (pY701), STAT1 (9172), and heat shock protein 90 were from Cell Signaling Technology. Lamin B was from Santa Cruz Biotechnology (Dallas, TX).
Confocal microscopy and quantification of nuclear localization of STAT proteins
Formaldehyde fixed (4%) (Electron Microscopy Sciences, Hatfield, PA) splenocytes (± IFN-α prestimulation for 30 min) were air-dried on the superfrost microscopic glass slides and blocked with 4% normal goat serum before incubating overnight with primary Abs—rabbit monoclonal STAT1 (D1K9Y; Cell Signaling Technology) and hamster CD11c (Bio-Legend). Species-matched secondary Abs (conjugated with flurochrome dye Alexa 594 or Alexa 488) were added for 2 h. The slides were washed, air-dried, and mounted directly by Vectashield antifade mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Tissue sections treated with no primary Abs were used as controls.
Images were taken with a Zeiss confocal microscope (Nikon Instruments, Melville, NY), and 10–15 fields/condition were analyzed with ImageJ. An algorithm was developed and used in ImageJ to quantify nuclear STAT1 intensity in cells that were positive for STAT1 and CD11c. CD11c+ cells were defined as cells with a ring of CD11c signal, and nuclear area was defined by DAPI signal. Total 100–150 CD11c+ cells were analyzed to quantify the nuclear localization of STAT1.
Quantitative gene expression analysis
Quantitative PCR
CD11c+ splenocytes were bead-enriched by negative selection using biotin Abs specific for CD3, CD19, NKp46, and Ly6c, with anti-biotin magnetic beads, according to the manufacturer instruction (Miltenyi Biotec, San Diego, CA). RNA was then extracted immediately after enrichment using Qiagen mini prep (Molecular Research Center, Cincinnati, OH). RNA was reverse-transcribed into cDNA (Life Technologies), and the cDNA was used as a template for PCR. Quantitative PCR assay was performed using The QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific) for Irf7, Oas3, Ifit1, Ifna1, Ifna2, Ifna4, Ifnb, and Socs1 genes using commercially available TaqMan primers/probes from Life Technologies. The housekeeping gene Hprt was used for normalization, and the data presented as relative gene expression.
NanoString
The mRNA-containing lysate from the sorted cells (pDCs) was incubated with a custom panel of 450 bar-coded probes specific for genes associated with DC development and function (34) (NanoString Technologies, Seattle, WA). Samples were run by the Laboratory of Molecular Technology at the National Cancer Institute (Frederick, MD). Raw data were normalized using the nSOLVER Analysis Software (NanoString Technologies), according to company protocol. mRNA counts were normalized by subtracting CodeCount = geo.mean, Background = mean.2sd, and SampleContent = housekeeping.geo.mean. Following normalization, differential expression of the genes was determined using a nonparametric Student t test. For pDC nanostring data, batch effects were removed using ComBat in the SVA package for R.
Statistical analyses
Homoscedastic two-tailed t tests were performed for most analyses. The p values < 0.05, 0.01, and 0.001 were considered as *, **, and *** levels of significance, respectively. Error bars are represented by the SD unless noted otherwise. Data that are emphasized as not significant are noted by ns.
Results
Prediabetic NOD mice and DCs produce more type 1 IFN in response to CpG than B6
To learn more about type 1 IFN responses in chronic autoimmunity, we first focused on in vivo responses in 8- to 10-wk-old prediabetic NOD mice. For this study, CpG was chosen to study the TLR9-mediated activation of DCs because this type 1 IFN-inducing signal mimics innate signals that contribute to diabetes pathogenesis (7, 35, 36). First, we measured serum levels of IFN-α and IFN-β from NOD and B6 mice 6 h post-CpG i.v. injection. We found that CpG stimulation induced significantly higher levels of serum IFN-α and IFN-β in prediabetic NOD mice compared with B6 (Fig. 1A). Furthermore, to determine how DCs directly respond to CpG stimulation ex vivo, CD11c+ DCs were enriched from spleen and stimulated with CpG for 6 h. Again, NOD DCs secreted more IFN-α and IFN-β in response to CpG stimulation (Fig. 1B). BM cells cultured with Flt3L differentiate into cDCs and pDCs, and these Flt3L BMDCs from NOD mice also produced more IFN-α in response to CpG compared with B6 Flt3L BMDCs (Fig. 1C). Similarly, type 1 IFN gene expression in CD11c+ DCs taken from in vivo CpG-stimulated NOD mice was higher than B6 mice (Fig. 1D). Without CpG stimulation, IFN-α was not detected above background with the sensitivity of currently available assays (data not shown), but biologically relevant responses to type 1 IFN can occur below this level (37). Taken together, these data suggest that even long after initiation of autoimmunity, NOD DCs produce more IFN-α and IFN-β upon CpG stimulation that may reflect the ongoing presence of low chronic type 1 IFN.
FIGURE 1.
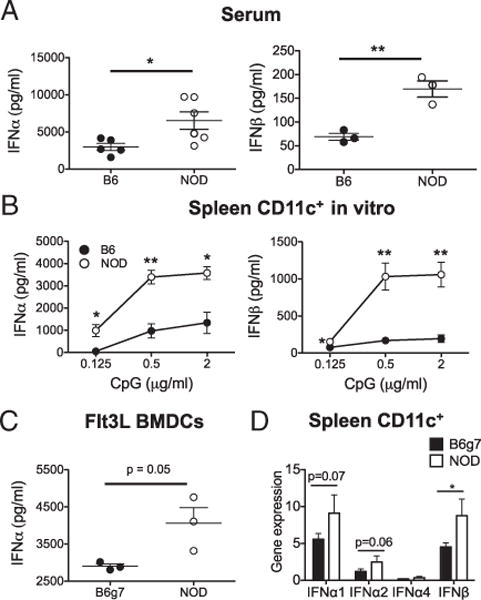
NOD mice show higher expression of type 1 IFN in response to CpG. Type 1 IFN levels were measured 6 h post-CpG stimulation by ELISA in serum samples of NOD and B6 mice 6 h post CpG treatment (A), culture supernatant of enriched-splenic DC culture (B), and culture supernatant of Flt3L-BMDC (C). (D) Type 1 IFN gene expression in CD11c+-enriched population measured by quantitative PCR. Data presented in this paper are from one of at least two to four independent experiments. Comparisons were made between NOD and B6 (A and B) or B6g7 (C and D). *p < 0.05, **p < 0.01.
The global response to CpG stimulation is muted in NOD cDCs
To elucidate how the DCs from NOD mice respond to CpG in relation to this increased type 1 IFN, NOD and B6 mice were treated with CpG for 12 h, and spleen cDC subsets (CD8+ and CD11b+) were isolated for gene expression studies (sorting gates shown in Supplemental Fig. 1A). PCA plots showed that biological replicate samples of DCs clustered within a group, but both stimulation with CpG and strain origin separated the samples. The shift in gene expression after CpG stimulation was larger in B6 DCs than NOD (Fig. 2A). A heat map generated from the microarray data corresponded with PCA results, showing a differential gene expression profile in CD8+ cDCs and CD11b+ cDCs between NOD and B6 mice (Supplemental Fig. 1B). To further characterize the differential response, we categorized genes as up-or downregulated in response to CpG with an FDR of 0.1, using the unstimulated control for each strain as the baseline to focus on genes induced with CpG. As shown by the Venn diagrams, although a core response was shared by both strains, more genes were upregulated (>500) or downregulated (~300) uniquely in B6 cDCs compared with NOD cDCs (~100) (Fig. 2B), showing an overall muted response in both NOD cDCs subsets. Even within the set of genes induced in both strains, the induction was usually lower in NOD cDCs compared with B6, as illustrated by focusing on the genes with the biggest upregulation in control B6 CD8+ cDCs: most are induced less in NOD (Fig. 2C). Next, pathway analysis on the set of genes induced (up or down with FDR 0.1) by CpG in B6 but not NOD DCs showed that genes induced by CpG in B6 but not NOD DCs were significantly enriched (log p > 1.3 z-score > 0.5) in 14 pathways, such as downregulation of apoptosis signaling and of NFAT in regulating immune response (data not shown). Confirming the lower NOD response, a parallel analysis of genes uniquely induced in NOD CD8+ cDCs identified no pathways with both significant p values and z-scores.
FIGURE 2.
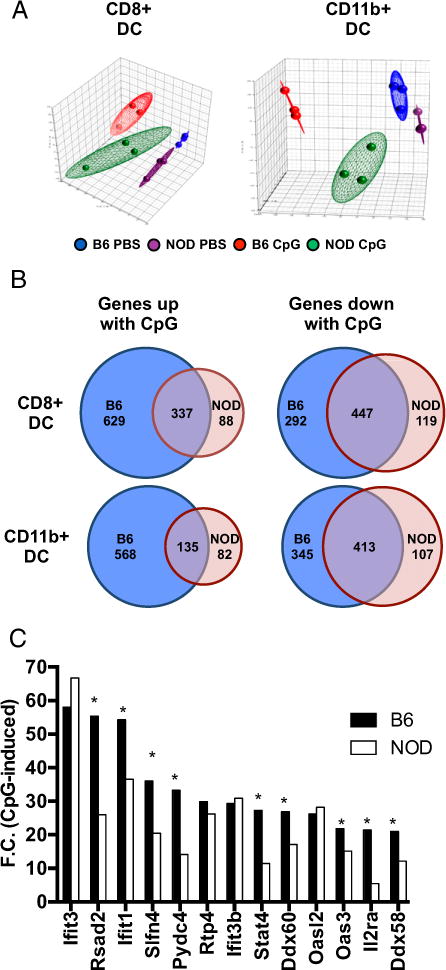
The global response of cDCs to CpG stimulation is muted in NOD. NOD or B6 mice were treated with CpG i.v. After 12 h, splenic DCs were isolated and sorted into CD8+ cDC, and CD11b+ cDC. Sorted DC subsets were analyzed by Affymetrix gene array. (A) PCA plot for CD8+ and CD11b+ cDCs. (B) Venn diagrams show numbers of genes that were upregulated and/or downregulated in DC subsets after CpG stimulation. Genes were categorized as up- or downregulated in response to CpG with an FDR of 0.1, using the unstimulated control for each strain as the baseline to focus on genes induced with CpG. (C) Fold-change comparison of gene expression (top 13 genes selected on the B6 strain) after CpG treatment between B6 and NOD CD8+ cDC. Each sample was pooled from three mice, and each group contained three samples for comparison. Comparison between NOD and B6 in (C) was performed by Student t test. *p < 0.05.
Gene expression was also assessed in CpG-stimulated pDCs. Because the number of pDCs decreases in the spleen after in vivo CpG stimulation, a custom NanoString DC panel was used that requires fewer cells than microarray analysis (gated as shown in Supplemental Fig. 1C). In contrast to the overall muted response to CpG in NOD cDCs, the differential response in NOD pDCs is mixed. Among the 25 genes most induced in B6g7 pDCs, 16 showed differential induction in B6g7 versus NOD, but unlike the cDCs (microarray analysis), some of these genes are induced more in NOD (Supplemental Fig. 1D, Supplemental Table I). We hypothesized that cDC response to CpG is dominated by responses downstream of the IFNAR, but pDC CpG responses will likely represent more of a mix between the direct TLR9 response and IFNAR response, which may account for a more complex pDC response. Therefore, despite increased type 1 IFN protein, the response to CpG in NOD cDCs is reduced compared with B6 cDCs, but the pDC response is mixed.
Impaired innate responsiveness in NOD DCs inhibits induction of costimulatory proteins
Fitting with this overall lower gene induction, expression of co-stimulatory genes such as CD80 and CD86 were lower in NOD DCs after CpG stimulation (fold-change NOD/B6: −1.9, p = 2.5 × 10−7 and 21.4, p = 6.0 × 10−3, respectively). Although expression of CD80 and CD86 are needed for full activation of effector T cells, they are also necessary for Treg maintenance, and NOD mice lacking CD80 and CD86 have accelerated diabetes development (38). Changes in DC activation were evaluated at the protein level by measuring expression of CD86 and CD80 in cDCs 12 h after in vivo CpG stimulation in NOD and B6 mice. CD86 expression was upregulated less following CpG stimulation in NOD DCs compared with the B6 DCs. This lower level of induced CD86 expression in NOD mice was observed in both cDC subsets and pDCs isolated from spleen, sLNs, or pLNs (Fig. 3A). We observed similar results for CD80, especially in CD8+ cDCs and pDCs (Fig. 3B). Therefore, CD86 protein expression is one key example of the overall muted NOD DC CpG response. These data highlight the inability of NOD DCs to respond fully to CpG stimulation that may alter activation of both Tregs that inhibit diabetes and pathogenic T cells.
FIGURE 3.
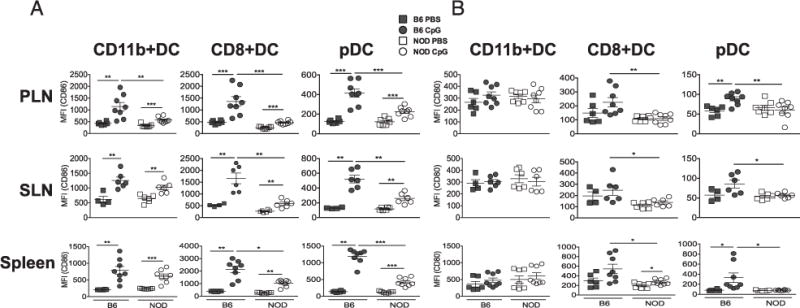
The costimulatory molecules CD86/CD80 are induced less in NOD DCs. NOD or B6 mice were treated with CpG i.v., and 12 h post-treatment, cells from the indicated lymphoid organs (spleen, pLNs, and sLNs) were stained for costimulatory molecule expression and DC subset markers (CD8+ cDC, CD11b+ cDC, and pDC subsets). CD86 expression (A) and CD80 expression (B) in DC subsets. Data presented as geometric mean fluorescence intensity (MFI). Data were pooled from three independent experiments. NOD and B6 strains after CpG stimulation were compared by Student t test. *p < 0.05, **p < 0.01, ***p < 0.001.
Some inflammatory proteins are higher in NOD DCs primarily because of increased production by moDCs
Despite this attenuated innate response, NOD DCs clearly exhibit strong proinflammatory responses in many contexts (39, 40). We previously showed that CD40 expression is higher on NOD cDCs (20). In this tudy, we find that CpG-induced CD40 expression was significantly higher in NOD DC subsets compared with B6 DCs, indicating the less tolerogenic DC function in NOD mice (Fig. 4A). Therefore, we were specifically interested in measuring other proinflammatory proteins to see how NOD DCs respond to stimulation at the protein level. To measure cytokine production, culture supernatants collected from splenic CD11c+ DC culture and serum samples of NOD and B6 mice after CpG-stimulation were tested for TNF-α, and IL-12p70 by ELISA. NOD mice produced higher amount of TNFα but less IL-12p70 than B6 DCs (Fig. 4B). Therefore, although the majority of the CpG response is lower in NOD, including CD86, CD80, and IL-12p70, some key proteins are induced more in NOD DCs, including IFN-α, TNF-α, and CD40.
FIGURE 4.
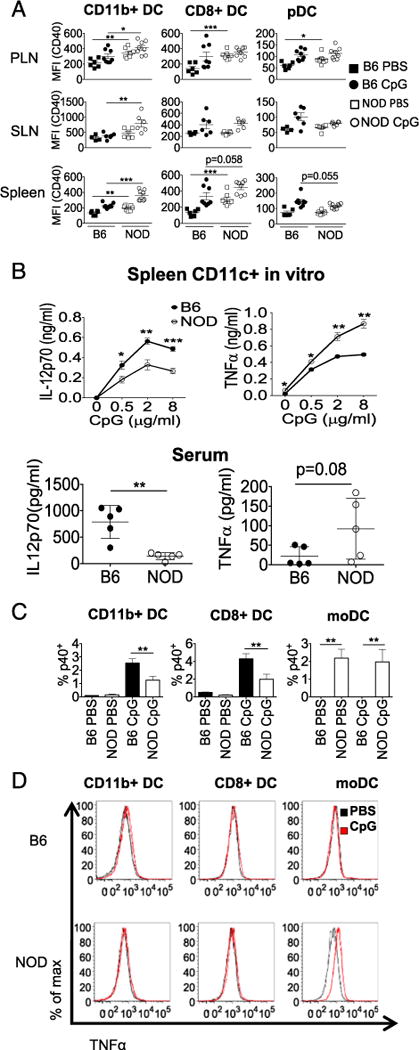
NOD cDCs express higher CD40, and NOD moDCs are the source of increased IL-12 and TNF. NOD or B6 mice were treated with CpG i.v., and 12 h posttreatment, cells from the indicated lymphoid organs (spleen, pLNs, and sLNs) were stained for CD40, TNFR superfamily member, and DC subset markers (CD8+ cDC, CD11b+ cDC, and pDC subsets) (A). Spleen CD11c+ DCs were bead-enriched from NOD and B6 mice and stimulated with the indicated concentration of CpG in vitro. IL-12p70 and TNF-α were measured in the culture supernatant by ELISA 12 h post-CpG stimulation (B). Six hours post-CpG injection, serum samples were collected for IL-12p70 and TNF-α analysis (B). Intracellular cytokine levels (IL-12p40 and TNF-α) were measured in CD8+ cDC, CD11b+ cDC, and moDC flow cytometry 12 h post-CpG stimulation (C and D). Data were either pooled from two to three independent experiments (A and B) or presented one representative experiment from three repeated experiments (C and D). *p < 0.05, **p < 0.01, ***p < 0.001.
To determine which cells were making these proinflammatory cytokines, a more specific gating scheme was used to separate the CD11b+ cDCs from the moDCs, inflammatory monocytes that have upregulated MHC class II (32). Most IL-12 was produced by cDCs, with NOD cDCs making less than B6 (Fig. 4B). However, moDCs from NOD but not B6 were positive for IL-12p40, even without activation (Fig. 4C). TNF-α is one signature cytokine of activated moDCs (41), and indeed, intracellular cytokine staining of DCs with this gating identified the moDCs as the primary producers of TNF-α (Fig. 4D). NOD moDCs have higher levels of TNF-α staining compared with B6 moDCs. Therefore, although NOD cDCs show a muted CpG response, NOD moDCs make more proinflammatory cytokines.
CD40 and CD86 induction in NOD cDCs is MyD88 dependent, but impaired NOD CD86 induction is independent of autoimmune B and T cell responses
Because CpG can signal via receptors other than TLR9, such as cytoplasmic receptors that signal via STING, we tested whether the response was dependent on TLR signaling adaptor MyD88. CD86 was used to represent the important lower NOD DC gene responses to CpG that dominated the gene expression analysis. CD40 was used to represent the smaller subset of genes that are more upregulated after CpG treatment in NOD compared with B6. The upregulation of both markers as well as IFN-α production (data not shown) in response to CpG was MyD88 dependent and therefore likely TLR9 mediated (Fig. 5A).
FIGURE 5.
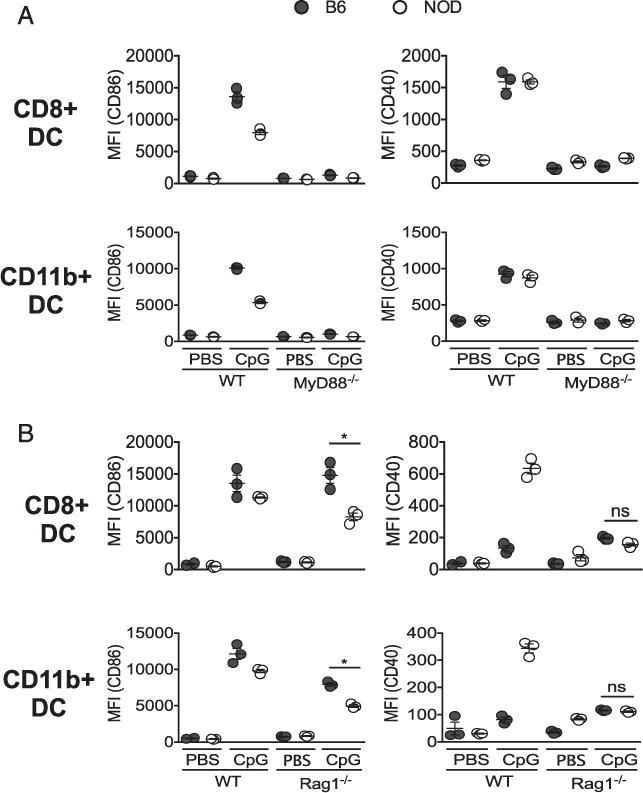
Impaired CD86 induction in NOD mice is MyD88 dependent but independent of autoimmune B and T cell responses. CpG was given i.v. to age-matched wild-type (WT) and MyD88−/− (A) or Rag1−/− NOD and B6 mice (B), and 12 h posttreatment, splenic CD8+ and CD11b+ cDCs were analyzed for CD86 and CD40 expression by flow cytometry. Data represent one independent experiment repeated two to three times with three mice per group. Each dot represents an individual mouse. Values are geometric mean fluorescence intensity (MFI). *p < 0.05.
Next, to investigate whether the impaired response to CpG stimulation in NOD mice occurs as a result of ongoing adaptive inflammation driven by T and B cells that directly induce diabetes pathogenesis, we examined costimulatory molecule expression in age-matched NOD.Rag1−/− and B6.Rag1−/− following CpG stimulation in vivo. We found that CD86 expression in CpG-treated NOD.Rag1−/− was significantly lower than CpG-treated B6.Rag1−/−, matching the CD86 expression pattern in CpG-treated NOD or B6 mice (Fig. 5B, left panel). However, unlike CD40 expression in NOD, NOD.Rag1−/− and B6.Rag1−/− showed comparable levels of CD40 in DC subsets (Fig. 5B, right panel), suggesting that the level of CD40 protein expression on NOD DCs may be dependent on the pathogenic T and B cell environment. Therefore, although some proinflammatory costimulation, such as CD40, is dependent on the adaptive autoimmune inflammation in NOD, the overall muted CpG response, as exemplified by CD86 expression, is intrinsic to the DCs and independent of the autoimmune process.
CD86 expression in NOD cDCs is more dependent on type 1 IFN pathway than CD40
Because NOD DCs make more IFN-α in response to CpG but have an overall lower response, this lower induction of gene expression may be downstream of the IFNAR rather than because of lower TLR9 signal induction. To assess this, first, Flt3L BMDCs were treated with anti-IFNAR Ab, and then, the expression of CD86 and CD40 was measured 12 h after CpG stimulation. As shown in Fig. 6A (left panel), IFNAR blocking greatly reduced the expression of CD86 in both NOD and B6 mice. In contrast, CD40 expression was only partially reduced by IFNAR blocking and was still higher in NOD compared with B6 DCs (Fig. 6A, right panel). Next, response of spleen cDCs to in vivo CpG stimulation and to acute blocking of IFNAR in vivo was tested. As in vitro, CD40 expression remained high in NOD cDCs compared with B6 cDCs, whereas CD86 expression was reduced close to basal levels in both strains of mice (Fig. 6B). Therefore, CD40 expression may be associated with direct TLR signaling and interaction with activated B and T cells, whereas blocking IFNAR yields similar CD86 expression in NOD and B6 DCs, suggesting NOD mice have a deficiency in type 1 IFN signaling.
FIGURE 6.
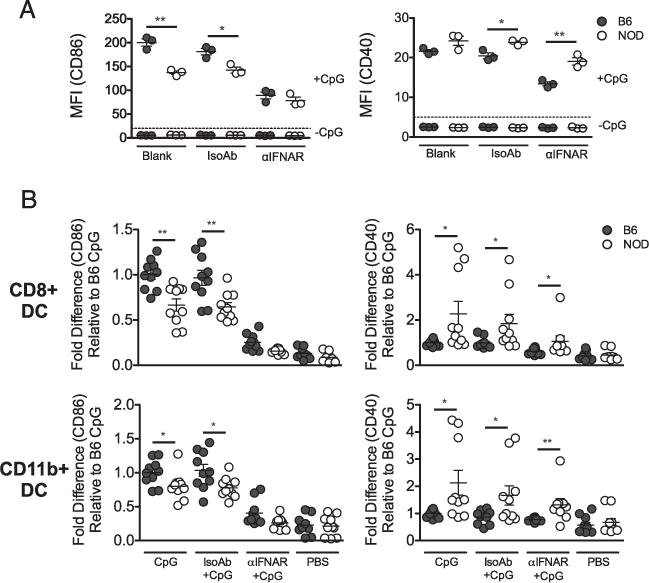
CD86 expression in NOD mice is more dependent on signaling downstream of IFNAR than CD40. (A) BMDCs were cultured with Flt3L, and cultures were blocked with anti-IFNAR Ab for 24 h before stimulation with CpG in vitro. CD86 (left panel) and CD40 (right panel) expression were measured by flow cytometry on cells gated on CD11c+ DCs. (B) CpG was given i.v., and anti-IFNAR Ab was injected i.p. in mice 24 h before i.v. CpG stimulation. Twelve hours post-CpG treatment, spleen cells from NOD and B6g7 mice were gated on CD8+ cDC and CD11b+ cDC subsets, and CD86 and CD40 were measured by flow cytometry. Data were pooled from three independent experiments. Data represent fold change compared with CpG-B6g7. Each dot represents an individual mouse. Values are geometric mean fluorescence intensity (MFI). *p < 0.05, **p < 0.01.
Impaired response to CpG in NOD cDCs is consistent with altered IFNAR signaling
The impaired CpG response in NOD cDCs could be due to either defects in TLR9 signaling or IFNAR signaling. To determine whether the observed muted CpG response was specific to TLR9 activation, mice were stimulated with polyinosinic-polycytidylic acid [poly(I:C)], a TLR3 agonist that also induces type 1 IFN. The poly(I:C)-induced response is very similar to the CpG response: NOD mice produced more IFN-β, but cDCs expressed less CD86 protein, and gene expression of IFN response genes was lower (Supplemental Fig. 2). The common element in the CpG and poly(I:C)-induced responses is type 1 IFN because TLR3 signaling is TRIF-dependent and distinct from TLR9.
Pathway analysis of genes up/downregulated with CpG in CD8+ cDCs from our microarray data demonstrate higher enrichment of several pathways in B6 DCs compared with NOD DCs, including the Ingenuity Pathway Analysis JAK/STAT cluster. The lower signal in NOD DCs for this pathway primarily was due to lack of MEK activation and ERK-induced NF-κB activation; both are found in the same arm of the pathway, which has been shown to enhance transcription of ISGF3-dependent genes (Supplemental Fig. 3A) (42–44). To determine the contribution of this MEK-mediated pathway in the differential IFN DC response observed between NOD and B6, we first stimulated spleen cells in vitro with CpG and measured pERK levels by flow cytometry in different DC subsets. pERK was significantly higher in B6 CD8+ cDCs compared with NOD (Supplemental Fig. 3B). Interestingly, when MEK inhibitor was used, IFN-α–induced CD86 expression was significantly reduced as observed in the Flt3L BMDCs (Supplemental Fig. 3C), consistent with a role for this pathway in augmenting IFN gene expression. Because these proteins are also activated directly downstream of TLR9, reduced MEK/ERK activation in NOD cDCs may contribute to the impaired response to CpG and IFN.
To focus more on transcription downstream of IFNAR, we were interested in gene expression induced by ISGF3, a complex of activated STAT1, STAT2, and IFN regulatory factor (IRF)9 that is the main transcription factor for IFN-induced genes (45). We found transcripts associated with ISGF3 binding sites are significantly enriched within the set of genes induced in B6 cDCs but not NOD cDCs, consistent with impaired type 1 IFN signal in NOD DCs (p = 0.001; Table I). Taken together, these data suggest that the reduced NOD cDC response to CpG may be due to impaired IFNAR response.
Altered IFNAR signaling in NOD cDCs because of decreased nuclear localization of STAT1
Next, IFNAR expression was measured on the two cDC populations, pDCs and moDCs, to assess this pathway at the protein level. Both NOD CD8+ and CD11b+ cDCs expressed slightly lower levels of IFNAR, whereas NOD pDCs and moDCs expressed similar levels as B6 DCs (Fig. 7A). Despite this, cDCs from NOD and B6g7 mice showed similar IFN-α–induced levels of pSTAT1 measured by flow cytometry, and moDCs and pDCs from NOD mice expressed more sustained pSTAT1 (Fig. 7B). Although canonical IFNAR signal is via phosphorylation of STAT1 and STAT2, STAT4 can also be activated by IFN (46). pSTAT2 induction was similar in B6 and NOD cDCs (data not shown), but pSTAT4 was lower in NOD cDCs (Fig 7B). Therefore, the lower type 1 IFN response in NOD cDCs is not simply because of the pSTAT1 levels observed by flow cytometry. The lack of enrichment of ISGF3-induced genes suggests the impairment is due to signals downstream of STAT1 and STAT2 activation but prior to ISGF3 binding.
FIGURE 7.
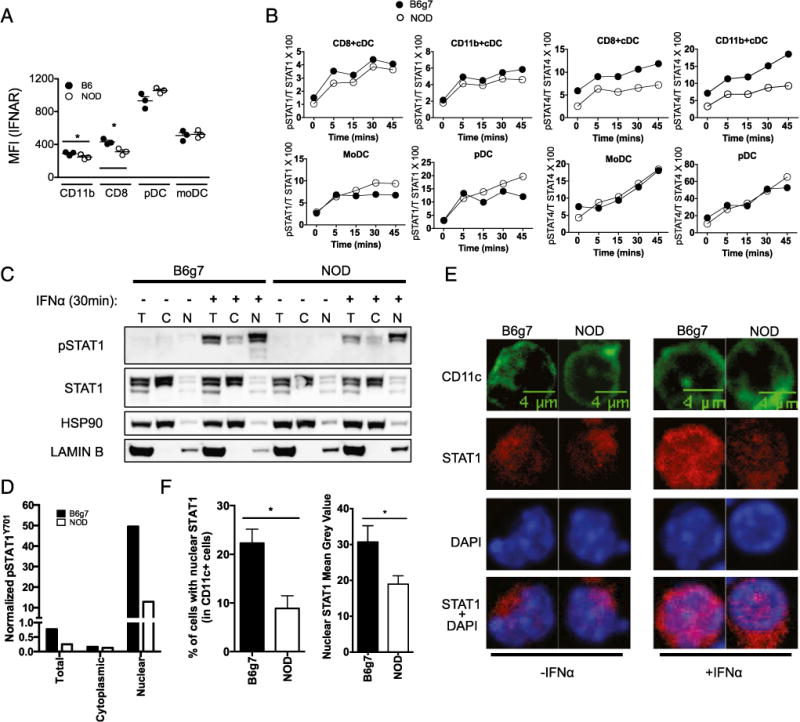
NOD DCs have altered IFN-α–induced signaling. (A) Comparison between NOD and B6 (A) or B6g7 (B–F) is shown at different levels of IFNAR signaling pathway. Expression of IFNAR on DC subsets was determined by flow cytometry. Data show mean fluorescence intensity (MFI) of cell surface IFNAR. (B) Intracellular levels of pSTAT1 and pSTAT4 expression in the indicated DC subsets (MHC Class IIhi gating applied) were measured by flow cytometry after IFN-α stimulation of collagenase-digested spleen cells. Data presented in this paper showed geometric mean MFI as the percentage of total STAT1 or STAT4. (C and D) Representative Western blot showing levels of pSTAT1 and STAT1 in the indicated cell fraction with and without IFN-α stimulation. Quantification of pSTAT1 was normalized to the respective fraction loading control (Lamin B for nucleus [N], heat shock protein 90 [HSP90] for cytoplasm [C], and total [T]). (E) Confocal microscopy of spleen cells with DAPI (blue), STAT1 (red), and CD11c+ DC (green). (F) Percentage of CD11c+ cells with nuclear STAT1 (left panel) and quantification of the amount of nuclear STAT1 within CD11c+ cells (right panel). Data represent one independent experiment of two to three repeat experiments. Mean + SEM is represented in (F). *p < 0.05.
A key step in the elicitation of signal from IFNAR is the nuclear localization of the ISGF3 complex, which is mediated primarily by pSTAT1 (26). Lower ISGF3 transcriptional activity in NOD DCs could result from inability of activated pSTAT1 and the other components of ISGF3 to localize to or be retained in the nucleus. To test this, NOD and B6g7 spleen cells were activated with IFN-α for 30 min, and cell nuclei were separated from the cytoplasmic fraction. Western blots analysis for pSTAT1 and total STAT1 was performed in both nuclear and cytoplasmic fractions. We observed decreased pSTAT1 in the nuclear fraction in NOD compared with B6g7 (Fig. 7C, 7D). Similarly, nuclear localization of STAT1 was quantified in NOD or B6g7 CD11c+ cells following IFN-α stimulation for 30 min using confocal microscopy (Fig. 7E). Among the CD11c+ cells, B6g7 mice showed significantly higher percentage of cells with STAT1 in the nuclei than NOD mice, and the amount of STAT1 signal that overlapped with nuclear DAPI signal was lower in NOD CD11c+ cells (Fig. 7F). Therefore, these observations at the single-cell level suggest that NOD DCs make more type 1 IFN but display decreased IFNAR signal because of impaired nuclear localization of pSTAT1.
Discussion
In this study, we demonstrate differential innate activation in NOD cDCs characterized by higher IFN-α and CD40, but overall lower response to CpG including lower CD80 and CD86 compared with cDCs from B6 mice. This lower CD86 in the NOD background is dependent on IFNAR signaling but not adaptive autoimmune inflammation as blocking IFNAR equalizes CpG-induced CD86 expression between the strains, but NOD.Rag1−/− mice still have lower CD86 expression on cDCs compared with B6.Rag1−/−. In contrast, CD40 differential expression was dependent on the autoimmune B and T cell responses. Therefore, some innate activation is secondary to adaptive autoimmunity, but some innate alterations may be intrinsic to the APCs.
Although initially counterintuitive for autoimmune NOD mice to display a lower innate immune response, cDCs contribute both to pathogenesis and tolerance induction in NOD mice, and many tolerance mechanisms require some intermediate level of maturation (47). For example, maintenance of Tregs is optimal with semimature cDCs, and expression of CD80 and CD86 is necessary for DC-mediated expansion of Tregs, and NOD mice lacking CD80 and CD86 develop diabetes more rapidly (38, 48, 49). Reduced DC inflammatory tone could impair some immune tolerance mechanisms. This fits with the hygiene hypothesis, which suggests a lack of early exposure to pathogens may lead to more autoimmunity. Indeed, administration of TLR ligands such as LPS or poly(I:C) can block diabetes in NOD mice (50, 51). In contrast, CD40 is a pathogenic signal that is dependent on the adaptive inflammatory response. DCs expressing low levels of CD40 induce Treg generation but with high level of CD40 suppress Treg induction (52). Previously, we have shown blocking CD40 restores tolerance by lowering the number of effector T cells and IFN-γ production (20). Therefore, the particular combination of lower intrinsic responsiveness paired with higher levels of select pathways, including CD40, could contribute to the imbalance between effector and Tregs.
We measured parameters associated with IFNAR signal, including IFNAR, and levels of the activated forms of STAT1 and STAT2, after IFN-α stimulation, and found that NOD mice express lower IFNAR on cDCs, but pSTAT1 and pSTAT2, as measured by phosphoflow, are similar to B6 DCs. Yet, despite induction of pSTAT1 and pSTAT2, gene transcripts associated with ISGF3 binding sites are not significantly enriched in NOD cDCs, consistent with a poor induction of downstream gene expression associated with IFNAR pathway in NOD mice. We show that this is due to impaired nuclear localization of IFN-α–activated STAT1, a component of ISGF3. The observed reduction of MEK activation and pSTAT4 activation in NOD cDCs could contribute to this defect as these pathways may normally amplify ISGF3 activation independent of STAT1 activation (44).
It is possible that impaired IFNAR signal in NOD DCs may be a result of low-level chronic type 1 IFN in NOD mice (46). The sensitivity of assays to detect IFN-α are not able to measure basal levels in sera, but CpG-stimulated IFN-α and IFN-β is higher in NOD than B6. Consistent with our results, a recent study in humans with T1D reported lower expression of IFN-inducible genes even in the presence of high levels of type 1 IFN (53). Viral infections may precipitate development of T1D in islet autoantibody– positive (prediabetic) individuals, which could account for the seasonal differences in onset of T1D (54, 55). CpG-induced changes in prediabetic NOD cDCs might model viral-induced changes in APC function that are relevant for the failure of immune regulation that precipitates β cell destruction and diabetes onset.
Previous studies on NOD mice have shown several alterations in DC phenotype, including increased NF-κB signaling (14). However, most functional studies of NOD DCs use BM GM-CSF–induced DCs that may be similar to moDCs, which our data suggest function quite differently from cDCs (56). Consistent with these prior studies using in vitro–derived moDCs (41, 57–59), we find moDCs directly from the spleen of NOD mice are hyperactive, with higher TNF-α production. Other studies focus on primary cDCs from the pancreas or pLN that are measuring effects of the local autoimmune inflammation, which is clearly relevant to disease pathogenesis, but in this study, we focus on cDCs distal to the autoimmune target that would likely present Ag in the context of immunotherapy in which exogenous self Ag is administered. Therefore, measuring the NOD spleen DC phenotype and function is relevant for designing Ag-specific immunotherapy for autoimmunity, and to our knowledge, this study is the first to comprehensively measure innate responses of cDCs in the context of prediabetic NOD mice.
Defective DC activation in NOD mice because of a lack of proper regulation of IFN pathway may be sufficient for inducing pathogenic responses but may not be enough for optimal development of tolerogenic responses. Taken together, these data offer new insights into the state of innate immunity and key APCs in prediabetic NOD mice, a potential intervention time for Ag-specific immunotherapy that would use these APCs.
Supplementary Material
Acknowledgments
We thank Alice Franks (Diabetes, Endocrinology, and Obesity Branch, NIDDK) for help with mouse husbandry and NIDDK/National Heart, Lung, and Blood Institute flow core and Dr. Phil McCoy for flow cytometry support. We thank the National Heart, Lung, and Blood Institute Bioinformatics Core Facility including Xujing Wang, and the NIDDK Genomics Core, Weiping Chen and Chithra Keembiyehetty, for gene array support and microarray data analysis. We also thank Dr. Giorgio Trinchieri and Dr. Brian Kelsall for feedback and critical reading of the manuscript.
This work was supported by the intramural research programs of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Heart, Lung, and Blood Institute and by a collaborative research agreement with Janssen Research and Development.
Abbreviations
- B6
C57BL/6
- B6g7
B6.H2g7
- BM
bone marrow
- cDC
conventional DC
- DC
dendritic cell
- DE
differentially expressed
- FDR
false-discovery rate
- Flt3L
Flt3 ligand
- IFNAR
IFN-α/β receptor
- IRF
IFN regulatory factor
- ISGF3
IFN-stimulated gene factor 3
- moDC
monocyte-derived DC
- NIDDK
National Institute of Diabetes and Digestive and Kidney Diseases
- PCA
principal component analysis
- pDC
plasmacytoid DC
- PFA
paraformaldehyde
- pLN
pancreatic lymph node
- poly(I:C)
polyinosinic-polycytidylic acid
- sLN
skin LN
- T1D
type 1 diabetes
- Treg
regulatory T cell
Footnotes
ORCIDs: 0000-0002-4496-8385 (M.J.R.); 0000-0003-4466-2391 (Y.Z.); 0000-0002-0270-3688 (K.B.R.); 0000-0002-5501-8553 (C.H.-I.); 0000-0001-8785-7653 (Y.C.); 0000-0003-3738-379X (K.V.T.).
The data presented in this article have been submitted to the National Center for Biotechnology Information’s Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE75883) under accession number GSE75883.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Grieco FA, Vendrame F, Spagnuolo I, Dotta F. Innate immunity and the pathogenesis of type 1 diabetes. Semin Immunopathol. 2011;33:57–66. doi: 10.1007/s00281-010-0206-z. [DOI] [PubMed] [Google Scholar]
- 2.Pearson JA, Wong FS, Wen L. The importance of the non obese diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun. 2016;66:76–88. doi: 10.1016/j.jaut.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 4.Lieberman SM, DiLorenzo TP. A comprehensive guide to antibody and T-cell responses in type 1 diabetes. Tissue Antigens. 2003;62:359–377. doi: 10.1034/j.1399-0039.2003.00152.x. [DOI] [PubMed] [Google Scholar]
- 5.André I, Gonzalez A, Wang B, Katz J, Benoist C, Mathis D. Checkpoints in the progression of autoimmune disease: lessons from diabetes models. Proc Natl Acad Sci USA. 1996;93:2260–2263. doi: 10.1073/pnas.93.6.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, Lehuen A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. 2013;19:65–73. doi: 10.1038/nm.3042. [DOI] [PubMed] [Google Scholar]
- 8.Calderon B, Carrero JA, Miller MJ, Unanue ER. Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response. Proc Natl Acad Sci USA. 2011;108:1567–1572. doi: 10.1073/pnas.1018975108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferris ST, Carrero JA, Mohan JF, Calderon B, Murphy KM, Unanue ER. A minor subset of Batf3-dependent antigen-presenting cells in islets of Langerhans is essential for the development of autoimmune diabetes. Immunity. 2014;41:657–669. doi: 10.1016/j.immuni.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Darrasse-Jèze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, Masilamani RF, Dustin ML, Rudensky A, Liu K, Nussenzweig MC. Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med. 2009;206:1853–1862. doi: 10.1084/jem.20090746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rönnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 2006;54:408–420. doi: 10.1002/art.21571. [DOI] [PubMed] [Google Scholar]
- 12.Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological β cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med. 2003;198:1527–1537. doi: 10.1084/jem.20030966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feili-Hariri M, Morel PA. Phenotypic and functional characteristics of BM-derived DC from NOD and non-diabetes-prone strains. Clin Immunol. 2001;98:133–142. doi: 10.1006/clim.2000.4959. [DOI] [PubMed] [Google Scholar]
- 14.Poligone B, Weaver DJ, Jr, Sen P, Baldwin AS, Jr, Tisch R. Elevated NF-κB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J Immunol. 2002;168:188–196. doi: 10.4049/jimmunol.168.1.188. [DOI] [PubMed] [Google Scholar]
- 15.Helft J, Böttcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, Reis e Sousa C. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+MHCII+ macrophages and dendritic cells. Immunity. 2015;42:1197–1211. doi: 10.1016/j.immuni.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 16.Guilliams M, Malissen B. A death notice for in-vitro-generated GM-CSF dendritic cells? Immunity. 2015;42:988–990. doi: 10.1016/j.immuni.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 17.Prasad SJ, Goodnow CC. Cell-intrinsic effects of non-MHC NOD genes on dendritic cell generation in vivo. Int Immunol. 2002;14:677–684. doi: 10.1093/intimm/dxf034. [DOI] [PubMed] [Google Scholar]
- 18.Saxena V, Ondr JK, Magnusen AF, Munn DH, Katz JD. The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J Immunol. 2007;179:5041–5053. doi: 10.4049/jimmunol.179.8.5041. [DOI] [PubMed] [Google Scholar]
- 19.Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14:571–578. doi: 10.1038/nri3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Price JD, Beauchamp NM, Rahir G, Zhao Y, Rieger CC, Lau-Kilby AW, Tarbell KV. CD8+ dendritic cell-mediated tolerance of autoreactive CD4+ T cells is deficient in NOD mice and can be corrected by blocking CD40L. J Leukoc Biol. 2014;95:325–336. doi: 10.1189/jlb.0113013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikolic T, Bouma G, Drexhage HA, Leenen PJ. Diabetes-prone NOD mice show an expanded subpopulation of mature circulating monocytes, which preferentially develop into macrophage-like cells in vitro. J Leukoc Biol. 2005;78:70–79. doi: 10.1189/jlb.1104662. [DOI] [PubMed] [Google Scholar]
- 22.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med. 2004;199:1467–1477. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 24.Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 25.Ng D, Gommerman JL. The regulation of immune responses by DC derived type I IFN. Front Immunol. 2013;4:94. doi: 10.3389/fimmu.2013.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Leung S, Qureshi S, Darnell JE, Jr, Stark GR. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon-α. J Biol Chem. 1996;271:5790–5794. doi: 10.1074/jbc.271.10.5790. [DOI] [PubMed] [Google Scholar]
- 27.Hata N, Sato M, Takaoka A, Asagiri M, Tanaka N, Taniguchi T. Constitutive IFN-α/β signal for efficient IFN-α/β gene induction by virus. Biochem Biophys Res Commun. 2001;285:518–525. doi: 10.1006/bbrc.2001.5159. [DOI] [PubMed] [Google Scholar]
- 28.Scheler M, Wenzel J, T€uting T, Takikawa O, Bieber T, von Bubnoff D. Indoleamine 2,3-dioxygenase (IDO): the antagonist of type I interferon-driven skin inflammation? Am J Pathol. 2007;171:1936–1943. doi: 10.2353/ajpath.2007.070281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, Cutler AJ, Doecke JD, Flint S, McKinney EF, et al. A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes. 2014;63:2538–2550. doi: 10.2337/db13-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-α initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA. 2008;105:12439–12444. doi: 10.1073/pnas.0806439105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guerrero AD, Dong MB, Zhao Y, Lau-Kilby A, Tarbell KV. Interleukin-2-mediated inhibition of dendritic cell development correlates with decreased CD135 expression and increased monocyte/macrophage precursors. Immunology. 2014;143:640–650. doi: 10.1111/imm.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong MB, Rahman MJ, Tarbell KV. Flow cytometric gating for spleen monocyte and DC subsets: differences in autoimmune NOD mice and with acute inflammation. J Immunol Methods. doi: 10.1016/j.jim.2015.08.015. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biburger M, Trenkwald I, Nimmerjahn F. Three blocks are not enough—blocking of the murine IgG receptor FcgRIV is crucial for proper characterization of cells by FACS analysis. Eur J Immunol. 2015;45:2694–2697. doi: 10.1002/eji.201545463. [DOI] [PubMed] [Google Scholar]
- 34.Fonseca DM, Hand TW, Han SJ, Gerner MY, Glatman Zaretsky A, Byrd AL, Harrison OJ, Ortiz AM, Quinones M, Trinchieri G, et al. Microbiota-dependent sequelae of acute infection compromise tissue-specific immunity. Cell. 2015;163:354–366. doi: 10.1016/j.cell.2015.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia CQ, Peng R, Chernatynskaya AV, Yuan L, Carter C, Valentine J, Sobel E, Atkinson MA, Clare-Salzler MJ. Increased IFN-α-producing plasmacytoid dendritic cells (pDCs) in human Th1-mediated type 1 diabetes: pDCs augment Th1 responses through IFN-α production. J Immunol. 2014;193:1024–1034. doi: 10.4049/jimmunol.1303230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Filippi CM, von Herrath MG. Viral trigger for type 1 diabetes: pros and cons. Diabetes. 2008;57:2863–2871. doi: 10.2337/db07-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gough DJ, Messina NL, Clarke CJ, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36:166–174. doi: 10.1016/j.immuni.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 39.Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci. 2013;1281:16–35. doi: 10.1111/j.1749-6632.2012.06826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Welzen-Coppens JM, van Helden-Meeuwsen CG, Leenen PJ, Drexhage HA, Versnel MA. Reduced numbers of dendritic cells with a tolerogenic phenotype in the prediabetic pancreas of NOD mice. J Leukoc Biol. 2012;92:1207–1213. doi: 10.1189/jlb.0312168. [DOI] [PubMed] [Google Scholar]
- 41.Hespel C, Moser M. Role of inflammatory dendritic cells in innate and adaptive immunity. Eur J Immunol. 2012;42:2535–2543. doi: 10.1002/eji.201242480. [DOI] [PubMed] [Google Scholar]
- 42.David M, Petricoin E, III, Benjamin C, Pine R, Weber MJ, Larner AC. Requirement for MAP kinase (ERK2) activity in interferon α- and interferon β-stimulated gene expression through STAT proteins. Science. 1995;269:1721–1723. doi: 10.1126/science.7569900. [DOI] [PubMed] [Google Scholar]
- 43.Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999;18:5601–5608. doi: 10.1093/emboj/18.20.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R, Young PR, Fish EN, Platanias LC. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274:30127–30131. doi: 10.1074/jbc.274.42.30127. [DOI] [PubMed] [Google Scholar]
- 45.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 46.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarbell KV, Yamazaki S, Steinman RM. The interactions of dendritic cells with antigen-specific, regulatory T cells that suppress autoimmunity. Semin Immunol. 2006;18:93–102. doi: 10.1016/j.smim.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 48.Yamazaki S, Iyoda T, Tarbell K, Olson K, Velinzon K, Inaba K, Steinman RM. Direct expansion of functional CD25+CD4+ regulatory T cells by antigen-processing dendritic cells. J Exp Med. 2003;198:235–247. doi: 10.1084/jem.20030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lesage S, Goodnow CC. Organ-specific autoimmune disease: a deficiency of tolerogenic stimulation. J Exp Med. 2001;194:F31–F36. doi: 10.1084/jem.194.5.f31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caramalho I, Rodrigues-Duarte L, Perez A, Zelenay S, Penha-Gonçalves C, Demengeot J. Regulatory T cells contribute to diabetes protection in lipopolysaccharide-treated non-obese diabetic mice. Scand J Immunol. 2011;74:585–595. doi: 10.1111/j.1365-3083.2011.02627.x. [DOI] [PubMed] [Google Scholar]
- 51.Serreze DV, Hamaguchi K, Leiter EH. Immunostimulation circumvents diabetes in NOD/Lt mice. J Autoimmun. 1989;2:759–776. doi: 10.1016/0896-8411(89)90003-6. [DOI] [PubMed] [Google Scholar]
- 52.Martin S, Agarwal R, Murugaiyan G, Saha B. CD40 expression levels modulate regulatory T cells in Leishmania donovani infection. J Immunol. 2010;185:551–559. doi: 10.4049/jimmunol.0902206. [DOI] [PubMed] [Google Scholar]
- 53.Panarina M, Kisand K, Alnek K, Heilman K, Peet A, Uibo R. Interferon and interferon-inducible gene activation in patients with type 1 diabetes. Scand J Immunol. 2014;80:283–292. doi: 10.1111/sji.12204. [DOI] [PubMed] [Google Scholar]
- 54.Kimpimäki T, Kupila A, Hämäläinen AM, Kukko M, Kulmala P, Savola K, Simell T, Keskinen P, Ilonen J, Simell O, Knip M. The first signs of β-cell autoimmunity appear in infancy in genetically susceptible children from the general population: the Finnish Type 1 Diabetes Prediction and Prevention Study. J Clin Endocrinol Metab. 2001;86:4782–4788. doi: 10.1210/jcem.86.10.7907. [DOI] [PubMed] [Google Scholar]
- 55.Lönnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, Savola K, Muona P, Simell T, Koskela P, Hyöty H. Enterovirus infection as a risk factor for β-cell autoimmunity in a prospectively observed birth cohort: the Finnish Diabetes Prediction and Prevention Study. Diabetes. 2000;49:1314–1318. doi: 10.2337/diabetes.49.8.1314. [DOI] [PubMed] [Google Scholar]
- 56.Boudaly S, Morin J, Berthier R, Marche P, Boitard C. Altered dendritic cells (DC) might be responsible for regulatory T cell imbalance and autoimmunity in nonobese diabetic (NOD) mice. Eur Cytokine Netw. 2002;13:29–37. [PubMed] [Google Scholar]
- 57.Morel PA, Vasquez AC, Feili-Hariri M. Immunobiology of DC in NOD mice. J Leukoc Biol. 1999;66:276–280. doi: 10.1002/jlb.66.2.276. [DOI] [PubMed] [Google Scholar]
- 58.Palucka KA, Taquet N, Sanchez-Chapuis F, Gluckman JC. Dendritic cells as the terminal stage of monocyte differentiation. J Immunol. 1998;160:4587–4595. [PubMed] [Google Scholar]
- 59.Moser EK, Hufford MM, Braciale TJ. Late engagement of CD86 after influenza virus clearance promotes recovery in a FoxP3+ regulatory T cell dependent manner. PLoS Pathog. 2014;10:e1004315. doi: 10.1371/journal.ppat.1004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.