

Abstract
Middle East respiratory syndrome (MERS) is an emerging infectious disease associated with a relatively high mortality rate of approximately 40%. MERS is caused by MERS coronavirus (MERS-CoV) infection, and no specific drugs or vaccines are currently available to prevent MERS-CoV infection. MERS-CoV is an enveloped virus, and its envelope protein (S protein) mediates membrane fusion at the plasma membrane or endosomal membrane. Multiple proteolysis by host proteases, such as furin, transmembrane protease serine 2 (TMPRSS2), and cathepsins, causes the S protein to become fusion competent. TMPRSS2, which is localized to the plasma membrane, is a serine protease responsible for the proteolysis of S in the post-receptor-binding stage. Here, we developed a cell-based fusion assay for S in a TMPRSS2-dependent manner using cell lines expressing Renilla luciferase (RL)-based split reporter proteins. S was stably expressed in the effector cells, and the corresponding receptor for S, CD26, was stably coexpressed with TMPRSS2 in the target cells. Membrane fusion between these effector and target cells was quantitatively measured by determining the RL activity. The assay was optimized for a 384-well format, and nafamostat, a serine protease inhibitor, was identified as a potent inhibitor of S-mediated membrane fusion in a screening of about 1,000 drugs approved for use by the U.S. Food and Drug Administration. Nafamostat also blocked MERS-CoV infection in vitro. Our assay has the potential to facilitate the discovery of new inhibitors of membrane fusion of MERS-CoV as well as other viruses that rely on the activity of TMPRSS2.
INTRODUCTION
Middle East respiratory syndrome (MERS), an emerging acute respiratory syndrome, was first described in 2012 (1, 2). The causative agent of the disease is MERS coronavirus (MERS-CoV), a member of the family of betacoronaviruses, and studies have supported the hypothesis that MERS-CoV infection has zoonotic origins involving bats and camels (3–6). The mortality rate is around 40%, with most fatalities occurring in patients with underlying medical conditions (7, 8). The absence of specific antiviral drugs and vaccines makes it difficult to control the spread of MERS, as revealed in the outbreak in South Korea in 2015 (9–11).
MERS-CoV is an enveloped virus, and its envelope protein (S protein) recognizes CD26 (also known as dipeptidyl peptidase [DPP4]) as the host receptor (12). The virus may enter the host cells via membrane fusion either at the plasma membrane or in the endosomes after endocytosis. In both pathways, the S protein undergoes additional proteolytic processing by the auxiliary host proteases to become fusion competent (13). The involvement of TMPRSS2 at the plasma membrane and cathepsins in endosomes has been demonstrated in several studies (13–17). This proteolytic processing step is essential and appears to occur only after S binds to CD26, providing another parameter for host range specificity. A previous study showed that TMPRSS2 inhibition was more effective than cathepsin inhibition in the prevention of MERS-CoV infection in vitro (16). Thus, MERS-CoV may rely on the direct membrane fusion pathway at the plasma membrane rather than on endocytic entry.
Fusion inhibitors halt the viral life cycle at its first step and minimize viral damage to the host. Here, we developed a cell-based membrane fusion assay for the MERS-CoV S protein to screen potential fusion inhibitors. We employed a pair of split reporter proteins called dual split proteins (DSPs), which have been used to analyze membrane fusion in several viruses (18–21). DSPs are the chimeric split proteins between green fluorescent protein (GFP) and Renilla luciferase (RL), and membrane fusion can be monitored by either GFP or RL signals (22). Although the membrane fusion can be detected by image analysis of the GFP signals, easier and more quantitative monitoring of membrane fusion can be achieved by measuring the RL activities with a luminometer using a membrane-permeant substrate for RL.
We optimized the DSP assay for high-throughput screening (HTS) of inhibitors against S-mediated membrane fusion and screened a library containing about 1,000 U.S. Food and Drug Administration (FDA)-approved drugs. From this screening, we identified nafamostat, a serine protease inhibitor, as a potent inhibitor of S-mediated membrane fusion. Based on this result, we further tested several other clinically approved inhibitors of serine proteases and found that nafamostat was the most potent inhibitor among the evaluated compounds. We also demonstrated the ability of nafamostat to block MERS-CoV infection in vitro.
MATERIALS AND METHODS
Cell lines and transient transfection.
A pair of previously described 293FT-based reporter cell lines that constitutively express individual split reporters (DSP1-7 and DSP8-11 proteins) (23) were used in this study and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and 1 μg/ml puromycin. For establishment of stable cell lines expressing MERS coronavirus S (MERS-S), CD26, and TMPRSS2, recombinant pseudotype retroviruses were produced using plat-E cells with a vesicular stomatitis virus (VSV)-G-expressing plasmid (24). 293FT-derived reporter cells infected with pseudotype viruses were selected with 1 μg/ml puromycin, 10 μg/ml blasticidin, and 300 μg/ml hygromycin for at least 1 week. These bulk selected cells were used to perform fusion assays. Calu3 (ATCC HTB-55) cells, which were derived from human lung epithelial cells, were used as target cells for the fusion and viral infection assays. Vero/TMPRSS2 cells were derived from Vero cells and expressed TMPRSS2 constitutively, as described previously (25). Briefly, to establish Vero/TMPRSS2 cells, Vero cells were cotransfected with a pCA7 vector carrying TMPRSS2 (pCA7-TMPRSS2) and pCXN2 carrying the neo gene. A Geneticin-resistant TMPRSS2-expressing clone (Vero/TMPRSS2) was selected (25).
Construction of expression vectors.
A synthetic DNA corresponding to the S gene of a MERS coronavirus (EMC 2012) (26) was generated by Taihe Gene (Beijing, China). We codon optimized the first 129 nucleotides to human genes to enhance the expression level of S to a level that was detectable by immunoblotting (data not shown). For establishment of stable cell lines expressing MERS-S, CD26, and TMPRSS2, coding regions of MERS-S and CD26 genes were cloned into a pMXs-internal ribosome entry site (IRES)-blasticidin retroviral vector (24), and TMPRSS2 was cloned into the pMXs-IRES-hygromycin vector.
DSP assay in a 384-well format.
One day before the DSP assay, effector cells expressing MERS-S and DSP8-11 and target cells expressing CD26, TMPRSS2, and DSP1-7 were seeded in 12-well cell culture plates (2 × 105 cells/500 μl) and 100-mm-diameter cell culture dishes (3 × 106 cells/10 ml), respectively. Two hours before the DSP assay, cells were treated with 6 μM EnduRen (Promega, Madison, WI, USA), a substrate for Renilla luciferase, to activate EnduRen. One microliter of each FDA-approved chemical (n = 1) or protease inhibitor (n = 3) dissolved in dimethyl sulfoxide (DMSO) or phosphate-buffered saline [PBS(−)] was added to the 384-well plates (Greiner Bioscience, Frickenhausen, Germany) using a 12-stage workstation (Biotech, Tokyo, Japan). Twenty microliters of DMEM containing 10% FBS and 6 μM EnduRen was added to dilute the chemicals. Next, 40 μl of each single cell suspension (effector and target cells) was added to the wells using a Multidrop dispenser (Thermo Scientific, Waltham, MA, USA). After incubation at 37°C for 4 h, the RL activity was measured using a PHERAStar Plus microplate reader (BMG Labtech, Cary, NC, USA).
Protease inhibitors and drug library.
Gabexate mesylate (Tokyo Chemical Industry, Tokyo, Japan), nafamostat mesylate (Tokyo Chemical Industry), camostat mesilate (Wako, Tokyo, Japan), sivelestat sodium tetrahydrate (LKT Laboratories, St. Paul, MN, USA), rivaroxaban (Adooq Bioscience, Irvine, CA, USA), telaprevir (Adooq Bioscience), and simeprevir (TRC, Toronto, Canada) were dissolved in DMSO at a concentration of 10 mM. Ulinastatin (Mochida Pharmaceutical Co., Ltd., Tokyo, Japan) was dissolved in PBS(−), which lacked Mg2+ and Ca2+. The FDA-approved drug library (L1300) was purchased from Selleck (Houston, TX, USA) and diluted in DMSO at a concentration of 100 μM. The names of the tested drugs are listed in the Table S1 in the supplemental material.
MERS-CoV infection assay.
The MERS-CoV entry assay was performed as described previously (16). Briefly, the target Calu3 cells were pretreated with the respective inhibitors for 1 h, and EMC2012 coronavirus was then added at a multiplicity of infection (MOI) of 10. The viruses and cells were incubated for 6 h, the cells were then lysed using Isogen (Nippon Gene), and the viral RNAs were measured by real-time PCR, as described previously (16). The effects of the respective inhibitors were evaluated by virus infection assays, as described previously. Briefly, the MERS-CoV (MOI = 0.01) was added to Calu3 cells and incubated for 2 h. The culture medium was then replaced with fresh medium containing inhibitors. The culture was maintained for 24 h, and the virus released into the culture supernatant was titrated using Vero/TMPRSS2 cells, as described previously (16).
RESULTS
Establishment of a 293FT cell-based membrane fusion assay system for MERS-CoV S using the split protein reporter.
To establish a cell-based DSP fusion assay for MERS-S protein, we used previously described 293FT cell lines that constitutively express a reporter protein called DSP as the effector and target cells (23). The DSP assay uses a pair of chimeric split reporter proteins between GFP and RL (22). The GFP domains self-associate and facilitate association of split RL domains. By this mechanism, membrane fusion between the effector and target cells by cocultivation can be monitored by both GFP and RL signals generated from the reassociated DSPs (Fig. 1). However, the RL mode provides a more quantitative measurement of membrane fusion than the GFP mode, since the latter requires more sophisticated image analysis for quantitation.
FIG 1.
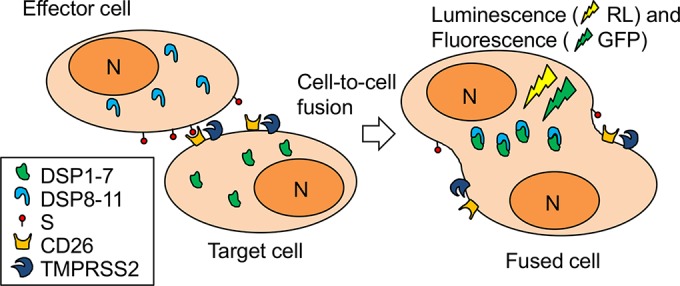
Cell-based membrane fusion assay for the MERS-S protein using the dual split protein (DSP) reporter. Membrane fusion mediated by the MERS-S protein was monitored by coculturing the effector and target cells expressing the respective split reporter proteins (dual split proteins [DSPs]). DSPs are split chimeric proteins of GFP and Renilla luciferase (RL) and recover both GFP (fluorescence) and RL (luminescence) signals by their reassociation upon mixture of cellular contents during membrane fusion. The effector cells express MERS-S protein, and the target cells express CD26 and TMPRSS2. DSP1-7 and DSP8-11, DSPs that reassociate themselves; S, the S protein of MERS-CoV; N, nucleus.
We attempted to establish stable cell lines by transducing necessary components using a retroviral pseudotype vector. We first generated effector cells by introducing the partially codon-optimized MERS-CoV S gene derived from EMC2012 into 293FT cells constitutively expressing DSP8-11. As for the target cells, we used 293FT cells constitutively expressing DSP1-7. The CD26 or TMPRSS2 gene was transduced to establish 293FT/DSP1-7/CD26 or 293FT/DSP1-7/TMPRSS2 cells, respectively. Consistent with previous studies, we found that target cells expressing either CD26 or TMPRSS2 alone were not sufficient to induce membrane fusion (Fig. 2). Finally, we established 293FT/DSP1-7/CD26/TMPRSS2, which expressed DSP1-7, CD26, and TMPRSS2 in a constitutive manner, by transducing TMPRSS2 into 293FT/DSP1-7/CD26 cells. This yielded target cells that could fuse with the effector cells upon cocultivation (Fig. 2). Both the effector and target cells were maintained with respective selection reagents in bulk, and no single cell cloning was performed.
FIG 2.
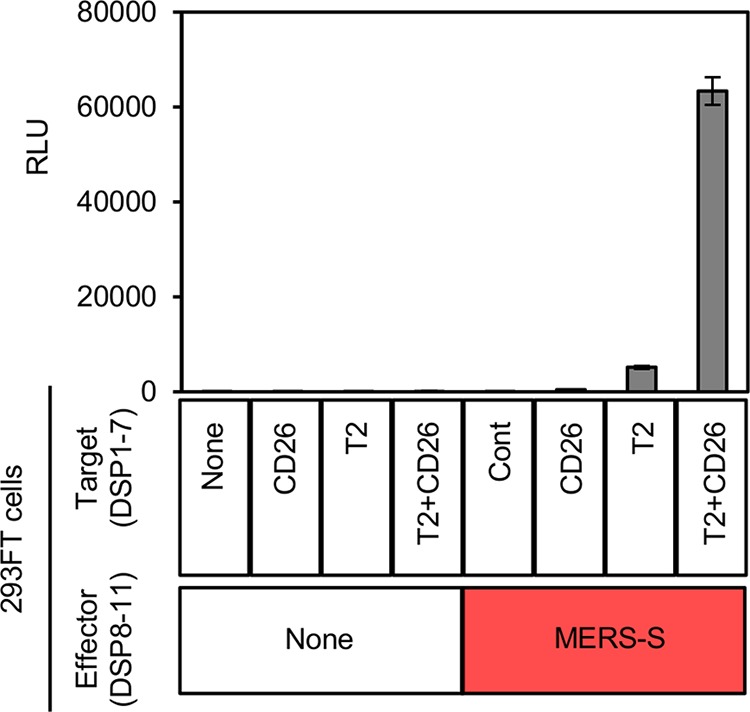
Required components for membrane fusion mediated by MERS-S. The effector 293FT cells expressing DSP8-11 were transduced with MERS-S. The target 293FT cells expressing DSP1-7 were transduced with CD26 and TMPRSS2 separately or simultaneously. Different combinations of these effector and target cells were cocultured, and the resulting RL activity was measured (shown in relative light units [RLU]). The names of the transduced proteins are indicated below the bar graph (None, no proteins other than the DSP reporters; Cont, control).
Optimization of the 293FT-based DSP assay for HTS.
To adapt our DSP assay to an HTS format, several parameters were adjusted, including the numbers of effector and target cells and the duration of cocultivation. The concentration of the substrate for RL, EnduRen, was reduced to 6 μM. Several parameters related to assay stability were examined (see Fig. S1 in the supplemental material). For the negative-control experiments, 293FT/DSP1-7 cells were cocultured with 293FT/DSP8-11/MERS-CoV S cells (n = 16) or 293FT/DSP1-7/CD26/TMPRSS2 cells were cocultured with 293FT/DSP8-11 cells (n = 16). For the positive-control experiment, 293FT/DSP1-7/CD26/TMPRSS2 cells were cocultured with 293FT/DSP8-11/MERS-CoV S cells in the absence of any chemicals (n = 132) or in the presence of 1% DMSO (n = 88), 2% DMSO (n = 66), or 1 μM camostat with 1% DMSO (n = 66). This experiment was performed on a 384-well plate at the same time. Statistical parameters were tested between all negative-control experiments (n = 32) and each positive-control experiment as described in a previous report (27). The Z′ factor value was >0.5 even in the presence of 2% DMSO. This result showed good reproducibility of our assay for HST (27).
Screening of the FDA-approved drug library using the 293FT-based DSP assay.
To identify potential inhibitors of MERS-S-mediated membrane fusion, we first performed a high-throughput DSP assay using 1,017 FDA-approved drugs at a concentration of 1 μM. Primary screening indicated that three compounds inhibited the DSP activity by more than 80% compared with the solvent-only control containing DMSO (Fig. 3A). One of the three compounds, nafamostat mesylate (nafamostat), a serine protease inhibitor, reduced DSP activity to only 1.66% of that of the control (more than 98% inhibition). We next examined the effects of each drug on the RL activity by administering each drug to cells coexpressing DSP1-7 and DSP8-11 (Fig. 3B). If a drug showed inhibition in the latter assay, the drug was therefore not a specific inhibitor of membrane fusion but a nonspecific inhibitor of RL itself. Nafamostat showed no inhibition of the RL activity when applied to cells coexpressing both DSP1-7 and DSP8-11 (Fig. 3B). Therefore, we concluded that nafamostat specifically inhibited DSP activity by interfering with MERS-S-mediated membrane fusion, most likely by inhibiting proteolysis of S by TMPRSS2, a serine protease. Notably, this screening assay indicated that the RL activity of our DSP assay was quite insensitive to nonspecific inhibition by many of the tested drugs. Direct inhibition of RL by 55% was observed with only one drug, fenoprofen calcium (Fig. 3B).
FIG 3.

The results of the high-throughput screening (HTS) of 1,017 FDA-approved drugs in the DSP assay for MERS-S. Results of screening of 1,017 drugs using the HTS DSP assay are shown. (A) The vertical axis shows the reading of the DSP activity (RL activity) for the tested drugs (1 μM). The RL values were normalized to the control, which contained DMSO only. The horizontal axis shows the identification number arbitrarily assigned to each drug. Each dot represents an individual drug. The dotted line indicates 20% of the control value. The value determined for the most active drug, nafamostat (1.66% of the control value), is indicated with a red arrow and red dot. (B) The effect of each drug on RL activity itself. To rule out the nonspecific direct inhibitory effect of each drug on the RL activity itself, each drug (1 μM) was applied to the cells coexpressing both DSP1-7 and DSP8-11, which harbored reassociated active DSP. The results are shown as in panel A. The value for nafamostat (106.1% of the control value) is indicated with a red arrow and red dot.
Evaluation of the effects of several protease inhibitors on MERS-S-mediated membrane fusion.
As shown in Fig. 3, we found that nafamostat, a serine protease inhibitor, potently inhibited MERS-S-mediated membrane fusion. On the basis of this finding, we tested several additional protease inhibitors not included in the first screening. We selected protease inhibitors currently in clinical use in Japan, i.e., gabexate mesylate, camostat mesilate (camostat), sivelestat sodium tetrahydrate, rivaroxaban, telaprevir, ulinastatin, and simeprevir. Ulinastatin was dissolved in PBS(−), and all other inhibitors were dissolved in DMSO. Nafamostat was also included as a comparison. The results are shown in Fig. 4A. Gabexate mesylate, sivelestat sodium tetrahydrate, and ulinastatin showed no inhibition of DSP activity, even at a higher concentration. Telaprevir and simeprevir showed some inhibition of DSP activity at a high concentration. As shown in a previous study (16), camostat showed 50% inhibition at 1 μM. The half-maximal inhibitory concentration (IC50) of nafamostat was 0.1 μM, about 10 times lower than that of camostat.
FIG 4.

Effects of several serine protease inhibitors on MERS-S-mediated membrane fusion, as examined by the DSP assay. Several clinically used serine protease inhibitors were evaluated by the DSP assay for their effects on MERS-S-mediated membrane fusion. (A) The effect of each drug on the coculture fusion assay using DSP as a reporter. Drugs were tested at different concentrations, and the additional proteins other than reporters (DSPs) transduced into the effector and target cells are indicated below the graph. none, no additional protein. The relative cell fusion value is represented as the DSP value (RL activity measured in RLU) normalized to that of the control assay performed with DMSO alone. Gabe, gabexate mesylate; Nafa, nafamostat; Camo, camostat mesilate; Sive, sivelestat sodium tetrahydrate; Riva, rivaroxaban; Tela, telaprevir; Ulina, ulinastatin; Sime, simeprevir. (B) The effect of each drug on RL measurement. Each drug was added to cells coexpressing DSP1-7 and DSP8-11 to evaluate its direct inhibitory effects on RL. The relative DSP signal is indicated in the vertical axis by setting the control value with DSP alone as 100%. (C) The DSP assay system using Calu3 cells as target cells. DSP1-7 was constitutively expressed in Calu3 cells. Using 293FT-derived effector cells expressing DSP8-11 and MERS-S (indicated as MERS-S), the DSP assay was performed in the presence of nafamostat or camostat. Effector cells expressing only DSP8-11 (indicated as “None”) were used as a control.
Next, we evaluated the possibility of nonspecific inhibition of the RL activity by each inhibitor (Fig. 4B). Because simeprevir showed an inhibitory profile similar to that seen in coculture assays, simeprevir directly inhibited RL itself rather than membrane fusion. The other drugs did not affect the RL activity and were likely to interfere with some of the steps involved in S-mediated membrane fusion, most likely, processing of the S protein by TMPRSS2 owing to the activities of these compounds as serine protease inhibitors. Based on this result, nafamostat was the best inhibitor among the drugs we evaluated.
Evaluation of several protease inhibitors using Calu3-based target cells.
Next, we used Calu3 cells as target cells and established an assay system similar to the 293FT cell-based assay. Calu3 cells are derived from human lung epithelium and endogenously express CD26 and TMPRSS2. The lung epithelium is a likely target of MERS-CoV infection in vivo, and Calu3 cells may represent a more suitable physiological target for MERS-S-mediated membrane fusion (16). Calu3 cells were transduced with a retroviral vector harboring DSP1-7. After the selection with puromycin, we obtained Calu3/DSP1-7 cells and evaluated the effects of nafamostat and camostat in these cells using DSP assays. The results showed that nafamostat was more efficient at inhibiting DSP than camostat, which was similar to the results of the 293FT-based assay (Fig. 4C). However, Calu3 cells as target cells were about 100-fold more sensitive to the inhibitors than 293FT cells. Indeed, the IC50 for nafamostat in this system was about 1 nM.
Effects of nafamostat and camostat on MERS-CoV infection.
Our DSP assays in 293FT and Calu3 cells identified nafamostat as a potent inhibitor of MERS-S-mediated membrane fusion (Fig. 4). This inhibition was most likely a result of inhibition of TMPRSS2 on the plasma membrane. Because MERS-CoV may infect cells via a TMPRSS2-independent endocytotic pathway, we evaluated the effects of nafamostat on MERS-CoV infection using an in vitro virus infection assay (Fig. 5). We measured the inhibition of MERS-CoV entry and the production of progeny viruses by nafamostat. MERS-Cov (EMC2012) infection in Calu3 was tested together with camostat as a comparison. The amount of internalized viral RNA in the cells preincubated with respective inhibitors was measured and quantified. Reduction of internalized viral RNA was observed for both camostat and nafamostat, but nafamostat was more effective than camostat and reduced viral entry by 100-fold at a concentration of as low as 1 nM (Fig. 5A). Next, we evaluated the effects of inhibitors on viral replication. The inhibitors were added after the virus had been incubated with Calu3 cells, and the amount of progeny viruses released into the culture medium was measured by titration using Vero/TMPRSS2 cells. The amount of progeny virus decreased on day 1, and nafamostat was significantly more effective than camostat (Fig. 5B).
FIG 5.

Inhibition of MERS-CoV infection by nafamostat in vitro. Data represent the results of inhibition of viral entry by administration of the serine protease inhibitors camostat and nafamostat. (A) Calu3 cells pretreated with different concentrations of individual inhibitors were challenged with MERS-CoV (EMC2012) at an MOI of 10, and the amount of internalized viral RNA was quantified by real-time PCR. The figure is representative of the results of duplicate experiments. The asterisks indicate the statistically significant differences determined by Student's t test (*, P < 0.001; **, P < 0.01; ***, P < 0.05). n.s., no significant difference. (B) Effects of inhibitors on viral replication. After viral infection, inhibitors were added at different concentrations, and the amount of progeny viruses released into the culture medium was quantified using Vero/TMPRSS2 cells. The figure shows representative results from day 1. Statistical analysis was performed using Student's t test (**, P < 0.01). n.s., no significant difference.
DISCUSSION
In this study, we established a DSP-based cell-cell fusion assay system to measure MERS-S-mediated membrane fusion. We performed a small-scale screening to test whether our DSP assay can be used in a drug screening. This fusion assay did not use live viruses and could be finished within several hours. Since it does not require the use of live viruses, our cell-based DSP assay can be performed in a conventional laboratory. Candidate drugs identified in the DSP assay can be further evaluated in a virus infection assay, since there may be potential differences between cell-cell and virus-cell membrane fusions.
We used the RL mode of the DSP reporter because it could provide a more quantitative reading than the GFP mode without image analyses. Furthermore, the luminescent signals of the RL mode were less likely to suffer from the nonspecific noise of natural color or possible fluorescence emitted from the test drugs in the GFP mode (data not shown). The potential false positivity resulting from nonspecific inhibition of RL itself was easily ruled out by testing of particular drugs in cells coexpressing the two DSPs simultaneously. Our results indicated that the DSP assay was highly stable, and nonspecific inhibition was rarely observed.
In this study, we first utilized 293FT cells constitutively expressing either DSP1-7 or DSP8-11 to determine the necessary components required for the assay system (MERS-S, CD26, and TMPRSS2). Because 293FT cells are easily transfectable, these cells could be conveniently used to express candidate components by transient transfection (data not shown). Consistent with previous studies, we found that both CD26 and TMPRSS2 were necessary in the target cells (12, 14, 16). However, in contrast to assays of infection by intact viruses, this cell-based assay measured fusion only at the cell surface and could not address the contribution of fusion after endocytosis. While this may be considered a shortcoming, we found it useful to effectively isolate two entry pathways for MERS-CoV and specifically address the contribution of TMPRSS2 at the plasma membrane. Since the proteolytic processing of S may depend on other unidentified cellular proteases, we may need to address this issue in our future study.
We also tested the DSP assay by establishing Calu3-based target cells. Calu-3 cells may be more relevant target cells than 293FT cells; however, the overall patterns in the 293FT-based assays and Calu3-based assays were similar. Interestingly, the fusion was more readily blocked in Calu3 cells than in 293FT cells, which could be explained by differences in the expression levels of CD26, TMPRSS2, and DSP. Notably, we currently cannot rule out the possibility of the involvement of other cellular factors. This suggests that the more sensitive Calu3-based assay may be useful for reducing the concentrations of the test drugs in a much larger scale of screening in the future.
Next, we adapted our DSP assay to an HTS format by establishing stable cell lines expressing the necessary components in effector and target cells. We also established cell lines stably expressing self-reassociated DSPs to exclude nonspecific inhibitors that block the RL activity of the DSPs. Our screening of 1,017 drugs identified nafamostat, a serine protease inhibitor, as a potential hit. Moreover, among the tested inhibitors, we found that nafamostat exhibited the strongest inhibitory activity. Consistent with a previous study, camostat was shown to inhibit membrane fusion (16); however, its efficacy was weaker than that of nafamostat. In vitro infection assays confirmed these findings.
Because MERS-S requires proteolytic activation by cellular proteases after receptor binding (13), we assumed that the inhibitory mechanism involved suppression of the TMPRSS2 activity. The efficacy of nafamostat in live virus infection was less potent than in our DSP assay. This may have been due to the relatively high MOI used in our assay to facilitate the detection of the internalized viral RNA or to the presence of an alternative endocytic entry mechanism of MERS-CoV that does not rely on TMPRSS2. The alternative endocytic pathway uses other proteases, such as cathepsins. It is hard to estimate the exact contribution of each entry pathway in real infections in vivo; however, our previous results showed that MERS-CoV depends primarily on the direct fusion pathway in vitro (16). Therefore, TMPRSS2 inhibitors against MERS may be expected to exhibit efficacy in vivo. However, other assay systems are required to screen potential drugs targeted at proteases other than TMPRSS2.
In this study, we screened drugs and reagents already available in clinical use. Such a screening may be relevant to emerging diseases, such as MERS, because screening of chemical leads and development of effective medicines through animal and human trials are highly time-consuming and costly; these factors may prevent prompt responses to the spread of emerging viruses.
As discussed above, nafamostat is expected to manifest its anti-MERS-CoV activity through inhibition of the host protease, TMPRSS2. Targeting host proteins rather than viral proteins has the advantage of avoiding treatment failure due to mutations of the target. However, we cannot rule out possible drug-resistant mutants because RNA viruses are known to generate quasispecies that contain a population of related but distinct mutants, some of which may develop drug resistance against inhibitors targeting the host protein. Targeting of host proteins may cause unwanted side effects during antiviral application, particularly when the antivirals are administered systemically. Unwanted side effects during the treatment of MERS may be reduced by topical introduction into the respiratory tract through a nebulizer. Other than therapeutic use in humans, treatment of potential carrier animals, such as camels or bats, with nafamostat or related compounds may be a possible intervention method.
Although our screening clearly showed the efficacy of the use of clinically available nafamostat against MERS-S-mediated membrane fusion in vitro, further pharmacokinetic or toxicity studies will be required to determine whether nafamostat can be used to treat MERS in human. Nafamostat is currently indicated for systemic use in the treatment of pancreatitis and disseminated intravascular coagulation. The anticoagulatory activity as implicated in the latter indication may cause unwanted side effects when it is applied to MERS. The feasibility of clinical application of nafamostat is beyond the scope of our current study; however, either nafamostat or its derivatives have potential to be used in the treatment of MERS.
TMPRSS2 proteolytically activates a wide range of viral envelope proteins (14, 16, 25, 28–35) and has been shown to be critical for the in vivo activation of influenza viruses (36–38). Therefore, it will be interesting to examine the efficacy of nafamostat against viruses other than MERS-CoV. Further chemical derivatization of compounds using nafamostat as a lead compound is also possible. We expect that these findings may be useful for combatting MERS. Further large-scale screenings using DSP assays are warranted to identify more-effective compounds against MERS-S, and these assays may be applicable to identification of potential drugs to treat other viral diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kunito Yoshiike for critical reading of the manuscript. We thank Editage for English language editing. We thank Ron A. M. Fouchier and Bart L. Haagmans of Erasmus Medical Center for providing MERS-CoV.
M.Y., X.L., J.I., and Z.M. conceived and designed the experiments. M.Y., X.L., and S.M. performed the experiments. M.Y., X.L., S.M., M.T., Y.K., J.I., and Z.M. analyzed the data. M.Y., S.M., M.T., J.I., and Z.M. wrote the paper.
This research was (partially) supported by the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from Ministry of Education, Culture, Sport, Science & Technology in Japan and by the Japan Agency for Medical Research and Development (AMED).
Funding Statement
This research is partially supported by the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from the Ministry of Education, Culture, Sport, Science & Technology in Japan and the Japan Agency for Medical Research and Development (AMED).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01043-16.
REFERENCES
- 1.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 2.Mackay IM, Arden KE. 2015. MERS coronavirus: diagnostics, epidemiology and transmission. Virol J 12:222. doi: 10.1186/s12985-015-0439-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdel-Moneim AS. 2014. Middle East respiratory syndrome coronavirus (MERS-CoV): evidence and speculations. Arch Virol 159:1575–1584. doi: 10.1007/s00705-014-1995-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Tawfiq JA, Memish ZA. 2014. What are our pharmacotherapeutic options for MERS-CoV? Expert Rev Clin Pharmacol 7:235–238. doi: 10.1586/17512433.2014.890515. [DOI] [PubMed] [Google Scholar]
- 5.Lu G, Wang Q, Gao GF. 2015. Bat-to-human: spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol 23:468–478. doi: 10.1016/j.tim.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reusken CB, Raj VS, Koopmans MP, Haagmans BL. 2016. Cross host transmission in the emergence of MERS coronavirus. Curr Opin Virol 16:55–62. doi: 10.1016/j.coviro.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Tawfiq JA, Assiri A, Memish ZA. 2013. Middle East respiratory syndrome novel corona MERS-CoV infection. Epidemiology and outcome update. Saudi Med J 34:991–994. [PubMed] [Google Scholar]
- 8.Alsahafi AJ, Cheng AC. 10 February 2016. The epidemiology of Middle East Respiratory Syndrome (MERS) coronavirus in the Kingdom of Saudi Arabia, 2012–2015. Int J Infect Dis doi: 10.1016/j.ijid.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Momattin H, Mohammed K, Zumla A, Memish ZA, Al-Tawfiq JA. 2013. Therapeutic options for Middle East respiratory syndrome coronavirus (MERS-CoV)–possible lessons from a systematic review of SARS-CoV therapy. Int J Infect Dis 17:e792–e798. doi: 10.1016/j.ijid.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma C, Wang L, Tao X, Zhang N, Yang Y, Tseng CT, Li F, Zhou Y, Jiang S, Du L. 2014. Searching for an ideal vaccine candidate among different MERS coronavirus receptor-binding fragments–the importance of immunofocusing in subunit vaccine design. Vaccine 32:6170–6176. doi: 10.1016/j.vaccine.2014.08.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh YH. 2015. Middle East respiratory syndrome coronavirus (MERS-CoV) nosocomial outbreak in South Korea: insights from modeling. PeerJ 3:e1505. doi: 10.7717/peerj.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, Zhang Y, Zhang W, Yuan Y, Bao J, Zhang B, Shi Y, Yan J, Gao GF. 2013. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 500:227–231. doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Millet JK, Whittaker GR. 2015. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res 202:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Kramer-Kuhl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, von Hahn T, Simmons G, Hofmann H, Pohlmann S. 2013. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol 87:5502–5511. doi: 10.1128/JVI.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian Z, Dominguez SR, Holmes KV. 2013. Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS-CoV) in virus entry and syncytia formation. PLoS One 8:e76469. doi: 10.1371/journal.pone.0076469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shirato K, Kawase M, Matsuyama S. 2013. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 87:12552–12561. doi: 10.1128/JVI.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millet JK, Whittaker GR. 2014. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci U S A 111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondo N, Miyauchi K, Meng F, Iwamoto A, Matsuda Z. 2010. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J Biol Chem 285:14681–14688. doi: 10.1074/jbc.M109.067090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813–820. doi: 10.1093/protein/gzs051. [DOI] [PubMed] [Google Scholar]
- 20.Atanasiu D, Saw WT, Gallagher JR, Hannah BP, Matsuda Z, Whitbeck JC, Cohen GH, Eisenberg RJ. 2013. Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J Virol 87:11332–11345. doi: 10.1128/JVI.01700-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saw WT, Matsuda Z, Eisenberg RJ, Cohen GH, Atanasiu D. 2015. Using a split luciferase assay (SLA) to measure the kinetics of cell-cell fusion mediated by herpes simplex virus glycoproteins. Methods 90:68–75. doi: 10.1016/j.ymeth.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakane S, Matsuda Z. 2015. Dual Split Protein (DSP) assay to monitor cell-cell membrane fusion. Methods Mol Biol 1313:229–236. doi: 10.1007/978-1-4939-2703-6_17. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Li X, Nakane S, Liu S, Ishikawa H, Iwamoto A, Matsuda Z. 2014. Co-expression of foreign proteins tethered to HIV-1 envelope glycoprotein on the cell surface by introducing an intervening second membrane-spanning domain. PLoS One 9:e96790. doi: 10.1371/journal.pone.0096790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitamura T, Koshino Y, Shibata F, Oki T, Nakajima H, Nosaka T, Kumagai H. 2003. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp Hematol 31:1007–1014. doi: 10.1016/S0301-472X(03)00260-1. [DOI] [PubMed] [Google Scholar]
- 25.Shirogane Y, Takeda M, Iwasaki M, Ishiguro N, Takeuchi H, Nakatsu Y, Tahara M, Kikuta H, Yanagi Y. 2008. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J Virol 82:8942–8946. doi: 10.1128/JVI.00676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473-12. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 28.Böttcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 80:9896–9898. doi: 10.1128/JVI.01118-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. 2010. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84:12658–12664. doi: 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrara F, Molesti E, Böttcher-Friebertshäuser E, Cattoli G, Corti D, Scott SD, Temperton NJ. 2012. The human transmembrane protease serine 2 is necessary for the production of group 2 influenza A virus pseudotypes. J Mol Genet Med 7:309–314. [PMC free article] [PubMed] [Google Scholar]
- 31.Abe M, Tahara M, Sakai K, Yamaguchi H, Kanou K, Shirato K, Kawase M, Noda M, Kimura H, Matsuyama S, Fukuhara H, Mizuta K, Maenaka K, Ami Y, Esumi M, Kato A, Takeda M. 2013. TMPRSS2 is an activating protease for respiratory parainfluenza viruses. J Virol 87:11930–11935. doi: 10.1128/JVI.01490-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann-Winkler H, Thiel V, Pohlmann S. 2013. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol 87:6150–6160. doi: 10.1128/JVI.03372-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baron J, Tarnow C, Mayoli-Nussle D, Schilling E, Meyer D, Hammami M, Schwalm F, Steinmetzer T, Guan Y, Garten W, Klenk HD, Bottcher-Friebertshauser E. 2013. Matriptase, HAT, and TMPRSS2 activate the hemagglutinin of H9N2 influenza A viruses. J Virol 87:1811–1820. doi: 10.1128/JVI.02320-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esumi M, Ishibashi M, Yamaguchi H, Nakajima S, Tai Y, Kikuta S, Sugitani M, Takayama T, Tahara M, Takeda M, Wakita T. 2015. Transmembrane serine protease TMPRSS2 activates hepatitis C virus infection. Hepatology 61:437–446. doi: 10.1002/hep.27426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garten W, Matrosovich M, Matrosovich T, Eickmann M, Vahhabzadeh A. 2004. Cleavage of influenza virus hemagglutinin by host cell proteases. Int Congr Ser 1263:218–221. doi: 10.1016/j.ics.2004.01.040. [DOI] [Google Scholar]
- 36.Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pohlmann S, Schughart K. 2013. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog 9:e1003774. doi: 10.1371/journal.ppat.1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakai K, Ami Y, Tahara M, Kubota T, Anraku M, Abe M, Nakajima N, Sekizuka T, Shirato K, Suzaki Y, Ainai A, Nakatsu Y, Kanou K, Nakamura K, Suzuki T, Komase K, Nobusawa E, Maenaka K, Kuroda M, Hasegawa H, Kawaoka Y, Tashiro M, Takeda M. 2014. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J Virol 88:5608–5616. doi: 10.1128/JVI.03677-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarnow C, Engels G, Arendt A, Schwalm F, Sediri H, Preuss A, Nelson PS, Garten W, Klenk HD, Gabriel G, Böttcher-Friebertshäuser E. 2014. TMPRSS2 is a host factor that is essential for pneumotropism and pathogenicity of H7N9 influenza A virus in mice. J Virol 88:4744–4751. doi: 10.1128/JVI.03799-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.