

Abstract
The molecular determinants and mechanisms involved in leukocyte trafficking across the blood-nerve barrier (BNB) in the Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP) variant of Guillain-Barré syndrome are incompletely understood. Prior work using a flow-dependent in vitro human BNB model demonstrated a crucial role for αM-integrin (CD11b)-intercellular adhesion molecule-1 interactions in AIDP patient leukocyte trafficking. The aim of this study is to directly investigate the biological relevance of CD11b in AIDP pathogenesis. Immunohistochemistry was performed on three AIDP patient sural nerve biopsies to evaluate endoneurial leukocyte CD11b expression. A severe murine experimental autoimmune neuritis (sm-EAN) model was utilized to determine the functional role of CD11b in leukocyte trafficking in vivo and determine its effect on neurobehavioral measures of disease severity, electrophysiological assessments of axonal integrity and myelination and histopathological measures of peripheral nerve inflammatory demyelination. Time-lapse video microscopy and electron microscopy were employed to observe structural alterations at the BNB during AIDP patient leukocyte trafficking in vitro and in situ respectively. Large clusters of endoneurial CD11b+ leukocytes associated with demyelinating axons were observed in AIDP patient sural nerves. Leukocyte CD11b expression was upregulated during sm-EAN. 5mg/kg of a function-neutralizing monoclonal rat-anti mouse CD11b antibody administered after sm-EAN disease onset significantly ameliorated disease severity, as well as electrophysiological and histopathological parameters of inflammatory demyelination compared to vehicle- and isotype antibody-treated mice. Consistent with in vitro observations of leukocyte trafficking at the BNB, electron micrographs of AIDP patient sural nerves demonstrated intact electron-dense endoneurial microvascular intercellular junctions during paracellular mononuclear leukocyte transmigration. Our data supports a crucial pathogenic role of CD11b in AIDP leukocyte trafficking, providing a potential therapeutic target for demyelinating variants of Guillain-Barré syndrome.
Keywords: acute inflammatory demyelinating polyradiculoneuropathy, blood-nerve barrier, CD11b, experimental autoimmune neuritis, Guillain-Barré syndrome, integrin
INTRODUCTION
Guillain-Barré syndrome (GBS) is the most common acute peripheral nervous system disorder. Several variants exist based on clinical, electrophysiological and pathological characteristics. Its most common variant, acute inflammatory demyelinating polyradiculoneuropathy (AIDP) is pathologically characterized by monocyte/macrophage-induced demyelination associated with intense T- and B-lymphocyte infiltration into peripheral nerves and nerve roots [23,28,32]. GBS pathogenesis involves a complex interplay of systemic immune activation (which may be mediated in genetically susceptible individuals via molecular mimicry) with T-lymphocyte activation with polarization, polyclonal B-lymphocyte maturation and immunoglobulin synthesis, and a tissue-restricted immune response characterized by hematogenous leukocyte trafficking across the blood-nerve barrier (BNB), complement-mediated lysis, complement- and antibody-dependent cellular cytotoxicity, cytokine-mediated injury, and Schwann cell innate and adaptive immune response modulation [5,11,14,23,31,40,41,28].
Current GBS disease-modifying treatments include intravenous immunoglobulin (IVIg) and plasma exchange [22]. Despite significant advances in molecular biology and genetics that provide insights into GBS pathogenesis, no new treatments or significant improvements in patient outcomes have occurred over the last 20 years [23,28]. Possible barriers to progress in the field include dearth of patient nerve biopsies for extensive exploratory studies, unavailability of human BNB models to study pathogenic inflammatory mechanisms in vitro and limitations in animal models required to verify pathogenic mechanisms in vivo before clinical trials are planned [14].
Inflammatory leukocyte migration across microvessels is a sequential, coordinated process (i.e. multistep paradigm) involving specific selectins, chemokines and adhesion molecules on endothelial cells, and selectin counterligands, chemokine receptors, integrins and matrix metalloproteases expressed by leukocytes [28,13,36,17,25]. Observational studies using GBS patient nerve biopsies demonstrated increased expression of specific pro-inflammatory cytokines, chemokines and chemokine receptors, adhesion molecules and matrix metalloproteases [28,12,19,3,20,21]. A pathogenic role for chemokine receptor CCR2 has been suggested in AIDP based on in situ nerve biopsy expression data and the attenuation of inflammatory demyelination following gene knockout and pharmacologic blockade in a representative animal model, severe murine experimental autoimmune neuritis (sm-EAN) [19,39]. The molecular determinants and signaling mechanisms of pathogenic leukocyte trafficking at the BNB, as well as BNB structural changes that occur during transmigration are incompletely elucidated. The recent isolation and characterization of human endoneurial endothelial cells and development of in vitro BNB models provides an avenue to study these mechanisms [38,27,26,1,9,7].
Integrins are heterodimeric receptors made up of non-covalently associated α and β subunits. These molecules are actively involved in immune regulation and inflammatory responses, as well as other cellular processes. Integrins, activated by endothelial membrane-bound chemokines, membrane-expressed platelet-activating factor, or locally secreted chemoattractants, have been implicated in the pathogenesis of multiple sclerosis, inflammatory bowel disease and psoriasis [36,17,25]. Using a flow-dependent in vitro BNB model, prior studies proposed a crucial role for αM-integrin (also known as CD11b)-intercellular adhesion molecule-1 (ICAM-1) interactions in untreated AIDP patient mononuclear leukocyte trafficking [37]. The purpose of this study is to determine whether αM-integrin is expressed in AIDP patient nerves in situ, evaluate its role in pathogenic leukocyte trafficking in vivo using a representative animal model and gain insights into BNB structural changes during transmigration.
METHODS
Patient material
Archived frozen sural nerve biopsies from three untreated AIDP patients and three age- and sex-matched normal controls stored in optimum cutting temperature compound at −80°C were obtained from the Shin J. Oh Muscle and Nerve Histopathology Laboratory, Department of Neurology, University of Alabama at Birmingham. Peripheral blood mononuclear leukocytes were isolated from whole heparinized blood from patients with clinical, electrophysiological and supportive cerebrospinal fluid evidence of AIDP using density gradient centrifugation and cryopreserved in liquid nitrogen, as previously published [29,37]. The study was approved by the Institutional Review Board, with an exemption obtained to use archived pathological specimens for research. Written informed consent was obtained from each subject providing blood.
Severe murine experimental autoimmune neuritis
SJL/J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Sm-EAN was induced in 8–12 week old, non-pregnant female mice using purified bovine peripheral nerve myelin emulsified in complete Freund adjuvant, with pertussis toxin and recombinant mouse interleukin-12 serving as co-adjuvants, as previously described [34,39]. Four independent experiments using a total of 83 age-matched mice were conducted to determine the effect of CD11b antibody blockade on sm-EAN and establish a dose-response relationship, followed by comparative studies using vehicle (phosphate buffered saline [PBS])- and isotype antibody-treated mice as negative controls, and human IVIg (gold standard GBS treatment) as a positive control. Mice were treated daily via intraperitoneal injection with a commercially available function-neutralizing rat anti-mouse CD11b monoclonal antibody (clone M1/70, Bio X Cell, West Lebanon, NH; 0.5–25 mg/kg), rat IgG2b isotype antibody, anti-KLH (clone LTF-2, Bio X Cell; 0.5–25 mg/kg), PBS and 400 mg/kg human IVIg (Carimune® nanofiltered, CSL Behring AG, Bern, Switzerland) on days 13–17 post-induction, about a week following the earliest signs of disease and during the most rapid disease progression phase [34,39]. Each experimental mouse was evaluated for neurobehavioral signs of weakness using a 6-point semi-quantitative Neuromuscular Severity Scale (NMSS) [34,39] and weighed daily for 28 days. Research studies received approval from the Institutional Animal Care and Use Committee, and were conducted in accordance with the United States Policy on Humane Care and Use of Laboratory Animals.
Flow cytometry of mouse leukocytes
In order to determine CD11b expression profile and kinetics relevant to pathogenic leukocyte trafficking in sm-EAN, flow cytometry was performed on whole blood obtained from the submandibular facial vein via lancet [6]. A total of 19 sm-EAN and 21 age-matched normal controls were studied at baseline and on days 13 and 30 days post-induction. In a subset of mice, blood was obtained 30 minutes following a single intraperitoneal administration of the monoclonal rat anti-mouse CD11b antibody used in sm-EAN experiments. Erythrocytes were lysed and leukocytes fixed with Lyse/fix buffer (Becton Dickinson) for 10 minutes. Following a wash, leukocytes were suspended in flow cytometry buffer (10% fetal bovine serum + 0.1% sodium azide in 1X PBS) with added Fc block reagent (Becton Dickinson) and incubated on ice for 5 minutes. Following centrifugation with supernatant discard, leukocytes were incubated with a cocktail of fluorochrome-conjugated primary antibodies diluted in flow cytometry buffer. The suspension was incubated on ice for 30 minutes in the dark, washed three times and further suspended into 200 μL flow cytometry buffer for analyses.
The following specific rat anti-mouse monoclonal antibodies were used for flow cytometry: PE-conjugated CD11b (clone 3A33, SouthernBiotech, Birmingham, AL: 1 μg/mL), FITC-conjugated CD3 (T-lymphocytes, clone:145-2C11, BioLegend, San Diego, CA: 5 μg/mL), Brilliant Violet 421-conjugated NK-1.1 (natural killer cells, clone: PK136, BioLegend: 5 μg/mL), APC-conjugated CD115 (clone:AFS98, monocytes: CD115+; neutrophils: CD115-, eBioscience, San Diego, CA: 2 μg/mL), and PerCP-Cy5.5-conjugated CD19 (B-lymphocytes, clone:1D3, BD Biosciences, San Jose, CA: 2 μg/mL). Cell sorting was performed using an LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ). At least 5,000 cells were analyzed per sample following an initial leukocyte gating strategy based on forward and side scatter characteristics using FlowJo® V10 (FlowJo LLC, Ashland, OR). Data were collected as % positive of total leukocytes and CD11b+ leukocytes, with mean fluorescence intensity for each marker.
Peripheral nerve electrophysiology and sciatic nerve procurement
Bilateral dorsal caudal tail nerve (DCTN) and sciatic nerve motor electrophysiology studies were performed on experimental mice in the prone position under ketamine-xylazine anesthesia on day 28 post-induction (at expected maximal severity) as previously described [35,34,30]. Electrophysiology studies were performed using a portable Keypoint® v5.11 electrodiagnostic system (Alpine Biomed Corporation, Fountain Valley, CA) with waveforms displayed on a Tecra S3 LCD monitor (Toshiba America, Irvine, CA). Distal and proximal compound motor action potential (CMAP) amplitudes (in mV), conduction velocity (in m/s) and distal and proximal CMAP total waveform durations (in ms) were measured or deduced for each nerve studied.
Following nerve electrophysiology, prone anesthetized mice were euthanized by cervical disarticulation. The sciatic nerves of each experimental mouse were immediately harvested and placed on filter paper cards to maintain longitudinal alignment. Nerve specimens were placed in molds filled with Tissue-Tek® optimum cutting temperature compound (Sakura Finetek USA Inc., Torrance, CA) and immediately stored at −80°C for future cryostat sectioning or in 3% glutaraldehyde in 0.1M phosphate buffer for 14–16 hours, followed by storage in 0.1M phosphate buffer at room temperature until further processed for plastic embedding, as previously published [30,34,39].
Indirect immunohistochemistry of peripheral nerves
Indirect fluorescent immunohistochemistry was performed on consecutive 20 μm thick axial and longitudinal cryostat sections of three AIDP and control sural nerves to detect and compare endoneurial leukocyte CD11b expression. This was also performed on serial 10 μm thick axial cryostat sections of six mouse sciatic nerves per experimental group to determine CD11b expression and characterize inflammatory infiltrates, as well as qualitatively determine demyelination and axonal loss between experimental groups, using published protocols [30,34,39]. Sections were fixed in acetone at −20°C, washed and air-dried prior to blocking with 10% normal goat serum in 1X PBS. All primary and secondary antibodies were diluted in 2% normal goat serum in 1X PBS.
The following primary antibodies were used: mouse anti-human CD11b IgG1 (clone: ICRF44, BioLegend: 20 μg/mL), rat anti-mouse CD11b IgG2b (clone M1/70, Bio X Cell: 20 μg/mL), polyclonal rabbit anti-human/mouse S100β IgG (Schwann cell marker; DAKO North America Inc. Carpinteria, CA: 20 μg/mL), polyclonal rabbit anti-mouse neurofilament-H IgG (axonal marker, Santa Cruz Biotechnology, Santa Cruz, CA: 4 μg/mL), rat anti-mouse F4/80 IgG2b (monocyte/macrophage marker, clone A3-1, AbD Serotec, Raleigh, NC: 20 μg/mL), rat anti-mouse CD3ε IgG2b (T-lymphocyte marker, clone C363.29B, Southern Biotech: 10 μg/mL) and rat anti-mouse CD19 IgG2a (B-lymphocyte marker, clone 6D5, Southern Biotech: 10 μg/mL). The following secondary antibodies were used: goat anti-mouse IgG (H+L) Alexa Fluor® 594 conjugate (Life technologies, Carlsbad, CA: 4 μg/mL), goat anti-rabbit IgG (H+L) Alexa Fluor® 488 conjugate (Life technologies: 4 μg/mL), goat anti-rabbit IgG (H+L) TXRD or FITC and goat anti-Rat IgG (H+L) TXRD or FITC (all from Southern Biotech: 2 μg/mL).
All sections were stained with 0.45 μM 4′, 6-diamidino-2-phenylindole (DAPI) for 5 minutes to detect nuclei and mounted with ProLong® Gold antifade mounting medium (Life technologies). Photomicrographs were taken using an Eclipse Ci-S Upright epifluorescent microscope with a D5-Qi2 camera (Nikon Instruments Inc., Melville, NY). Automated quantification of CD11b+ leukocytes, monocytes/macrophages, T-lymphocytes and B-lymphocytes per section was performed on merged photomicrographs using the NIS-Elements AR software (Nikon). Four serial sections from the distal sciatic nerve separated by 30–50 μm were analyzed per experimental mouse. Data were expressed as number of cells/section to eliminate errors in cross-sectional area that may occur with tissue processing ex vivo and variable increases in endoneurial area that may occur with edema associated with inflammation.
Sciatic nerve morphometric analysis
Glutaraldehyde-fixed sciatic nerves from 6 mice per experimental group were post-fixed in 1% osmium tetroxide for 2 hours and embedded in epoxy resin. 1 μm semi-thin sections stained with 1% toluidine blue were digitally photographed following light microscopy using an Eclipse Ci-S upright epifluorescent microscope with a D5-Qi2 camera (Nikon) in order to quantify the degree of demyelination (% total demyelinated area per section), as previously published [34,30,39]. Briefly, the demyelinated areas were visually identified, manually traced, quantified and added up then divided by the total cross-sectional area for each section. Four distal sections separated by 40 μm were analyzed per experimental mouse. Representative color photomicrographs were subsequently obtained using an Axioskop epifluorescent microscope equipped with an Axiocam MRc 5 digital camera (Carl Zeiss Microscopy, Jena, Germany).
In vitro blood-nerve barrier leukocyte trafficking model
Untreated AIDP patient leukocyte trafficking across the BNB in vitro was studied in real time using a flow-dependent leukocyte trafficking assay. Intravital microscopy is not currently possible in rodent peripheral nerves. A parallel plate flow chamber was attached to cytokine-treated confluent primary endoneurial endothelial cells cultured on rat-tail collagen-coated CellBIND® petri dishes, coupled to time-lapse video microscopy as previously described [37,7,26]. Videos were cropped, magnified and analyzed to determine the route of leukocyte transmigration at the BNB using the NIS-Elements AR software program (Nikon).
Electron microscopy
In order to determine BNB structural changes during pathogenic leukocyte trafficking in situ, ultramicroscopy of two untreated AIDP patient sural nerves was performed from archived epoxy resin-embedded blocks initially sectioned (1 μm thick) and stained with 1% toluidine blue and a basic fuchsin counterstain, using well-established protocols [30,34,38]. Digital light photomicrographs and ultramicrographs were generated to detect leukocyte adhesion and transmigration at the BNB and determine changes in endoneurial endothelial intercellular tight junctions at those sites.
Statistical analyses
Investigator-blinded data analyses were performed for all parameters evaluated using the GraphPad Prism® 6 statistical program (GraphPad Software, Inc., La Jolla, CA). Mann-Whitney U-test or the Wilcoxon-Kruskall’s Rank Sum Test was used to determine statistically significant differences between non-parametric variables while one- or two-tailed unpaired Student’s/Welch’s t-test (or analysis of variance for multiple comparisons) was used for parametric variables based on the Shapiro-Wilk test of normality (including measures of skew and kurtosis). Means are displayed, with variations of the mean depicted as standard errors. Statistical significance is defined as a p-value < 0.05.
RESULTS
In situ CD11b expression in AIDP
Previously, we demonstrated the differential expression αM-integrin (CD11b) on untreated AIDP patient leukocyte subpopulations and its critical role in leukocyte transmigration at the BNB in vitro [37]. Clusters of CD11b+ leukocytes (which are predominantly monocytes/macrophages) associated with focal demyelination were seen within the endoneurium of AIDP patient sural nerves (Fig 1a–i) but not in controls (Fig 1j–l). Dispersed, single CD11b+ leukocytes were also observed in some sections. This observation suggests potential relevance of CD11b in AIDP pathogenesis.
Figure 1. CD11b leukocyte expression in AIDP.

Digital indirect fluorescent photomicrographs from three untreated AIDP patients (AIDP1: a–c; AIDP2: d–f; AIDP3: g–i) show leukocyte clusters (DAPI, blue) associated with myelinated axons (S100β, green; a, d, g) with CD11b expression (red; b, e, h) co-localizing with focal demyelinating leukocytes, as demonstrated by the merged images (c, f, i). A normal control nerve is shown for comparison (j–l). Scale bars = 50 μm for a–i, 100 μm for j–l.
Kinetics of leukocyte CD11b in severe murine experimental autoimmune neuritis
Flow cytometry data demonstrated a mean 4.12-fold increase in circulating CD11b+ leukocytes on day 13 following sm-EAN induction compared to controls (16.37% vs. 3.98%) with a uniform relative increase in T-cells (1.32% vs. 1.68%), B-cells (3.29% vs 3.60%), natural killer cells (0.34% vs. 0.73%) and monocytes (27.22% vs. 35.49%), with slight decrease in neutrophils (67.83% vs. 57.30%). By day 30 (at expected peak maximal severity), there was still a 2.20-fold increase in CD11b+ leukocytes in circulating blood between sm-EAN and controls (12.06% vs. 5.50%), with a 3-fold relative increase in CD3+ T-cells (4.31% vs 1.43%), 22-fold decrease CD19+ B-cells (0.06% vs. 1.37%), 3.26-fold decrease in natural killer cells (0.98% vs. 3.19%) and 2.63-fold decrease in monocytes (12.24% v. 32.22%), implying selective infiltration of specific CD11b+ leukocyte subpopulations into peripheral nerves in sm-EAN. This data demonstrates upregulation in leukocyte CD11b expression in a representative mouse model of AIDP supporting a possible pathogenic role in this disorder.
Mouse Neuromuscular Severity Scores
Administration of a rat anti-mouse CD11b monoclonal antibody daily for 5 consecutive days following disease onset reduced NMSS in a cohort sm-EAN affected mice in a dose-dependent manner, with the maximum effect observed with a minimum effective dose of 5 mg/kg (data not shown). CD11b antagonism resulted in improvement in NMSS to near baseline levels compared to progressive worsening seen in PBS- and isotype antibody-treated mice, with statistically significant differences noted from day 15 onwards (Fig 2). The mean NMSS of CD11b antibody-treated mice was 1.2 on day 28 post-induction compared to 4.9 and 4.3 in PBS- and isotype-treated controls respectively. Cohorts of mice had the same mean NMSS at the start of treatment (3.3–3.8) with no differences between the groups. Compared to human IVIg treatment, statistically significant differences in NMSS were observed from day 21 post-induction, with a mean NMSS of 3.6 in these mice on day 28 (Fig 2). There was no significant difference in daily weights between the experimental groups (data not shown). These data demonstrate a crucial role of CD11b in sm-EAN pathogenesis during the disease effector phase.
Figure 2. Effect of CD11b antagonism on neuromuscular severity scores (NMSS) in severe murine experimental autoimmune neuritis.
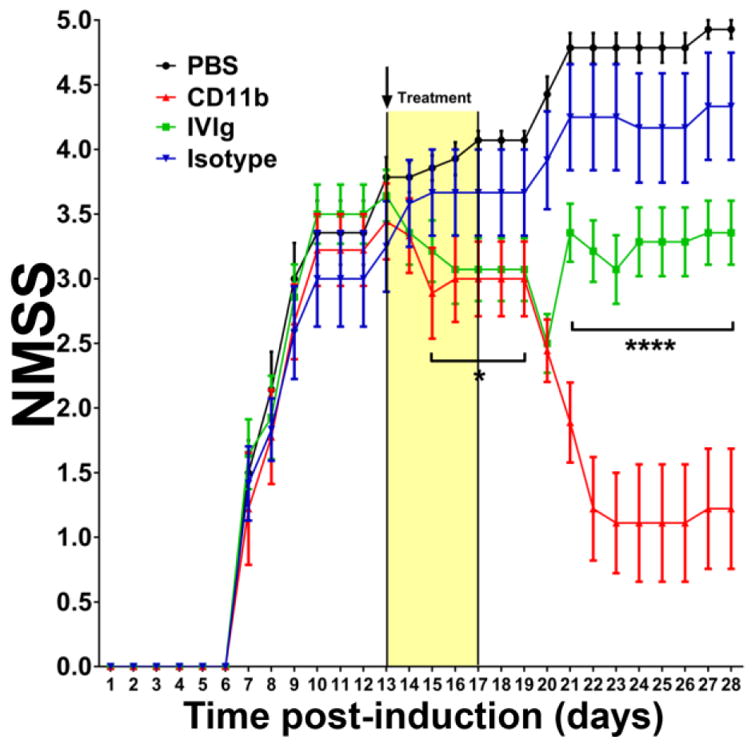
5 mg/kg rat-anti mouse CD11b antibody administered from days 13 (black arrow) to 17 post induction (Yellow treatment bar) following disease onset significantly reduced disease severity with improvement in motor function close to normal levels 9 days after treatment initiation that persisted up to 28 days post-induction (red line). Statistically significant differences in NMSS were observed from day 15 post-induction for CD11b antibody-treated mice relative to vehicle- (PBS, black line) and isotype antibody- (blue line) treated mice, with no difference compared to human IVIg-treated mice (green line) initially. Statistically significant differences in NMSS between CD11b antibody-treated and human IVIg-treated mice were seen from day 21 post-induction that persisted until study completion. N=48, with 4 independent cohorts studied. * p<0.05, **** p<0.0001 relative to CD11b antibody treatment.
Mouse motor nerve electrophysiology
CMAP amplitude is a measure of axonal integrity, conduction velocity measures myelination status of the fastest firing axons and total waveform duration measures the degree of synchrony in myelinated axon conduction. DCTN is affected earlier than the sciatic nerve and its electrophysiological parameters better correlate with NMSS in sm-EAN [34]. CD11b antagonism resulted in a statistically significant increase in mean DCTN CMAP amplitudes (Fig 3a), increase in mean conduction velocity (Fig 3b), and reduced total distal CMAP duration (Fig 3c) in sm-EAN mice compared to PBS- and isotype antibody-treated controls. Although a statistically significant difference was observed in mean sciatic nerve CMAP amplitudes compared to PBS-treated mice only (Fig 3d), a statistically significant increase in conduction in mean conduction velocity and reduced mean distal CMAP duration following CD11b inhibition was observed in sm-EAN compared to vehicle (PBS) and isotype-treated controls (Fig 3e–f). Compared to human IVIg treatment, CD11b antagonism resulted in increased DCTN and sciatic nerve conduction velocities and sciatic nerve distal CMAP amplitude durations, indicative of reduced demyelination (Fig 3a–f). These data support an important role of CD11b in pathogenic axonal loss and demyelination in sm-EAN. The partial effect of human or mouse isotype immunoglobulins on CMAP amplitudes may indicate a protective role against complement- or antibody-mediated axonal degeneration by macrophages that occurs during inflammatory demyelination. Representative DCTN and sciatic nerve motor electrophysiology waveforms from PBS- and CD11b antibody-treated sm-EAN mice are shown to demonstrate the improved CMAP amplitudes and reduced total CMAP durations (Fig 3g–j).
Figure 3. Effect of CD11b antagonism on motor electrophysiology in severe murine experimental autoimmune neuritis.

Bar histograms of mean motor electrophysiology parameters at day 28 post-induction (expected maximal disease severity) from the dorsal caudal tail nerves (DCTN, a–c) and the sciatic nerves (ScN, d–f) comparing CD11b antibody treatment with vehicle (PBS)-, human IVIg- and isotype antibody-treated mice is shown. Statistically significant increased distal compound motor unit action potential (CMAP) amplitudes (a) and conduction velocities (b and e) with decreased distal CMAP durations (c and f) comparing CD11b antibody-treated to PBS- and isotype-treated controls indicated preserved axonal integrity and protection against demyelination in this model. There was a statistically significant difference in ScN distal CMAP amplitude compared to PBS-treated mice (d); near significance was observed compared to isotype controls (p=0.06). Compared to human IVIg-treated mice, CD11b antagonism resulted in statistically significant increased conduction velocities (b and e) and decreased ScN distal CMAP duration (f) with no difference in the other parameters implying better protection against demyelination with similar preservation in axonal integrity. N= 48, with 4 independent cohorts studied. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 and ns = not significant relative to CD11b antibody treatment.
Motor electrophysiology waveforms from PBS- and CD11b antibody-treated mice are shown (g–j) to illustrate increased CMAP amplitudes and reduced durations following CD11b antagonism. In each figure, the upper right number indicates the sensitivity (y-axis) in mV per division, while the upper left number indicates the sweep speed (x-axis) in ms per division. The numbers just above each waveform on the far right indicates the stimulus current needed to generate a supramaximal response in mA. The upper waveform (green) represents the distal response while the lower waveform (red) represents the proximal response stimulating 6–12 mm away.
Mouse sciatic nerve inflammation
CD11b antagonism significantly reduced mean number of CD11b + leukocytes, total F4/80+ monocyte/macrophages and total CD3+ T-lymphocytes per sciatic nerve section in sm-EAN compared to PBS- and isotype antibody-treated mice. A significant difference in the total number of CD19+ B-lymphocytes compared to vehicle-treated but not isotype-treated mice at expected peak severity was observed (Fig 4a–d). CD11b antagonism resulted in mean reductions in CD11b+, F4/80+, CD3+ and CD19+ cells of 89%, 87%, 91% and 94% relative to PBS-treated mice, and 76%, 70%, 72% and 86% relative to isotype-treated mice. Statistically significant reductions in mean number of CD11b+ leukocytes (70%) and CD3+ T-lymphocytes (76%) were observed following CD11b inhibition compared to IVIg-treated sm-EAN mice with a 50% reduction in F4/80+ monocyte/macrophages that did not reach significance (p=0.24). These data demonstrate that CD11b inhibition attenuates pathogenic peripheral nerve inflammation mediated by monocytes/macrophages and T-lymphocytes in sm-EAN, supporting a crucial role in disease pathogenesis. Our previous work had shown a minimum pathogenic F4/80+ monocyte/macrophage infiltration threshold in sm-EAN using CCR2 heterozygous and knockout mice [39] above which inflammatory demyelination would similarly occur. This may explain the similarity in clinical course despite the significant differences in cell counts between PBS- and isotype-treated mice. Due to the patchy nature of inflammation, it is also possible that more proximal segments of the sciatic nerves had more leukocyte infiltration than the distal segments evaluated.
Figure 4. Effect of CD11b antagonism on inflammation in severe murine experimental autoimmune neuritis.
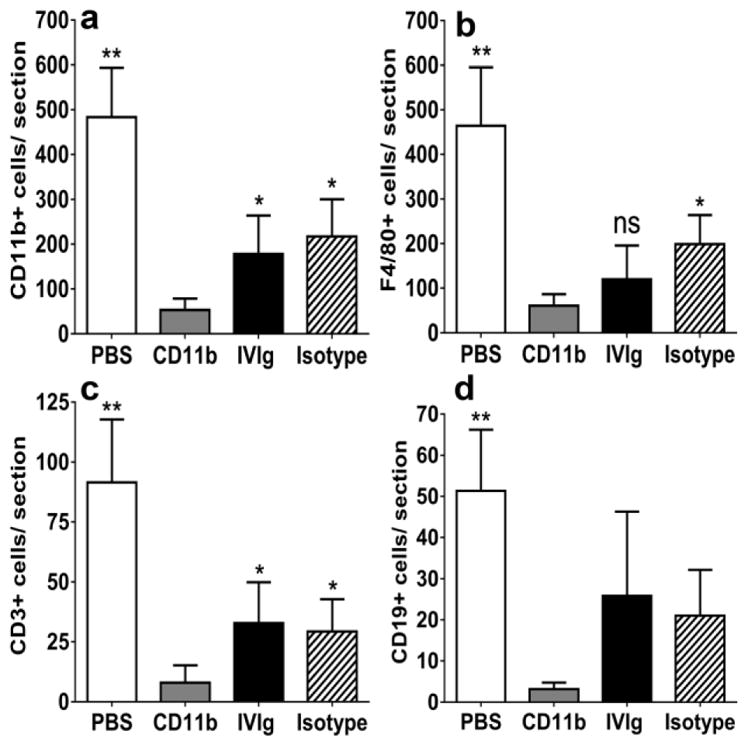
Bar histograms of mean numbers of CD11b+ leukocytes (a), and total F4/80+ monocytes/macrophages (b), CD3+ T-lymphocytes (c) and CD19+ B-lymphocytes (d) per sciatic nerve section on day 28 post-induction (maximum expected disease severity) are shown. Counts from 4 axial cross-sections per mouse (imaged at 400X magnification) were averaged to obtain a representative count for each mouse studied and mean counts from six mice per experimental group were compared. Statistically significant reductions in CD11b+, F4/80+ and CD3+ leukocytes were observed following CD11b antibody treatment compared to vehicle (PBS)- and isotype antibody-treated controls (a–c), with a significant reduction in CD19+ leukocytes observed with PBS-treated mice only (d). These data imply a robust inhibitory effect on CD11b+ leukocyte infiltration (upregulated in this model based on flow cytometry data), as well as the known effectors of inflammatory demyelination (macrophages and T-lymphocytes) in sm-EAN. Compared to human IVIg-treated mice, CD11b antibody treatment demonstrated statistically significantly reduced total CD11b+ leukocytes and CD3+ T-lymphocytes (a and c) with a 50% reduction in F4/80+ monocytes/macrophages that did not achieve significance (b) probably due to the wide variations in inflammation seen within the same nerve and between nerves of IVIg-treated mice in this model. N=24 axial cross-sections per experimental group. * p <0.05, ** p<0.01 and ns = not significant relative to CD11b antibody treatment.
The precise mechanism of human IVIg or mouse isotype antibody effect in sm-EAN is unknown; however partial efficacy may be related to modulation of complement-mediated axonal injury induced by membrane attack complex or antibody-dependent cellular cytotoxicity via FcγR blockade. Human IVIg-treated mice demonstrated reduced F4/80+ counts compared to PBS and isotype-treated mice (Fig 4b). This study did not evaluate phenotypic shifts from pro-inflammatory M1 to anti-inflammatory M2 macrophages, pro-inflammatory CD4+ Th1/Th17 to anti-inflammatory Th2 or increase in regulatory CD4+ CD25+ FoxP3+ T lymphocytes or alterations in pro-inflammatory and anti-inflammatory/regulatory B-cells that may explain improved clinical course with human IVIg not seen with isotype-treated mice [5,23,28,32]. Representative photomicrographs depicting differences in sciatic nerve inflammation between CD11b antagonism and PBS, human IVIg and isotype-treated sm-EAN mice are shown in Fig 5.
Figure 5. CD11b and inflammatory leukocyte expression in severe murine experimental autoimmune neuritis.

Representative indirect fluorescent photomicrographs from the sciatic nerves of sm-EAN affected mice at expected maximal severity demonstrate intense infiltrates of CD11b+ leukocytes and F4/80+ monocytes/macrophages (red) with scattered T- and B-lymphocyte infiltrates (green) in vehicle (PBS)-treated mice (a–d). Administration of 5 mg/kg rat anti-mouse CD11b antibody following disease onset significantly attenuated leukocyte infiltration (e–h) compared to PBS-treated, human IVIg-treated (i–l) and isotype antibody-treated (m–p) mice. White arrows indicate leukocyte marker-positive cells. Scale bars = 50 μm (a, b, e, f, i, j, m, n) and 75 μm (c, d, g, h, k, l, o, p).
Mouse sciatic nerve inflammatory demyelination
Sciatic nerve inflammatory demyelination with secondary axonal loss is characteristic of sm-EAN [34]. Qualitative analyses demonstrated reduced demyelination (more retained honeycomb and less disorganized endoneurial S100β expression) following CD11b antagonism compared to PBS-, human IVIg- and isotype antibody-treated mice (Figs 6a–d), consistent with the motor electrophysiology and inflammatory leukocyte quantification data. Focal axonal loss associated with endoneurial leukocyte infiltrates was also less commonly seen in CD11b antibody-treated sm-EAN mice (Figs 6e–h). Quantitative morphometric evaluation of demyelination performed on semi-thin plastic-embedded sciatic nerve sections at expected peak severity demonstrated statistically significantly reductions in mean % demyelinated area per section following CD11b antagonism compared to PBS-, isotype antibody- and human IVIg-treated mice (4.5% vs. 19.8%, 8.2% and 9.0% respectively) (Fig 6i). Representative photomicrographs demonstrating normal myelinated axons in CD11b-treated mice, foci of inflammatory cells with completely demyelinated or thinly myelinated axons in vehicle- and isotype-treated mice and multiple thinly myelinated regenerating clusters in human IVIg-treated mice are shown (Fig 6j–m). These thinly myelinated regenerating clusters were seen in demyelinated sections of human IVIg-treated mice probably contribute to the improved clinical course compared to isotype-treated mice that lack regeneration despite similar degrees of demyelination, as shown in Fig 6i.
Figure 6. Effect of CD11b antagonism on inflammatory demyelination in severe murine experimental autoimmune neuritis.

Representative indirect fluorescent photomicrographs from the sciatic nerves of sm-EAN affected mice at expected maximal severity demonstrate multiple foci of inflammatory cells (blue) associated with endoneurial architecture disruption (altered honeycomb appearance of myelinated axons with S100β staining [green]), indicative of demyelination in vehicle (PBS)-treated mice (a). The honeycomb appearance was restored in CD11b antibody-treated mice (b) with partial restoration of endoneurial architecture with residual leukocyte clusters seen in human IVIg-treated mice (c). Isotype-treated mice demonstrated similar patterns of inflammatory demyelination seen in PBS-treated mice with some preserved areas (d). Focal axonal loss (depicted by lack of heavy chain neurofilament staining [red]) associated with leukocyte infiltrates (blue) seen in PBS-treated mice (e) were not readily seen in CD11b antibody-treated mice (f). Small foci of inflammatory axonal loss were observed in human IVIg-treated mice (g), while isotype-treated mice demonstrated more diffuse foci similar to those observed in PBS-treated mice (h). White arrows indicate leukocyte foci. Scale bars = 50 μm.
Bar histograms indicating the mean demyelinated area per plastic-embedded, toluidine-blue-stained, semi-thin sciatic nerve section from sm-EAN mice at expected peak severity show a statistically significant reduction following CD11b antibody treatment compared with PBS-, isotype- and human IVIg-treated mice (i), supporting the motor electrophysiological data. * p<0.05 and ** p<0.01 relative to CD11b antibody treatment. Representative digital light photomicrographs show a region of mononuclear leukocyte infiltration with demyelination and axonal loss in a PBS-treated sm-EAN mouse (white arrows, j) with relative paucity of demyelination observed in CD11b-treated mice (k). Clusters of thinly myelinated small-diameter axons (indicative of regeneration) with residual mononuclear leukocytes (white asterisk) were seen in human IVIg-treated mice (l) while thinly myelinated large-diameter axons (indicative of demyelination) and multiple foci of mononuclear leukocyte infiltrates were seen in isotype-treated mice (white asterisk, M). Scale bars = 50 μm.
CD11b mechanism in inflammatory demyelination
The neurobehavioral, electrophysiological and histopathological data support a functional role of CD11b in sm-EAN pathogenesis during the effector phase, as well as the efficacy of CD11b antagonism in acute inflammatory demyelination in vivo. Flow cytometry data demonstrated complete depletion of circulating CD11b+ leukocytes in two-thirds of sm-EAN mice following intraperitoneal administration of a single 5mg/kg dose of the function-neutralizing anti-CD11b antibody on day 13 post-induction. Mice with partial leukocyte depletion had subpopulation profiles similar to untreated mice, implying preferential depletion of pathogenic leukocytes (data not shown). Flow cytometry data at day 30 coupled with the histopathological data support CD11b+ leukocyte transmigration into peripheral nerves in sm-EAN. These data, coupled with in situ observations of CD11b+ leukocytes associated with demyelination in AIDP patient sural nerves, as well as data published from AIDP patient leukocyte trafficking at the BNB in vitro [37] support the hypothesis that CD11b mediates pathogenic hematogenous monocyte and T-lymphocyte adhesion at the BNB in AIDP.
Human blood-nerve barrier structure during transmigration
AIDP leukocyte trafficking using the flow-dependent, cytokine stimulated in vitro BNB model involved the sequential process of rolling, arrest, firm adhesion and transmigration via the paracellular route (between endoneurial endothelial cells) [37], as shown in Supplementary video 1. Transmigration of CD11b+ leukocytes at the BNB was seen in AIDP sural nerves (Fig 7a–c). Semi-thin plastic-embedded sections from the same AIDP patients demonstrated demyelination with mononuclear leukocyte adhesion and transmigration at the BNB (Fig 7d–f). Ultramicrographs demonstrate mononuclear leukocyte adhesion and diapedesis with intact electron dense junctions between endoneurial endothelial cells (consistent with tight junctions) within the inflammatory milieu consisting of monocytes/macrophages, T-lymphocytes and B-lymphocytes (Figs 7g–j). Interestingly, electron-dense junctions were observed between infiltrating leukocytes and endoneurial endothelial cells without discernible intercellular gaps (Figs 7k–l). These observations imply that pathogenic leukocyte trafficking in AIDP occurs across endoneurial microvessels with structurally intact BNB via the paracellular route, with retention of electron-dense leukocyte-endothelial cell contacts during diapedesis.
Figure 7. Characterization of leukocyte transmigration at the BNB in AIDP.

Digital indirect fluorescent photomicrographs of leukocytes (DAPI, blue) extravasating from an endoneurial microvessel within an area of focal demyelination (disruption in continuous S100β staining [green], a) in an AIDP patient sciatic nerve biopsy demonstrated CD11b expression (b), verified by co-localization in the merged image (c). Digital light photomicrographs of toluidine-blue-stained, basic fuchsin-counterstained semi-thin plastic-embedded sections demonstrated numerous thinly myelinated large-diameter axons (black arrows) consistent with demyelination as expected in AIDP (d) with several endoneurial microvessels demonstrating leukocyte extravasation (black asterisk, e). Higher magnification of one of these microvessels (f) shows perivascular lymphocytes (black asterisk) and monocytes (black arrows). Ultramicrographs of endoneurial microvessels show leukocyte adhesion and diapedesis via the paracellular route with preserved electron dense intercellular contacts between endothelial cells (black arrows, g–j), providing evidence of an intact BNB during inflammation in AIDP. Monocyte transmigration (h), endoneurial macrophages with and without myelin debris (h and i), perivascular T-lymphocytes and a B-lymphocyte (i) are also shown. Electron-dense leukocyte-endothelial cell junctions formed by infiltrating leukocytes and endoneurial endothelial cells were observed (black arrows, k and l) without discernible intercellular gaps. BL= B-lymphocyte, EC= endothelial cell, Lc= Leukocyte, Mac= macrophage, Mo= monocyte, P= pericyte, TL= T-lymphocyte. Scale bars = 20 μm (a–c), 200 μm (d), 100 μm (e), 50 μm (f), 2 μm (g), 5 μm (h and i), 1 μm (j and l) and 0.5 μm (k).
DISCUSSION
In this study, we provide evidence supporting a crucial pathogenic role for αM-integrin (CD11b) in AIDP mononuclear leukocyte trafficking. CD11b+ leukocytes were detected in untreated AIDP patient sural nerves (absent in unaffected controls), associated with focal demyelination. Using a representative murine AIDP model, a specific leukocyte-depleting, function-neutralizing anti-CD11b antibody significantly abrogated the neurobehavioral, electrophysiological and histopathological hallmarks of acute inflammatory demyelination with secondary axonal loss. This was consistent with published data implicating CD11b-ICAM-1 signaling in untreated AIDP patient mononuclear leukocyte adhesion and transmigration at the BNB in vitro. [37] In addition to deducing an important molecular determinant of pathogenic leukocyte trafficking in AIDP, this study also provides a potential target for therapeutic intervention. Furthermore, we demonstrate CD11b+ leukocyte trafficking across the BNB in situ, intact endoneurial endothelial intercellular tight junctions within the inflamed nerve, and electron-dense leukocyte-endothelial cell junctions during paracellular diapedesis.
The leukocyte adhesion cascade has been pathogenically implicated in demyelinating GBS based on studies demonstrating increased soluble selectins and cellular adhesion molecules, deemed to be shed by activated endothelial cells, in the sera of patients during the course of illness compared to healthy controls [4,8,18]. In a single study, maximum levels of soluble-ICAM-1 and soluble-vascular cell adhesion molecule-1 (VCAM-1) were observed during the plateau phase (maximal clinical severity) with a gradual decline during recovery [4]. The same study demonstrated increased GBS patient lymphocyte adhesion to bound recombinant human ICAM-1 and VCAM-1 in an adhesion assay and transmigration across a fibronectin layer [4], without flow incorporation or shear forces needed for rapid integrin signaling [25,36,17,2].
ICAM-1, a surface protein and member of the immunoglobulin superfamily, is expressed by endothelial microvessels in GBS patient nerves [21]. Increased ICAM-1 expression by human primary endoneurial endothelial cells has been described in vitro following cytokine treatment using concentrations compatible with inflammatory disorders [37]. Antibody-mediated ICAM-1 blockade significantly reduced untreated AIDP patient flow-dependent adhesion to the BNB in vitro to basal levels [37], supporting its importance to pathogenic leukocyte trafficking in GBS. ICAM-1 binds to activated leukocyte integrins CD11a or αL-integrin (expressed on all leukocyte subpopulations) and CD11b (expressed predominantly on monocytes/macrophages, natural killer cells, neutrophils, and a subset of B- and T-lymphocytes) [25,36,37,17]. CD11b was the integrin responsible for ICAM-1-dependent AIDP patient mononuclear leukocyte adhesion, with monocytes/macrophages being the most prevalent leukocyte subpopulation trafficking at the BNB [37], consistent with in situ observations in patient nerve biopsies and representative AIDP animal models [24,10,14,28,34].
BNB compromise has been hypothesized to contribute to GBS pathogenesis primarily based on observed fenestrations in endoneurial microvessels early on in the disease course of rat EAN [15]. These fenestrations could provide a passive entry route for systemically generated autoantibodies resulting in axonal injury, demyelination or both via complement-mediated and macrophage-dependent cytotoxicity [5,14,23,28,32,31,40,11]. We demonstrated intact electron-dense endoneurial microvessel intercellular junctions at sites of leukocyte trafficking in AIDP patient nerves in this study, implying that the BNB is structurally intact during inflammation. This observation supports published observations that failed to demonstrate a reduction in BNB transendothelial resistance following treatment of confluent cultures with physiological cytokine concentrations for 24 hours in vitro [37]. AIDP leukocyte trafficking at the BNB is an active coordinated process, supported by published in vitro video microscopy under flow conditions [37,7,28] (also shown in this study), and the widely accepted multi-step paradigm [13,28,36,17,25].
We clearly demonstrate paracellular transmigration of AIDP patient leukocytes at the BNB in situ and in vitro. Interestingly, these infiltrating leukocytes formed electron-dense intercellular contacts with adjacent endoneurial endothelial cells during diapedesis, as observed in other microvessels during inflammation [16,33,17]. Junctional adhesion molecules (JAM)-A, B and C, Platelet-endothelial cell adhesion molecule-1 (PECAM-1) and CD99 are possible molecular components of these electron dense contacts that facilitate diapedesis via homophillic (i.e. the same molecule on both the leukocyte and endothelial cell) interactions. JAM-A is expressed by the BNB without significant change in expression or distribution following physiologic cytokine treatment in vitro [37,38]. Next generation whole transcriptome sequencing demonstrated JAM-A, JAM-C and PECAM-1 expression by endoneurial endothelial cells and AIDP peripheral blood mononuclear leukocytes, with JAM-B expressed only by endoneurial endothelial cells in vitro (unpublished observations). Although CD99 was not expressed on these cells in vitro, expression of CD99 molecule like 2 (expressed at endothelial cell contracts and implicated in leukocyte extravasation in mice) was observed (unpublished observations). A single study evaluating in vitro monocyte blood-brain barrier transmigration without flow showed transient focal loss of adherens and tight junction proteins during PECAM-1 and CD99-dependent paracellular transmigration without alternations in transendothelial electrical resistance [33]. This study further supports our inferences on BNB structural changes and retention of restrictive barrier properties during AIDP patient leukocyte trafficking. Further studies are planned to determine the precise determinants of leukocyte diapedesis at the BNB in AIDP using the in vitro human BNB and sm-EAN as experimental models.
Despite known limitations of in vitro leukocyte trafficking assays and animal models of AIDP [7,34,37,14], we propose a crucial pathogenic role for CD11b in AIDP leukocyte BNB trafficking, recognizing that we had access to a limited number of patient nerve biopsies. CD11b antagonism is a plausible therapeutic approach worth considering for early stage clinical trials based on our proof-of-concept data. Modulating pathogenic leukocyte trafficking could occur following systemic drug administration without need for high BNB penetrance. Further work is needed to completely elucidate the molecular determinants and signaling mechanisms involved in AIDP leukocyte trafficking, as well as mechanisms of active transport of systemically generated pathogenic autoantibodies across the intact BNB or a contributory role of functional BNB disruption in this disorder.
Supplementary Material
A compressed two minute video generated via real-time time-lapse digital microscopy demonstrates leukocytes from an untreated AIDP patient (phase bright) rolling and undergoing arrest and firm adhesion between cytokine stimulated confluent BNB-forming endoneurial endothelial cells (dark spindle-shaped background cells) under a continuous flow velocity of 1 mm/s in vitro. Diapedesis, detected by a change in leukocyte phase from bright to dark, was observed via the paracellular route with slight endothelial cell displacement, similar to in situ ultrastructural observations in AIDP patient sural nerve biopsies. Leukocytes were attracted to sites of firm adhesion and active transmigration, consistent with inflammatory cell clusters seen in AIDP patient biopsies. Frame rate = 10 frames per second (10X normal, compressing 20 minutes in real time).
Acknowledgments
Special thanks to past and current employees of the Shin J Oh Muscle and Nerve Histopathology Laboratory, University of Alabama at Birmingham for archived peripheral nerves and processed slides from AIDP patients and controls. Special thanks to EMLABS Inc. for performing the electron microscopy work. This project was supported by National Institutes of Health (NIH) grant R21 NS078226 (2012–2015) and institutional funds from the University of Alabama at Birmingham. The funding sources had no involvement in the conduct of the research, manuscript preparation, data collection/analyses or decision to submit this work for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
AUTHORSHIP
C.D. and E.E.U. were involved in study conception and experimental design, and all authors (C.D., S.P.P., E.S.H., and E.E.U.) participated in data acquisition, analysis and interpretation, and in the drafting of the manuscript, figures and supplementary video.
POTENTIAL CONFLICTS OF INTEREST
E.E.U. has received royalties from Baylor Licensing Group for simian virus-40 large T-antigen immortalized human endoneurial endothelial cells and from Springer Science + Business Media for an edited book on laboratory protocols that describes the flow-dependent in vitro BNB assay. C.D., S.P.P. and E.S.H. have nothing to disclose.
References
- 1.Abe M, Sano Y, Maeda T, Shimizu F, Kashiwamura Y, Haruki H, Saito K, Tasaki A, Kawai M, Terasaki T, Kanda T. Establishment and characterization of human peripheral nerve microvascular endothelial cell lines: a new in vitro blood-nerve barrier (BNB) model. Cell Struct Funct. 2012;37:89–100. doi: 10.1247/csf.11042. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi E, Molteni R, Pardi R, Dubini G. Microfluidics for in vitro biomimetic shear stress-dependent leukocyte adhesion assays. J Biomech. 2013;46:276–283. doi: 10.1016/j.jbiomech.2012.10.024. [DOI] [PubMed] [Google Scholar]
- 3.Chiang S, Ubogu EE. The role of chemokines in Guillain-Barre syndrome. Muscle Nerve. 2013;48:320–330. doi: 10.1002/mus.23829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Creange A, Chazaud B, Sharshar T, Plonquet A, Poron F, Eliezer MC, Raphael JC, Gherardi RK. Inhibition of the adhesion step of leukodiapedesis: a critical event in the recovery of Guillain-Barre syndrome associated with accumulation of proteolytically active lymphocytes in blood. J Neuroimmunol. 2001;114:188–196. doi: 10.1016/s0165-5728(00)00465-3. [DOI] [PubMed] [Google Scholar]
- 5.Dalakas MC. Pathogenesis of immune-mediated neuropathies. Biochim Biophys Acta. 2015;1852:658–666. doi: 10.1016/j.bbadis.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Golde WT, Gollobin P, Rodriguez LL. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Anim (NY) 2005;34:39–43. doi: 10.1038/laban1005-39. [DOI] [PubMed] [Google Scholar]
- 7.Greathouse KM, Palladino SP, Dong C, Helton ES, Ubogu EE. Modeling leukocyte trafficking at the human blood-nerve barrier in vitro and in vivo geared towards targeted molecular therapies for peripheral neuroinflammation. J Neuroinflammation. 2016;13:3. doi: 10.1186/s12974-015-0469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hadden RD, Karch H, Hartung HP, Zielasek J, Weissbrich B, Schubert J, Weishaupt A, Cornblath DR, Swan AV, Hughes RA, Toyka KV Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial G. Preceding infections, immune factors, and outcome in Guillain-Barre syndrome. Neurology. 2001;56:758–765. doi: 10.1212/wnl.56.6.758. [DOI] [PubMed] [Google Scholar]
- 9.Kanda T. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J Neurol Neurosurg Psychiatry. 2013;84:208–212. doi: 10.1136/jnnp-2012-302312. [DOI] [PubMed] [Google Scholar]
- 10.Kiefer R, Kieseier BC, Stoll G, Hartung HP. The role of macrophages in immune-mediated damage to the peripheral nervous system. Prog Neurobiol. 2001;64:109–127. doi: 10.1016/s0301-0082(00)00060-5. [DOI] [PubMed] [Google Scholar]
- 11.Kieseier BC, Kiefer R, Gold R, Hemmer B, Willison HJ, Hartung HP. Advances in understanding and treatment of immune-mediated disorders of the peripheral nervous system. Muscle Nerve. 2004;30:131–156. doi: 10.1002/mus.20076. [DOI] [PubMed] [Google Scholar]
- 12.Kieseier BC, Tani M, Mahad D, Oka N, Ho T, Woodroofe N, Griffin JW, Toyka KV, Ransohoff RM, Hartung HP. Chemokines and chemokine receptors in inflammatory demyelinating neuropathies: a central role for IP-10. Brain. 2002;125:823–834. doi: 10.1093/brain/awf070. [DOI] [PubMed] [Google Scholar]
- 13.Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. 2007;17:243–250. doi: 10.1111/j.1750-3639.2007.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer zu Horste G, Hartung HP, Kieseier BC. From bench to bedside--experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nat Clin Pract Neurol. 2007;3:198–211. doi: 10.1038/ncpneuro0452. [DOI] [PubMed] [Google Scholar]
- 15.Mizisin AP, Weerasuriya A. Homeostatic regulation of the endoneurial microenvironment during development, aging and in response to trauma, disease and toxic insult. Acta Neuropathol. 2011;121:291–312. doi: 10.1007/s00401-010-0783-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:327–334. doi: 10.1016/s1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 17.Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Musso AM, Zanusso GL, Bonazzi ML, Tomelleri G, Bonetti B, Moretto G, Vio M, Monaco S. Increased serum levels of ICAM-1, ELAM-1 and TNF-alpha in inflammatory disorders of the peripheral nervous system. Ital J Neurol Sci. 1994;15:267–271. doi: 10.1007/BF02339236. [DOI] [PubMed] [Google Scholar]
- 19.Orlikowski D, Chazaud B, Plonquet A, Poron F, Sharshar T, Maison P, Raphael JC, Gherardi RK, Creange A. Monocyte chemoattractant protein 1 and chemokine receptor CCR2 productions in Guillain-Barre syndrome and experimental autoimmune neuritis. J Neuroimmunol. 2003;134:118–127. doi: 10.1016/s0165-5728(02)00393-4. [DOI] [PubMed] [Google Scholar]
- 20.Pollard JD, Baverstock J, McLeod JG. Class II antigen expression and inflammatory cells in the Guillain-Barre syndrome. Ann Neurol. 1987;21:337–341. doi: 10.1002/ana.410210404. [DOI] [PubMed] [Google Scholar]
- 21.Putzu GA, Figarella-Branger D, Bouvier-Labit C, Liprandi A, Bianco N, Pellissier JF. Immunohistochemical localization of cytokines, C5b-9 and ICAM-1 in peripheral nerve of Guillain-Barre syndrome. J Neurol Sci. 2000;174:16–21. doi: 10.1016/s0022-510x(99)00328-7. [DOI] [PubMed] [Google Scholar]
- 22.Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barre syndrome. Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group. Lancet. 1997;349:225–230. [PubMed] [Google Scholar]
- 23.Rinaldi S. Update on Guillain-Barre syndrome. J Peripher Nerv Syst. 2013;18:99–112. doi: 10.1111/jns5.12020. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt B, Toyka KV, Kiefer R, Full J, Hartung HP, Pollard J. Inflammatory infiltrates in sural nerve biopsies in Guillain-Barre syndrome and chronic inflammatory demyelinating neuropathy. Muscle Nerve. 1996;19:474–487. doi: 10.1002/(SICI)1097-4598(199604)19:4<474::AID-MUS8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 25.Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. 2005;7:151–185. doi: 10.1146/annurev.bioeng.7.060804.100423. [DOI] [PubMed] [Google Scholar]
- 26.Ubogu EE. Chemokine-dependent signaling pathways in the peripheral nervous system. Methods Mol Biol. 2013;1013:17–30. doi: 10.1007/978-1-62703-426-5_2. [DOI] [PubMed] [Google Scholar]
- 27.Ubogu EE. The molecular and biophysical characterization of the human blood-nerve barrier: current concepts. J Vasc Res. 2013;50:289–303. doi: 10.1159/000353293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ubogu EE. Inflammatory neuropathies: pathology, molecular markers and targets for specific therapeutic intervention. Acta Neuropathol. 2015;130:445–468. doi: 10.1007/s00401-015-1466-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ubogu EE, Callahan MK, Tucky BH, Ransohoff RM. Determinants of CCL5-driven mononuclear cell migration across the blood-brain barrier. Implications for therapeutically modulating neuroinflammation. J Neuroimmunol. 2006;179:132–144. doi: 10.1016/j.jneuroim.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Ubogu EE, Yosef N, Xia RH, Sheikh KA. Behavioral, electrophysiological, and histopathological characterization of a severe murine chronic demyelinating polyneuritis model. J Peripher Nerv Syst. 2012;17:53–61. doi: 10.1111/j.1529-8027.2012.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van den Berg B, Walgaard C, Drenthen J, Fokke C, Jacobs BC, van Doorn PA. Guillain-Barre syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. 2014;10:469–482. doi: 10.1038/nrneurol.2014.121. [DOI] [PubMed] [Google Scholar]
- 32.Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barre syndrome. Lancet. 2016 doi: 10.1016/S0140-6736(16)00339-1. [DOI] [PubMed] [Google Scholar]
- 33.Winger RC, Koblinski JE, Kanda T, Ransohoff RM, Muller WA. Rapid remodeling of tight junctions during paracellular diapedesis in a human model of the blood-brain barrier. J Immunol. 2014;193:2427–2437. doi: 10.4049/jimmunol.1400700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xia RH, Yosef N, Ubogu EE. Clinical, electrophysiological and pathologic correlations in a severe murine experimental autoimmune neuritis model of Guillain-Barre syndrome. J Neuroimmunol. 2010;219:54–63. doi: 10.1016/j.jneuroim.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 35.Xia RH, Yosef N, Ubogu EE. Dorsal caudal tail and sciatic motor nerve conduction studies in adult mice: technical aspects and normative data. Muscle Nerve. 2010;41:850–856. doi: 10.1002/mus.21588. [DOI] [PubMed] [Google Scholar]
- 36.Yonekawa K, Harlan JM. Targeting leukocyte integrins in human diseases. J Leukoc Biol. 2005;77:129–140. doi: 10.1189/jlb.0804460. [DOI] [PubMed] [Google Scholar]
- 37.Yosef N, Ubogu EE. alpha(M)beta(2)-integrin-intercellular adhesion molecule-1 interactions drive the flow-dependent trafficking of Guillain-Barre syndrome patient derived mononuclear leukocytes at the blood-nerve barrier in vitro. J Cell Physiol. 2012;227:3857–3875. doi: 10.1002/jcp.24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yosef N, Xia RH, Ubogu EE. Development and characterization of a novel human in vitro blood-nerve barrier model using primary endoneurial endothelial cells. J Neuropathol Exp Neurol. 2010;69:82–97. doi: 10.1097/NEN.0b013e3181c84a9a. [DOI] [PubMed] [Google Scholar]
- 39.Yuan F, Yosef N, Lakshmana Reddy C, Huang A, Chiang SC, Tithi HR, Ubogu EE. CCR2 Gene Deletion and Pharmacologic Blockade Ameliorate a Severe Murine Experimental Autoimmune Neuritis Model of Guillain-Barre Syndrome. PLoS One. 2014;9:e90463. doi: 10.1371/journal.pone.0090463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuki N, Hartung HP. Guillain-Barre syndrome. N Engl J Med. 2012;366:2294–2304. doi: 10.1056/NEJMra1114525. [DOI] [PubMed] [Google Scholar]
- 41.Zhang HL, Zheng XY, Zhu J. Th1/Th2/Th17/Treg cytokines in Guillain-Barre syndrome and experimental autoimmune neuritis. Cytokine Growth Factor Rev. 2013;24:443–453. doi: 10.1016/j.cytogfr.2013.05.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A compressed two minute video generated via real-time time-lapse digital microscopy demonstrates leukocytes from an untreated AIDP patient (phase bright) rolling and undergoing arrest and firm adhesion between cytokine stimulated confluent BNB-forming endoneurial endothelial cells (dark spindle-shaped background cells) under a continuous flow velocity of 1 mm/s in vitro. Diapedesis, detected by a change in leukocyte phase from bright to dark, was observed via the paracellular route with slight endothelial cell displacement, similar to in situ ultrastructural observations in AIDP patient sural nerve biopsies. Leukocytes were attracted to sites of firm adhesion and active transmigration, consistent with inflammatory cell clusters seen in AIDP patient biopsies. Frame rate = 10 frames per second (10X normal, compressing 20 minutes in real time).