

ABSTRACT
Lanthanides are utilized by microbial methanol dehydrogenases, and it has been proposed that lanthanides may be important for other type I alcohol dehydrogenases. A triple mutant strain (mxaF xoxF1 xoxF2; named MDH-3), deficient in the three known methanol dehydrogenases of the model methylotroph Methylobacterium extorquens AM1, is able to grow poorly with methanol if exogenous lanthanides are added to the growth medium. When the gene encoding a putative quinoprotein ethanol dehydrogenase, exaF, was mutated in the MDH-3 background, the quadruple mutant strain could no longer grow on methanol in minimal medium with added lanthanum (La3+). ExaF was purified from cells grown with both calcium (Ca2+) and La3+ and with Ca2+ only, and the protein species were studied biochemically. Purified ExaF is a 126-kDa homodimer that preferentially binds La3+ over Ca2+ in the active site. UV-visible spectroscopy indicates the presence of pyrroloquinoline quinone (PQQ) as a cofactor. ExaF purified from the Ca2+-plus-La3+ condition readily oxidizes ethanol and has secondary activities with formaldehyde, acetaldehyde, and methanol, whereas ExaF purified from the Ca2+-only condition has minimal activity with ethanol as the substrate and activity with methanol is not detectable. The exaF mutant is not affected for growth with ethanol; however, kinetic and in vivo data show that ExaF contributes to ethanol metabolism when La3+ is present, expanding the role of lanthanides to multicarbon metabolism.
IMPORTANCE ExaF is the most efficient PQQ-dependent ethanol dehydrogenase reported to date and, to our knowledge, the first non-XoxF-type alcohol oxidation system reported to use lanthanides as a cofactor, expanding the importance of lanthanides in biochemistry and bacterial metabolism beyond methanol dehydrogenases to multicarbon metabolism. These results support an earlier proposal that an aspartate residue near the catalytic aspartate residue may be an indicator of rare-earth element utilization by type I alcohol dehydrogenases.
INTRODUCTION
Methylotrophy is the capability of organisms to metabolize reduced carbon compounds lacking carbon-carbon bonds as the sole source of carbon and energy (1). The genus Methylobacterium is comprised of aerobic facultative methylotrophs that can metabolize single-carbon compounds, such as methanol and methylamine, as well as multicarbon substrates like ethanol, acetate, ethylamine, pyruvate, and succinate (2, 3). Members of the genus Methylobacterium are wide-spread plant epiphytes (4, 5) that utilize their metabolic flexibility to gain an advantage in the phyllosphere, an oligotrophic environment with transient substrate availability (6, 7).
Methanol dehydrogenase (MDH) is an essential enzyme for the methylotrophic metabolism of methanol and methane (8). In Gram-negative methylotrophic bacteria, MDHs are soluble, periplasmic proteins with pyrroloquinoline quinone (PQQ) as the prosthetic group (9, 10). The best studied PQQ-containing MDHs are α2β2 tetramers consisting of the MxaF and MxaI proteins (11–14) that contain calcium (Ca2+) in the active site (15, 16). Studies have provided evidence for the physiological role of a second type of PQQ-dependent MDH, XoxF, which has ∼50% amino acid identity to MxaF from MxaFI-type MDHs (17). Metagenomic and environmental proteomics studies have demonstrated that xoxF is more widespread than mxaF in environmental samples (18–21). Phylogenetic analysis of putative PQQ-containing MDHs has shown that XoxF-type MDHs are genetically diverse with at least five distinct clades, and it has been suggested that MxaFI-type MDHs represent a minor fraction of these MDHs (8, 22). It has been further proposed that MxaFI-type MDHs may be the result of a second evolutionary event, with an ancestral XoxF-type MDH prototype (22). Together, these suppositions suggest that XoxF-type MDHs may be the primary MDHs for methylotrophy. For the few that have been studied to date, production of functional XoxF-type MDHs requires the presence of lanthanides in the growth medium (23–27) and all reported XoxF-type MDHs that have been biochemically characterized thus far contain a lanthanide such as cerium (Ce3+) or lanthanum (La3+), rather than Ca2+ in the active site (25, 27). Limited kinetic studies of XoxF-type MDHs have shown an increased efficiency when oxidizing methanol compared to that of MxaFI-type MDHs (summarized in reference 22). Functional MxaFI-type MDHs may be produced in the absence of lanthanides in organisms that have both XoxF- and MxaFI-type MDHs, although the number of studies reporting this is limited (28–30).
The genome of the model methylotroph Methylobacterium extorquens AM1 contains genes encoding three known PQQ-containing MDH homologs: one MxaFI-type MDH and two XoxF-type MDHs (12). The MxaFI-type MDH is required for growth with methanol in the absence of exogenous lanthanides (25, 29). Of the two XoxF-type MDHs, a catalytic role has been demonstrated only for XoxF1, not for XoxF2 (25), although genetic studies demonstrate the importance of xoxF2 for growth with methanol in the absence of xoxF1 (29, 31). XoxF1 and XoxF2 share ∼90% amino acid identity and are required for functional expression of the mxaFI genes (31). Vu et al. (29) demonstrated that when lanthanides were present in the growth medium, a triple mutant strain (mxaF xoxF1 xoxF2; referred to subsequently as MDH-3) of M. extorquens AM1 that lacks all known MDHs was still able to grow in methanol medium but with a severe reduction in growth rate (29). This finding suggested that an additional lanthanide-dependent enzyme or pathway that can oxidize methanol exists in M. extorquens AM1. This supposition was confirmed by detection of methanol oxidation activity in cell extracts from the MDH-3 mutant strain grown in methanol medium with exogenous La3+ (29).
Aside from the aforementioned MDHs, the genome of M. extorquens AM1 contains genes encoding two additional predicted PQQ-dependent ADHs. One is annotated as a putative general type I ADH encoded by the open reading frame META1_4973 (12). This general type I ADH does not contain the second aspartate residue proposed for lanthanide coordination (22). The second enzyme is a possible quinoprotein ethanol dehydrogenase (QEDH) annotated as ExaA, encoded by META1_1139. Biochemical studies with ExaA homologs purified from Rhodopseudomonas and Pseudomonas strains demonstrated that ExaA enzymes are calcium-dependent dehydrogenases capable of oxidizing a broad range of alcohols, including methanol (32–35). Like the MxaFI-type MDH, in vitro activity assays require a high pH (∼pH 9) and ammonia or a primary amine as an activator (32, 35–37). Despite the methanol-oxidizing capability of these enzymes, only low growth yields with methanol have been reported under aerobic conditions, calling into question whether or not ExaA is capable of supporting growth with methanol for the MDH-3 mutant of M. extorquens AM1. Intriguingly, however, ExaA from M. extorquens AM1 contains an aspartate residue proposed to be properly positioned for lanthanide coordination.
We examined the predicted ExaA and type I ADH from M. extorquens AM1 as potential lanthanide-utilizing alcohol dehydrogenases. In this work, we report that the ExaA quinoprotein ethanol dehydrogenase, and not the type I ADH, is responsible for methanol oxidation in the MDH-3 mutant strain. ExaA was purified and biochemically characterized. Unlike other ExaA homologs reported, ExaA from M. extorquens AM1 utilizes La3+ rather than Ca2+ as a cofactor. Due to its La3+-dependent nature, we propose to rename ExaA in M. extorquens AM1 as ExaF to distinguish it from other ExaA homologs. We show that ExaF is functionally an ethanol dehydrogenase, with secondary activities with formaldehyde, methanol, and acetaldehyde. Our in vitro data suggest that ExaF may catalyze the sequential oxidations of ethanol to acetaldehyde and then to acetate. We identify ExaF as the newest member of the lanthanide-containing enzymes, demonstrating for the first time that lanthanides are utilized by enzymes other than XoxF-type MDHs and showing that lanthanides are relevant beyond single-carbon microbial metabolism.
MATERIALS AND METHODS
Chemicals.
Potassium phosphate trihydrate was purchased from Acros Organics (New Jersey). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Waltham, MA).
Bacterial strains and plasmids.
M. extorquens AM1 strains and Escherichia coli strains and plasmids used in this work are described in Table 1. Null mutations in pqqF, exaF, and META1_4973 were generated using the allelic-exchange suicide plasmid pCM184 (38) with the wild-type strain as a recipient. The kanamycin (Km) resistance cassette in the mxaF xoxF1 xoxF2::Km mutant strain described in a previous study (29) was deleted using the cre expression vector pCM157 as described previously (38). META1_4973 and exaF were individually mutated in the MDH-3 mutant background to generate the MDH-3 META1_4973::Km and MDH-3 exaF::Km strains (Table 2). Diagnostic colony PCR was used to confirm the Km insertion and deletion. The Km resistance cassette was removed from the MDH-3 exaF::Km strain as described above, and the quadruple mutant was complemented using pNG265 (see Tables 3 and 4).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| TOP10 | Competent cells for cloning | Invitrogen |
| S17-1 | Helper strain for conjugation | 57 |
| M. extorquens | ||
| AM1 | Rifamycin-resistant derivative | 58 |
| AM1 exaF::Km | Deletion mutant | This study |
| AM1 META1_4973::Km | Deletion mutant | This study |
| AM1 pqqF::Km | Deletion mutant | This study |
| AM1 MDH-3 | ΔmxaF ΔxoxF1 ΔxoxF2 deletion mutant | This study |
| AM1 MDH-3 exaF::Km | Deletion mutant | This study |
| AM1 MDH-3 exaF | Deletion mutant | This study |
| AM1 MDH-3 META1_4973::Km | Deletion mutant | This study |
| Plasmids | ||
| pNG265 | Pxox1-exaF-hexahistidine tag | This study |
| pNG267 | Pxox1-hexahistidine tag | This study |
| pNG271 | Pmxa-exaF-hexahistidine tag | This study |
| pLB01 | Pxox1-xoxF1-hexahistidine tag | This study |
| pAWP78 | IncP-based broad-host-range replicating vector | Addgene |
| pCM184 | Kmr Apr Tcr allelic-exchange vector | 38 |
| pHV23 | pCM184::xoxF; donor for xoxF1::Km | This study |
| pHV24 | pCM184::exaF; donor for exaF::Km | This study |
| pHV25 | pCM184::META1_4973; donor for META1_4973::Km | This study |
TABLE 2.
Primers used in this study
| Construct or mutation | Primer | Sequence (5′–3′) |
|---|---|---|
| Constructs | ||
| pLB01 | pAWP_backboneFor | TTGTCGGGAAGATGCGTGATCTG |
| pAWP_backboneRev | CAGCTCACTCAAAGGCGGTAATAC | |
| Pxox1_xoxF1For | CGTATTACCGCCTTTGAGTGAGCTGCTGAATTTAGCAGGCAAGTTTCCTG | |
| Pxox1_xoxF1_6xhisRev | ATCAGATCACGCATCTTCCCGACAATTAGTGGTGATGGTGATGATGACGACCTTCGATGTTGTTCGGCAGCGAGAAGACCG | |
| pNG265 | pLB01_backboneRev | GGATTCCTCCGACAAGTCTTATCCG |
| exaFFor | TAAGACTTGTCGGAGGAATCCATGAGAATGCGGAACCATTTCCTG | |
| exaFRev | ATCAGATCACGCATCTTCCCGACAATTAGTGGTGATGGTGATGATGACGACCTTCGATTCGGGAGGCGAGCGCCTTC | |
| pNG267 | Forward | CTTGTCGGAGGAATCCATCGAAGGTCGTCATCATCACCA |
| Reverse | GGATTCCTCCGACAAGTCTTATCCG | |
| pNG271 | backboneFor | TTGTCGGGAAGATGCGTGATCTG |
| backboneRev | ATCAGATCACGCATCTTCCCGACAATTAGTGGTGATGGTGATGATGACGACCTTCGATTCGGGAGGCGAGCGCCTTC | |
| PmxaFor | TACCGCCTTTGAGTGAGCTGCCCGCTTGGTCGG | |
| PmxaRev | CAGCTCACTCAAAGGCGGTAATAC | |
| Null mutations | ||
| exaF | exaF_1139ULEcoRI | GTGAATTCCCTCCGACAGCCTACTGATGAAC |
| exaF_1139URKpnI | CAGGTACCGTCTTGGCGTCGTTGAGGATGTC | |
| exaF_1139DLHpaI | CAGTTAACGCCGTTCGTGTCCAACATCAACTG | |
| exaF_1139DRSacI | TAGAGCTCGTTGATGCCAGCCGTCGTCTTGG | |
| META1_4973 | adh_4973ULEcoRI | TGAATTCAGCAGAGTGACACGCCGCACGAAG |
| adh_4973URKpnI | CAGGTACCACGAACCTTGGACCGGCTTGTC | |
| adh_4973DLHpaI | TGTTAACCGAGATCCTCTGGCGCTTCCAGT | |
| adh_4973DRSacI | TGAGCTCTCCACTACATCCATTACGGCTGG | |
| pqqF | pqqF_2330ULEcoRI | TAGAATTCACCGTCCCGCCATCACCTATCGC |
| pqqF_2330URKpnI | ATGGTACCGTGATTCAGGCCTCGCTGCGGTC | |
| pqqF_2330DLHpaI | TAGTTAACGGTGACCGGCTACCTGACCAAGG | |
| pqqF_2330DRSacI | TAGAGCTCGTAGCGCAGGCTGGCGATCATCT | |
| xoxF1 | xoxF_1740ULEcoRI | TAGAATTCGCCCCGATGGCAGGAATTAAAGT |
| xoxF_1740URKpnI | ATGGTACCCTGCTTCGGCTCGTACTTCCACA | |
| xoxF_1740DLHpaI | TAGTTAACGCCTGTTCTACGTCCCCACCAAC | |
| xoxF_1740DRSacI | TAGAGCTCCTGCACCGACGAGACCAAGAAGA |
TABLE 3.
Growth rate constants of M. extorquens strains grown with methanol as the carbon source
| Strain | Growth rate constant (h−1) for strain grown ina: |
|
|---|---|---|
| CH3OH (no La) | CH3OH (+La) | |
| Wild-type strain AM1 | 0.147 (1.6) | 0.152 (1.5) |
| AM1 MDH-3 | No growth | 0.042 (1.5) |
| AM1 pqqF::Km | No growth | No growth |
| AM1 META1_4973::Km | 0.151 (1.6) | 0.158 (1.5) |
| AM1 exaF::Km | 0.161 (1.6) | 0.152 (1.5) |
| AM1 MDH-3 META1_4973::Km | No growth | 0.042 (1.5) |
| AM1 MDH-3 exaF::Km | No growth | No growth |
| AM1 MDH-3 exaF/pNG265 | No growth | 0.039 (0.3) |
Results are the mean from three or four biological replicates. Final growth yields determined by light scattering at OD600 are shown in parentheses. Variances in growth rates and yields among replicates for each strain are <10%.
TABLE 4.
Growth rate constants of M. extorquens strains grown with ethanol as the carbon source
| Strain | Growth rate constant (h−1) for strain grown ina: |
|
|---|---|---|
| C2H6O (no La) | C2H6O (+La) | |
| Wild-type strain AM1 | 0.085 (1.3) | 0.095 (0.9) |
| AM1 MDH-3 | No growth | 0.097 (1.6) |
| AM1 exaF::Km | 0.080 (1.4) | 0.080 (0.8) |
| AM1 MDH-3 exaF::Km | No growth | No growth |
| AM1 MDH-3 exaF/pNG265 | No growth | 0.061 (1.4) |
Results are the mean from three or four biological replicates. Final growth yields determined by light scattering at OD600 are given in parentheses. Variances in growth rates and yields among replicates for each strain are <10%.
Protein production constructs were generated using the mxa and xox1 promoter regions, exaF, and sequence encoding 6 histidines. The PCR products were assembled using Gibson Assembly master mix (39, 40) (New England BioLabs, Ipswich, MA). Primers used for plasmid construction are listed in Table 2 and were designed to generate insert fragments with 20-bp overlapping regions to the respective backbone fragments. Briefly, the promoterless, kanamycin-selectable IncP cloning vector pAWP78 (Addgene, Cambridge, MA) was amplified as a linear PCR fragment using the pAWP_backbone primers. The lanthanide-inducible promoter region of xoxF1 (Pxox1) (29) was amplified from M. extorquens AM1 wild-type genomic DNA using the pLB01 primers. The pLB01 reverse primer contains a hexahistidine sequence and Factor Xa protease cleavage site for tag removal. The two fragments were joined together, generating plasmid pLB01. pLB01 was used as the template DNA to PCR amplify the pAWP78 backbone DNA with Pxox1 using the pAWP78_backboneFor and the pLB01_backboneRev primers. The gene encoding the putative quinoprotein ethanol dehydrogenase (exaF) was amplified from M. extorquens AM1 genomic DNA using the exaF forward and reverse primers. The reverse primer contains a hexahistidine sequence and Factor Xa protease cleavage site. The backbone and insert fragments were assembled to generate pNG265. Using pNG265 as the template DNA, a linear DNA fragment of pAWP78 with Pxox1 was generated using the pNG267 forward and reverse primers. The fragment was recircularized using assembly methods to generate the “empty” plasmid pNG267. Using pNG265 as the template DNA, a backbone DNA fragment was generated containing the promoterless, linearized plasmid. The mxa promoter region (41) was PCR amplified using the Pmxa forward and reverse primers. The backbone and insert fragments were assembled, generating pNG271.
E. coli TOP10 cells (Sigma-Aldrich, St. Louis, MO) were used for cloning and plasmid propagation as reported previously (42). All plasmids were verified by colony PCR and sequencing. Mating of plasmids into M. extorquens AM1 was performed using E. coli S17-1 (Sigma-Aldrich, St. Louis, MO) cells for conjugal transfer as reported previously (41).
Growth conditions.
E. coli cultures were grown in culture tubes with Luria-Bertani broth purchased from BD (Franklin Lakes, NJ). For M. extorquens AM1 cultures, glassware was prepared as reported in reference 29. M. extorquens AM1 cultures were grown in minimal medium (43) in borosilicate culture tubes, polystyrene round-bottom tubes, or shake flasks at 30°C with 15 mM succinic acid, 34 mM ethanol, or 125 mM methanol added as the growth substrate. A 2 μM concentration of LaCl3 was added to the growth medium when stated. When antibiotics were necessary, kanamycin (Km) was added to a final concentration of 50 μg/ml.
For purification of enzyme, 200-ml cultures were grown in shake flasks with succinate to late exponential phase and then transferred to 10 liters of fresh minimal medium with methanol and kanamycin in a 10-liter New Brunswick Celligen/BioFlo 115 bioreactor (Edison, NJ). For purification in the presence of La3+, 20 μM LaCl3 was added to the medium. Cultures were grown at 30°C with 2 standard liters-per-minute (SLPM) airflow and the agitation rate set at 1,000 rpm.
Phenotypic analysis.
Growth curve measurements were performed as follows: glassware was cleaned of residual lanthanides as described previously (29). Briefly, 3 ml of minimal medium plus 125 mM methanol was inoculated with the wild-type strain grown to mid-exponential phase. Cultures were grown to the maximal optical density at 600 nm (OD600) and discarded, and the glassware was washed and sterilized. Next, 2 ml of minimal medium was inoculated from isolated colonies into the lanthanide-free borosilicate culture tubes with 15 mM succinate as the growth substrate. Cultures were incubated at 30°C with shaking at 200 rpm until they reached mid-exponential phase (∼OD600 1.0), and then 0.12 to 0.2 ml of preculture was used to inoculate 3 to 6 ml of minimal medium in a new culture tube with 125 mM methanol or 34 mM ethanol as the growth substrate. A 2 μM concentration of La3+ was added for Ca2+-plus-La3+ conditions. Tube cultures were shaken at 200 rpm at 30°C, and the OD600 was monitored over time using an Ultraspec 10 cell density meter (Amersham Biosciences, Little Chalfont, UK) or a Spectronic 20D spectrophotometer (Milton Roy Company, Warminster, PA) (29). Three or four biological replicates were measured for each condition.
Purification of ExaF.
Cultures were grown to a final OD600 of 3.2 to 4.0 in 2.8-liter baffled flasks with 1.5 liter of methanol minimal medium, both with and without 20 μM LaCl3, and cells were harvested by centrifugation using a Sorvall RC6+ centrifuge (Thermo Fisher Scientific, Waltham, MA) at 21,000 × g at 4°C for 10 min. The supernatant was discarded, and cell pellets were flash frozen with liquid nitrogen and stored at −80°C until needed. Cell pellets were resuspended in a double volume of 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM imidazole and disrupted using a French pressure cell (Aminco, Haverhill, MA) at 20,000 lb/in2. Cell debris was removed by ultracentrifugation using a Sorvall WX Ultra 80 centrifuge (Thermo Fisher Scientific, Waltham, MA) for 1 h at 4°C at 100,000 × g. Protein was purified by metal ion affinity chromatography (IMAC) as follows. The supernatant was loaded onto an equilibrated 2 ml Ni-nitrilotriacetic acid (NTA) Superflow resin (Qiagen, Hilden, Germany). A wash step of 5 column volumes was performed with buffer containing 50 mM imidazole, and elution was performed using buffer containing 400 mM imidazole. One-milliliter fractions were collected and analyzed using a 4 to 12% SDS-PAGE gel (Life Technologies, Carlsbad, CA). Pure fractions were pooled, concentrated with an Amicon 10-kDa ultracentrifugal unit (Millipore, Billerica, MA), and desalted on a PD-10 column (GE Healthcare, Pittsburgh, PA) that was equilibrated with 25 mM Tris-HCl (pH 8.0), 150 mM NaCl.
The hexahistidine tag was removed using Factor Xa protease (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Enzyme used for kinetic measurements was reconstituted by combining the purified enzyme with an equimolar concentration of LaCl3 and 20 μM PQQ and incubating the mixture overnight at 4°C. The reconstituted enzyme was desalted and concentrated as mentioned above.
Methanol dehydrogenase activity measurements.
Methanol dehydrogenase activity was measured by monitoring the phenazine methosulfate (PMS)-mediated reduction of 2,6-dichlorophenol-indophenol (DCPIP) (ε600 = 21 mM−1 cm−1) (44). Initial assays were performed as described by Anthony and Zatman (45) with the modifications reported by Vu et al. (29). The assay protocol was then modified as follows: 100 mM Tris-HCl (pH 9.0) was combined with either 100 μM CaCl2 or 100 μM LaCl3, 5 mM methylamine or 15 mM NH4Cl, 10 mM PQQ, 100 μM DCPIP, 100 μM PMS, and 6.25 to 12.5 μl of cell extract (concentrations ranging from 4 to 30 mg/ml) or 0.4 to 1.0 μg of pure enzyme. When cell extracts were used, the reaction mixture was incubated for 5 to 10 min at 30°C to mitigate any methanol-independent reduction of DCPIP. Methanol-independent reduction of DCPIP was not observed with pure enzyme. The reaction was initiated by adding the assay mixture to 10 μl of 1 M methanol in a microplate well, resulting in a final reaction volume of 185 μl. The decrease in absorbance of DCPIP was monitored in a BioTek EpochII microplate reader (BioTek, VT). Methanol dehydrogenase activity was also monitored using cytochrome c isoform 1 from Saccharomyces cerevisiae as the final electron acceptor and monitoring its reduction at 550 nm under oxic conditions. For this assay, 3.5 to 30 μg of pure enzyme was added to a final assay volume of 1 ml. Protein concentrations were measured using the bicinchoninic acid assay (Sigma-Aldrich, St. Louis, MO) (46) according to the manufacturer's instructions.
Enzyme kinetics.
Kinetic parameters for ExaF were determined using the second assay detailed above with various substrate concentrations. This assay was used due to the minimal or complete absence of substrate-independent reduction of DCPIP. If background activity was detected, it was subtracted from enzyme activity measurements. Data were fitted by nonlinear regression, and kinetic parameters were calculated according to the Michaelis-Menten equation using Prism GraphPad 6 (GraphPad Software, La Jolla, CA).
Mr determination.
The molecular weight of the purified protein sample was determined by size exclusion chromatography-multiangle light scattering (SEC-MALS) using a Superdex 200 Increase 10/300 GL column (Bio-Rad, Hercules, CA) with 25 mM Tris-HCl buffer (pH 8) containing 150 mM sodium chloride and a miniDAWN TREOS multiangle light scattering detector with an Optilab T-rEX refractometer (Wyatt, Santa Barbara, CA). Data analysis was performed using Astra software according to the manufacturer's specifications (Wyatt, Santa Barbara, CA).
Identification of cofactor and metal content.
The absorbance spectrum of the enzyme as purified was recorded in a 1-cm-path-length cuvette at room temperature using a UV-2600 UV-Vis spectrophotometer (Shimadzu, Columbia, MD). The PQQ concentration and content were calculated using the molar absorption coefficient of 9,620 M−1 · cm−1 (47). The metal content of the enzyme as purified was determined by inductively coupled plasma mass spectrometry (ICP-MS) at the Michigan State University Laser Ablation ICP-MS Facility using a Thermo Fisher Scientific ICAP Q ICP-MS (Waltham, MA) in collision cell mode.
ExaF homology model.
The amino acid sequence for ExaF was submitted to the Phyre2 Web portal (48) and analyzed in intensive mode for structural homology. The template with the highest homology, PDB 1FLG, was chosen for further analysis. Overlay of our ExaF homology model with PDB 1FLG was performed using PyMOL (49).
RESULTS
ExaF supports growth with methanol in the presence of lanthanum.
Growth studies demonstrated that the MDH-3 mutant strain, deficient in the three known MDHs, is still capable of growth in methanol minimal medium when lanthanides are exogenously supplied (29) (Fig. 1; Table 3). Besides the genes known to encode MDH homologs (mxaF, xoxF1, and xoxF2), the M. extorquens AM1 genome contains genes encoding a putative quinoprotein alcohol dehydrogenase (META1_4973) and a possible quinoprotein ethanol dehydrogenase (ExaF; META1_1139). Null mutations were constructed for each putative quinoprotein in the MDH-3 mutant strain background, and growth in methanol medium with La3+ was measured (Fig. 1; Table 3). The additional loss of META1_4973 encoding the putative type I ADH showed no additional decrease in growth rate from the MDH-3 mutant strain (Table 3). In contrast, a quadruple mutant lacking the predicted quinoprotein ethanol dehydrogenase (exaF; META1_1139) eliminated growth, indicating that ExaF is responsible for the unknown methanol oxidation activity reported by Vu et al. (29) (Fig. 1; Table 3). The MDH-3 exaF mutant was complemented for growth with both methanol and ethanol using pNG265. The mutant was not complemented for growth with either substrate using pNG271 (Tables 3 and 4). The strain lacking only exaF did not exhibit a growth defect compared to the wild-type strain in the presence of La3+. These results suggest that while ExaF can contribute to methanol growth with La3+, it is not required in the wild-type strain, and the methanol oxidation capacity of the MDH-3 mutant may be due to a promiscuous activity of ExaF using methanol as an alternative substrate. To demonstrate that ExaF requires PQQ as a cofactor, a gene encoding an essential PQQ synthesis protein, pqqF (META1_2330) (50, 51), was mutated. The pqqF mutant was unable to grow in methanol medium with La3+, suggesting that PQQ is essential for all La3+-dependent methanol oxidation enzymes in M. extorquens AM1, including ExaF (Table 3).
FIG 1.
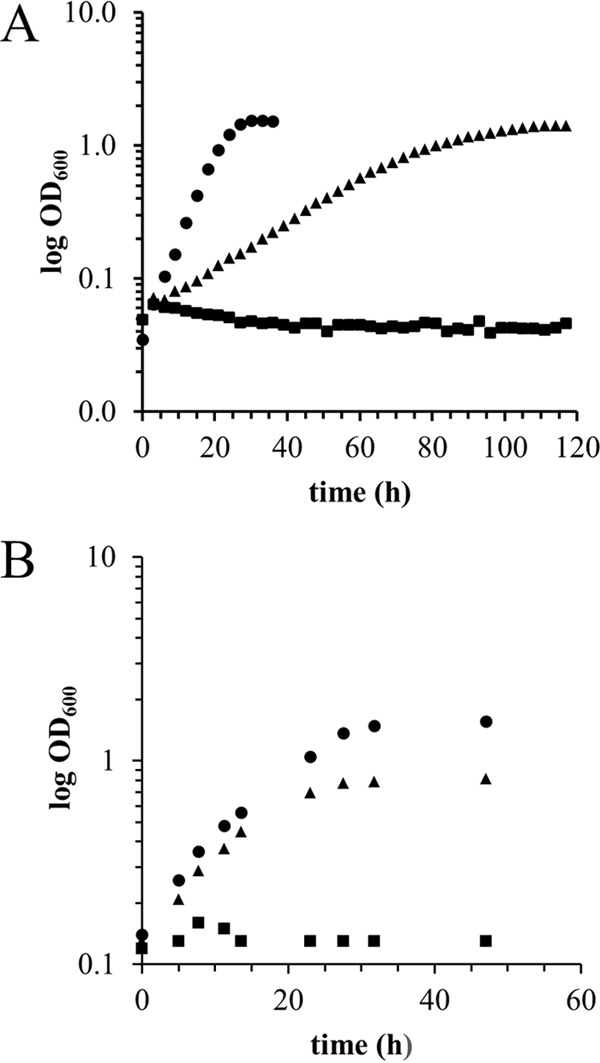
La3+-dependent methylotrophic growth with methanol and ethanol. Optical density was monitored over time for cultures grown in minimal medium with 125 mM methanol (A) or 34 mM ethanol (B) as the growth substrate. The growth medium was supplemented with 2 μM LaCl3. Represented strains are wild-type M. extorquens AM1 (circles), MDH-3 (triangles), and MDH-3 exaF::Km (squares). The results are representative of three biological replicate cultures for each strain.
MDH activity of ExaF in cell extracts.
MDH activity was measured in cell extracts from the MDH-3 mutant strain grown with 125 mM methanol and La3+ (29) but found to be very low (9 nmol · mg−1 · min−1 compared to 81 nmol · mg−1 · min−1 for the wild-type strain) when the traditional DCPIP-linked assay was used as described by Anthony and Zatman (45). By modifying the DCPIP-linked MDH assay (see Materials and Methods), including the use of methylamine rather than ammonium as the activator, we detected 5-fold-higher activity of ExaF in cell extracts of the MDH-3 mutant strain (45 ± 2 nmol · mg−1 · min−1) than what was previously reported (29). This finding indicates that methylamine is a better activator for ExaF in vitro than ammonium. With our modifications to the assay, we detected minimal to no methanol-independent reduction of DCPIP in the “no-substrate” control assay preparations with PMS as an artificial electron acceptor and methylamine as an activator, making it an improved protocol for measuring the enzyme kinetics for ExaF.
Purification of ExaF.
As growth of the MDH-3 mutant with methanol as a substrate is dependent on the addition of exogenous La3+ to the medium, biochemical investigation of ExaF was performed to directly test the metal specificity.
His-tagged (C-terminal) ExaF was expressed in wild-type M. extorquens AM1 from the La3+-inducible promoter Pxox1 in the presence of La3+ (29, 30) or from Pmxa in the absence of La3+ in minimal methanol medium. MDH activity was not detectable in extracts from cultures lacking La3+ when methylamine was added as the activator for the assay. ExaF-His6 from both cultures, with (ExaF-La) and without (ExaF-Ca) La3+, was purified (Fig. 2A), and the histidine tag was cleaved. Purified ExaF-La had a specific activity for methanol of 6.5 ± 0.2 μmol · mg−1 · min−1 as measured following the reduction of DCPIP. A 40% decrease in activity was observed for ExaF-La when ammonium replaced methylamine as the activator. Specific activities of 6 ± 2 (pH = 7) and 37 ± 4 (pH = 9) nmol · mg−1 · min−1 for ExaF-La were measured following the reduction of cytochrome c. Purified ExaF-Ca did not exhibit any oxidation activity toward methanol. ExaF-La was stable when kept on ice at 4°C for at least 72 h, retaining >90% activity. After 7 days under the same conditions, the enzyme retained >50% of the initial activity. ExaF-La that had been frozen with 10% glycerol and stored at −80°C was no longer active after 12 h, but activity could be recovered by incubating the thawed enzyme with an equimolar concentration of LaCl3 and 20 μM PQQ.
FIG 2.
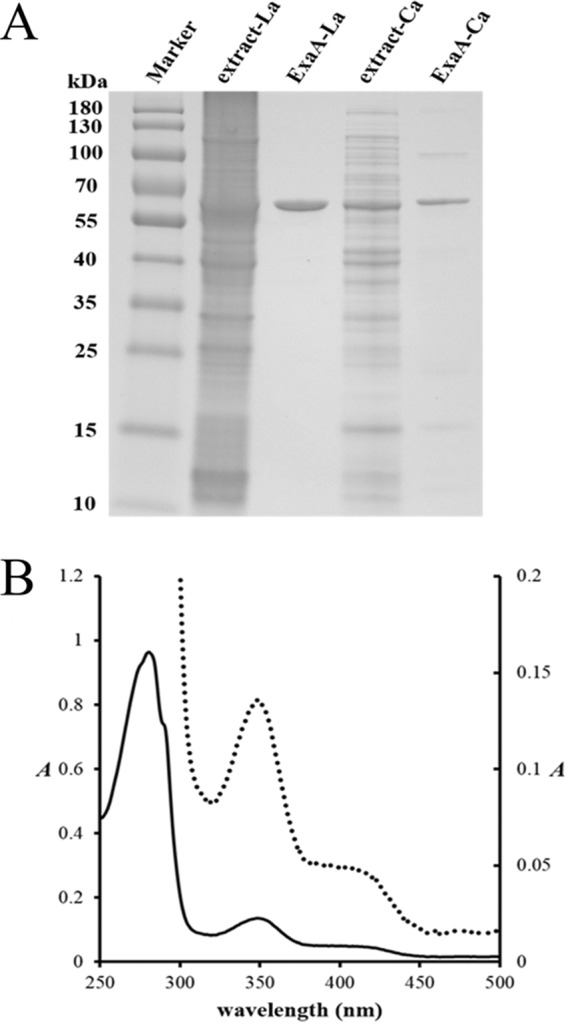
Production, purification, and UV-Vis analysis of ExaF. (A) SDS-PAGE analysis of ExaF produced with or without La3+. ExaF was produced in wild-type M. extorquens AM1 using either pNG265 with the La-inducible xox1 promoter or pNG271 with the mxa promoter, which is constitutive in the absence of exogenous lanthanides. Cells were grown in methanol minimal medium with (pNG265) or without (pNG271) La3+ and purified by IMAC. Lanes (left to right): protein marker, cell extract with La3+, purified ExaF from culture grown with La3+, cell extract without La3+, and purified ExaF from culture grown without La3+. (B) UV-visible spectrum of purified ExaF. Enzyme (0.9 mg/ml) was prepared in 25 mM Tris-HCl, pH 8.0. The solid line corresponds to the left axis. The dotted line corresponds to the right axis.
Analytical characterization of ExaF.
ExaF exhibited an apparent molecular mass of ∼61 kDa for the denatured protein (Fig. 2A), consistent with the expected value based on the gene sequence. The native molecular mass of ExaF was determined by SEC-MALS to be 126.4 ± 2.3 kDa, corresponding to a dimer. The UV-visible absorption spectrum of the purified enzyme (14.2 μM) is consistent with spectra of other PQQ-containing enzymes (32–34), with a broad shoulder and maximum absorbance from 345 to 350 nm in addition to transitions near 280 nm from the aromatic residues (Fig. 2B). Using the molar extinction coefficient of 9,620 M−1 · cm−1 (47) for PQQ, the concentration of bound quinone was 13.2 μM, indicating a 1:1 molar ratio to protomer of enzyme. The pronounced absorbance at 345 nm was suggestive of a metal ion in the enzyme active site (52, 53). ICP-MS measurements of the purified enzyme from culture grown with La3+ determined 1.3 mol of La3+ per mol of ExaF protomer, indicating a 1:1 ratio of La3+ to protomer of enzyme.
Enzyme kinetics of ExaF with one-carbon substrates.
The capacity to generate formate as the end product of methanol oxidation has been suggested for XoxF-MDH (22, 27). Our initial biochemical and phenotypic studies suggested that ExaF may exhibit a similar catalytic property, thus warranting further biochemical investigation of the enzyme. To gain further insight into the catalytic properties of ExaF with one-carbon substrates, we measured kinetic parameters of ExaF with methanol and formaldehyde. Vmax and Km values are reported in Fig. 3C. The maximal reaction velocity of ExaF-La for formaldehyde was 23% greater than for methanol as the substrate. The Km for formaldehyde was in the micromolar range and ∼100 times lower than the Km for methanol. Thus, kcat/Km determinations show that ExaF-La is 100 times more efficient at oxidizing formaldehyde than methanol. Activity was not detected for ExaF-Ca with methanol. The kinetic measurements indicate that ExaF-La can catalyze the oxidation of formaldehyde and support the possibility that ExaF-La is capable of oxidizing methanol to formate in vivo. Interestingly, ExaF-La is also an efficient formaldehyde dehydrogenase (Fig. 3C), suggesting a possible role in formaldehyde metabolism.
FIG 3.

ExaF kinetic parameters with one- and two-carbon substrates. Michaelis-Menten plots are shown for methanol and formaldehyde (A) and ethanol and acetaldehyde (B). Kinetic parameters are given for one-carbon substrates (C) and two-carbon substrates (D).
Enzyme kinetics with two-carbon substrates.
The low efficiency of ExaF methanol oxidation suggests that methanol is not the primary substrate of this enzyme. Because ExaF is a homolog of the quinoprotein ethanol dehydrogenases, we investigated the kinetic properties of ExaF with the two-carbon substrates, ethanol and acetaldehyde. Vmax and Km values for ExaF-La are reported in Fig. 3D. The Vmax with ethanol was 6.3 U · mg−1, similar to the maximal velocity with methanol; however, the Km for ethanol was determined to be 0.9 μM, compared to 14, 11, and 163 μM for homologous enzymes from Pseudomonas strains (32, 37, 54), making it the lowest value ever reported for a quinoprotein ethanol dehydrogenase. When limiting concentrations of ethanol were added to the assay, approximately two times the molar equivalent of DCPIP was reduced (∼230%), indicating that ethanol was being oxidized to the level of acetic acid. The Vmax with acetaldehyde was 2.7 U · mg−1, 3-fold lower than the maximal velocity observed with formaldehyde, while the Km for acetaldehyde was over 4-fold higher than that of formaldehyde. In comparison, ExaF-Ca oxidation activities with ethanol (7 nmol · mg−1 · min−1) or acetaldehyde (24 nmol · mg−1 · min−1) as the substrate for this form of the enzyme were considerably lower. Taken together, the kinetic analyses demonstrate that ExaF is a highly efficient, lanthanide-dependent ethanol dehydrogenase.
Phenotypic analysis of ExaF-dependent growth with ethanol.
Because our kinetic studies strongly indicated that ethanol is its primary substrate, we assessed the role of ExaF in ethanol metabolism. Growth rate constants were determined for the wild-type, MDH-3, and the MDH-3 exaF::Km strains using ethanol as their sole carbon source (Table 4; Fig. 1B). The wild-type strain grew similarly in the presence and absence of La3+; however, the MDH-3 mutant strain grew only when exogenous La3+ was added to the growth medium. Additional loss of exaF::Km in the MDH-3 mutant background eliminated this growth, suggesting a contribution by ExaF during ethanol growth when La3+ is available. The single exaF mutant did not exhibit a growth defect compared to the wild-type strain with or without exogenous La3+, indicating that either XoxF-type MDH (in the presence of La3+) or MxaFI-type MDH (in the absence of La3+) can support growth with ethanol as well.
DISCUSSION
A role for lanthanides in microbial metabolism has only recently been discovered, and its study continues to uncover surprises. Prior to this work, XoxF-type MDHs were the only enzymes known to contain lanthanides in their active sites (27). ExaF (sometimes referred to as QEDH) homologs from P. aeruginosa, Rhodopseudomonas, and Acinetobacter have all been reported to contain Ca2+ (32, 33, 35, 37, 55, 56). We were unable to detect oxidation activity for ExaF from M. extorquens AM1 in cell extracts from culture grown without La3+. Pure ExaF-Ca did not exhibit activity with methanol and exhibited minimal activity with ethanol. ICP-MS measurements confirmed that ExaF preferentially binds La3+, and kinetic analyses showed that ExaF oxidizes ethanol and methanol at similar rates but that it has a clear substrate preference for ethanol (Fig. 3A and C). Interestingly, activity following the reduction of cytochrome c at neutral pH was detected under oxic conditions, providing insights to design an activity assay independent of redox dyes. Compared to the other quinoprotein ethanol dehydrogenases, ExaF is a remarkably efficient enzyme (Fig. 4). ExaF is most similar to the quinoprotein ethanol dehydrogenase from P. aeruginosa. Their amino acid sequences are 57% identical, and structural homology is over 90% with a root mean square deviation of 0.246 (Fig. 5). However, of the quinoprotein ethanol dehydrogenases, only QEDH from Pseudomonas putida has an aspartate residue positioned for lanthanide coordination.
FIG 4.
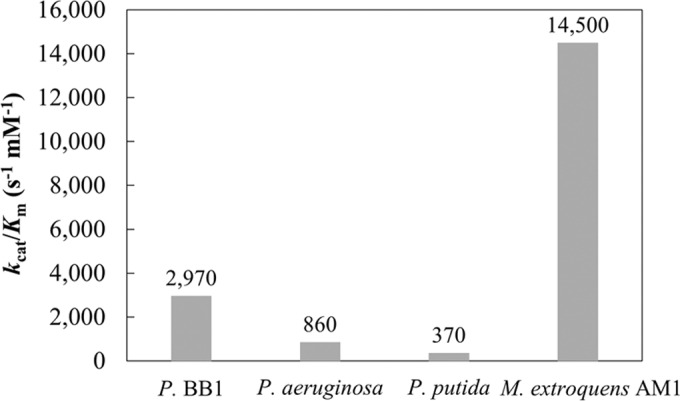
Catalytic efficiencies of quinoprotein ethanol dehydrogenases reported as kcat/Km. Enzymes are represented as follows: P. BB1, QEDH (54); P. aeruginosa, QEDH (32); P. putida, QEDH (37); M. extorquens AM1, ExaF (QEDH) (this study). Values are for ethanol as the substrate.
FIG 5.
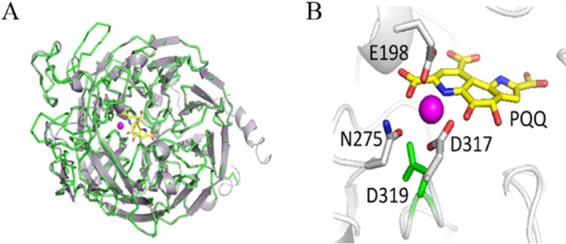
ExaF homology model. (A) Predicted structure of ExaF (shown in cartoon view; gray) using the crystal structure of the quinoprotein ethanol dehydrogenase QEDH from P. aeruginosa (green; PDB 1FLG) (59) as the template. (B) Expanded view of the catalytic site of ExaF. PQQ is shown in yellow. La3+ metal ligand is shown in magenta. The La3+ position was modeled after the Ca2+ in the QEDH structure. Amino acids predicted to coordinate with La3+ are numbered according to their positions in the structure. The aspartate residue (D319) that has been suggested to be necessary for lanthanide coordination is highlighted in green, next to the predicted catalytic aspartate (D317).
It has been proposed that La3+ and lanthanides in general may function as more potent Lewis acids than Ca2+, generating a stronger polarization of PQQ for catalysis (22). It has also been proposed that incorporation of a lanthanide into the active site may modify the kinetic properties of the enzyme, as suggested by two XoxF-type MDHs that demonstrate relatively higher formaldehyde oxidation efficiencies than those of MxaF-type MDHs (22, 27). Formaldehyde is a highly toxic intermediate that is integral to methylotrophy. ExaF can also oxidize formaldehyde with relatively high efficiency, suggesting a possible contribution by ExaF to methylotrophy. This result contrasts ExaF with other reported QEDH enzymes that are generally inefficient catalysts for formaldehyde oxidation (32, 33, 35, 56).
Our phenotypic analyses show that in the MDH-3 mutant strain, ExaF supports growth with methanol when exogenous La3+ is added to the medium. The kinetic analysis with methanol indicates that ExaF is not an efficient methanol dehydrogenase, suggesting that the methanol oxidation detected in vivo in the MDH-3 mutant strain is due to a promiscuous activity of ExaF. This low activity is a likely explanation for the lower growth rate with methanol observed for the MDH-3 mutant. It is unclear at this time why growth yields are 2-fold higher in the MDH-3 strain than in the wild type when grown in medium with ethanol and La3+. One possibility is that loss of the MDHs affects expression of exaF, as XoxF has previously been shown to affect expression of xoxF and mxaF (31). If exaF expression is upregulated in the MDH-3 mutant, this may allow for increased growth yields, as ethanol may be converted to acetate, preventing the loss of valuable carbon as volatile acetylaldehyde. Alternatively, the metabolic network may be altered in the MDH-3 mutant, redirecting carbon and nutrient flow. A systems biology approach would be helpful in identifying the cause of yield difference.
The lack of a growth rate defect in the MDH-3 strain grown with ethanol as a sole carbon source demonstrates the contribution of ExaF to ethanol oxidation in vivo. Therefore, we add ExaF to the lanthanide-utilizing enzymes and demonstrate that the role of lanthanides in microbial metabolism extends beyond XoxF-type MDHs and one-carbon metabolism. It is unclear, however, whether or not ExaF is the primary dehydrogenase utilized to oxidize ethanol when La3+ is available, since the exaF single mutant does not exhibit a growth defect with ethanol compared to the wild-type strain. It is likely that XoxF also contributes to ethanol growth with La3+, although kinetic studies of XoxF1 from M. extorquens AM1 are needed to determine its capacity for oxidizing substrates other than methanol. Further studies involving genetic and phenotypic growth analyses might determine which enzyme, if either, is the predominant catalyst for ethanol oxidation in vivo in the presence of La3+. Alternatively, both ExaF and XoxF may be produced to provide more robust alcohol oxidation capacity than a single dehydrogenase. Methylobacterium species are often associated with the aerial parts of the plant, or the phyllosphere, which is considered a dynamic and oligotrophic environment. Some phyllosphere metabolites can be utilized for growth by these facultative methylotrophic plant epiphytes and include carbohydrates, amino acids, and organic volatile compounds, such as methanol, ethanol, and acetate. The metabolic capability for methanol metabolism in Methylobacterium confers a fitness advantage in situ (6, 7). In the presence of La3+, the XoxF-type MDH is the primary methanol dehydrogenase; however, its capacity for ethanol oxidation is unknown. Producing an arsenal of enzymes to utilize multiple substrates in a nutrient-poor environment such as the phyllosphere may confer a metabolic advantage as well.
ACKNOWLEDGMENTS
We thank R. Hausinger for his careful and insightful reading of the manuscript, as well as for allowing us access to analytical instruments used in this work. We thank L. Bolerjack for assistance with Gibson Assembly. We also thank F. Yarza and R. Crisostomo for assistance in conducting growth curve analyses and Daniel Parrell for his assistance with the enzyme homology model.
Funding to support the contributions of N.M.G. and N.C.M.-G. came from Michigan State University startup funds. E.S. and H.N.V. are supported by a San José State University Research, Scholarship and Creative Activity grant and a California State University Program for Education and Research in Biotechnology (CSUPERB) New Investigator Grant. G.A.S. is supported by an SJSU Undergraduate Research Grant.
REFERENCES
- 1.Anthony C. 1982. The biochemistry of methylotrophs. Academic Press, London, United Kingdom. [Google Scholar]
- 2.Dunstan PM, Anthony C. 1972. Microbial metabolism of C1 and C2 compounds. Biochem J 128:107–115. doi: 10.1042/bj1280107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunstan PM, Anthony C, Drabble WT. 1972. Microbial metabolism of C 1 and C 2 compounds. The involvement of glycollate in the metabolism of ethanol and of acetate by Pseudomonas AM1. Biochem J 128:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corpe W, Rheem S. 1989. Ecology of the methylotrophic bacteria on living leaf surfaces. FEMS Microbiol Lett 62:243–249. doi: 10.1111/j.1574-6968.1989.tb03698.x. [DOI] [Google Scholar]
- 5.Vorholt JA. 2012. Microbial life in the phyllosphere. Nat Rev Microbiol 10:828–840. doi: 10.1038/nrmicro2910. [DOI] [PubMed] [Google Scholar]
- 6.Sy A, Timmers ACJ, Knief C, Vorholt JA. 2005. Methylotrophic metabolism is advantageous for Methylobacterium extorquens during colonization of Medicago truncatula under competitive conditions. Appl Environ Microbiol 71:7245–7252. doi: 10.1128/AEM.71.11.7245-7252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abanda-Nkpwatt D, Müsch M, Tschiersch J, Boettner M, Schwab W. 2006. Molecular interaction between Methylobacterium extorquens and seedlings: growth promotion, methanol consumption, and localization of the methanol emission site. J Exp Bot 57:4025–4032. doi: 10.1093/jxb/erl173. [DOI] [PubMed] [Google Scholar]
- 8.Chistoserdova L. 2011. Modularity of methylotrophy, revisited. Environ Microbiol 13:2603–2622. doi: 10.1111/j.1462-2920.2011.02464.x. [DOI] [PubMed] [Google Scholar]
- 9.Anthony C. 1996. Quinoprotein-catalysed reactions. Biochem J 320:697–711. doi: 10.1042/bj3200697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anthony C. 1986. Bacterial oxidation of methane and methanol. Adv Microb Physiol 27:113–110. doi: 10.1016/S0065-2911(08)60305-7. [DOI] [PubMed] [Google Scholar]
- 11.Chistoserdova L, Chen S-W, Lapidus A, Lidstrom ME. 2003. Methylotrophy in Methylobacterium extorquens AM1 from a genomic point of view. J Bacteriol 185:2980–2987. doi: 10.1128/JB.185.10.2980-2987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vuilleumier S, Chistoserdova L, Lee M-C, Bringel F, Lajus A, Zhou Y, Gourion B, Barbe V, Chang J, Cruveiller S, Dossat C, Gillett W, Gruffaz C, Haugen E, Hourcade E, Levy R, Mangenot S, Muller E, Nadalig T, Pagni M, Penny C, Peyraud R, Robinson DG, Roche D, Rouy Z, Saenampechek C, Salvignol G, Vallenet D, Wu Z, Marx CJ, Vorholt JA, Olson MV, Kaul R, Weissenbach J, Médigue C, Lidstrom ME. 2009. Methylobacterium genome sequences: a reference blueprint to investigate microbial metabolism of C1 compounds from natural and industrial sources. PLoS One 4:e5584. doi: 10.1371/journal.pone.0005584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chistoserdova L, Lapidus A, Han C, Goodwin L, Saunders L, Brettin T, Tapia R, Gilna P, Lucas S, Richardson PM, Lidstrom ME. 2007. Genome of Methylobacillus flagellatus, molecular basis for obligate methylotrophy, and polyphyletic origin of methylotrophy. J Bacteriol 189:4020–4027. doi: 10.1128/JB.00045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ward N, Larsen Ø, Sakwa J, Bruseth L, Khouri H, Durkin AS, Dimitrov G, Jiang L, Scanlan D, Kang KH, Lewis M, Nelson KE, Methé B, Wu M, Heidelberg JF, Paulsen IT, Fouts D, Ravel J, Tettelin H, Ren Q, Read T, DeBoy RT, Seshadri R, Salzberg SL, Jensen HB, Birkeland NK, Nelson WC, Dodson RJ, Grindhaug SH, Holt I, Eidhammer I, Jonasen I, Vanaken S, Utterback T, Feldblyum TV, Fraser CM, Lillehaug JR, Eisen JA. 2004. Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol 2:e303. doi: 10.1371/journal.pbio.0020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia ZX, Dai WW, Xiong JP, Hao ZP, Davidson VL, White S, Mathews FS. 1992. The three-dimensional structures of methanol dehydrogenase from two methylotrophic bacteria at 2.6-Å resolution. J Biol Chem 267:22289–22297. [PubMed] [Google Scholar]
- 16.Ghosh M, Anthony C, Harlos K, Goodwin MG, Blake C. 1995. The refined structure of the quinoprotein methanol dehydrogenase from Methylobacterium extorquens at 1.94 Å. Structure 3:177–187. doi: 10.1016/S0969-2126(01)00148-4. [DOI] [PubMed] [Google Scholar]
- 17.Harms N, Ras J, Koning S, Reijnders WNM, Stouthamer AH, van Spanning RJM. 1996. Genetics of C1 metabolism regulation in Paracoccus denitrificans, p 126–132. In Lidstrom ME, Tabita FR (ed), Microbial growth on C1 compounds. Kluwer Academic Publishers, Dordrecht, The Netherlands. [Google Scholar]
- 18.Chistoserdova L. 2011. Methylotrophy in a lake: from metagenomics to single-organism physiology. Appl Environ Microbiol 77:4705–4711. doi: 10.1128/AEM.00314-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chistoserdova L, Lidstrom M. 2013. Aerobic methylotrophic prokaryotes, p 267–285. In Rosenberg E, DeLong EF, Thompson F, Lory S, Stackebrandt E (ed), The prokaryotes, 4th ed Springer, Berlin, Germany. [Google Scholar]
- 20.Sowell SM, Abraham PE, Shah M, Verberkmoes NC, Smith DP, Barofsky DF, Giovannoni SJ. 2011. Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J 5:856–865. doi: 10.1038/ismej.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalyuzhnaya MG, Lapidus A, Ivanova N, Copeland AC, McHardy AC, Szeto E, Salamov A, Grigoriev IV, Suciu D, Levine SR, Markowitz VM, Rigoutsos I, Tringe SG, Bruce DC, Richardson PM, Lidstrom ME, Chistoserdova L. 2008. High-resolution metagenomics targets specific functional types in complex microbial communities. Nat Biotechnol 26:1029–1035. doi: 10.1038/nbt.1488. [DOI] [PubMed] [Google Scholar]
- 22.Keltjens JT, Pol A, Reimann J, Op Den Camp HJM. 2014. PQQ-dependent methanol dehydrogenases: rare-earth elements make a difference. Appl Microbiol Biotechnol 98:6163–6183. doi: 10.1007/s00253-014-5766-8. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt S, Christen P, Kiefer P, Vorholt JA. 2010. Functional investigation of methanol dehydrogenase-like protein XoxF in Methylobacterium extorquens AM1. Microbiology 156:2575–2586. doi: 10.1099/mic.0.038570-0. [DOI] [PubMed] [Google Scholar]
- 24.Hibi Y, Asai K, Arafuka H, Hamajima M, Iwama T, Kawai K. 2011. Molecular structure of La3+-induced methanol dehydrogenase-like protein in Methylobacterium radiotolerans. J Biosci Bioeng 111:547–549. doi: 10.1016/j.jbiosc.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 25.Nakagawa T, Mitsui R, Tani A, Sasa K, Tashiro S, Iwama T, Hayakawa T, Kawai K. 2012. A catalytic role of XoxF1 as La3+-dependent methanol dehydrogenase in Methylobacterium extorquens strain AM1. PLoS One 7:e50480. doi: 10.1371/journal.pone.0050480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu ML, Wessels HJCT, Pol A, Op den Camp HJM, Jetten MSM, van Niftrik L, Keltjens JT. 2015. XoxF-type methanol dehydrogenase from the anaerobic Methanotroph “Candidatus Methylomirabilis oxyfera.” Appl Environ Microbiol 81:1442–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pol A, Barends TRM, Dietl A, Khadem AF, Eygensteyn J, Jetten MSM, Op den Camp HJM. 2014. Rare earth metals are essential for methanotrophic life in volcanic mudpots. Environ Microbiol 16:255–264. doi: 10.1111/1462-2920.12249. [DOI] [PubMed] [Google Scholar]
- 28.Skovran E, Martinez-Gomez NC. 2015. Just add lanthanides. Science 348:862–863. doi: 10.1126/science.aaa9091. [DOI] [PubMed] [Google Scholar]
- 29.Vu HN, Subuyuj GA, Vijayakumar S, Good NM, Martinez-Gomez NC, Skovran E. 2016. Lanthanide-dependent regulation of methanol oxidation systems in Methylobacterium extorquens AM1 and their contribution to methanol growth. J Bacteriol 198:1250–1259. doi: 10.1128/JB.00937-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farhan Ul Haque M, Kalidass B, Bandow N, Turpin EA, Dispirito AA, Semrau JD. 2015. Cerium regulates expression of alternative methanol dehydrogenases in Methylosinus trichosporium OB3b. Appl Environ Microbiol 81:7546–7552. doi: 10.1128/AEM.02542-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skovran E, Palmer AD, Rountree AM, Good NM, Lidstrom ME. 2011. XoxF is required for expression of methanol dehydrogenase in Methylobacterium extorquens AM1. J Bacteriol 193:6032–6038. doi: 10.1128/JB.05367-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Görisch H, Rupp M. 1989. Quinoprotein ethanol dehydrogenase from Pseudomonas. Antonie Van Leeuwenhoek 56:35–45. doi: 10.1007/BF00822582. [DOI] [PubMed] [Google Scholar]
- 33.Groen B, Frank J, Duine JA. 1984. Quinoprotein alcohol dehydrogenase from ethanol-grown Pseudomonas aeruginosa. Biochem J 223:921–924. doi: 10.1042/bj2230921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mutzel A, Görisch H. 1991. Quinoprotein ethanol dehydrogenase: preparation of the apo-form and reconstitution with pyrroloquinoline quinone and Ca2+ or Sr2+ ions. Agric Biol Chem 55:1721–1726. doi: 10.1080/00021369.1991.10870854. [DOI] [Google Scholar]
- 35.Bamforth CW, Quayle JR. 1978. The dye-linked alcohol dehydrogenase of Rhodopseudomonas acidophila. Comparison with dye-linked methanol dehydrogenases. Biochem J 169:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rupp M, Görisch H. 1988. Purification, crystallisation and characterization of quinoprotein ethanol dehydrogenase from Pseudomonas aeruginosa. Biol Chem Hoppe Seyler 369:431–439. doi: 10.1515/bchm3.1988.369.1.431. [DOI] [PubMed] [Google Scholar]
- 37.Toyama H, Fujii A, Matsushita K, Shinagawa E, Ameyama M, Adachi O. 1995. Three distinct quinoprotein alcohol dehydrogenases are expressed when Pseudomonas putida is grown on different alcohols. J Bacteriol 177:2442–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marx CJ, Lidstrom ME. 2002. Broad-host-range cre-lox system for antibiotic marker recycling in Gram-negative bacteria. Biotechniques 33:1062–1067. [DOI] [PubMed] [Google Scholar]
- 39.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 40.Gibson DG. 2011. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marx CJ, Lidstrom ME. 2001. Development of improved versatile broad-host-range vectors for use in methylotrophs and other Gram-negative bacteria. Microbiology 147:2065–2075. doi: 10.1099/00221287-147-8-2065. [DOI] [PubMed] [Google Scholar]
- 42.Good NM, Martinez-Gomez NC, Beck DAC, Lidstrom ME. 2015. Ethylmalonyl coenzyme A mutase operates as a metabolic control point in Methylobacterium extorquens AM1. J Bacteriol 197:727–735. doi: 10.1128/JB.02478-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delaney NF, Kaczmarek ME, Ward LM, Swanson PK, Lee M-C, Marx CJ. 2013. Development of an optimized medium, strain and high-throughput culturing methods for Methylobacterium extorquens. PLoS One 8:e62957. doi: 10.1371/journal.pone.0062957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armstrong JM. 1964. The molar extinction coefficient of 2,6-dichlorophenol indophenol. Biochim Biophys Acta 86:194–197. doi: 10.1016/0304-4165(64)90180-1. [DOI] [PubMed] [Google Scholar]
- 45.Anthony C, Zatman LJ. 1967. The microbial oxidation of methanol. The prosthetic group of the alcohol dehydrogenase of Pseudomonas sp. M27: a new oxidoreductase prosthetic group. Biochem J 104:960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. 1985. Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 47.Duine JA, Frank J, Jongejan JA. 1987. Enzymology of quinoproteins. Adv Enzymol Relat Areas Mol Biol 59:169–212. [DOI] [PubMed] [Google Scholar]
- 48.Kelley L, Mezulis S, Yates C, Wass M, Sternberg M. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA. [Google Scholar]
- 50.Morris CJ, Biville F, Turlin E, Lee E, Ellermann K, Fan WH, Ramamoorthi R, Springer AL, Lidstrom ME. 1994. Isolation, phenotypic characterization, and complementation analysis of mutants of Methylobacterium extorquens AM1 unable to synthesize pyrroloquinoline quinone and sequences of pqqD, pqqG, and pqqC. J Bacteriol 176:1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Springer AL, Ramamoorthi R, Lidstrom ME. 1996. Characterization and nucleotide sequence of pqqE and pqqF in Methylobacterium extorquens AM1. J Bacteriol 178:2154–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodwin MG, Anthony C. 1996. Characterization of a novel methanol dehydrogenase containing a Ba2+ ion at the active site. Biochem J 318:673–679. doi: 10.1042/bj3180673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goodwin MG, Avezoux A, Dales SL, Anthony C. 1996. Reconstitution of the quinoprotein methanol dehydrogenase from inactive Ca2+-free enzyme with Ca2+, Sr2+ or Ba2+. Biochem J 319:839–842. doi: 10.1042/bj3190839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dijkstra M, Van Den Tweel W, De Bont J, Frank J, Duine J. 1985. Monomeric and dimeric quinoprotein alcohol dehydrogenase from alcohol-grown Pseudomonas BB1. J Gen Microbiol 131:3163–3169. [Google Scholar]
- 55.Chattopadhyay A, Förster-Fromme K, Jendrossek D. 2010. PQQ-dependent alcohol dehydrogenase (QEDH) of Pseudomonas aeruginosa is involved in catabolism of acyclic terpenes. J Basic Microbiol 50:119–124. doi: 10.1002/jobm.200900178. [DOI] [PubMed] [Google Scholar]
- 56.Duine JA, Frank J. 1981. Quinoprotein alcohol dehydrogenase from a non-methylotroph, Acinetobacter calcoaceticus. J Gen Microbiol 122:201–209. [DOI] [PubMed] [Google Scholar]
- 57.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 58.Nunn DN, Lidstrom ME. 1986. Isolation and complementation analysis of 10 methanol oxidation mutant classes and identification of the methanol dehydrogenase structural gene of Methylobacterium sp. strain AM1. J Bacteriol 166:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keitel T, Diehl A, Knaute T, Stezowski JJ, Höhne W, Görisch H. 2000. X-ray structure of the quinoprotein ethanol dehydrogenase from Pseudomonas aeruginosa: basis of substrate specificity. J Mol Biol 297:961–974. doi: 10.1006/jmbi.2000.3603. [DOI] [PubMed] [Google Scholar]