

Summary
Eicosanoid lipids play important roles in cellular signaling as second messengers in inflammation, immune response, vascular tone, and the central nervous system. Biosynthesis of eicosanoid lipids proceeds via hydrolysis of esterified arachidonic acid from phospholipids followed by the oxidation of the released arachidonic acid by a variety of enzymes including cyclooxygenases (COX). Herein, we demonstrate the remarkable ability of COX-2, but not COX-1, to directly oxidize 2-arachidonoyl-lysolipids resulting in the generation of a previously unknown class of eicosanoid-lysolipids, and provide evidence that intracellular lipases can release eicosanoids from their eicosanoid-lysolipid precursors. Importantly, genetic ablation of a phospholipase, iPLA2g, significantly reduced the amounts of these eicosanoid-lysolipids in murine hepatic tissue and fibroblasts. Furthermore, calcium-stimulation of wild-type murine lung fibroblasts produced robust increases in these eicosanoid-lysolipids which were markedly attenuated in iPLA2g-/- fibroblasts. Collectively, these results identify an iPLA2g-initiated pathway generating a new class of lipid metabolites with potential signaling functions resulting from the direct COX-2 catalyzed oxidation of 2-arachidonoyl-lysolipids.
Graphical abstract
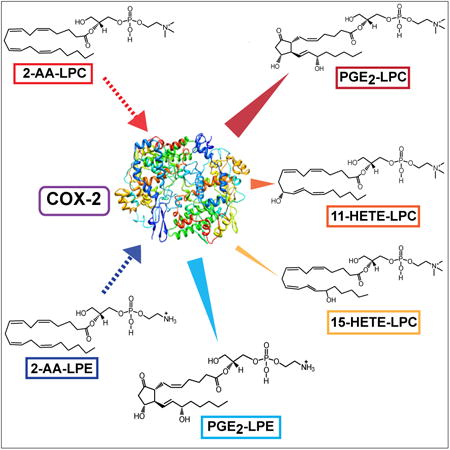
Introduction
The biosynthesis of prostaglandins from arachidonic acid (AA) is initiated by prostaglandin endoperoxide synthase (PGHS), more commonly referred to as cyclooxygenase (COX) (Funk, 2001; Smith et al., 2000; Marnett et al., 2002; Smith et al., 2011). In humans, two COX isoforms, designated COX-1 and COX-2, are present that are encoded by distinct genes which are differentially regulated (Meade et al., 1993; Herschman et al., 1994). Cyclooxygenases play crucial roles in generating eicosanoid lipid mediators that have multiple diverse effects on cellular signaling, inflammation, vascular tone, and ion channel function as well as numerous other physiologic and pathophysiologic processes (Dubois et al., 1998; Smith et al., 2000; Marnett et al., 2002; Ricciotti and FitzGerald, 2011). Both COX-1 and COX-2 are bifunctional heterodimers that oxidize arachidonic acid to eicosanoid products through two discrete reactions that are catalyzed by functionally interacting active sites within each enzyme (Kurumbail et al., 2001). First, two molecules of diatomic oxygen are added to arachidonic acid in the cyclooxygenase reaction resulting in the generation of the endoperoxide PGG2. The second reaction catalyzed by PGHS is a peroxidase reaction, which reduces the 15-hydroperoxy substituent of PGG2 to the 15-hydroxy endoperoxide PGH2. The resultant PGH2 is a branch point metabolite for the generation of a rich repertoire of signaling eicosanoids (e.g., prostaglandins, thromboxane, and prostacyclin) orchestrated by the integrated actions of multiple downstream enzymes that collectively regulate physiological processes in health, but precipitate pathological alterations in disease (Dogne et al., 2005; Ricciotti and FitzGerald, 2011).
Initially, it was generally believed that COX-1 was constitutively expressed in nearly all cell types while COX-2 was expressed specifically in immune cells promoting inflammation (Dubois et al., 1998; Smith et al., 2000; Marnett et al., 2002). Accordingly, it was assumed that COX-2 specific inhibition would ameliorate the pathologic sequelae of pain and inflammation without the toxic side effects (e.g., gastritis, mucosal ulceration) typically associated with the use of non-selective COX-1/COX-2 inhibitors. Substantial efforts were devoted to the development of COX-2 specific inhibitors such as Vioxx (Rofecoxib), Celebrex (Celecoxib), and selective NSAIDS. However, treatment of patients with COX-2 specific inhibitors resulted in an unacceptable incidence of cardiovascular events including sudden death (Cannon and Cannon, 2012; Bhala et al., 2013; Hennekens et al., 2008; Conaghan et al., 2012). The unanticipated mortality associated with use of COX-2 specific inhibitors fueled multiple mechanistic studies that collectively demonstrated that the cardiovascular mortality resulting from COX-2 specific inhibition was due to the combined contributions of the absence of COX-2 reaction products in a variety of cell types (e.g., endothelial cells, platelets, renal cells and cardiac myocytes) (Wang et al., 2009; Streicher et al., 2010; Ricciotti and FitzGerald, 2011; Yu et al., 2012). However, the precise molecular mechanisms leading to the untoward effects of COX-2 specific inhibition remain incompletely understood (Anwar et al., 2015).
Previously, we identified and characterized a novel membrane-associated calcium-independent phospholipase A2γ (iPLA2γ, also known as PNPLA8) that is present in myocardium and multiple other tissues (e.g., heart, brain, kidney, lung) (Mancuso et al., 2000). This enzyme catalyzes both the sn-1 and the sn-2 hydrolysis of phospholipids containing oleic acid at the sn-2 position. In sharp contrast, when polyunsaturated aliphatic constituents (e.g., arachidonic acid) are present at the sn-2 position, iPLA2γ catalyzes the highly regioselective cleavage of phospholipids at the sn-1 position resulting in the generation of 2-acyl-lysolipids (Yan et al., 2005). Thus, the regiospecificity of iPLA2γ is dependent upon the nature of the sn-2 aliphatic constituent. This unanticipated regiospecificity of iPLA2γ identified a previously unappreciated signaling pathway resulting in the generation of 2-arachidonoyl-lysophosphatidylcholine (2-AA-LPC) which serves as a central branch point metabolite in multiple signaling cascades (Moon et al., 2012). Since the active site of COX-2 possesses a substantially larger steric volume in comparison to that of COX-1(Smith et al., 2000; Marnett et al., 2002), we hypothesized that the active site of COX-2 could accommodate 2-arachidonoyl-lysolipids generated from iPLA2γ resulting in their direct regio- and stereospecific oxidation to generate novel classes of signaling molecules which we have termed “eicosanoid-lysolipids”.
Herein, we report the ability of COX-2 to efficiently and stereospecifically oxidize the arachidonoyl moiety of both 2-AA-LPC and 2-arachidonoyl-lysophosphatidylethanolamine (2-AA-LPE) into a variety of eicosanoid-lysolipids including PGE2-LPC, PGE2-LPE, 11(R)-HETE-LPC, 15(R)-HETE-LPC (major), and 15(S)-HETE-LPC (minor). We provide further evidence that these moieties identified in vitro are natural products by identifying their presence in fibroblast cell cultures, hepatic tissue and human myocardium. Finally, we demonstrate that acute calcium stimulation of cultured fibroblasts results in the robust production of these metabolites which can be inhibited by genetic ablation of iPLA2γ. Collectively, these results identify a new class of eicosanoid-lysolipid metabolites that are generated through a novel signaling pathway by the sequential actions of iPLA2γ mediated hydrolysis of arachidonate-containing phospholipids and direct COX-2 catalyzed oxidation of the arachidonoyl-lysolipid products.
Results
Cyclooxygenase-2, but not cyclooxygenase-1, efficiently catalyzes the oxidation of 2-AA-LPC and 2-AA-LPE to novel eicosanoid-lysolipids
To identify novel eicosanoid-lysolipids produced by COX-2 from 2-AA-LPC by high resolution high mass accuracy (HRAM) mass spectrometry, we first generated a virtual database (VDB) of accurate masses of polyunsaturated lysolipids and their oxidized derivatives. This VDB was comprised of the mass of the starting material (2-AA-LPC or 2-AA-LPE) plus the addition of 1 to 6 oxygen atoms with varying degrees of unsaturation in the acyl chain (Table S1). Next, we synthesized 2-AA-LPC from 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine using Candida rugosa lipase as previously described (Creer and Gross, 1985), purified the resultant 2-AA-LPC by HPLC and incubated it with purified recombinant human COX-2. Analysis of the reaction products by LC-MS using HRAM mass spectrometry demonstrated that COX-2 catalyzed the generation of multiple novel metabolites whose accurate mass corresponded to oxidized eicosanoid-lysolipids predicted in the virtual database. Representative extracted ion chromatograms of the observed products from COX-2 mediated oxidation of 2-AA-LPC are shown in Figure 1A. Multistage tandem fragmentation analyses (MSn) (n=2-4) identified multiple informative fragment ions (Figure S1) which in conjunction with the chromatographic elution profiles led to their provisional assignments as PGE2-LPC, 15-HETE-LPC, and 11-HETE-LPC. Proposed fragmentation pathways of the eicosanoid-lysolipids leading to the observed accurate mass product ions are shown in Figure S2. To substantiate the proposed assignments, we hydrolyzed the sn-2 acyl chain of the eicosanoid-lysolipids by treatment with purified recombinant cytosolic phospholipase A2α (cPLA2α) (Pete and Exton, 1996; Pete et al., 1996), derivatized the released oxidized non-esterified fatty acid with N-(4-aminomethylphenyl)-pyridinium (AMPP), separated the resultant AMPP-eicosanoids by RPHPLC, and analyzed the products by HRAM mass spectrometry (Liu et al., 2013). The elution times and fragmentation patterns of the AMPP-derivatized eicosanoids released by cPLA2α from the COX-2 generated eicosanoid-lysolipids were indistinguishable from authentic standards (Figure 2) thereby confirming the proposed structural assignments. In sharp contrast, incubation of 2-AA-LPC with purified recombinant COX-1 (Figure 1A, red line) resulted in the generation of only diminutive amounts of eicosanoid-lysolipids. Control incubations (without COX-1 or COX-2) did not exhibit measurable amounts of the observed eicosanoid-lysolipids (Figure 1A, black line). Thus, the production of PGE2-LPC and 11-HETE-LPC by COX-2 occurs rapidly, but COX-1 only slowly catalyzes the oxidation of 2-AA-LPC (∼5% of that by COX-2).
Figure 1. Cyclooxygenases mediate oxidation of 2-arachidonoyl-lysolipids (2-AA-LPC and 2-AA-LPE) to produce novel eicosanoid-lysolipids.

Extracted ion chromatograms of COX-1 and COX-2 oxidized eicosanoid-lysolipid products utilizing either 2-AA-LPC (A) or 2-AA-LPE (B) as substrate. Purified recombinant COX-1 (2 μg) or COX-2 (2 μg) was incubated with 2-AA-LPC (10 μM) or 2-AA-LPE (10 μM) in 100 mM Tris-HCl buffer (pH 8.0) at 30°C for 10 min. The reaction was terminated by addition of methanol and acidified to pH 4 with glacial acetic acid. The reaction products were then purified by solid phase extraction following separation on a C18 HPLC column prior to analysis utilizing an LTQ-Orbitrap mass spectrometer with a mass resolution of 30,000 at m/z=400 in the positive ion mode. The extracted ion chromatograms (with 3 ppm mass window) for the identified metabolic products of PGE2-LPC (m/z 592.3245), 11-HETE-LPC (m/z 560.3347), and 15-HETE-LPC (m/z 560.3347) for 2-AA-LPC as well as PGE2-LPE (m/z 550.2726) and 11-HETE-LPE (m/z 518.2877) for 2-AA-LPE are shown.
Figure 2. Identification of eicosanoid-lysolipids generated by COX-2 catalyzed oxidation of 2-AA-LPC and 2-AA-LPE.

COX-2 generated eicosanoid-lysolipids were treated with cPLA2α and the resultant released eicosanoid products were chemically derivatized with AMPP, separated on a C18 HPLC column and analyzed by mass spectrometry. Panel-I, standard PGE2 elution time (A) and its MS/MS product ion spectrum (B); Panel-II, elution time (C) and MS/MS product ion spectrum (D) for the product generated by cPLA2α mediated hydrolysis of the COX-2 oxidation of 2-AA-LPC; Panel-III, elution time (E) and MS/MS product ion spectrum (F) for the product generated by cPLA2α mediated hydrolysis of the COX-2 oxidation of 2-AA-LPE; Panel-IV, elution times (G) for 15-HETE (11.69 min) and 11-HETE (12.08 min) standards along with their MS/MS product ion spectra H for 15-HETE and I for 11-HETE, respectively; Panel-V, elution times (J) and MS/MS product ion spectra (K and L) for the products generated by cPLA2α mediated hydrolysis of the COX-2 oxidized products of 2-AA-LPC.
Next, we sought to determine whether 2-AA-LPE could also serve as substrate for COX-1 and/or COX-2. First, 2-AA-LPE was synthesized by acid hydrolysis of 1-O-1′-(Z)-octadecenyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine and purified by RPHPLC. Incubation of the resultant 2-AA-LPE with purified recombinant COX-2 resulted in the generation of predominantly PGE2-LPE along with small amounts of 11-HETE-LPE (Figure 1B). Similar to results obtained with 2-AA-LPC, incubation of COX-1 with 2-AA-LPE produced only diminutive amounts of oxidized products. Collectively, these results demonstrate the ability of COX-2 to oxidize 2-AA-lysolipids to novel classes of eicosanoid-lysolipid products that have the potential to either serve directly as signaling metabolites or indirectly as precursors for a wide variety of downstream lipid 2nd messengers.
The affinity of the COX-2 for 2-AA-lysolipds is a critical determinant of its activity in intact cells. Accordingly, additional experiments were performed to determine the Km values of COX-2 for the formation of eicosanoid-lysolipid products (Table S2). The Km values for formation of PGE2-LPC and 11-HETE-LPC were found to be almost identical (11.7 μM and 12.3 μM, respectively) whereas the Km value for formation of 15-HETE-LPC was 2-3-fold higher (30.0 μM) in accordance with the lower yield of this minor product at a constant substrate concentration. The difference in Km values for these COX-2 products of 2-AA-LPC indicate that there are different stereoelectronic relationships leading to its binding at the active site which after removal of the 13-proS hydrogen, alternatively favoring either cyclization or oxygenation leading to the observed formation of PGE2-LPC, 11-HETE-LPC, or 15-HETE-LPC, respectively.
Determination of the stereochemistry of COX-2 generated eicosanoid-lysolipids
The absolute stereochemistry of the COX-2 generated eicosanoid-lysolipid products were determined after hydrolysis by recombinant cPLA2α, derivatization of the released eicosanoids by AMPP, and separation of enantiomers by chiral chromatography with analysis by HRAM mass spectrometry of both molecular ions and their fragmentation products. Through comparisons with authentic chiral standards, the results demonstrated that the 11-HETE produced by COX-2 was exclusively 11(R)-HETE (Figure 3A-C) while the 15-HETE produced was comprised of ∼67% 15(S)-HETE and ∼33% 15(R)-HETE (Figure 3D-F). Since PGE2 contains four chiral centers, the PGE2 stereoisomers potentially present in the eicosanoid-lysolipids released by treatment with human recombinant cPLA2α are diastereomers with differing physical properties that can be separated by RPHPLC. The released PGE2 from either PGE2-LPC or PGE2-LPE was derivatized with AMPP, separated by RPHPLC and analyzed by HRAM mass spectrometry. The resultant product was identified to be exclusively 9-oxo-11R,15S-dihydroxy-5Z,13E-prostadienoic acid by comparisons with authentic PGE2 diastereomer standards (Figure 3G-I).
Figure 3. Chiral-phase and reverse-phase HPLC-MS analyses of eicosanoids produced following cPLA2α mediated hydrolysis of PGE2-LPC, PGE2-LPE, 11-HETE-LPC and 15-HETE-LPC generated by COX-2 catalyzed oxidation of 2-AA-LPC and 2-AA-LPE.

COX-2 generated eicosanoid-lysolipids were treated with cPLA2α and the resultant eicosanoid products were derivatized with AMPP, purified by SPE and analyzed by chiral-phase (for 11-HETE and 15-HETE stereoisomers) HPLC-MS and reverse-phase (for PGE2 stereoisomers) HPLC-MS analyses. A, Standards of racemic 11(R/S)-HETE; B, Standard of 11(R)-HETE; C, 11-HETE produced by cPLA2α mediated hydrolysis of the COX-2 oxidized products of 2-AA-LPC; D, Standards of racemic 15(R/S)-HETE; E, Standard of 15(S)-HETE; F, 15-HETE produced by cPLA2α mediated hydrolysis of the COX-2 oxidized products of 2-AA-LPC; G, Standards of PGE2 stereoisomers indicated as follows: 15(R)-PGE2 (1), 8-iso-PGE2 (2), PGE2 (9-oxo-11R,15S-dihydroxy-5Z,13E-prostadienoic acid) (3), and 11β-PGE2 (4); H, PGE2 produced by cPLA2α mediated hydrolysis of the COX-2 oxidized products of 2-AA-LPC; I, PGE2 produced by cPLA2α mediated hydrolysis of the COX-2 oxidized products of 2-AA-LPE.
Differential effects of aspirin on the production of eicosanoid-lysolipid products
Previous work has demonstrated that aspirin acetylates Ser-516 of COX-2 (Lecomte et al., 1994; Wennogle et al., 1995), thereby completely abolishing the ability of the enzyme to form prostaglandins and concomitantly increasing the production of 11-HETE and 15-HETE (Xiao et al., 1997). To identify the effects of aspirin and resultant Ser-516 acetylation on the formation of eicosanoid-lysolipids, COX-2 was preincubated with aspirin prior to incubation with 2-AA-LPC or 2-AA-LPE. Analysis of reaction products by LC-MS demonstrated that PGE2-LPC and PGE2-LPE production were almost completely inhibited by aspirin (Figure S3). Furthermore, the formation of 11-HETE-LPC was also markedly inhibited (>90%) by aspirin pretreatment. In sharp contrast, the formation of 15-HETE-LPC was almost completely unaffected and was the most abundant product after pre-incubation of COX-2 with aspirin utilizing 2-AA-LPC as substrate. Collectively, these results demonstrate the highly specific effects of aspirin on the profile of eicosanoid-lysolipid molecular species produced by COX-2 mediated oxidation of 2-AA-lysolipids.
Increased production of oxidized 2-AA-lisolipids by COX-2 utilizing large unilamellar vesicles (LUVs) in comparison to small unilamellar vesicles (SUVs)
To further examine the activity of COX-2 in the oxidation of 2-AA-LPC and 2-AA-LPE as guests in a host bilayer, COX-2 was incubated with SUVs or LUVs comprised of DOPC/DOPE/2-AA-LPC (molar ratio: 35/10/5) or DOPC/DOPE/2-AA-LPE (molar ratio: 35/10/5). Remarkably, the rates of production of PGE2-LPC, 11-HETE-LPC, and 15-HETE-LPC by COX-2 mediated oxidation of 2-AA-LPC present in large unilamellar vesicles were three to four-fold higher than in small unilamellar vesicles (Figure S4 A). Similarly, although less pronounced, the rate of oxidation of 2-AA-LPE by COX-2 to form PGE2-LPE was 50% higher in LUVs than SUVs (Figure S4 B). Collectively, these results demonstrate the ability of COX-2 to preferentially oxidize 2-AA-lysolipids in physiologic membrane bilayers with large radii of curvature (e.g., mimicking the plasma membrane) in comparison to small physically constrained membranes.
Eicosanoid-lysolipids are natural products in murine liver and their abundance is decreased by genetic ablation of iPLA2γ
To determine if the newly identified eicosanoid-lysolipids are natural products, we first examined murine hepatic tissue. Eicosanoid-lysolipids from freshly isolated hepatic tissue were extracted by a modified Bligh and Dyer procedure, enriched by solid phase extraction, and further purified by C18 HPLC column chromatography. The collected eicosanoid-lysolipids were hydrolyzed by purified recombinant cPLA2α and their resultant eicosanoid products were charge-switch derivatized with AMPP, separated by chiral chromatography and analyzed by LC-MS/MS by comparisons with authentic stereospecific standards. The results clearly identified the presence of 11(R)-HETE, 15(R/S)-HETE as well as the naturally occurring cyclooxygenase PGE2 product (i.e. 9-oxo-11R,15S-dihydroxy-5Z,13E-prostadienoic acid) in mouse hepatic tissues (Figure 4A-C). Next, we quantified the eicosanoid-lysolipids in hepatic tissues from wild-type and germline iPLA2γ-/- mice that we previously generated and characterized (Mancuso et al., 2007). Genetic ablation of iPLA2γ resulted in a dramatic reduction of 2-AA-LPC and 2-AA-LPE (Figure 4D). Furthermore, the amounts of PGE2-LPC, PGE2-LPE, 11-HETE-LPC and 15-HETE-LPC were also markedly decreased in iPLA2γ-/- mice in comparison to their WT littermates (Figure 4E). The levels of free eicosanoids PGE2, 11-HETE and 15-HETE were also decreased in hepatic tissue from the iPLA2γ-/- mouse in comparison to their WT littermates (Table S3). To exclude the possibility that alterations in COX-2 expression were responsible for the observed decrease in eicosanoid-lysolipids, we measured COX-2 protein content by Western blot analysis. No significant differences in COX-2 protein levels in hepatic tissues from WT vs. iPLA2γ-/- mice were observed (Figure 4F).
Figure 4. Eicosanoid-lysolipids present in wild-type (WT) and iPLA2γ-/-(KO) murine hepatic tissue.
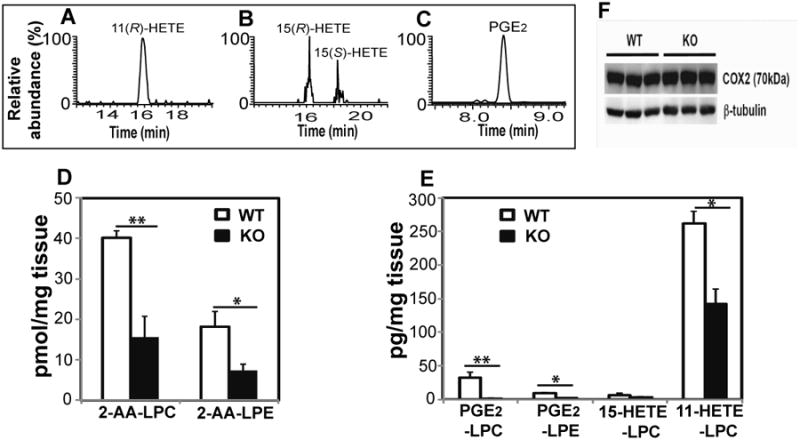
Hepatic tissue was isolated from wild-type (WT) and iPLA2γ knock-out (KO) mice and flash frozen in liquid nitrogen. The extracts were purified by solid-phase extraction and the isolated eicosanoid-lysolipids were hydrolyzed by purified recombinant cPLA2α followed by charge-switch derivatization with AMPP, chiral chromatographic separation, and compared with authentic stereospecific standards. A-C, Stereochemical determination of eicosanoid-lysolipid products present in WT hepatic tissue; D, Content of 2-AA-LPC and 2-AA-LPE in hepatic tissue from WT and KO mice; E, Content of PGE2-LPC, PGE2-LPE, 11-HETE-LPC, and 15-HETE-LPC in hepatic tissue from WT and KO mice. Data are expressed as mean values ± SEM of six replicates. Statistical analyses were performed using an unpaired two-tailed Student t test; P<0.05 was considered significant. *P<0.05 and **P<0.01; F, Western analysis of COX-2 expression in WT and KO mouse liver using β-tubulin as a loading control.
The production of eicosanoid-lysolipids is regulated by calcium-mediated activation of iPLA2γ
To determine whether the generation of eicosanoid-lysolipids was dynamically regulated by increases in intracellular calcium ion, wild-type murine lung fibroblasts (MLF) were grown in culture and treated with either 1.0 μM A23187 or vehicle alone. Calcium influx induced a robust increase in both 2-AA-LPC production and eicosanoid-lysolipid generation (Figure 5). To mechanistically identify whether iPLA2γ was the enzymatic source of the 2-AA-lysolipids produced, we used cultured lung fibroblasts from the germline iPLA2γ-/- mice we generated (Mancuso et al., 2007). Ablation of iPLA2γ markedly decreased the amount of calcium-stimulated eicosanoid-lysolipid production in iPLA2γ-/- fibroblasts relative to their WT littermate controls (Figure 5). Since calcium activates iPLA2γ to produce 2-AA-lysolipids (Moon et al., 2012), these results suggest that eicosanoid-lysolipid production is initiated by iPLA2γ and that the 2-AA-lysolipid products are oxidized by COX-2 to generate eicosanoid-lysolipids in intact cells.
Figure 5. Biosynthesis of 2-AA-LPC, PGE2-LPC, 11-HETE-LPC and 15-HETE-LPC in murine lung fibroblasts (MLF) from wild-type (WT) and iPLA2γ-/-(KO) mice stimulated with calcium ionophore.

A , Endogenous 2-AA-LPC production from MLF cells from WT and KO mice stimulated with A23187 (1μM) or vehicle alone at 37°C for 15 min.; B, Endogenous production of PGE2-LPC, 11-HETE-LPC and 15-HETE-LPC in MLF cells from WT and KO mice stimulated with or without (vehicle alone) A23187 calcium ionophore; C, Quantitation of PGE2-LPC, 11-HETE-LPC and 15-HETE-LPC released into the extracellular media of MLF cells from WT and KO mice stimulated with or without (vehicle alone) A23187 calcium ionophore. Data are expressed as mean values ± SEM of six replicates. Statistical analyses were performed using unpaired two-tailed Student t test; P<0.05 was considered significant. *P<0.05 and **P<0.01.
Eicosanoid-lysolipids are present in human myocardium
Since the increased mortality from selective COX-2 inhibition in humans and mice results largely from cardiovascular dysfunction (Hennekens et al., 2008; Conaghan et al., 2012; Wang et al., 2009; Streicher et al., 2010), we sought to identify the presence of eicosanoid-lysolipids in normal human myocardium of cardiac donors that were unmatched for transplantation. Freshly isolated human ventricular myocardium from non-matched deidentified donors was rapidly frozen in liquid nitrogen, pulverized into a fine powder, extracted, and the eicosanoid-lysolipids were isolated following C18 HPLC column separation. The collected eicosanoid-lysolipids were hydrolyzed by purified recombinant cPLA2α followed by charge-switch derivatization with AMPP, chiral chromatography and high resolution LC-MS/MS. Comparisons with authentic stereospecific standards clearly demonstrated the presence of PGE2 (i.e. 9-oxo-11R,15S-dihydroxy-5Z,13E-prostadienoic acid), 15(R/S)-HETE, and 11(R)-HETE with minor amounts of 11(S)-HETE (∼5%) (Figure 6A-C). Substantial amounts of 2-AA-LPC and 2-AA-LPE along with their downstream metabolites PGE2-LPC, PGE2-LPE, 11-HETE-LPC, and 15-HETE-LPC were detected (Figure 6D and E). The levels of the free eicosanoids PGE2, 11-HETE and 15-HETE were also quantified and the results are summarized in Table S3. Since cardiac electrophysiologic function has been previously shown to be modulated by lysolipids as well as by multiple eicosanoids (Gross et al., 1982; Gubitosi-Klug et al., 1995; Wang et al., 2009; Streicher et al., 2010; Ricciotti and FitzGerald, 2011; Cedars et al., 2009; Jenkins et al., 2009), these results suggest that selective inhibition of COX-2 may alter the profile of eicosanoid-lysolipids in myocardium thereby contributing to the hemodynamic and electrophysiologic dysfunction observed after selective COX-2 inhibition.
Figure 6. Eicosanoid-lysolipid molecular species present in human myocardium.

Freshly isolated human ventricular myocardium from non-matched donors was rapidly frozen in liquid nitrogen, pulverized into a fine powder and eicosanoid-lysolipids were extracted, purified by solid-phase extraction, and the isolated eicosanoid-lysolipids were hydrolyzed by purified recombinant cPLA2α followed by charge-switch derivatization of the released eicosanoids with AMPP. The derivatized eicosanoids were separated by chiral HPLC and analyzed by mass spectrometry through comparisons with authentic stereospecific standards. A-C, Stereochemical determination of eicosanoid-lysolipid products present in human myocardium; D, Content of 2-AA-LPC and 2-AA-LPE present in human myocardium; E, Content of PGE2-LPC, PGE2-LPE, 11-HETE-LPC, and 15-HETE-LPC present in human myocardium. Data are expressed as mean values ± SEM of six replicates.
Identification of 15-lipoxygenase-mediated production of 15-hydroxyeicosatetraenoic acid-lysophosphatidylcholine
Since the amount of 15-HETE-LPC produced in calcium-stimulated murine lung fibroblasts greatly exceeded that of other COX-2 oxidation products of 2-AA-LPC and decreased substantially in fibroblasts from germline iPLA2γ K/O mice, these results suggested that 2-AA-LPC was potentially a 15-lipoxygenase substrate. To address this possibility, we incubated purified human 15-lipoxygenase with 2-AA-LPC for 10 min and the reaction products were analyzed by high resolution LC-MS/MS. Remarkably, human 15-lipoxygenase robustly oxidized 2-AA-LPC generating 15-hydroperoxyeicosatetraenoic acid lysophosphatidylcholine (15-HpETE-LPC) which was subsequently reduced by triphenylphosphine to 15-HETE-LPC (Figure S5). These results demonstrate the suitability of 2-AA-LPC to serve as substrate for both cyclooxygenases and lipoxygenases and further suggest that 2-AA-lysophospholipids can be oxidized in addition by a multiplicity of oxidases (both known and unknown) to greatly expand the repertoire of lipid 2nd messengers in cells through these novel metabolic and signaling pathways.
Discussion
The present work identifies the previously unrecognized ability of cyclooxygenase-2 to directly oxidize arachidonoyl-lysolipids resulting in the production of new classes of eicosanoid-lysolipids. In this study, we identified the COX-2-dependent oxidation of 2-AA-LPC and 2-AA-LPE, determined the regiochemistry and stereochemistry of the reaction products, and demonstrated the presence of these metabolites in multiple cells and tissues. Moreover, through genetic ablation of iPLA2γ, we have shown that production of these eicosanoid-lysolipid metabolites is largely dependent upon the calcium-mediated activation of iPLA2γ to generate 2-arachidonoyl-lysolipid substrates for COX-2. Upon calcium ionophore stimulation, production of PGE2-LPC, 11-HETE-LPC, and 15-HETE-LPC was dramatically increased in both WT MLF cells and in the extracellular medium of these cells. A substantial portion of the 15-HETE-LPC produced by MLF cells in the current study was likely generated by 15-lipoxygenase using 2-AA-LPC as substrate. Importantly, eicosanoid-lysolipid production was significantly lower after calcium stimulation in MLF cells from iPLA2γ-/- mice in comparison to their WT counterparts. Collectively, these findings establish a sequential process initiated by calcium-mediated activation of iPLA2γ to produce 2-arachidonoyl-lysolipids followed by their subsequent COX-2 mediated oxidation to generate eicosanoid-lysolipids. Additionally, these studies demonstrate that the resultant eicosanoid-lysolipids can be directly hydrolyzed by a variety of intracellular phospholipases (e.g., cPLA2α) resulting in the liberation of non-esterified eicosanoids that have previously been demonstrated to possess potent biological effects in inflammation, vasoregulation, osmoregulation, and hemostasis amongst many other physiologic and pathophysiologic processes (Dubois et al., 1998; Smith et al., 2000; Marnett et al., 2002; Ricciotti and FitzGerald, 2011).
Recent studies have provided strong evidence that COX-2 inhibition in multiple cell types (e.g., endothelial cells, platelets, kidneys and cardiac myocytes) contributes to the increased risk of cardiovascular events due to disruption of the production of the physiologic repertoire of signaling lipids in a cell-type and context-dependent manner. Previous studies have demonstrated the requirement for COX-2 products for the normal physiologic function of endothelial cells, smooth muscle cells, platelets, and cardiac myocytes (Ricciotti and FitzGerald, 2011). For example, in myocardium, cardiac myocyte specific COX-2 knockout results in electrophysiologic alterations (Wang et al. 2009). Similarly, endothelial specific COX-2 knockout leads to a marked reduction in prostacyclin production in endothelial cells (Tang et al., 2014). These effects may be further exacerbated under conditions of myocardial stress (e.g., ischemia/reperfusion) where activation of phospholipases results in production of excessive amounts of fatty acids and lysolipids which have been demonstrated to compromise ion channel kinetics and electrophysiologic function (Streicher et al., 2010; Ricciotti and FitzGerald, 2011; Yu et al., 2012; Cedars et al., 2009; Jenkins et al., 2009; Liu et al., 2007). Sudden death from specific COX-2 inhibition likely results from lethal changes in cardiac function caused by alterations in the profile of intact eicosanoid-lysolipids or their hydrolyzed products that regulate ion channel function, responses to stress, and inflammation.
In addition to the well established oxidation of non-esterified arachidonic acid (Funk, 2001; Smith et al., 2000; Marnett et al., 2002; Smith et al., 2011), previous work has shown that COX-2, but not COX-1, can effectively oxidize 2-arachidonoyl glycerol (Kozak et al., 2000) which is consistent with the current results utilizing 2-AA-LPC and 2-AA-LPE as substrates. Recent structural studies determining the position of 1-AA-glycerol within the active site of murine COX-2 have revealed that the glycerol moiety is positioned at the opening of the cyclooxygenase channel near Arg-120 and Tyr-355 (Vecchio and Malkowski, 2011). Interestingly, the 2,3-dihydroxypropyl group of 1-AA-glycerol is not stabilized by either ionic or hydrogen bonding interactions with the polar side chains of Arg-120, Arg-513, or Glu-524 as has been previously proposed (Kozak et al., 2001). Perhaps one or more of these residues may participate in binding of the polar head group of 2-AA-LPC or 2-AA-LPE to the enzyme. In addition, work by Malkowski and coworkers has identified rotamer conformations of Leu-531 in COX-2 which increase the volume of the opening of the cyclooxygenase channel thereby allowing a wider range of substrates to be oxidized by COX-2 relative to COX-1 (Vecchio and Malkowski, 2011). Details at the atomic level of the binding of 2-AA-lysolipids to COX-2 await similar structural crystallographic and solution state NMR studies to determine the stereoelectronic relationships and molecular interactions of 2-AA-lysolipid substrates within the active site.
An interesting aspect of the current results is the functional significance of parallel pathways for the generation of free eicosanoids vs. eicosanoid-lysolipids for COX-2 mediated signaling. There are several potential reasons underlying the existence of parallel reaction pathways to generate signaling metabolites. First, eicosanoid-lysolipids are likely endogenous ligands for specific receptors that may have higher affinities than their eicosanoids counterparts due to electrostatic interactions with the quaternary amine polar head group. Second, eicosanoid-lysolipids could serve as latent signaling precursors for the orchestrated spatio-temporal release of free eicosanoids by intracellular lipases. Since the off rate of lysolipids from membrane bilayers greatly exceeds that of non-esterified fatty acids, the ability of oxidized lysophospholipids to readily dissociate from cellular membranes would facilitate inter-organelle communication from their sites of formation to their sites of action or for further metabolism to novel complex signaling moieties (e.g., further oxidation, conjugation to glutathione, etc.). Third, since lysolipids are transported by a lysolipid transporter (Tarling et al., 2013; Contreras et al., 2010), it is tempting to speculate that eicosanoid-lysolipids can utilize the same transporter for the delivery of specific eicosanoids to neighboring cells to allow transcellular lipid 2nd messenger signaling. Indeed, it seems likely that the identified metabolites represent only a small fraction of those arising from the oxidation of sn-2 polyunsaturated fatty acid-lysolipids (e.g., oxidized linoleic, arachidonic, docosahexaenoic acid-lysolipid adducts). Fourth, we point out that intracellular lysophospholipases C and lysophospholipases D (autotaxin) have previously been shown to hydrolyze arachidonoyl-LPC suggesting that the repertoire of eicosanoid-based lipid signaling can be further diversified by removal of the polar head group in mammalian cells (Di Marzo et al., 1996; Tokumura et al., 2002) (Figure 7). In this regard, polyunsaturated lysophosphatidic acids have been demonstrated to be selectively generated from autotaxin mediated cleavage of their lysophosphatidylcholine precursors in asthmatic patients following allergen challenge (Ackerman et al., 2016). Thus, eicosanoid-lysolipids represent not only a new class of dynamically regulated signaling molecules, but in addition likely serve as precursors for eicosanoid-glycerols or eicosanoid-lysophosphatidic acids whose relative amounts and biologic effects are likely cell type and context dependent (Di Marzo et al., 1996; Tokumura et al., 2002). Collectively, these results provide a new perspective on lipid 2nd messenger generation identifying eicosanoid-lysolipids as natural products derived from AA-containing lysophospholipids that likely possess receptor-mediated signaling functions that modulate inflammation, cardiovascular effects and ion channel kinetics in myocardium, peripheral vessels and other tissues.
Figure 7. Oxidation of 2-arachidonoyl-lysolipids (2-AA-LPC and 2-AA-LPE) to eicosanoid-lysolipids and subsequent metabolism to eicosanoid-glycerols and eicosanoid-lysophosphatidic acids.

Eicosanoid-lysolipids generated from 2-AA-LPC and 2-AA-LPE by multiple oxygenases (e.g., COX, LOX and P450) can be hydrolyzed by intracellular lysophospholipases C (LPLC) and lysophospholipases D (LPLD) to produce eicosanoid-glycerols and eicosanoid-lysophosphatidic acids (LPA), respectively. Thus, eicosanoid-lysolipids represent not only a new class of dynamically regulated signaling molecules, but in addition likely serve as precursors for further metabolism to mediate a wide variety of lipid signaling cascades. PG: prostaglandin; HETE: hydroxyeicosatetraenoic acid; EET: epoxyeicosatrienoic acid.
Significance
Calcium-activated iPLA2γ catalyzes the regiospecific hydrolysis of mitochondrial membrane lipids containing polyunsaturated fatty acids at the sn-2 position to generate 2-arachidonoyl-lysolipids which serve as a signaling node in multiple lipid 2nd messenger cascades. The current results indicate that COX-2, but not COX-1, effectively oxidizes 2-arachidonoyl-lysophosphatidylcholine (2-AA-LPC) to generate prostaglandin E2-LPC (PGE2-LPC), 11-hydroxyeicosatetraenoic acid-LPC (11-HETE-LPC) and lesser amounts of 15-hydroxyeicosatetraenoic acid-LPC (15-HETE-LPC). In addition, COX-2 oxidizes 2-arachidonoyl-lysophosphatidylethanolamine (2-AA-LPE) to predominantly PGE2-LPE, but not 11-HETE-LPE or 15-HETE-LPE. Resolution of the PGE2 diastereomers demonstrated that PGE2-LPC and PGE2-LPE were exclusively 9-oxo-11R,15S-dihydroxy-5Z,13E-prostadienoic acid-LPC and 9-oxo-11R,15S-dihydroxy-5Z,13E-prostadienoic acid-LPE respectively. Furthermore, 11-HETE-LPC was exclusively 11(R)-HETE-LPC while 15-HETE-LPC was comprised of ∼67% 15(S)-HETE-LPC and ∼33% 15(R)-HETE-LPC. These eicosanoid-lysolipids are natural products since they are present in cultured murine fibroblasts, murine liver and fresh human myocardium in high enantiomeric excess. The pathway for their synthesis was identified through reverse genetics using tissue and cultures of fibroblasts from germline iPLA2γ knockout mice. Since the amounts of these eicosanoid-lysolipid molecular species in hepatic tissue and fibroblast cultures from iPLA2γ-/- mice were markedly reduced in comparison to their wild type counterparts, these results demonstrate that iPLA2γ initiates the production of 2-AA-lysolipids that are subsequently oxidized to generate these novel metabolites. This notion is substantiated by demonstration that calcium-stimulation of wild type murine lung fibroblasts produced robust increases in eicosanoid-lysolipids which are markedly attenuated by genetic ablation of iPLA2γ. Collectively, these results identify novel signaling pathways employing direct oxidation of arachidonoylated-lysolipids by COX-2 to generate multiple previously unknown lipid 2nd messengers that may either serve directly as novel signaling molecules or be subjected to further metabolism to produce bioactive molecules for a wide variety of lipid signaling cascades.
Experimental Procedures
Cyclooxygenase enzymatic assays
(1) Kinetic analysis of the cyclooxygenase mediated oxidation of 2-AA-LPC or 2-AA-LPE. Purified COX-1 or COX-2 was preincubated with 1 μM hematin in 400 μl of 100 mM Tris-HCl buffer (pH 8.0) containing 10% glycerol for 5 min at room temperature. The substrate 2-AA-LPC or 2-AA-LPE (10 μM) was then added to initiate the reaction followed by incubation at 30°C for various times. The reaction was then terminated by adding 80 μl of methanol acidified to pH 4 with glacial acetic acid and applied to a Strata-X solid phase extraction cartridge previously preconditioned with 3 ml of methanol and equilibrated with 3 ml of 20% methanol/80% H2O. After washing with H2O, the reaction products were eluted with methanol and analyzed by LC-MSn methods as described below. (2) Inhibition of COX-2 mediated oxidation of 2-AA-LPC or 2-AA-LPE by aspirin. Purified human COX-2 was diluted in 400 μl of 100 mM Tris-HCl buffer pH 8.0 and treated with 2 mM aspirin at 37°C for 30 min. The buffer was then supplemented with hematin (1 μM) for 5 min at room temperature, and 2-AA-LPC or 2-AA-LPE (10 μM) was then added. After incubation at various times at 30°C, reactions were terminated with acidified methanol and the products were extracted by solid phase extraction as described above and analyzed by LC-MSn as described below.
15-Lipoxygenase enzymatic assay
Purified 15-LOX (2 μg) was incubated with 2-AA-LPC (10 μM) in 400 μl of 50 mM Tris-HCl buffer (pH 7.2) containing 10% glycerol for 10min in room temperature (23°C). The reaction was then terminated by addition of 80 μl of methanol acidified to pH 4 with glacial acetic acid and applied to a Strata-X solid phase extraction cartridge previously preconditioned with 3 ml of methanol and equilibrated with 3 ml of 20% methanol/80% H2O. After washing with H2O, the reaction products were eluted with methanol and analyzed by LC-MSn methods as described below.
Reverse phase liquid chromatography-tandem mass spectrometry (LC-MSn)
LC-MSn analyses were performed using an LTQ-Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA) equipped with a Surveyor HPLC system (Thermo Scientific, San Jose, CA). Detailed instrumentation settings were provided in Supplemental Information.
Identification of the eicosanoid products generated through hydrolysis of eicosanoid-lysolipids by cPLA2α
The eicosanoids-lysolipid products generated by COX-2 were incubated with purified cPLA2α (5 μg) in 200 μl of HEPES buffer (10 mM HEPES, pH 7.4, 100 mM KCl, 0.5 mM CaCl2, and 10% glycerol) for 15 min at 37°C. Reactions were stopped by addition of 40 μl methanol followed by acidification of the reaction mixture to pH 4 with glacial acetic acid. Terminated reactions were immediately applied to a Strata-X solid phase extraction and the products were derivatized with AMPP as previously described (Liu et al., 2013). Samples were then analyzed by LC-MSn methods as described above in the section “Reverse phase liquid chromatography-tandem mass spectrometry” and in the section “Chiral phase chromatography-tandem mass spectrometry” that was described in details in Supplemental Information.
Human myocardium and murine hepatic tissue sample preparation and analysis of eicosanoids and eicosanoid-lysolipids
Human myocardial tissue was obtained through the Washington University Translational Cardiovascular Tissue Core (WUTCT) in accordance with protocols approved by WUTCT Advisory Committee at Washington University School of Medicine. All animal breeding, care and euthanasia were performed according to protocols approved by the Animal Studies Committee at Washington University School of Medicine. Detailed procedures for tissue preparation and quantitation were provided in Supplemental Information.
Endogenous eicosanoid-lysolipid production in mouse lung fibroblasts (MLF) from wild-type and iPLA2γ-/- mice
Mouse lung fibroblasts were isolated from wild type (WT) or iPLA2γ-/- mice as previously described (Seluanov et al., 2010). Primary cells were cultured in minimum essential media (MEM) containing 15% heat-inactivated fetal bovine serum, 1X MEM nonessential amino acids, 1 mM sodium pyruvate, 100 units/ml of penicillin, and 100 μg/ml of streptomycin on 10 cm tissue culture dishes. Experiments were performed by using MLF cells between the second and fourth passage. At ∼90% confluence, MLF cells were placed in serum-free MEM alone for 3 hrs prior to incubation in fresh serum-free MEM containing either DMSO (0.1%) vehicle alone or 1.0 μM A23187 for 15 min. The medium was then collected and extracted by Strata-X solid phase extraction as described above. After removal of the media, cells were washed with phosphate buffered saline (PBS), harvested in 1 ml ice-cold PBS by using a cell scraper, and then immediately transferred to 2 ml CHCl3/CH3OH (1:1, v/v) and vortexed. Following phase separation by centrifugation at 15,000g for 15 min, the CHCl3 layer was evaporated under a nitrogen stream and resuspended in 100 μl of 80% CH3OH in H2O for LC-MS/MS analysis as described above in the section “Reverse phase liquid chromatography-tandem mass spectrometry”.
Supplementary Material
Highlight.
COX-2 directly oxidizes 2-arachidonoyl-lysophospholipids to eicosanoid-lysolipids.
Lipases (e.g., cPLA2α) release eicosanoids from eicosanoid-lysolipid precursors.
Genetic ablation of iPLA2γ reduces the amounts of eicosanoid-lysolipids.
Calcium-stimulation robustly increases the production of eicosanoid-lysolipids.
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grants RO1HL118639, RO1DK100679 and R01HL133178.
Footnotes
Author Contributions: X.L. and R.W.G. designed studies. X.L., S.H.M. and C.M.J. conducted the experiments. X.L., S.H.M., C.M.J and R.W.G analyzed the data. H.F.S. generated and provided iPLA2γ knockout mice. X.L., C.M.J and R.W.G. prepared the manuscript. X.L., S.H.M., C.M.J., H.F.S., and R.W.G. proofread the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackerman SJ, Park GY, Christman JW, Nyenhuis S, Berdyshev E, Natarajan V. Polyunsaturated lysophosphatidic acid as a potential asthma biomarker. Biomark Med. 2016;10:123–135. doi: 10.2217/bmm.15.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar A, John Anwar I, Delafontaine P. Elevation of cardiovascular risk by non-steroidal anti-inflammatory drugs. Trends Cardiovasc Med. 2015;25:726–735. doi: 10.1016/j.tcm.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, Farkouh ME, FitzGerald GA, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–779. doi: 10.1016/S0140-6736(13)60900-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon CP, Cannon PJ. COX-2 Inhibitors and Cardiovascular Risk. Science. 2012;336:1386–1387. doi: 10.1126/science.1224398. [DOI] [PubMed] [Google Scholar]
- Cedars A, Jenkins CM, Mancuso DJ, Gross RW. Calcium Independent Phospholipases in the Heart: Mediators of Cellular Signaling, Bioenergetics and Ischemia-induced Electrophysiologic Dysfunction. J Cardiovasc Pharmacol. 2009;53:277–289. [PMC free article] [PubMed] [Google Scholar]
- Conaghan PG. A turbulent decade for NSAIDs: update on current concepts of classification, epidemiology, comparative efficacy, and toxicity. Rheumatol Int. 2012;32:1491–1502. doi: 10.1007/s00296-011-2263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras FX, Sanchez-Magraner L, Alonso A, Goni FM. Transbilayer (flip-flop) lipid motion and lipid scrambling in membranes. FEBS Lett. 2010;584:1779–1786. doi: 10.1016/j.febslet.2009.12.049. [DOI] [PubMed] [Google Scholar]
- Creer MH, Gross RW. Separation of isomeric lysophospholipids by reverse phase HPLC. Lipids. 1985;20:922–928. doi: 10.1007/BF02534778. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L, Sugiura T, Waku K. Potential biosynthetic connections between the two cannabimimetic eicosanoids, anandamide and 2-arachidonoyl-glycerol, in mouse neuroblastoma cells. Biochem Biophys Res Commun. 1996;227:281–288. doi: 10.1006/bbrc.1996.1501. [DOI] [PubMed] [Google Scholar]
- Dogne JM, Hanson J, Pratico D. Thromboxane, prostacyclin and isoprostanes: therapeutic targets in atherogenesis. Trends Pharmacol Sci. 2005;26:639–644. doi: 10.1016/j.tips.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Gross RW, Corr PB, Lee BI, Saffitz JE, Crafford WA, Jr, Sobel BE. Incorporation of radiolabeled lysophosphatidyl choline into canine Purkinje fibers and ventricular muscle. Electrophysiological, biochemical, and autoradiographic correlations. Circ Res. 1982;51:27–36. doi: 10.1161/01.res.51.1.27. [DOI] [PubMed] [Google Scholar]
- Gubitosi-Klug RA, Yu SP, Choi DW, Gross RW. Concomitant acceleration of the activation and inactivation kinetics of the human delayed rectifier K+ channel (Kv1.1) by Ca(2+)-independent phospholipase A2. J Biol Chem. 1995;270:2885–2888. doi: 10.1074/jbc.270.7.2885. [DOI] [PubMed] [Google Scholar]
- Hennekens CH, Borzak S. Cyclooxygenase-2 inhibitors and most traditional nonsteroidal anti-inflammatory drugs cause similar moderately increased risks of cardiovascular disease. J Cardiovasc Pharmacol Ther. 2008;13:41–50. doi: 10.1177/1074248407312990. [DOI] [PubMed] [Google Scholar]
- Herschman HR. Regulation of prostaglandin synthase-1 and prostaglandin synthase-2. Cancer Metastasis Rev. 1994;13:241–256. doi: 10.1007/BF00666095. [DOI] [PubMed] [Google Scholar]
- Jenkins CM, Cedars A, Gross RW. Eicosanoid signaling pathways in the heart. Cardiovasc Res. 2009;82:240–249. doi: 10.1093/cvr/cvn346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak KR, Prusakiewicz JJ, Rowlinson SW, Schneider C, Marnett LJ. Amino Acid Determinants in Cyclooxygenase-2 Oxygenation of the Endocannabinoid 2-Arachidonylglycerol. J Biol Chem. 2001;276:30072–30077. doi: 10.1074/jbc.M104467200. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Rowlinson SW, Marnett LJ. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J Biol Chem. 2000;275:33744–33749. doi: 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- Kurumbail RG, Kiefer JR, Marnett LJ. Cyclooxygenase enzymes: catalysis and inhibition. Curr Opin Struct Biol. 2001;11:752–760. doi: 10.1016/s0959-440x(01)00277-9. [DOI] [PubMed] [Google Scholar]
- Lecomte M, Laneuville O, Ji C, DeWitt DL, Smith WL. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J Biol Chem. 1994;269:13207–13215. [PubMed] [Google Scholar]
- Liu SJ. Inhibition of L-type Ca2+ channel current and negative entropy induced by arachidonic acid in adult rat ventricular myocytes. Am J Physiol Cell Physiol. 2007;293:C1594–C1604. doi: 10.1152/ajpcell.00284.2007. [DOI] [PubMed] [Google Scholar]
- Liu X, Moon SH, Mancuso DJ, Jenkins CM, Guan S, Sims HF, Gross RW. Oxidized fatty acid analysis by charge switch derivatization, selected reaction monitoring, and accurate mass quantitation. Anal Biochem. 2013;442:40–50. doi: 10.1016/j.ab.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, et al. Genetic ablation of calcium-independent phospholipase A2γ leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J Biol Chem. 2007;282:34611–34622. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso DJ, Jenkins CM, Gross RW. The Genomic Organization, Complete mRNA Sequence, Cloning, and Expression of a Novel Human Intracellular Membrane-associated Calcium-independent Phospholipase A2. J Biol Chem. 2000;275:9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- Marnett LJ, DuBois RN. COX-2: a target for colon cancer prevention. Annu Rev Pharmacol Toxicol. 2002;42:55–80. doi: 10.1146/annurev.pharmtox.42.082301.164620. [DOI] [PubMed] [Google Scholar]
- Meade EA, Smith WL, DeWitt DL. Expression of the murine prostaglandin (PGH) synthase-1 and PGH synthase-2 isozymes in Cos-1 cells. J Lipid Mediators. 1993;6:119–129. [PubMed] [Google Scholar]
- Moon SH, Jenkins CM, Liu X, Guan S, Mancuso DJ, Gross RW. Activation of mitochondrial calcium-independent phospholipase A2γ (iPLA2γ) by divalent cations mediating arachidonate release and production of downstream eicosanoids. J Biol Chem. 2012;287:14880–14895. doi: 10.1074/jbc.M111.336776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pete MJ, Exton JH. Purification of a Lysophospholipase from Bovine Brain That Selectively Deacylates Arachidonoyl-substituted Lysophosphatidylcholine. J Biol Chem. 1996;271:18114–18121. doi: 10.1074/jbc.271.30.18114. [DOI] [PubMed] [Google Scholar]
- Pete MJ, Wu DW, Exton JH. Subcellular fractions of bovine brain degrade phosphatidylcholine by sequential deacylation of the sn-1 and sn-2 positions. Biochim Biophys Acta. 1996;1299:325–332. doi: 10.1016/0005-2760(95)00225-1. [DOI] [PubMed] [Google Scholar]
- Ricciotti E, FitzGerald GA. Prostaglandins and Inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluanov A, Vaidya A, Gorbunova V. Establishing Primary Adult Fibroblast Cultures from Rodents. J Vis Exp. 2010;44:2033. doi: 10.3791/2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:149–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Smith WL, Urade Y, Jakobsson PJ. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem Rev. 2011;111:5821–5865. doi: 10.1021/cr2002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicher JM, Kamei K, Ishikawa TO, Herschman H, Wang Y. Compensatory hypertrophy induced by ventricular cardiomyocyte-specific COX-2 expression in mice. J Mol Cell Cardiol. 2010;49:88–94. doi: 10.1016/j.yjmcc.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SY, Monslow J, Todd L, Lawson J, Pure E, FitzGerald GA. Cyclooxygenase-2 in Endothelial and Vascular Smooth Muscle Cells Restrains Atherogenesis in Hyperlipidemic Mice. Circulation. 2014;129:1761–1769. doi: 10.1161/CIRCULATIONAHA.113.007913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarling EJ, de Aguiar Vallim TQ, Edwards PA. Role of ABC transporters in lipid transport and human disease. Trends Endocrinol Metab. 2013;24:342–350. doi: 10.1016/j.tem.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- Vecchio AJ, Malkowski MG. The Structural Basis of Endocannabinoid Oxygenation by Cyclooxygenase-2. J Biol Chem. 2011;286:20736–20745. doi: 10.1074/jbc.M111.230367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Patelb VV, Ricciottia E, Zhouc R, Levinb MD, Gaod E, Yu Z, Ferrarib VA, Lub MM, Xu J, et al. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci USA. 2009;106:7548–7552. doi: 10.1073/pnas.0805806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennogle LP, Liang H, Quintavalla JC, Bowen BR, Wasvary J, Miller DB, Allentoff A, Boyer W, Kelly M, Marshall P. Comparison of recombinant cyclooxygenase-2 to native isoforms: aspirin labeling of the active site. FEBS Lett. 1995;371:315–320. doi: 10.1016/0014-5793(95)00930-8. [DOI] [PubMed] [Google Scholar]
- Xiao G, Tsai AL, Palmer G, Boyar WC, Marshall PJ, Kulmacz RJ. Analysis of hydroperoxide-induced tyrosyl radicals and lipoxygenase activity in aspirin-treated human prostaglandin H synthase-2. Biochemistry. 1997;36:1836–1845. doi: 10.1021/bi962476u. [DOI] [PubMed] [Google Scholar]
- Yan W, Jenkins CM, Han X, Mancuso DJ, Sims HF, Yang K, Gross RW. The Highly Selective Production of 2-Arachidonoyl Lysophosphatidylcholine Catalyzed by Purified Calcium-independent Phospholipase A2γ. J Biol Chem. 2005;280:26669–26679. doi: 10.1074/jbc.M502358200. [DOI] [PubMed] [Google Scholar]
- Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Pure E, et al. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4:132–154. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.