

Abstract
Background
Neural tube defects (NTDs) are one of the most common congenital birth defects, with myelomeningocele (MM) being the most severe form compatible with life. Recent studies show a link between mitochondrial folate one carbon metabolism and NTDs via the glycine cleavage system (GCS). We hypothesize that single nucleotide polymorphisms (SNPs) and novel variants in the coding regions of the GCS genes increase the risk for MM.
Methods
DNA was obtained from 96 subjects with MM born before the United States mandated folic acid fortification of grains in 1998. Primers were designed for PCR amplification and sequencing of all exons in the AMT gene, one of four genes in the GCS, followed by identification of SNPs and novel variants. An additional 252 MM subjects underwent whole exome sequencing to examine all four GCS genes (aminomethyltransferase, glycine dehydrogenase, glycine cleavage system protein-H, and dihydrolipoamide dehydrogenase).
Results
We identified six novel, heterozygous variants in the AMT gene with three predicted to be deleterious to AMT function (p.Val7Leu, p.Pro251Arg, and p.Val380Met). Five extremely rare, known heterozygous variants were found in the AMT gene and one in the GLDC gene. No novel variants in the exons of the other two GCS genes (DLD and GCSH) were identified.
Conclusions
We identified novel and rare, known variants in two of the four GCS genes that may contribute to the development of MM. Consistent with previous findings, the current study provides additional support that genetic variations in GCS genes contribute to the risk of NTDs.
Keywords: Aminomethyltransferase, myelomeningocele, folate metabolism, glycine cleavage system
INTRODUCTION
One of the most common congenital birth defects in the United States is the neural tube defect (NTD). The neural tube forms early in pregnancy with closure complete about one month after conception (Botto et al., 1999; Blencowe et al., 2010). Failure of complete closure results in an NTD. The open neural tube defect is the most common form and can present in two ways. A cephalad defect causes anencephaly, absence of a major portion of the forebrain and skull, resulting in an exposed and undeveloped cerebrum. Anencephaly is incompatible with life. A caudal defect causes spina bifida and can affect any portion of the spinal cord. The most common and severe form that is compatible with life is myelomeningocele (MM), leading to the spinal cord protruding through an opening (Au et al., 2010).
There are an estimated 3,000 pregnancies affected by NTDs per year, with slightly more than 1,500 live births resulting in spina bifida. The most serious, long term complications associated with MM are due to neurological problems and vary from patient to patient. Some of these include partial or complete paralysis and weakness of the body below the level of the defect, bowel and urinary dysfunction, hydrocephalus, orthopedic abnormalities, developmental delay, and learning disabilities (Sandler, 2010). The sum of these complications results in annual medical care and surgical costs totaling an estimated $200 million in the United States (Williams et al., 2015).
As there is no cure, research efforts have focused on determining the cause in hopes of finding ways to reduce the risk of developing MM. We have learned through research that there is not one specific cause but have determined that multiple factors influence the formation of an NTD (Au et al., 2010; Greene et al., 2011; Copp et al., 2013). In the general population, the risk of an NTD is about 0.1% (Detrait et al., 2005). However, the recurrence risk dramatically increases to 2-5% if a sibling is already affected, suggesting a familial inheritance (Detrait et al., 2005). Syndromes such as trisomies 13 and 18 are also associated with NTDs, suggesting a genetic effect (Au et al., 2010). Finally, environmental factors seem to play an important role as well, specifically maternal factors of folic acid consumption, obesity, insulin-dependent diabetes, and the use of anticonvulsant medications (Shaw et al., 1996; Hendricks et al., 2001; Au et al., 2010; Blomberg and Kallen 2010; Lupo et al., 2012; Ruggiero et al., 2015).
Since the early 1990s, maternal folate status has been known to be important in the risk for NTD formation (MRC Vitamin Study Research Group, 1991; Czeizel et al., 1992). In 1998, the United States mandated folic acid fortification of cereal and grain products to increase the consumption of folate by pregnant women (). Food fortification resulted in a decline in the prevalence rate of spina bifida by 31% (Boulet et al., 2008; Williams et al., 2015) but not eradication of the disease. The finding suggests that there are other factors involved in folate metabolism affecting neural tube closure; however, the potential mechanisms are still unclear. The pathways used for folate one-carbon metabolism operating in the cytosol have been widely studied and many gene variations have been found to be associated with increasing the risk of MM (Burren et al., 2008). More recent studies have shown a link between mitochondrial folate one-carbon metabolism and NTDs, specifically looking at the enzymes in the glycine cleavage system (GCS) (Narisawa et al., 2012).
Folate one-carbon metabolism produces one-carbon units to be used in the synthesis of purines, thymidylate, and methionine, components that are building blocks for RNA and DNA, neurotransmitters, membrane lipids, and proteins to name a few (Pai et al., 2015). All of the described reactions occur in the cytosol and obtain one-carbon units from the mitochondria. The GCS is a key pathway occurring in the mitochondria that helps in the production of the required one-carbon units. The GCS is part of the glycine and serine catabolism pathway and is composed of four enzymes: glycine dehydrogenase (GLDC), aminomethyltransferase (AMT), glycine cleavage system protein H (GCSH), and dihydrolipoamide dehydrogenase (DLD) (Kikuchi, 1973). The system is attached to the inner membrane of the mitochondria and is triggered in response to high concentrations of glycine.
The GCS plays an important role in the development of non-ketotic hyperglycinemia (NKH), an inherited inborn error of metabolism (Azize et al., 2014). Biallelic mutations of the AMT or GLDC genes result in a deficiency of the GCS and inability to break down glycine causing encephalopathy and often death (Kure et al., 2006). Mutations in the other two genes of the GCS, GCSH and DLD, have not been described in patients with NKH. The growing research on mitochondrial folate metabolism has shown a link between the GCS pathway and NTDs. An animal model study by Narisawa (2012) found that mice with functional Amt were viable, fertile, and without malformations. However, 87% of knockout mice without Amt genes developed NTDs and the GCS activity was undetectable, suggesting that AMT function is essential for GCS activity and necessary for successful neural tube closure. Pai (2015) used a mouse model to demonstrate the link between NTDs and NKH. They found that mice with homozygous mutations of Gldc or Amt caused NKH phenotypes and NTDs. The purpose of our study was to examine the occurrence and functional implication of sequence variations found within the coding regions of the glycine cleavage system genes in patients with non-syndromic MM.
METHODS
Patient samples
Our study population consisted of subjects with MM chosen from a cohort of non-syndromic MM subjects participating in various genetic studies in our laboratory. The study protocols were approved by the Institutional Review Board at McGovern Medical School in Houston, Texas. The patient characteristics have been described previously (Au et al., 2008). We examined 96 MM subjects including 48 Caucasians of European descent and 48 Hispanics of Mexican descent born before mandatory folic acid fortification in the United States (Aneji et al., 2012). They were selected randomly from the larger study cohort after enrollment across three sites (Texas, California, and Canada).
DNA Collection
As previously described (Au et al., 2008; Ruggiero et al., 2015), whole blood and saliva samples were obtained from MM patients and parents when available. The Puregene DNA Extraction Kit (Gentra Systems, Minneapolis, MN) was used to isolate DNA from whole blood, and the Oragene DNA Collection Kit (DNA Genotek, Ontario, Canada) was used for saliva samples.
Polymerase Chain Reaction Amplification
Polymerase chain reaction (PCR) and nested-sequencing primers (available upon request) were designed for each of the nine AMT exons using the reference genomic sequence of AMT (NM_000481) from the University of Santa Cruz Genome Browser (UCSC Genome Browser). Flanking regions up to 100 base pairs on either side of each exon were included to examine the splicing donors and acceptors as well. The primers were synthesized by Integrated DNA Technologies USA (Coralville, IA). AMT exons were amplified by PCR with MyTaq Hot Start DNA Polymerase (Bioline USA Inc, Taunton, MA) using the MJ Research PTC-100 Thermal Cycler (MJ Research, Waltham, MA). PCR product sizes were examined by electrophoresis in a 1.4% agarose gel. The excess primers and nucleotides were removed by treating the amplified products with exonuclease I and shrimp alkaline phosphatase (United States Biochemicals, Affymetrix, Cleveland, OH) prior to sequencing.
Sequencing and Analysis
Sanger sequencing was utilized with the BigDye Terminator Protocol (Applied Biosystems, Foster City, CA) and nested-sequencing primers. The products were resolved on the ABI310 Genetic Analyzer (Life Technologies Inc, Grand Island, NY). The DNA sequences of the subjects were manually compared to the AMT reference sequence to identify single nucleotide polymorphisms (SNPs) and novel variants. The variants not reported by the Single Nucleotide Polymorphism Database (dbSNP 144) were considered novel and further confirmed by sequencing a fresh PCR preparation from both directions. When available, parental genomic DNA was also sequenced to determine if novel variants were de novo or inherited. The ethnically matched reference population was obtained from the 1000 Genomes Project.
Whole Exome Sequencing
An additional 500 subjects with MM and their parents were selected from the cohort described in Au (2008) to undergo whole exome sequencing (WES). The cohort consists of patients born either before or after mandated folate fortification in the United States. Whole exome sequencing (WES) using the Ion Proton System was performed following the standard workflow of the manufacturer (Thermofisher Scientific). Currently, 252 samples have been analyzed to identify sequence variations in each of the four genes comprising the glycine cleavage system. Variants not recorded in dbSNP 144 were considered novel.
RESULTS
Upon examination of the nine exons in the AMT gene in 96 MM subjects, we identified four novel, heterozygous variants (Table 1) in ten patients. Variant c.19G>C (Figure 1), a single base pair substitution changing valine to leucine, was found in exon 1 in three Caucasian subjects. Variant c.555C>T (Figure 2) results in a silent change of the proline codon in exon 6 and was observed in five Hispanic subjects. Variant c.631G>A (Figure 3), another single base pair substitution in exon 6, changed the glutamic acid codon to lysine in one Caucasian subject. The last variant, c.752C>G (Figure 4), changed the proline codon to arginine in exon 7 and was observed in one Hispanic subject.
Table 1.
Novel variants identified across the AMT gene in patients with myelomeningocele (MM)
| Chr3 (AMT) Location | A1/A2 | cDNA Variant | Significance | Caucasian MM A2/A1 | Hispanic MM A2/A1 | GERP |
|---|---|---|---|---|---|---|
| 49459865 | C/G | c.19G>C | p.Val7Leu | 3/93 | 0/96 | -9.74 |
| 49456834 | G/A | c.555C>T | p.Pro185 | 0/96 | 5/91 | 3.47 |
| 49456758 | C/T | c.631G>A | p.Glu211Lys | 1/95 | 0/96 | 3.64 |
| 49456529 | G/C | c.752C>G | p.Pro251Arg | 0/96 | 1/95 | 4.32 |
A1, reference allele; A2, variant allele; MM, myelomeningocele; AMT, aminomethyltransferase gene; GERP, Genomic Evolutionary Rate Profiling score.
Figure 1.
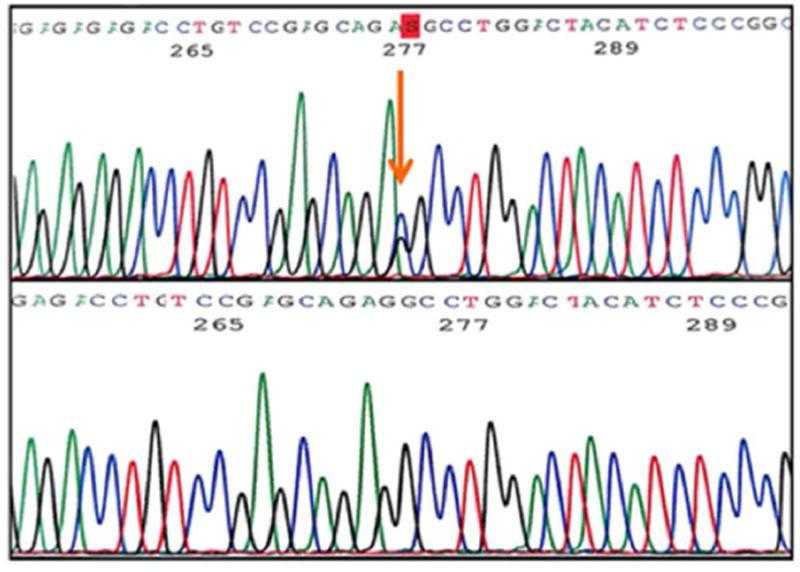
Exon 1 variant c.19G>C
Figure 2.
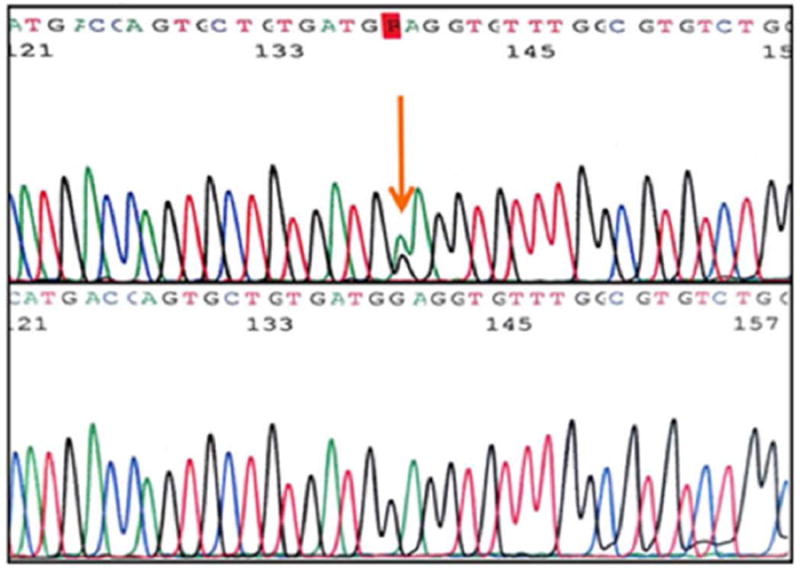
Exon 6 variant c.555C>T
Figure 3.
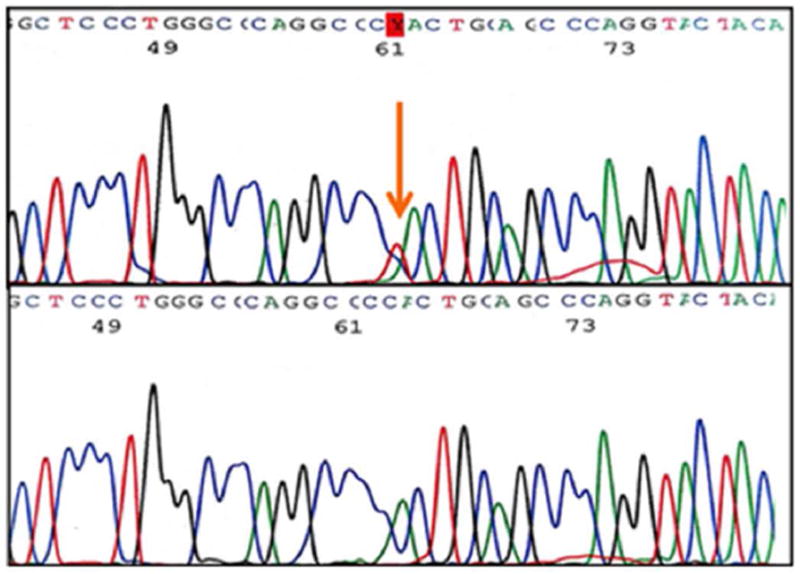
Exon 6 variant c.631G>A
Figure 4.
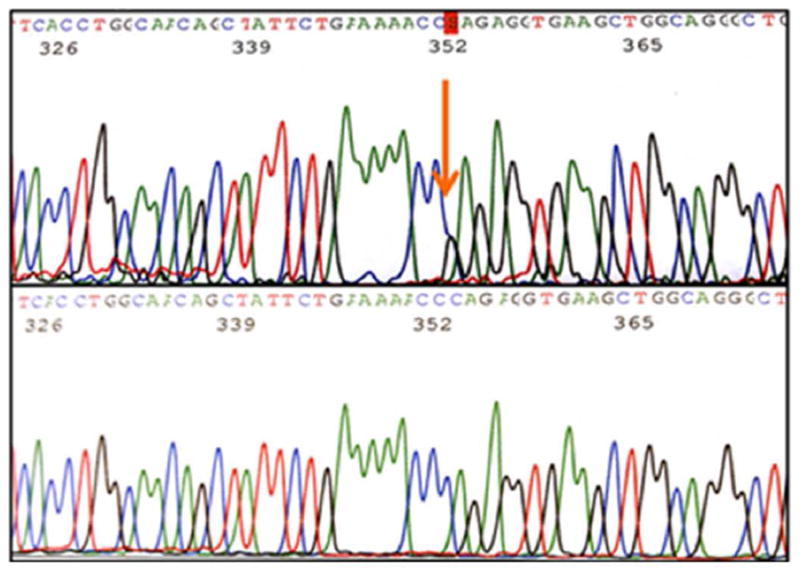
Exon 7 variant c.752C>G
The whole exome sequencing project provided data for an additional 252 MM subjects (121 Caucasian and 131 Hispanic Mexican), expanding the analysis to all four genes in the GCS. We identified an additional two novel and five extremely rare, known heterozygous variants in the AMT gene and one extremely rare, known variant in the GLDC gene (Table 2). A novel missense variant c.1138G>A, located in the AMT gene, changes the valine 380 codon to methionine (p.Val380Met) and is of interest due to its proximity to the splice donor site. The other novel AMT variant was found 519 bases upstream of the transcription start site of AMT mRNA in the core promoter region. The variant c.231G>A, occurring in exon two as a silent mutation, and four other noncoding variants were recently described as extremely rare SNPs with minor allele frequencies varying from zero to 0.03%.
Table 2.
Variants identified from Whole Exome Sequencing Project
| Chr3(AMT) Location | A1/A2 | cDNA Variant | Significance | Caucasian MM A2/A1 | Hispanic MM A2/A1 | GERP | Reference A2% |
|---|---|---|---|---|---|---|---|
| 49461781 | G/A | c.-238-670g>a | Promoter region | 1/241 | 0/262 | -3.52 | rs777617203 NA |
| 49460630 | G/A | c.-238-519g>a | Promoter region | 0/242 | 1/261 | 2.35 | Novel |
| 49459959 | G/C | c.-76G>C | 5’ -UTR | 1/241 | 0/262 | 3.01 | rs544461335 (0/0)¶ |
| 49459564 | G/A | c.231G>A | p.Ser77 | 0/242 | 1/261 | -8.91 | rs779002947 (0.0002)ˆ |
| 49456619 | G/C | c.697-35G>C | Intron region | 0/242 | 1/261 | -1.2 | rs746080712 (0.00003)ˆ |
| 49455047 | G/A | c.1138G>A; c.1127+1g>a |
p.Val380Met splice donor loss |
0/242 | 1/261 | 5.08 | Novel |
| 49454887 | G/T | c.*86G>T | 3’ –UTR New splice site | 0/242 | 1/261 | -0.41 | rs745685639 (0.00004)ˆ |
| Chr9 (GLDC) Location | A1/A2 | cDNA Variant | Significance | Caucasian MM A2/A1 | Hispanic MM A2/A1 | GERP | Reference A2% |
| 6602018 | G/A | c.1155+91C>T | Intron region | 0/241 | 1/262 | 1.26 | rs779753406 NA |
A1, reference allele; A2, variant allele; MM, myelomeningocele; AMT, aminomethyltransferase gene; GLDC, glycine dehydrogenase gene; GERP, Genomic Evolutionary Rate Profiling score. NA – population data not available.
1KGenomes A2 frequency for EUR/MXL.
data from EVA-ExAc [aggregated from multiple studies listed http://exac.broadinstitute.org/faq with a total of 60,706 individuals: African/African American (5,203), Latino (5,789), East Asian (4,327), South Asian (8,256), Finnish (3,307), Non-Finnish European (33,370), Other (454)]
In addition, only one variant was found in the GLDC gene, located in intron eight of an Hispanic Mexican MM subject. The variant was recently described in dbSNP without population frequency information. Interestingly, we did not find novel or rare exome variants in the other two GCS genes, DLD and GCSH, among the 252 MM subjects who were sequenced.
DISCUSSION
Current evidence suggests that the cause of NTDs is multifactorial, including environmental influences, ethnicity, and genetic variation. Because of the complexity of factors resulting in NTD formation, we are far from a cure or any preventive measures but instead strive to determine ways to decrease the risk of occurrence and further explore the many mechanisms involved in such a complicated disease process. Folate one-carbon metabolism is a key player in neural tube closure, specifically functionally deleterious mutations found in the glycine cleavage system genes in NTD cases, as it provides one-carbon units for DNA synthesis, methylation, and cell proliferation (Narisawa et al., 2012).
Mouse-model studies have shown that the genes encoding enzymes in the GCS have a significant role in NTDs. Narisawa (2012) identified multiple heterozygous variants in the Amt gene that resulted in decreased enzymatic activity of the GCS. Further evidence showed that GCS activity was required for neural tube closure by the presence of NTDs in mice lacking enzyme activity (Narisawa et al., 2012). Pai (2015) provided similar results for the Gldc gene in the mouse model, stating that it was a requirement for neural tube closure as well. In both of these studies, the NTDs consisted of anencephaly, exencephaly, and spina bifida.
In the current study, we provide additional genetic support that the GCS genes are likely involved in the development of myelomeningocele. Of the four novel variants found in the AMT gene, two are likely to be damaging to enzyme activity: c.519G>C (p.Val7Leu) and c.752C>G (p.Phe251Arg). Both affected codons are highly conserved evolutionarily with high positive GERP scores. In addition, AMT with either variant was considered damaging by SIFT, PROVEAN, and Polyphen2. In contrast, the other two novel variants, c.555C>T (p.Pro185) and c.631G>A (p.Glu211Lys), were predicted to be benign by in silico analyses. The exact biological significance for these variants to AMT remains to be validated experimentally.
All of the novel variants in the AMT gene displayed in Table 1 were re-sequenced for confirmation and some parental DNA was available for seven of the ten subjects found to have novel variants. Trios (DNA of subject, mother, and father) were available for two subjects, one with variant c.555C>T and the other with variant c.752C>G. For each of these, the variant was also found in the mother but not the father, suggesting maternal inheritance. Duos (DNA of subject and one parent) were available for five subjects. Four of these subjects had variant c.555C>T with maternal DNA available for three subjects and paternal DNA for one subject. In these duos, the variant was found in one mother but not in the remaining two mothers or the father. The other duo pertained to variant c.631G>A, which was found in the father but maternal DNA was not available. Therefore, it is difficult to determine a specific inheritance pattern but it is possible that these variants were inherited.
Both novel variants and extremely rare SNPs (alternate allele frequency between 0 and 0.03%) were identified from the whole exome sequencing project because the sequencing included promoters, exons, and introns. One novel variant (c.1138G>A) identified in an Hispanic subject changed the codon for valine 380 to methionine and is considered damaging to AMT by functional analysis programs. The nucleotide 1138G is highly conserved among vertebrates and has a high GERP score, implying its importance in evolution. The variant also changed the conserved splice donor motif “gt” to “at”, abolishing the splice donor site from exon 9 to exon 10 for AMT variant 4 (NM_00164712). The biological function of AMT variant 4 has not yet been identified so the significance of this change is unclear. The other variants found by WES are more frequent than reported in dbSNP144. Biological significance of these variants needs to be experimentally examined as well.
It is important to note that variants have only been found in the AMT and GLDC genes, but not GCSH or DLD genes, also part of the GCS. AMT is the smallest gene (5,901 bp with about 1,212 bp coding sequences) of the four GCS genes but was found to have the highest occurrence of novel sequence variants in subjects with MM. Prior studies examining the GCS genes have not found mutations in the DLD gene and only a handful in the GCSH gene in cases of non-ketotic hyperglycinemia (Kure et al., 2006), suggesting these two enzymes are extremely important to other cellular functions, not just folate metabolism. DLD, in particular, is an important catalyst in three other mitochondrial enzyme complexes in additional to the GCS: pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and branched-chain α-ketoacid dehydrogenase (Johnson et al., 1997). In a study examining the effect of DLD, heterozygous mice display only half of the wild-type activity levels for the associated mitochondrial complexes mentioned above (Johnson et al., 1997). Dld knockout mice die prenatally prior to the gastrulation phase, providing evidence that DLD is essential for embryo survival (Johnson et al., 1997). Based on these findings, mutations in these genes would not be identified in our study cohort because all MM subjects survive embryogenesis to birth.
There were several strengths to our study. We re-sequenced all exons of the AMT gene in 96 MM subjects from two ethnic groups with the highest prevalence of NTDs using the Sanger sequencing method, considered the gold standard. Our study population consisted of MM subjects born prior to the mandated folate fortification era; thus allowing us the potential to identify variants in patients that may not have shown the MM phenotype post-folate fortification rescue. We also had some parental DNA available from affected subjects with new variants to speculate the inheritance pattern of the discovered novel variants.
Limitations of our study include the small sample size; however, results from the WES study of 500 cases will provide a better perspective on the importance of GCS genes and myelomeningocele. Other limitations include the low rare allele frequency of SNPs, together with the small sample size, affecting the power of the study and the ability to establish significant associations of these SNPs with NTD risk. Lastly, although Sanger sequencing is the gold standard, its use does limit the number of samples we could examine due to the time commitment and high cost.
In conclusion, we provide evidence that damaging heterozygous variant alleles in GCS genes identified in MM cases may increase the risk of MM. Our findings, along with other studies, have shown that GCS genes play an important role in neural tube closure. However, further research is needed to examine the role of each GCS gene in the pathogenesis of NTDs and how the identified novel variants affect GCS enzyme activity.
Supplementary Material
Acknowledgments
We would like to thank Sarah Riosa, Do-Kyun Kim, and Michael Brown for their excellent technical support and the patients and their families for their participation. This study was supported by the NIH/NICHD grant (5R01HD073434) to Dr. Kit Sing Au and the Richard W. Mithoff Professorship Funds provided by Dr. Kathleen Kennedy (Richard W. Mithoff Professor of Pediatrics, Division of Neonatal-Perinatal Medicine, McGovern Medical School, Houston, Texas).
Footnotes
The authors report no conflicts of interest.
References
- Aneji CN, Northrup H, Au KS. Deep sequencing study of the MTHFR gene to identify variants associated with myelomeningocele. Birth Defects Res Part A Clin Mol Teratol. 2012;94:84–90. doi: 10.1002/bdra.22884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KS, Ashley-Koch A, Northrup H. Epidemiologic and genetic aspects of spina bifida and other neural tube defects. Dev Disabil Res Rev. 2010;16:6–15. doi: 10.1002/ddrr.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KS, Tran PX, Tsai CC, et al. Characteristics of a spina bifida population including North American Caucasian and Hispanic individuals. Birth Defects Res Part A Clin Mol Teratol. 2008;82:692–700. doi: 10.1002/bdra.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azize NA, Ngah WZ, Othman Z, et al. Mutation analysis of glycine decarboxylase, aminomethyltransferase and glycine cleavage system protein-H genes in 13 unrelated families with glycine encephalopathy. J Hum Genet. 2014;59:593–597. doi: 10.1038/jhg.2014.69. [DOI] [PubMed] [Google Scholar]
- Blencowe H, Cousens S, Modell B, et al. Folic acid to reduce neonatal mortality from neural tube disorders. Int J Epidemiol. 2010;39(Suppl 1):i110–i121. doi: 10.1093/ije/dyq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M, Kallen B. Maternal obesity and morbid obesity: the risk for birth defects in the offspring. Birth Defects Res Part A Clin Mol Teratol. 2010;88:35–40. doi: 10.1002/bdra.20620. [DOI] [PubMed] [Google Scholar]
- Botto L, Moore CA, Khoury MJ, et al. Neural Tube Defects. N Engl J Med. 1999;341:1509–1519. doi: 10.1056/NEJM199911113412006. [DOI] [PubMed] [Google Scholar]
- Boulet SL, Yang Q, Mai C, et al. Trends in the postfortification prevalence of spina bifida and anencephaly in the United States. Birth Defects Res Part A Clin Mol Teratol. 2008;82:527–532. doi: 10.1002/bdra.20468. [DOI] [PubMed] [Google Scholar]
- Burren KA, Copp AJ, Greene NDE, et al. Gene-environment interactions in the causation of neural tube defects: folate deficiency increases susceptibility conferred by loss of Pax3 function. Hum Mol Genet. 2008;17:3675–3685. doi: 10.1093/hmg/ddn262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Stanier P, Greene ND. Neural tube defects: recent advances, unsolved questions, and controversies. Lancet Neurol. 2013;12:799–810. doi: 10.1016/S1474-4422(13)70110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327:1832–1835. doi: 10.1056/NEJM199212243272602. [DOI] [PubMed] [Google Scholar]
- Detrait ER, George TM, Etchevers HC, et al. Human neural tube defects: developmental biology, epidemiology, and genetics. Neurotoxicol Teratol. 2005;27:515–524. doi: 10.1016/j.ntt.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene ND, Stanier P, Moore GE. The emerging role of epigenetic mechanisms in the etiology of neural tube defects. Epigenetics. 2011;6:875–893. doi: 10.4161/epi.6.7.16400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks KA, Nuno OM, Suarez L, et al. Effects of hyperinsulinemia and obesity on risk of neural tube defects among Mexican Americans. Epidemiology. 2001;12:630–635. doi: 10.1097/00001648-200111000-00009. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Yang HS, Magnuson T, et al. Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci. 1997;94:14512–14517. doi: 10.1073/pnas.94.26.14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi G. The glycine cleavage system: composition, reaction mechanism, and physiological significance. Mol Cell Biochem. 1973;1:169–187. doi: 10.1007/BF01659328. [DOI] [PubMed] [Google Scholar]
- Kure S, Aoki Y, Matsubara Y, et al. Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat. 2006;27:343–52. doi: 10.1002/humu.20293. [DOI] [PubMed] [Google Scholar]
- Lupo PJ, Canfield MA, Chapa C, et al. Diabetes and obesity related genes and the risk of neural tube defects in the National Birth Defects Prevention Study. Am J Epidemiol. 2012;176:1101–1109. doi: 10.1093/aje/kws190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338:131–137. [PubMed] [Google Scholar]
- Narisawa A, Matsubara Y, Kure S, et al. Mutations in genes encoding the glycine cleavage system predispose to neural tube defects in mice and humans. Hum Mol Genet. 2012;21:1496–503. doi: 10.1093/hmg/ddr585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai YJ, Copp AJ, Greene ND, et al. Glycine decarboxylase deficiency causes neural tube defects and features of non-ketotic hyperglycinemia in mice. Nat Commun. 2015;6:6388. doi: 10.1038/ncomms7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero JE, Northrup H, Au KS. Association of facilitated glucose transporter 2 gene variants with the myelomeningocele phenotype. Birth Defects Res Part A Clin Mol Teratol. 2015;103:479–87. doi: 10.1002/bdra.23358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler AD. Children with spina bifida: key clinical issues. Pediatr Clin North Am. 2010;57:879–892. doi: 10.1016/j.pcl.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Shaw GM, Velie EM, Schaffer D. Risk of neural tube defect affected pregnancies among obese women. JAMA. 1996;275:1093–1096. doi: 10.1001/jama.1996.03530380035028. [DOI] [PubMed] [Google Scholar]
- Williams J, Mai CT, Mulinare J, et al. Updated estimates of neural tube defects prevented by mandatory folic acid fortification – United States, 1995-2011. MMWR Morb Mortal Wkly Rep. 2015;64:1–5. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.