

Abstract
Ceftolozane-tazobactam has potent activity against Pseudomonas aeruginosa, a pathogen associated with cystic fibrosis (CF) acute pulmonary exacerbations (APE). Due to the rapid elimination of many antibiotics, CF patients frequently have altered pharmacokinetics. In this multicenter, open-label study, we described the population pharmacokinetics and safety of ceftolozane-tazobactam at 3 g every 8 h (q8h) in 20 adult CF patients admitted with APE. Population pharmacokinetics were determined using the nonparametric adaptive grid program in Pmetrics for R. A 5,000-patient Monte Carlo simulation was performed to determine the probability of target attainment (PTA) for the ceftolozane component at 1.5 g and 3 g of ceftolozane-tazobactam q8h across a range of MICs using a primary threshold exposure of 60% free time above the MIC (fT>MIC). In these 20 adult CF patients, ceftolozane and tazobactam concentration data were best described by 2-compartment models, and ceftolozane clearance (CL) was significantly correlated with creatinine clearance (r = 0.71, P < 0.001). These data suggest that ceftolozane and tazobactam clearance estimates in CF patients are similar to those in adults without CF (ceftolozane CF CL, 4.76 ± 1.13 liter/h; tazobactam CF CL, 20.51 ± 4.41 liter/h). However, estimates of the volume of the central compartment (Vc) were lower than those for adults without CF (ceftolozane CF Vc, 7.51 ± 2.05 liters; tazobactam CF Vc, 7.85 ± 2.66 liters). Using a threshold of 60% fT>MIC, ceftolozane-tazobactam regimens of 1.5 g and 3 g q8h should achieve PTAs of ≥90% at MICs up to 4 and 8 μg/ml, respectively. Ceftolozane-tazobactam at 3 g q8h was well tolerated. These observations support additional studies of ceftolozane-tazobactam for Pseudomonas aeruginosa APE in CF patients. (This study has been registered at ClinicalTrials.gov under identifier NCT02421120.)
INTRODUCTION
Cystic fibrosis (CF) is an autosomal recessive genetic disorder that affects multiple organ systems (1). Nearly 1,000 new cases of CF are diagnosed annually in the United States, and more than 75% of these diagnoses are made by the age of 2 (2). Understanding the importance of augmenting airway clearance, aggressively treating infection, and correcting nutrition deficits have collectively increased the median survival from 31 years in 2002 to 41 years in 2013, with approximately half of CF patients being 18 years or older (1, 2). Lungs are the most prominent affected organs, and 85% of CF mortality is attributed to lung disease (3). CF lung disease begins with inflammation of the pulmonary tissues along with impaired airway mucociliary clearance, which consequently results in chronic infection of the airways due to impaired bacterial clearance. Lung function in CF progressively declines with the occurrence of acute episodes of pulmonary exacerbation caused by the organisms chronically occupying the CF airway (3). Common organisms that inhabit the lungs of adult CF patients include Pseudomonas aeruginosa and Staphylococcus aureus, including drug-resistant strains (2). High rates of antimicrobial resistance are reported within these organisms as a consequence of repeated exposures to multiple antibiotic courses for the management of chronic infections (2, 4). Furthermore, some resistant phenotypes challenge antibiotic therapy through the formation of biofilms (production of mucoid alginate by the bacteria to improve survival), collectively making it difficult to eradicate a pathogen from the CF lung (4). Despite these challenges, intravenous antibiotics with antipseudomonal activity continue to be the mainstay of treatment for acute pulmonary exacerbations in adult CF patients; however, new agents are needed to address the development of resistance within these difficult-to-treat pathogens.
Ceftolozane is a novel, broad-spectrum cephalosporin antibiotic approved for the treatment of complicated urinary tract and complicated intra-abdominal infections (5). Currently, the antibiotic is also being studied for the treatment of ventilated nosocomial pneumonia (ClinicalTrials registration no. NCT02070757). Compared with other currently available cephalosporins, ceftolozane is intrinsically more stable against the multiple resistance mechanisms employed by P. aeruginosa, such as AmpC β-lactamase production and efflux (6–8). When ceftolozane is combined with tazobactam, an older β-lactamase inhibitor, the antimicrobial coverage of ceftolozane-tazobactam (Zerbaxa; Merck & Co., Inc.) broadens to include Gram-negative organisms producing some β-lactamases, including certain extended-spectrum β-lactamases. Given the potent activity of ceftolozane-tazobactam against P. aeruginosa, including multidrug resistant isolates from CF patients, this antibiotic could play a significant role in the treatment of CF acute pulmonary exacerbations. However, there is established evidence that patients with CF may have altered pharmacokinetics compared with those of non-CF patients (4). As a result, it would be prudent to understand the pharmacokinetics of ceftolozane in adult CF patients in order to select the appropriate dosing regimen in this special population, which might differ from the dosing used to treat infections in non-CF patients.
MATERIALS AND METHODS
Study design and participants.
This study was a prospective, multicenter, open-label pharmacokinetic trial (ClinicalTrials registration no. NCT02421120) of ceftolozane-tazobactam in 20 CF adults hospitalized for the treatment of an acute pulmonary exacerbation requiring intravenous antibiotic therapy. The study was reviewed and approved by the institutional review boards of all participating centers (Hartford Hospital IRB HHC-2015-0107, University of North Carolina IRB 15-1307, Drexel [St. Christopher's Hospital for Children] IRB 1506003732, and Indiana University IRB 1504405675), and informed consent was required of all participants. Inclusion criteria required that each participant was 18 years of age or older, had documented CF, and was hospitalized for an acute pulmonary exacerbation that required treatment with intravenous antibiotics as per their treating CF physician. If female, participants had to be nonpregnant and nonlactating. Participants were excluded if they had any of the following: history of any moderate or severe hypersensitivity or allergic reaction to any β-lactam antibiotic; prior (within 24 h of receiving the first dose of the study drug) or concomitant receipt of cefepime (the internal standard for the assay), piperacillin-tazobactam, or any anionic organic acids that are actively secreted through the renal tubule (i.e., probenecid, furosemide, thiazide diuretics, and angiotensin-converting enzyme [ACE] inhibitors); history of a lung transplant at any time in the past or any other organ transplantation within the last 6 months; moderate to severe renal dysfunction, defined as a creatinine clearance (CLCR) of <50 ml/min (as calculated by the Cockcroft-Gault equation using actual body weight), or requirement for continuous renal replacement therapy or hemodialysis; a hemoglobin level of less than 8 g/dl at baseline; any rapidly progressing disease or immediately life-threatening illness (defined as imminent death within 48 h in the opinion of the investigator); or planned or prior participation in any other interventional drug study within 30 days.
Antibiotic administration.
Participants received 4 to 6 doses of 3 g ceftolozane-tazobactam every 8 h (q8h) with each dose infused as a 60-min infusion, in addition to their standard antibiotic therapy. Commercially available prepackaged preparations of 1.5 g (lot SP1334-A; expiration date, April 2016) ceftolozane-tazobactam (Zerbaxa; Merck & Co., Inc., Kenilworth, NJ) were supplied by the manufacturer and stored according to prescribing recommendations. For each dose, two vials of ceftolozane-tazobactam (1,000 mg/500 mg/vial) were reconstituted with 10 ml of 0.9% sodium chloride for injection or sterile water for injection. The entire volume (∼11 ml) of each reconstituted vial was added to an infusion bag containing 80 ml of 0.9% sodium chloride for injection, for a total volume of approximately 102 ml. All doses were stored in a refrigerator (2°C to 8°C) until administration within 24 h.
Ceftolozane-tazobactam concentration determination.
Blood samples were collected no earlier than after the third dose of ceftolozane-tazobactam. Six samples per participant were collected at the following time points: 0 h (immediately before the start of the final dose) and 0 to 5 min, 15 to 30 min, 60 to 120 min, 180 to 240 min, and 360 to 420 min after the end of the infusion of the final dose. The exact times of sample collection were recorded for use in the pharmacokinetic analyses. All blood samples were placed on ice for no longer than 5 min and then immediately centrifuged at 4,000 × g for 15 min to collect the separated serum. The serum was divided equally into three cryovials for storage at −80°C until ceftolozane and tazobactam concentrations were determined at the Center for Anti-Infective Research and Development (Hartford, CT, USA). Ceftolozane and tazobactam concentrations in plasma were assessed using a validated high-performance liquid chromatography (HPLC) procedure determined using a previously published assay (9). The lower limit of quantification of the assay was 0.4 μg/ml for both analytes. For ceftolozane, the mean interday coefficients of variation for high (40 μg/ml) and low (0.6 μg/ml) check samples were 4.6% and 5.4%, respectively. The mean intraday coefficients of variation were 3.2% and 7.4%, respectively. For tazobactam, the mean interday coefficients of variation for high (40 μg/ml) and low (0.6 μg/ml) check samples were 4.8% and 5.9%, respectively. The mean intraday coefficients of variation were 2.7% and 3.2%, respectively.
Pharmacokinetic analyses.
Concentration data for both ceftolozane and tazobactam were modeled separately using the nonparametric adaptive grid program (NPAG) in Pmetrics for R (Laboratory of Applied Pharmacokinetics and Bioinformatics, Los Angeles, CA) (10). One- and two-compartment models were differentiated based on visual fit and the Akaike information criterion (AIC) (11). A multiplicative assay variance model was determined by fitting a polynomial to the plot of the interday assay standard deviation (SD) versus the measured ceftolozane and tazobactam concentrations, generating the formulas SD = γ × (0.0039 + 0.0469 × C) and SD = γ × (0.0425 + 0.0391 × T) where C is ceftolozane concentration, T is tazobactam concentration, and γ represents all environmental variability excluding the assay. Separate estimates for γ were determined for ceftolozane and tazobactam models. Once the base model was established for ceftolozane, linear regression (Sigma Plot version 12.5; Systat, Chicago, IL, USA) was used to characterize the relationship between clearance (CL) and volume of distribution parameters and participant covariates (age [years], height [cm], weight [kg], serum creatinine [mg/dl], creatinine clearance [CLCR] [ml/min]) as estimated by the Cockcroft-Gault equation using actual body weight, body mass index [BMI] [kg/m2], fat free mass [kg] [12], and lean body weight [kg] [13]). Covariates were analyzed as ratios and by allometric scaling. Statistically significant covariates were then incorporated into the population model and tested for superiority by the log-likelihood ratio test. A covariate model was not developed for tazobactam, since it contributes little to the microbiological activity of ceftolozane against P. aeruginosa.
Monte Carlo simulation.
A 5,000-patient Monte Carlo simulation (Crystal Ball; Oracle Corporation, Redwood Shores, CA) was conducted for the ceftolozane component of the ceftolozane-tazobactam 1.5-g and 3-g q8h dosage regimens with each dose infused over 60 min. Only ceftolozane was simulated due to its antipseudomonal activity. To determine the covariance relationship between CLCR and all pharmacokinetic parameter estimates, Pmetrics was utilized to recreate the covariance matrix while incorporating this variable. The mean, standard deviation, and covariance matrix for all pharmacokinetic parameters and CLCR were inserted into Crystal Ball to simulate an adult CF patient population that was similar to the 20 participants enrolled. All pharmacokinetic parameter estimates were assumed to follow log-Gaussian distributions, and the CLCR distribution followed a Gaussian shape and range that replicated the distribution for the 20 participants (mean, 117.7 ml/min; range, 62 to 197 ml/min). Concentrations were simulated at 15-min intervals for 7 doses. The probability of pharmacodynamic target attainment (PTA) was assessed over a range of MICs between 0.25 and 64 μg/ml in doubling dilutions. A protein binding estimate of 20% was used to correct total ceftolozane concentrations to unbound drug (14). The pharmacodynamic indices targeted included 39% free time above the MIC (fT>MIC), an exposure which demonstrated a 1-log kill against P. aeruginosa in a murine thigh infection model (15), 60% fT>MIC, an exposure commonly predictive of cephalosporin microbiological success in pneumonia and CFU reduction against Gram-negative bacteria (16–19), and 100% fT>MIC as the maximal achievable fT[mt]MIC threshold for the dosing interval. A PTA of ≥90% was considered optimal.
Safety and tolerability.
The safety and tolerability of ceftolozane-tazobactam at 3 g q8h were assessed throughout the treatment course until completion of the final blood sample. Safety was determined by assessment of adverse events reported by the participant or medical provider and any clinically significant changes in laboratory values (chemistry, hematology, and liver function tests) between the start and completion of therapy.
RESULTS
Participants.
Twenty-one adult CF patients were enrolled, and 20 completed the study. Demographic and clinical characteristics for the 20 participants are provided in Table 1. The bacteria isolated included P. aeruginosa (n = 14/20, 70%), S. aureus (n = 11/20, 55%; 8/11 [73%] were methicillin-resistant S. aureus [MRSA]), Escherichia coli (n = 2/20, 10%), Achromobacter xylosoxidans (n = 1/20, 5%), and Burkholderia cepacia (n = 1/20, 5%). One participant had no bacteria isolated at baseline, and a second did not have a specimen submitted for culture. Because this was not a treatment study, all participants received other intravenous antibiotics to treat their acute pulmonary exacerbation, with the most common regimens including a beta-lactam, typically ceftazidime or meropenem, plus an aminoglycoside.
TABLE 1.
Baseline characteristics of 20 CF adults
| Characteristic | Value (n = 20) |
|---|---|
| Age (yr), mean ± SD (range) | 25.4 ± 9.7 (18–53) |
| Wt (kg), mean ± SD (range) | 53.2 ± 8.2 (39.8–69.4) |
| Height (cm), mean ± SD (range) | 161.8 ± 8.29 (149.7–175.3) |
| Male gender, no. (%) | 6 (30) |
| Body mass index (kg/m2), mean ± SD (range) | 20.4 ± 3.32 (15.3–25.3) |
| Fat free mass (kg), mean ± SD (range) | 38.4 ± 6.6 (29.5–53.5) |
| Lean body wt (kg), mean ± SD (range) | 41.8 ± 5.6 (33.5–55.8) |
| Baseline CLCRa (ml/min), mean ± SD (range) | 117.7 ± 33.1 (62–197) |
Creatinine clearance calculated via the Cockcroft-Gault equation using actual body weight.
Ceftolozane pharmacokinetics.
A total of 111 ceftolozane concentrations were available for model development (Fig. 1). A two-compartment model fit the data best, as determined by a smaller AIC score of 700.90, versus 849.28 for the one-compartment model. Among all tested covariates, only CLCR (ml/min) was significantly associated with ceftolozane clearance (r = 0.71, P < 0.001). The final model produced an AIC of 699.11 and estimated clearance as a ratio of CLCR [CL (liters/h) = CLi + (CLs × CLCR)], where CLi represents the intercept estimate (i.e., nonrenal clearance) and CLs represents the slope estimate (i.e., proportion of CLCR including a unit correction responsible for renal clearance). The final parameter estimates from the population model are presented in Table 2. The final estimate for γ was 1.54, indicating little environmental variability. The observed versus population-predicted ceftolozane concentration and the observed versus individual-predicted ceftolozane concentration are provided in Fig. 2.
FIG 1.
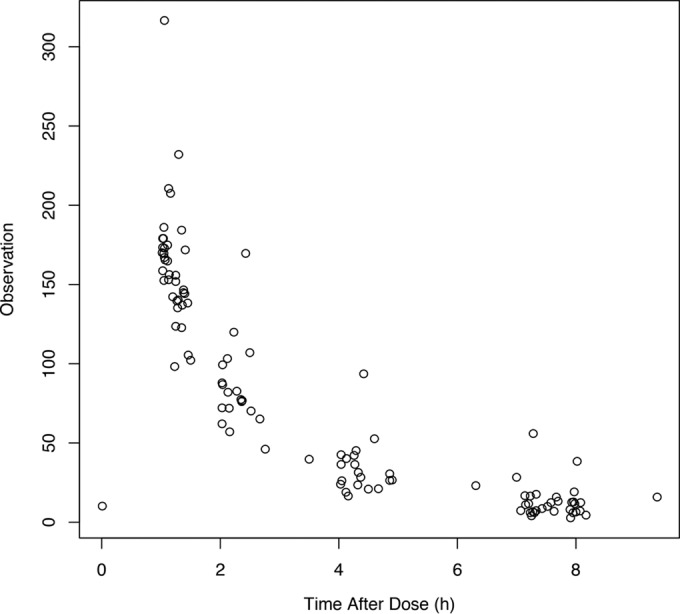
Observed ceftolozane concentrations in 20 CF adults.
TABLE 2.
Final ceftolozane population pharmacokinetic parameter estimates for 20 adults with CF
| Pharmacokinetic parametera | Mean | SD | Median |
|---|---|---|---|
| Observed | |||
| Vc (liters) | 7.51 | 2.05 | 8.11 |
| CLi (liters/h) | 2.00 | 0.85 | 2.21 |
| CLs (liters/h) | 0.02 | 0.01 | 0.02 |
| Kcp (h−1) | 0.87 | 1.98 | 0.23 |
| Kpc (h−1) | 0.66 | 0.46 | 0.48 |
| Calculated | |||
| CL (liters/h) | 4.76 | 1.13 | 4.84 |
| t1/2 (h) | 2.87 | 0.97 | 2.91 |
Vc, volume of central compartment; CLi and CLs, clearance intercept term and clearance slope term, respectively, where total body clearance (CL) = CLi + (CLs × CLCR) and CLCR is creatinine clearance (ml/min) calculated with the Cockcroft-Gault equation; Kcp and Kpc, intercompartment transfer constants; t1/2, half life.
FIG 2.

Observed versus population-predicted (left) and maximum a posteriori probability (MAP) Bayesian individual-predicted (right) (using median parameter estimates) ceftolozane concentrations for the final model.
Tazobactam pharmacokinetics.
For tazobactam, a total of 106 concentrations were available for model development. Notably, 36 (34%) of the tazobactam concentrations were undetectable (n = 32) or below the limit of detection (BDL) (n = 4). The majority (81%) of these undetectable or BDL samples were troughs, occurring at 0 h (immediately before the final dose) or 360 to 420 min after the end of the infusion. Samples that resulted in no peaks on the chromatogram were represented as zero during model fitting, while samples that were below the limit of detection but resulted in a visual peak on the chromatogram were represented as the estimated concentration. Modeling was performed with and without these BDL concentration data; no differences in parameter estimates were found, so all data were used to develop the final model. A two-compartment model fit the data best, as determined by a smaller AIC score of 383.50, versus 401.94 for the one-compartment model. The final parameter estimates from the population model are presented in Table 3. The final estimate for γ was 5.53. The observed versus population-predicted tazobactam concentration and the observed versus individual-predicted tazobactam concentration are provided in Fig. 3.
TABLE 3.
Final tazobactam population pharmacokinetic parameter estimates for 20 adults with CF
| Pharmacokinetic parametera | Mean | SD | Median |
|---|---|---|---|
| Observed | |||
| Vc (liters) | 7.85 | 2.66 | 8.42 |
| CL (liters/h) | 20.51 | 4.41 | 20.13 |
| Kcp (h−1) | 1.88 | 1.91 | 0.97 |
| Kpc (h−1) | 2.49 | 2.29 | 1.52 |
| Calculated | |||
| t1/2 (h) | 2.91 | 2.44 | 3.07 |
Vc, volume of central compartment; CL, clearance; Kcp and Kpc, intercompartment transfer constants; t1/2, half life.
FIG 3.

Observed versus population-predicted (left) and maximum a posteriori probability (MAP) Bayesian individual-predicted (right) (using median parameter estimates) tazobactam concentrations for final model.
Monte Carlo simulation.
The PTA results for 1.5 g and 3 g q8h as 60-min infusions are displayed in Fig. 4. The optimal PTA was achieved for the 1.5-g dose at fT>MIC targets of 39%, 60%, and 100% for MICs of ≤8, 4, and 2 μg/ml, respectively. The optimal PTA was achieved for the 3-g dose at fT>MIC targets of 39%, 60%, and 100% for MICs of ≤16, 8, and 4 μg/ml, respectively.
FIG 4.

Monte Carlo simulation results, showing the probability of the ceftolozane component of ceftolozane-tazobactam at 1.5 g (left) and 3 g (right) q8h as 60-min infusions achieving 39%, 60%, and 100% fT>MIC in adults with CF with a mean (range) CLCR of 117.7 (62 to 197) ml/min.
Safety and tolerability.
Ceftolozane-tazobactam was well tolerated in this study, with only one participant discontinuing therapy secondary to an allergic reaction after the first dose. Adverse events were reported in 6 participants and included chest tightness and tongue swelling (n = 1), erythema in upper extremities (n = 1), electrolyte abnormalities (n = 2), vaginal itching (n = 1), and elevated liver function test results (n = 1).
DISCUSSION
Ceftolozane-tazobactam has the potential to join the antibiotic armamentarium for the treatment of acute pulmonary exacerbations in cystic fibrosis patients. This antibiotic retains activity against P. aeruginosa isolates not susceptible to other commonly used antipseudomonal agents and has demonstrated excellent penetration into the epithelial lining fluid of healthy volunteers (8, 20, 21). The pharmacokinetics of ceftolozane-tazobactam have been studied in healthy volunteers, patients with renal dysfunction, and patients with active infections (5, 22). However, to our knowledge, this is the first pharmacokinetic study of ceftolozane and tazobactam in adults with CF during an acute pulmonary exacerbation.
The concentration data for both ceftolozane and tazobactam were best described by 2-compartment models. CLCR was found to be the most significant covariate on ceftolozane clearance, which is anticipated as ceftolozane is eliminated primarily via glomerular filtration (14, 23). The final equation for clearance in the population pharmacokinetic model of ceftolozane was CL = 2.00 + (0.02 × CLCR). In contrast to the case for other patient characteristics (e.g., body weight and height), this equation significantly improved the fit compared with the base model. These findings suggest that there is an ∼10% change in ceftolozane clearance for every 20% increase or decrease in CLCR. Similarly, a population pharmacokinetic study including healthy volunteers and non-CF patients observed clearance to change by ∼15% for every 20% change in CLCR (22). We did not identify any direct relationships with body size descriptors and ceftolozane clearance, which is also reflective of the findings by Chandorkar and colleagues (22). Importantly, mean clearance estimates for our patients and adults without CF with normal kidney function were similar (CF CL, 4.76 liters/h; non-CF CL, 5.11 liters/h) (22).
In contrast with Chandorkar and colleagues, we were not able to identify any significant relationship between actual body weight and volume of the central compartment (Vc) for ceftolozane with linear or allometric scaling (22). Other body weight descriptors (lean body weight, BMI, and fat free mass) were also not significantly associated with the volume of the central compartment in our CF population. We attribute this lack of finding to the small range of body weights in our study population, which may have prevented identification of a significant relationship. Despite this, these CF patients had smaller volume of the central compartment estimates than those without CF (CF Vc, 7.51 liters; non-CF Vc, 11.4 liters). This finding is further exemplified by greater ceftolozane maximum concentrations (Cmax) achieved in this CF population than in healthy volunteers receiving the same dose (23). Since ceftolozane diffuses into the extracellular fluid, these differences may be attributed to the lower body weight of the 20 CF patients studied (22, 23). These observed differences in the volume of the central compartment estimates between CF and non-CF patients are further exaggerated when active infection is considered. Chandorkar and colleagues (22) observed the average Vc to be 11.4 liters in healthy volunteers but 21% to 59% higher in infected patients with complicated urinary tract and intra-abdominal infections, respectively. Given that our CF patients were enrolled with an acute pulmonary exacerbation of their chronic infection, this makes the differences in volumes of distribution compared with those in non-CF infected patients (22) more profound (∼20%). Despite these observed differences, the observed half-life for ceftolozane (2.87 ± 0.97 h) in these CF patients was similar to estimates in healthy volunteers with normal kidney function (∼3 h) (23).
Similar observations were apparent with tazobactam. Clearance estimates in CF patients were similar to those in adults without CF (CF CL, 20.5 liters/h; non-CF CL, 18.0 liters/h), while the volume of the central compartment was approximately half of that in adults without CF (CF Vc, 7.9 liters; non-CF Vc, 14.2 liters) (22). Again, this is likely associated with the low body weights of the CF population, as tazobactam distributes mainly into extracellular space (24). While tazobactam has been used extensively in combination with piperacillin, this is the first data for tazobactam pharmacokinetics in CF patients.
Due to the morbidity and mortality associated with acute pulmonary infections, optimal antimicrobial treatment is prudent, considering that approximately 70% of the adult CF population is infected with P. aeruginosa (2, 25). The MIC50 and MIC90 of ceftolozane alone against P. aeruginosa isolates from CF patients have been reported as 0.5 and 2 μg/ml, respectively (20). A similar study in children with CF with multidrug-resistant strains demonstrated higher MIC50 and MIC90 values of 2 and 8 μg/ml, respectively (8). The Food and Drug Administration (FDA) and the European Committee on Antimicrobial Testing (EUCAST) currently define P. aeruginosa susceptibility to ceftolozane-tazobactam as an MIC of ≤4/4 μg/ml (26, 27). Due to high observed concentrations, presumably because of low body weight, the standard ceftolozane-tazobactam dosing regimen, 1.5 g q8h, was still able to obtain a PTA of >90% at MICs up to 8, 4, and 2 μg/ml using fT>MIC targets of 39%, 60%, and 100%, respectively. In contrast, the higher dose currently being studied for nosocomial pneumonia, 3 g q8h, obtained a PTA of >90% at MICs up to 16, 8, and 4 μg/ml for those pharmacodynamic thresholds, respectively. The ceftolozane fT>MIC required for successful treatment of a CF acute pulmonary exacerbation is not known; therefore, in the absence of these data, we have settled on the human data with cefepime and ceftazidime to guide our dosing recommendations (i.e., 60% fT>MIC). Considering the risk of antibiotic-resistant bacteria, we advocate for the higher dosing regimen for CF patients with normal kidney function until the pharmacodynamic threshold in CF pulmonary exacerbation can be determined (16, 19).
Ceftolozane-tazobactam at 3 g q8h was well tolerated in these adult CF patients. Adverse effects while receiving ceftolozane-tazobactam were mild, and most could be attributed to a coadministered medication. Notably, one participant did develop a type I hypersensitivity reaction with chest tightness and throat swelling while receiving the first dose. This participant was not noted to be allergic to any other cephalosporins or piperacillin-tazobactam but immediately had her infusion abated, was removed from the study, and was treated accordingly with supportive care, with full recovery. Electrolyte abnormalities (hypokalemia) and elevated liver function test (alanine aminotransferase [ALT] and aspartate transaminase [AST]) effects are reported in the ceftolozane-tazobactam package insert, based on a 1.5-g dose. Participants had these abnormalities at baseline, which were further exaggerated by ceftolozane-tazobactam administration.
There are some limitations to this study. Only 20 adult CF patients were enrolled, and most were homogenous in body size and kidney function. We cannot, therefore, extrapolate our conclusions on ceftolozane pharmacokinetics using this model to heavier CF patients or those with poor kidney function (<60 ml/min), although presumably, similar reductions in CLCR would require dosage adjustment as per current standard prescribing recommendations. We also did not enroll pediatric CF patients, a very important population for whom further study of ceftolozane-tazobactam pharmacokinetics is required. Finally, this study was not designed to be a clinical trial, so no outcome data were collected, and all participants were concurrently treated with other antibiotics targeting their acute pulmonary exacerbation. However, the observations in this study justify clinical assessments of ceftolozane-tazobactam at an optimized dosing regimen.
In conclusion, 2 population pharmacokinetic models were developed that accurately describe ceftolozane and tazobactam pharmacokinetics in adult CF patients with acute pulmonary exacerbation and suggest some differences in mean parameter estimates compared with those in adults without CF. CF patients demonstrate similar clearance rates but smaller volumes of the central compartment for both compounds, which could potentiate higher ceftolozane-tazobactam concentrations than seen in non-CF patients, as observed in our study. These higher concentrations did not affect the safety or tolerability of this dose and may be of benefit when treating resistant organisms as demonstrated by the Monte Carlo simulation. These observations support further development of ceftolozane-tazobactam for the treatment of CF acute pulmonary exacerbation due to P. aeruginosa.
ACKNOWLEDGMENTS
J.L.K. has received research funding from Merck & Co., Inc. (Kenilworth, NJ). D.P.N. has received research funding from and is a member of the speakers' bureau for Merck & Co., Inc. (Kenilworth, NJ). The remaining authors have no conflicts of interest to disclose.
We acknowledge Arlinda Carr, Christina Sutherland, Debora Santini, Dee Rendock, J. Samuel Pope, John McArdle, Angela Lehman, Colleen Sakon, Lisa Bendy, Laurie Varlotta, and all participants with CF and their families for assistance with the conduct of study.
REFERENCES
- 1.Elborn JS. 29 April 2016. Cystic fibrosis. Lancet doi: 10.1016/S0140-6736(16)00576-6. [DOI] [PubMed] [Google Scholar]
- 2.Cystic Fibrosis Foundation. 2014. Patient registry annual data report. Cystic Fibrosis Foundation, Bethesda, MD. [Google Scholar]
- 3.Flume P, Mogayzel PJ, Robinson K, Goss CH, Rosenblatt RL, Kuhn RJ, Marshall BC. 2009. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med 180:802–808. doi: 10.1164/rccm.200812-1845PP. [DOI] [PubMed] [Google Scholar]
- 4.Touw D. 1998. Clinical pharmacokinetics of antimicrobial drugs in cystic fibrosis. Pharm World Sci 20:149–160. doi: 10.1023/A:1008634911114. [DOI] [PubMed] [Google Scholar]
- 5.Zhanel G, Chung P, Adam H, Zelenitsky S, Denisuik A, Schweizer F, Lagacé-Wiens PR, Rubinstein E, Gin AS, Walkty A, Hoban DJ, Lynch JP III, Karlowsky JA. 2014. Ceftolozane/tazobactam: a novel cephalosporin/β-lactamase inhibitor combination with activity against multidrug-resistant gram-negative bacilli. Drugs 74:31–51. doi: 10.1007/s40265-013-0168-2. [DOI] [PubMed] [Google Scholar]
- 6.Takeda S, Nakai T, Wakai Y, Ikeda F, Hatano K. 2007. In vitro and in vivo activities of a new cephalosporin, FR264205, against Pseudomonas aeruginosa. Antimicrob Agents Chemother 51:826–830. doi: 10.1128/AAC.00860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moyá B, Beceiro A, Cabot G, Juan C, Zamorano L, Alberti S, Oliver A. 2012. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob Agents Chemother 56:4771–4778. doi: 10.1128/AAC.00680-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuti JL, Pettit RS, Neu N, Cies JJ, Lapin C, Muhlebach MS, Novak KJ, Nguyen ST, Saiman L, Nicolau DP. 2015. Microbiological activity of ceftolozane/tazobactam, ceftazidime, meropenem, and piperacillin/tazobactam against Pseudomonas aeruginosa isolated from children with cystic fibrosis. Diagn Microbiol Infect Dis 83:53–55. doi: 10.1016/j.diagmicrobio.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 9.Sutherland C, Nicolau DP. 4 April 2016. Development of an HPLC method for the determination of ceftolozane/tazobactam in biological and aqueous matrixes. J Chromatogr Sci doi: 10.1093/chromsci/bmw047. [DOI] [PubMed] [Google Scholar]
- 10.Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit 34:467–476. doi: 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akaike H. 1974. A new look at the statistical model identification. IEEE Trans Automat Contr 19:716–723. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
- 12.Cheymol G. 2000. Effects of obesity on pharmacokinetics implications for drug therapy. Clin Pharmacokinet 39:215–231. doi: 10.2165/00003088-200039030-00004. [DOI] [PubMed] [Google Scholar]
- 13.Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. 2005. Quantification of lean bodyweight. Clin Pharmacokinet 44:1051–1065. doi: 10.2165/00003088-200544100-00004. [DOI] [PubMed] [Google Scholar]
- 14.Ge Y, Whitehouse MJ, Friedland I, Talbot GH. 2010. Pharmacokinetics and safety of CXA-101, a new antipseudomonal cephalosporin, in healthy adult male and female subjects receiving single- and multiple-dose intravenous infusions. Antimicrob Agents Chemother 54:3427–3431. doi: 10.1128/AAC.01753-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lepak AJ, Reda A, Marchillo K, Van Hecker J, Craig WA, Andes D. 2014. Impact of MIC range for Pseudomonas aeruginosa and Streptococcus pneumoniae on the ceftolozane in vivo pharmacokinetic/pharmacodynamic target. Antimicrob Agents Chemother 58:6311–6314. doi: 10.1128/AAC.03572-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crandon JL, Bulik CC, Kuti JL, Nicolau DP. 2010. Clinical pharmacodynamics of cefepime in patients infected with Pseudomonas aeruginosa. Antimicrob Agents Chemother 54:1111–1116. doi: 10.1128/AAC.01183-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacVane SH, Kuti JL, Nicolau DP. 2014. Clinical pharmacodynamics of antipseudomonal cephalosporins in patients with ventilator-associated pneumonia. Antimicrob Agents Chemother 58:1359–1364. doi: 10.1128/AAC.01463-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Craig WA. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 26:1–10. doi: 10.1086/516284. [DOI] [PubMed] [Google Scholar]
- 19.Muller AE, Punt N, Mouton JW. 2013. Optimal exposures of ceftazidime predict the probability of microbiological and clinical outcome in the treatment of nosocomial pneumonia. J Antimicrob Chemother 68:900–906. doi: 10.1093/jac/dks468. [DOI] [PubMed] [Google Scholar]
- 20.Zamorano L, Juan C, Fernández-Olmos A, Ge Y, Cantón R, Oliver A. 2010. Activity of the new cephalosporin CXA-101 (FR264205) against Pseudomonas aeruginosa isolates from chronically-infected cystic fibrosis patients. Clin Microbiol Infect 16:1482–1487. doi: 10.1111/j.1469-0691.2010.03130.x. [DOI] [PubMed] [Google Scholar]
- 21.Chandorkar GL, Huntington JA, Gotfried MH, Rodvold KA, Umeh O. 2012. Intrapulmonary penetration of ceftolozane/tazobactam and piperacillin/tazobactam in healthy adult subjects. J Antimicrob Chemother 67:2463–2469. doi: 10.1093/jac/dks246. [DOI] [PubMed] [Google Scholar]
- 22.Chandorkar G, Xiao A, Mouksassi MS, Hershberger E, Krishna G. 2015. Population pharmacokinetics of ceftolozane/tazobactam in healthy volunteers, subjects with varying degrees of renal function and patients with bacterial infections. J Clin Pharmacol 55:230–239. doi: 10.1002/jcph.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller B, Hershberger E, Benziger D, Trinh M, Friedland I. 2012. Pharmacokinetics and safety of intravenous ceftolozane-tazobactam in healthy adult subjects following single and multiple ascending doses. Antimicrob Agents Chemother 56:3086–3091. doi: 10.1128/AAC.06349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sörgel F, Kinzig M. 1993. The chemistry, pharmacokinetics and tissue distribution of piperacillin/tazobactam. J Antimicrob Chemother 31:39–60. [DOI] [PubMed] [Google Scholar]
- 25.Mayer-Hamblett N, Rosenfeld M, Emerson J, Goss G, Aitken ML. 2002. Developing cystic fibrosis lung transplant referral criteria using predictors of 2-year mortality. Am J Respir Crit Care Med 166:1550–1555. doi: 10.1164/rccm.200202-087OC. [DOI] [PubMed] [Google Scholar]
- 26.Food and Drug Administration. 2014. Zerbaxa® (ceftolozane-tazobactam). http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206829lbl.pdf Accessed 24 June 2016.
- 27.European Committee on Antimicrobial Susceptibility Testing. 2016. Clinical breakpoints—bacterial (v6.0). http://www.eucast.org/clinical_breakpoints Accessed 27 May 2016.