

Abstract
Cytochrome P450 2B6 (CYP2B6) metabolizes clinically important drugs and other compounds. Its expression and activity vary widely among individuals, but quantitative estimation is hampered by the lack of safe and selective in vivo probes of CYP2B6 activity. Efavirenz, a nonnucleoside HIV-1 reverse transcriptase inhibitor, is mainly cleared by CYP2B6, an enzyme strongly inhibited in vitro by voriconazole. To test efavirenz metabolism as an in vivo probe of CYP2B6 activity, we quantified the inhibition of CYP2B6 activity by voriconazole in 61 healthy volunteers administered a single 100-mg oral dose of efavirenz with and without voriconazole administration. The kinetics of efavirenz metabolites demonstrated formation rate-limited elimination. Compared to control, voriconazole prolonged the elimination half-life (t1/2) and increased both the maximum concentration of drug in serum (Cmax) and the area under the concentration-time curve from 0 h to t (AUC0–t) of efavirenz (mean change of 51%, 36%, and 89%, respectively) (P < 0.0001) with marked intersubject variability (e.g., the percent change in efavirenz AUC0–t ranged from 0.4% to ∼224%). Voriconazole decreased efavirenz 8-hydroxylation by greater than 60% (P < 0.0001), whereas its effect on 7-hydroxylation was marginal. The plasma concentration ratio of efavirenz to 8-hydroxyefavirenz, determined 1 to 6 h after dosing, was significantly increased by voriconazole and correlated with the efavirenz AUC0–t (Pearson r = >0.8; P < 0.0001). This study demonstrates the mechanisms of voriconazole-efavirenz interaction, establishes the use of a low dose of efavirenz as a safe and selective in vivo probe for phenotyping CYP2B6 activity, and identifies several easy-to-use indices that should enhance understanding of the mechanisms of CYP2B6 interindividual variability. (This study is registered at ClinicalTrials.gov under identifier NCT01104376.)
INTRODUCTION
Hepatic cytochrome P450 (CYP) 2B6 (CYP2B6) was originally thought to play a minor role in human drug metabolism (1). Recent studies have revealed that hepatic CYP2B6 can account for 4 to 10% of the total hepatic CYP protein pool (2–4), as opposed to the <0.2% value reported in an earlier study (5). It is now considered to be the main enzyme responsible for the metabolism of many clinically important drugs and other compounds (1–4, 6, 7). Hepatic CYP2B6 protein expression and activity vary extensively (20- to 280-fold and 25- to 80-fold, respectively) among human liver tissues (3, 4, 8, 9). Polymorphisms in the CYP2B6 gene and a host of nongenetic factors, including exposure to inducers and inhibitors (2–4, 10), contribute to the marked variability reported in enzyme activity. This variability accounts for the interpatient differences observed in the pharmacokinetics and effects of clinically important CYP2B6 substrates, including efavirenz (EFV) (10), methadone (11), ketamine (12), propofol (13), cyclophosphamide (14), bupropion (15), artemisinin derivatives (16), and nevirapine (17).
Accurate assessment of hepatic CYP2B6 activity has been hampered by the lack of a selective and easy-to-use phenotyping probe. The hydroxylation of bupropion, predominantly catalyzed by CYP2B6 (18), was an early and frequently used in vitro and in vivo probe of CYP2B6 activity (19). However, its usefulness in vivo to quantitatively assess induction of drug interactions (20) and to phenotype genetic variants of CYP2B6 (21) has been limited due to the significant competition from non-CYP2B6 parallel elimination pathways (22, 23) and the complex disposition of bupropion and 4-hydroxybupropion (24–26). Quantifying S-bupropion hydroxylation was thought to improve bupropion utility as an in vivo CYP2B6 probe (27). However, substantial presystemic elimination (i.e., hepatic first pass) of S-bupropion by carbonyl reductases coupled with the complex disposition of (S,S)-hydroxybupropion was recently shown to compromise S-bupropion as a useful probe (25, 26).
Our previous work has established that CYP2B6 is the principal enzyme responsible for the metabolism of efavirenz (EFV) to 8-hydroxyefavirenz (8-OHEFV) and further downstream hydroxylation forming 8,14-dihydroxyefavirenz (di-OHEFV) (9, 28, 29). We showed that variants in the CYP2B6 gene lead to reduced EFV metabolism (9, 30–33). Several studies in HIV-1-infected individuals have demonstrated a strong association between these variants and EFV metabolism (reviewed in references 10 and 34). We have also recently reported that rifampin and EFV, both inducers of CYP2B6, increase the rate of EFV 8-hydroxylation in healthy volunteers (35, 36). The fraction of EFV metabolized via CYP2B6 hydroxylation is over 80% (28, 37). Thus, it was likely that efavirenz would be superior to other in vivo probes of CYP2B6 activity. Although EFV has been recommended by regulatory agencies in the United States and Europe (38, 39) as an in vivo probe of CYP2B6 activity, formal validation and the conditions for its use are lacking. Its long half-life (t1/2) (32) and the frequent adverse central nervous system (CNS) effects associated with therapeutic dosing (40) may hinder the full utility of efavirenz as an in vivo probe of CYP2B6 activity.
Voriconazole, an expanded-spectrum triazole antifungal, is currently recommended as first-line treatment for invasive aspergillosis in both adults and children. It is also widely used to treat other fungal infections, particularly in patients who are intolerant or have developed resistance to other antifungal drugs (41). However, voriconazole frequently alters the disposition of concomitantly administered drugs, potentially leading to loss of efficacy or increased risk for adverse effects of the affected drugs (42). In vitro and in vivo studies (42, 43) suggest that voriconazole alters the pharmacokinetics of coadministered drugs through inhibition of CYP2B6, CYP2C9, CYP2C19, and/or CYP3A4/5. In vitro, CYP2B6 is strongly inhibited by voriconazole (Ki, <0.5 μM) and is predicted to markedly inhibit CYP2B6 activity in vivo (43). Indeed, voriconazole has been shown to increase the exposure of EFV (44) and another CYP2B6 substrate, methadone (45), in healthy volunteers, but the disposition of the respective major metabolites of these drugs was not reported. Of note, the in vivo effects of voriconazole in healthy volunteers were less pronounced than what would be expected based on our in vitro data (43), perhaps due to a bidirectional interaction between EFV and voriconazole when the two drugs are chronically administered together (44). Thus, the precise mechanisms involved in these interactions and the capacity of voriconazole to inhibit CYP2B6 activity in vivo at noninducing doses of EFV remain to be established.
In the present study, we determined the disposition of a single 100-mg oral dose of efavirenz alone and after chronic administration of voriconazole to healthy volunteers. The main objectives were to test whether (i) voriconazole, which inhibits CYP2B6 in vitro, alters CYP2B6 activity in vivo; (ii) the disposition of a single oral dose of EFV would serve as a selective marker of CYP2B6 activity in vivo; and (iii) metabolic ratios (MRs) of EFV and its primary metabolites are reliable indices of CYP2B6 activity.
MATERIALS AND METHODS
Study subjects.
Male and nonpregnant female volunteers between the ages of 18 and 55 years, weighing ≥50 kg and within 32% of their ideal body weight, and determined to be healthy through preenrollment screening (medical history, physical exam, standard laboratory blood and urine test, and electrocardiogram [ECG]) were recruited for this study. The protocol was approved by the Indiana University School of Medicine Institutional Review Board. After volunteers were allowed to read the protocol and the study was carefully explained to them, each participant signed a written informed consent prior to participation. The trial was registered at http://www.clinicaltrials.gov (trial identifier NCT01104376). The study was conducted at the Clinical Research Center of Indiana University's Clinical Translational Sciences Institute, located at Indiana University Hospital.
At the initial screening, a blood sample (∼10 ml) was obtained from each subject and genomic DNA was extracted and saved for genotyping assay. Subjects were also requested to adhere to dietary and medication restrictions, including discontinuation of herbal supplements and nonprescription medications, and to refrain from tobacco or marijuana use beginning 1 week before the study and continuing until completion of the study. Subjects were excluded for significant health conditions such as heart, gastrointestinal (GI), liver, or kidney disease. Volunteers with laboratory results that were not within normal limits, including abnormal baseline ECG readings, or who had a history of or current psychiatric illness, eyesight disturbances (e.g., blurred vision), or alcohol or drug abuse were excluded, as were female subjects with a positive urine test for pregnancy and subjects with weight less than 50 kg (110 lb) or body mass index (BMI) greater than 32. Additional exclusion criteria included anyone who had a blood donation within the past 2 months or who had participated in another research study involving intensive blood sampling. Finally, Indiana University employees or students under the supervision of any of the investigators were also ineligible to participate in this study.
Study design.
This was a one-sequence, open-label study designed to characterize the disposition of EFV by examining the metabolism and pharmacokinetics of efavirenz administered alone and after treatment with voriconazole. Volunteers were studied in three phases with a total of two inpatient days (24 h each) and eight separate outpatient visits. The study design is depicted in Fig. S1 in the supplemental material.
In phase 1, subjects arrived at the Clinical Research Center in the morning after an overnight fast. A sterile indwelling catheter was inserted in a vein in one arm for blood sample collection. A urine pregnancy test was performed, if applicable. A predose blood sample and urine samples were obtained, and then a single 100-mg oral dose of EFV was administered with ∼200 ml water. As safety measures, vital signs and ECG were recorded before and every 6 h after EFV dosing. Participants were also monitored for potential side effects and encouraged to report immediately any unusual feelings. A standard meal was served 3 h after EFV dosing, and volunteers were otherwise allowed a regular diet. Blood samples (∼10 ml) were collected at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, and 24 h after EFV administration. Urine voided was collected periodically (0, 4, 8, 12, 16, and 24 h) after EFV administration. Following the 24-h blood sample, subjects were discharged with an appointment to return to the Indiana Clinical Research Center outpatient clinic for an additional 48-, 72-, 120-, and 168-hour blood sample collection. They were also supplied with containers to collect urine for 48 h. Plasma was separated by centrifugation at 3,000 rpm, and two 10-ml urine aliquots were saved after the volume was recorded. Plasma and urine samples were stored at −80°C until assayed for EFV, voriconazole, and their respective metabolites.
Phase 2 of the study began after the 168-h blood draw for phase 1 on day 8. Volunteers received two oral loading doses of voriconazole (400 mg in the morning and in the evening) followed by 200 mg orally twice daily from day 9 to day 16. Subjects were supplied with a diary to record the date and time of voriconazole ingestion and any side effects that they might experience.
For phase 3, volunteers were readmitted to the Clinical Research Center for a second time in the morning of day 10 after an overnight fast. A second urine pregnancy test was performed, if applicable. A sterile indwelling catheter was inserted again into an arm vein for blood sampling. A blood sample was obtained for a complete metabolic panel including liver enzymes to assess voriconazole safety. Baseline blood and urine samples were obtained as in phase 1. Individuals received a morning dose of 200 mg of voriconazole orally. Two blood samples (∼10 ml) were obtained at 20 min and 50 min after voriconazole dosing. One hour after the voriconazole administration, subjects ingested a single 100-mg oral dose of EFV with ∼200 ml water. A standard meal was allowed 3 h after EFV dosing. An evening dose of 200 mg of voriconazole orally was given while subjects were in the Clinical Research Center. Dosing with voriconazole (200 mg twice daily) continued for 6 days after discharge. All other procedures were the same as described in phase 1. Following collection of the final blood sample on the morning of day 17, exit exams that included a physical and a vision exam, an ECG, and a repeat of the screening laboratory tests were performed and the study diaries were collected. Day 17 marked the completion of the study.
Analysis of drugs and metabolites in plasma and urine.
A new high-performance liquid chromatography (HPLC)–tandem mass spectrometry (LC-MS/MS) method was developed to simultaneously measure EFV, voriconazole, and their respective major oxidative metabolites.
(i) Chemicals.
EFV, 8-OHEFV, 7-OHEFV, di-OHEFV, voriconazole, voriconazole N-oxide, deuterated efavirenz (EFV-d4), and nevirapine were purchased from Toronto Research Chemicals (Ontario, Canada). Formic acid, β-glucuronidase type H-5 from Helix pomata, and glycine were purchased from Sigma-Aldrich (St. Louis, MO). Methanol, ethyl acetate, sodium hydroxide, sodium chloride, and sodium azide were purchased from Fisher Scientific Company LLC (Hanover Park, IL). Sodium acetate was obtained from J. B. Baker (Phillipsburg, NJ). Water for LC-MS was prepared using a Nanopure Infinity UV system (Barnstead Thermolyne, Dubuque, IA). Other chemicals were of HPLC grade.
(ii) Extraction procedures.
Drugs and metabolites were extracted and assayed after enzymatic deconjugation with β-glucuronidase. A 500-μl amount of plasma was mixed with 500 μl of 0.2 M sodium acetate (pH 5), 25 μl of 600 mM sodium azide, and 50 μl of β-glucuronidase (1,000 U/ml). The mixture was incubated for 18 h on a shaker at 37°C. After deconjugation, 30 μl of 1 μg/ml EFV-d4 and 500 ng/ml nevirapine were added as internal standards. After 1.25 ml of 1 M glycine-1 M sodium hydroxide (pH 11.3) and 8 ml of ethyl acetate were added and mixed for 15 min, the sample was centrifuged (Allegra 6; Beckman Coulter, Brea, CA) at 3,600 rpm for 15 min at 0°C. The organic layer was evaporated to dryness in a Savant RVT5105 refrigerated vapor trap (Thermo Fisher Scientific Inc., Waltham, MA).
Deconjugation of drugs and metabolites in urine was performed as described for plasma. The dried plasma and urine samples were reconstituted in 120 μl of a 50:50 mixture of the two mobile phases. Standard curves were generated by addition of known concentrations of the analytes (10, 50, 100, 500, and 1,000 ng/ml) into blank plasma or urine. After the addition of internal standards, standards were extracted as described above. The standard curves were linear over the range of 10 to 1,000 ng/ml.
(iii) LC-MS/MS analysis.
Analysis was performed on an Applied Biosystems API 3200 triple-quadrupole mass spectrometer controlled by Analyst software version 1.5.1 (Applied Biosystems/MDS Sciex, Foster City, CA) on a PC running Windows XP software (Microsoft, Redmond, WA). This MS/MS was coupled to an HPLC system consisting of two LC-20AD pumps, an SIL-20AHT UFLC autosampler, a DGU-20A3 degasser, and a CBM-20A controller (Shimadzu, Columbia, MD). Chromatographic separation was achieved using a Phenomenex Luna C18 column (150 by 4.6 mm, 5-μm particle size) along with a Phenomenex Luna C18 4- by 3.00-mm guard column (Phenomenex, Torrance, CA). Mobile phase A consisted of methanol-formic acid (0.1% in water, 1/99, vol/vol), and mobile phase B contained methanol-formic acid (0.1% in water, 99/1, vol/vol) delivered in a gradient elution mode at a flow rate of 0.8 ml/min. The mobile phases were started at 50% mobile phase B, with a linear gradient to 90% B between 0.01 and 16 min, and then reequilibrated to initial conditions between 16.01 min and 20 min (50% B). Before and after each injection, the needle was washed with acetonitrile-water (75%/25%, vol/vol). MS optimization was achieved via adjustment of both the compound-dependent and instrument-dependent parameters for EFV, 8-OHEFV, 7-OHEFV, di-OHEFV, dihydroxyvoriconazole, and hydroxyvoriconazole (M3) in negative mode using EFV-d4 as an internal standard and, for voriconazole, voriconazole N-oxide and hydroxyvoriconazole (M2) with nevirapine as an internal standard in positive mode. Analytes were optimized at a source temperature of 500°C, under unit resolution for quadrupoles 1 and 3, and at a dwell time of 60 ms and a setting time of 700 ms. Optimal gas pressures were as follows: collision gas, 7 lb/in2; curtain gas, 25 lb/in2; ion source gas (i), 55 lb/in2; ion source gas (ii), 55 lb/in2. Ion spray voltage was 4,000 V in positive mode. In negative mode, optimal gas pressures for all analytes, including the internal standards, were as follows: collision gas, 9 lb/in2; curtain gas, 30 lb/in2; ion source gas (i), 40 lb/in2; ion source gas (ii), 55 lb/in2. Ion spray voltage was −3,500 V. The compound-dependent mass spectrometer parameters and the parent and fragment ions used in multiple reaction monitoring mode for quantification are listed in Table S1 in the supplemental material for both the positive and negative modes.
Pharmacokinetics analysis.
Pharmacokinetic parameters were estimated from plasma concentration-versus-time data by noncompartmental analysis, using PKSolver software (46). The area under the plasma concentration-versus-time curve (AUC) from time zero to the last quantifiable concentration (AUC0–t) was estimated using the linear/logarithmic trapezoidal rule for the absorption and elimination portions of the curve, respectively. The terminal elimination rate constant (λz) was estimated from the slope of the regression line fitted to the log plasma concentration-time data by the method of linear least squares. The AUC was extrapolated to infinity from the last measured concentration (CLAST) as the quotient of CLAST and λz. Partial AUCs were also calculated as needed. The elimination half-life (t1/2) was calculated as the quotient of 0.693 and λz. The maximum plasma concentration (Cmax) and the time to Cmax (tmax) were determined by visual inspection of the data. Plasma metabolic ratios (pMRs) over various time periods were computed for each metabolite as the quotient of the parent AUC and the respective metabolite AUC. Single plasma concentration ratios were also computed for individual time points between 0.5 and 168 h. The total amounts (milligrams) of EFV and metabolites recovered in urine (Ae) were obtained. The renal clearance (CLr) of parent drug and metabolites was calculated as the quotient of amount of parent drug or metabolites excreted unchanged in urine over the first 48 h (Ae0–48) and the plasma AUC0–48. The amounts of metabolite excreted in urine were corrected for molecular weight differences to yield molar mass excreted. The formation clearance of efavirenz to metabolite (CLf) was estimated as the quotient of the molar amount of metabolite recovered in urine (Ae0–48) and the AUC0–48 of EFV. The di-OHEFV product is a secondary metabolite derived from 8-OHEFV. The sum of the two metabolites excreted as 8-hydroxy over 48 h was used to compute total 8-hydroxylation, while the conversion of di-OHEFV from 8-OHEFV was obtained by dividing Ae of di-OHEFV/AUC of 8-OHEFV.
Statistical analysis.
Data are reported as means ± standard deviations (SDs) or ranges. Comparison of pharmacokinetic parameters among control and treatment phases was performed by paired t test using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA). Pearson's correlation analysis was performed using GraphPad. Differences were judged to be due to voriconazole treatment rather than chance variation when P was <0.05.
RESULTS
Pharmacokinetics data from 61 subjects who fully completed all phases of the study are presented in this paper. The demographic characteristics and safety profiles of EFV and voriconazole are shown in Table S2 in the supplemental material.
Certain pharmacokinetic properties of EFV and its metabolites are depicted in Fig. S2 in the supplemental material. In the control phase, plasma concentrations of 7-, 8-, and di-OHEFV declined in parallel with EFV plasma concentrations (see Fig. S2A) and the elimination half-lives of the metabolites were comparable to that of the parent drug (Table 1), indicating that the metabolites demonstrated formation rate-limited elimination. Similar trends were observed in the voriconazole-treated group (see Fig. S2B). EFV was the main circulating moiety followed by 8-, di-, and 7-OHEFV (see Fig. S2C). Compared to controls, the exposure of 8- and di-OHEFV relative to the exposure of EFV was considerably reduced by voriconazole, whereas that of 7-OHEFV remained similar between the groups (see Fig. S2D).
TABLE 1.
Pharmacokinetic parameters of EFV and its metabolites in healthy volunteers after a single 100-mg oral dose of EFV at baseline (control) and after treatment with voriconazole to steady statea
| Drug and PK parameter | Control | Voriconazole | % mean change | P value |
|---|---|---|---|---|
| EFV | ||||
| t1/2 (h) | 121 ± 78 | 165 ± 91 | 51 | <0.01 |
| tmax (range, h) | 2.5 (1–7) | 2.5 (1–10) | NS | |
| Cmax (ng/ml) | 809 ± 322 | 1,028 ± 388 | 36 | <0.001 |
| AUC0–t (ng/ml · h) | 22,675 ± 7,277 | 41,521 ± 13,829 | 89 | <0.001 |
| CLr (ml/min/kg) | 0.0125 ± 0.008 | 0.0106 ± 0.009 | −15 | |
| 8-OHEFV | ||||
| t1/2 (h) | 127.7 ± 76.9 | —b | ND | |
| tmax (range, h) | 2.5 (1.5–4.0) | 2.5 (1.5–12.0) | <0.5 | |
| Cmax (ng/ml) | 210.2 ± 96.2 | 91.0 ± 44.4 | −53 | <0.0001 |
| AUC0–t (ng/ml · h) | 7,213 ± 3,237 | 7,400 ± 3,182 | 9.1 | NS |
| CLr (ml/min/kg) | 25.6 ± 11.6 | 22.2 ± 16.1 | −3.1 | NS |
| 7-OHEFV | ||||
| t1/2 (h) | 102.8 ± 61.7 | —b | ND | |
| tmax (range, h) | 3 (1.5–12) | 3 (1–12) | NS | |
| Cmax (ng/ml) | 13.8 ± 15.5 | 13.5 ± 13.4 | 30 | NS |
| AUC0–t (ng/ml · h) | 716.2 ± 804 | 1,161 ± 1,150 | 103 | <0.0001 |
| CLr (ml/min/kg) | 138.7 ± 145.1 | 77.6 ± 74.3 | −21 | <0.0001 |
| Di-OHEFV | ||||
| t1/2 (h) | 112.8 ± 78.5 | —b | ND | |
| tmax (range, h) | 2.5 (1.5–10) | 3.0 (2–12) | <0.05 | |
| Cmax (ng/ml) | 90.5 ± 67.0 | 24.4 ± 17.1 | −68 | <0.0001 |
| AUC0–t (ng/ml · h) | 3,052 ± 2,343 | 2,321 ± 1,850 | −20 | <0.0001 |
| CLr (ml/min/kg) | 54.9 ± 30.9 | 51.0 ± 29.3 | −15 | NS |
Abbreviations: PK, pharmacokinetic; t1/2, terminal elimination half-life; tmax, time to maximum concentration (Cmax); AUC0–t, area under the plasma concentration-time curve where t is the last measured concentration (∼168 h); CLr, apparent renal clearance; NS, not significant; ND, not determined. Data are expressed as means ± SDs, except for tmax values, which are medians and ranges.
t1/2s of the metabolites were reliably estimated for the control phase as listed in the table; these values could not be reliably estimated in some subjects in the voriconazole group because the concentrations at the last sampling time were higher than the preceding concentrations.
The plasma concentration-versus-time profiles of EFV after oral administration alone and with voriconazole are shown in Fig. 1A. The corresponding pharmacokinetic parameters are summarized in Table 1. Compared to EFV alone, voriconazole markedly increased plasma concentrations of EFV (Fig. 1A). Accordingly, the Cmax and AUC0–168 (i.e., AUC0–t) of EFV were significantly increased and its elimination half-life (t1/2) was prolonged (Table 1). The changes varied widely among subjects (e.g., the mean AUC0–t change of EFV was 89% and ranged between 0.4 and ∼224%). The percent change in EFV exposure correlated significantly with steady-state exposure (AUC0–12) of voriconazole (r = 0.27, P = 0.04) and voriconazole N-oxide (r = 0.28, P = 0.03). Voriconazole also increased the apparent AUC0–∞ of EFV by 145% (range, −3% to +443%) (data not shown). Although this parameter could not be accurately estimated as the residual area beyond the last sampling time (168 h) was much greater than 10%, the data nevertheless indicate a larger effect of voriconazole on EFV exposure than is estimated using AUC0–t. The apparent renal clearance (CLr) and time to Cmax (tmax) of EFV were not significantly affected by voriconazole (Table 1).
FIG 1.
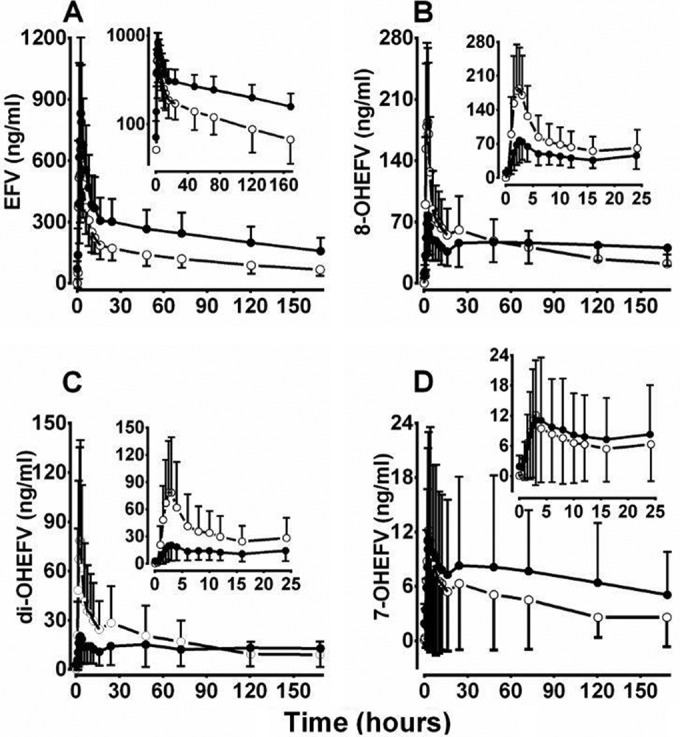
Time course of plasma concentrations of efavirenz (EFV) (A), 8-hydroxy-EFV (8-OHEFV) (B), di-OHEFV (C), and 7-OHEFV (D) in healthy volunteers after a single 100-mg oral dose alone (○) or after treatment with voriconazole to steady state (●). The insets for EFV represent log-transformed concentration-time profile of EFV (A) or plasma concentration-time profiles of metabolites from 0 to 24 h (B, C, and D).
The plasma concentration-versus-time profiles of 8-, di-, and 7-OHEFV after oral administration of EFV alone and with voriconazole are shown in Fig. 1. Voriconazole markedly reduced the concentrations of 8- and di-OHEFV at the earlier time points (Fig. 1B and C), whereas the effect on 7-OHEFV was increased at later time points (Fig. 1D). Accordingly, voriconazole significantly decreased the Cmax and partial AUCs of 8- and di-OHEFV (AUC0–12, AUC0–24, and AUC0–48) (all, P < 0.0001), except AUC0–t of 8-OHEFV, which did not reach a statistically significant level, and increased the partial AUCs of 7-OHEFV (Table 1; Fig. 2) (P, <0.05 to <0.0001). Voriconazole marginally but significantly changed the tmax of 8- and di-OHEFV (P < 0.05) and the CLr of 7-OHEFV (P < 0.0001) (Table 1); CLr values of 8- and di-OHEFV and Cmax and tmax of 7-OHEFV were comparable between the treatments (Table 1).
FIG 2.
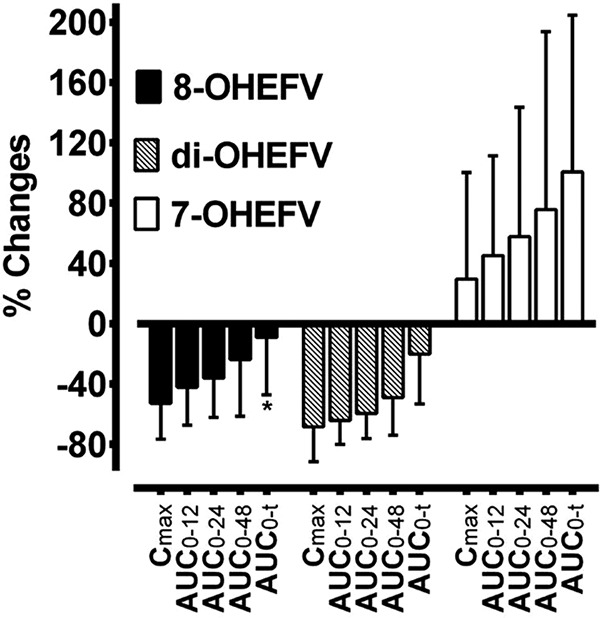
Percent changes in maximum concentration (Cmax) and partial area under the plasma concentration-versus-time curve (AUC) of efavirenz (EFV) metabolites in healthy volunteers after a single 100-mg oral dose of EFV without (control) and with voriconazole treatment to steady state. Percent changes of 8-hydroxy-EFV (8-OHEFV), 7-OHEFV, and di-OHEFV were calculated as 100 × (voriconazole − control)/control. Except for changes in AUC0–t of 8-OHEFV and Cmax of 7-OHEFV, which did not reach a statistically significant level, all other parameters were significantly altered by voriconazole (P < 0.05).
The composition of the 0- to 48-h urinary excretion products (Ae0–48, nanomoles) of EFV was analyzed (Table 2). EFV is mainly excreted as metabolites (greater than 99%; 8-OHEFV > di-OHEFV > 7-OHEFV >>>> EFV). Recovery of unchanged EFV was less than 0.02%. The total percentage of the dose recovered in urine as EFV and metabolites was generally small (on average, <10%). Compared to controls, voriconazole increased Ae0–48 of EFV and decreased Ae0–48 of 8- and di-OHEFV (P < 0.001); Ae0–48 of 7-OHEFV was marginally decreased by voriconazole. Assessment of the relative metabolite formation clearance (CLf) in control (and voriconazole) indicates that 49% (51.5%), 12% (19%), and 88% (80%) are accounted for on average by CLf of EFV to 8-OHEFV, 7-OHEFV, and total 8-hydroxylation, respectively (Table 2). The relative contribution of CLf of EFV to 7-OHEFV was increased ∼70% by voriconazole, possibly due to shunting of EFV metabolism to 7-hydroxylation because of inhibition of CYP2B6-mediated 8-hydroxylation. The CLf of 8-OHEFV to di-OHEFV was more than 2- to 4-fold higher than the CLf of EFV to 8-OHEFV in the control and voriconazole treatments, respectively. The formation of EFV to total 8-hydroxylation pathway would be underestimated by approximately 39% and ∼29% in the control and voriconazole groups, respectively, if sequential metabolism to di-OHEFV were not taken into account (Table 2). Voriconazole significantly reduced the CLf of EFV to 8-OHEFV, 7-OHEFV, and total 8-hydroxylation by 61.5%, 39.8%, and 66.5%, respectively (Table 2).
TABLE 2.
Urinary excretion parameters of EFV and metabolites in healthy volunteers after a single 100-mg oral dose of EFV alone (control) and after treatment with voriconazole to steady state
| Parameter | Mean ± SD for group: |
% mean change | P value | |
|---|---|---|---|---|
| Control | Voriconazole | |||
| Amt (Ae0–48, nmol) recovered in urine over 48 h | ||||
| EFV | 27.0 ± 12.8 | 36.0 ± 20.3 | 50.1 | <0.001 |
| % dose | 0.009 ± 0.004 | 0.012 ± 0.006 | <0.001 | |
| 8-OHEFV | 15,733 ± 6,362 | 9,334 ± 5,015 | −33.3 | <0.001 |
| % dose | 5.0 ± 2.0 | 3.0 ± 1.6 | <0.001 | |
| 7-OHEFV | 4,030 ± 2,459 | 3,363 ± 1,501 | 8.5 | <0.01 |
| % dose | 1.3 ± 0.8 | 1.1 ± 0.5 | <0.01 | |
| Di-OHEFV | 11,871 ± 4,803 | 5,088 ± 3,245 | −48.8 | <0.001 |
| % dose | 3.8 ± 1.5 | 1.6 ± 1.0 | <0.001 | |
| 8- + di-OHEFV | 27,603 ± 10,681 | 14,422 ± 8,021 | −41.1 | <0.001 |
| % dose | 8.7 ± 3.4 | 4.6 ± 2.5 | <0.001 | |
| Total % of dose | 10.0 ± 3.9 | 5.6 ± 2.6 | <0.001 | |
| CLfa of EFV to metabolite (ml/min) | ||||
| EFV to 8-OHEFV | 558 ± 272 | 219 ± 209 | −61.5 | <0.001 |
| % total CLf | 49.0 ± 6.4 | 51.5 ± 7.2 | 6.2 | <0.01 |
| EFV to 7-OHEFV | 141 ± 99 | 74 ± 34 | −39.8 | <0.001 |
| % total CLf | 12.4 ± 5.6 | 19.8 ± 8.4 | 70.1 | <0.001 |
| 8-OHEFV to di-OHEF | 1,505 ± 649 | 940 ± 784 | −37.1 | <0.001 |
| EFV to 8- + di-OHEFV | 996 ± 468 | 345 ± 349 | −66.5 | <0.001 |
| % total CLf | 87.6 ± 5.6 | 80.2 ± 8.4 | −9.0 | <0.001 |
CLf, 0- to 48-h formation clearance.
Plasma (AUC of EFV/AUC of metabolite) and urine (Ae0–48 of metabolite/Ae0–48 of EFV) metabolic ratios (MRs) were calculated as potential indices of CYP2B6 activity (Table 3). Plasma partial AUC MRs of 8- and di-OHEFV and their combination were substantially increased by voriconazole, with AUC0–12 and AUC0–24 ratios being most sensitive; urine MRs were significantly decreased (Table 3). Plasma and urine MRs involving 8-hydroxylation were significantly correlated with EFV exposure (plasma MRs, r ≥ 0.8 for 8-OHEFV and total 8-hydroxylation, r ≥ 0.74 for di-OHEFV; urine MRs, r = −0.43 to 0.47; all, P < 0.0001) (data not shown). No changes were observed in the plasma 7-OHEFV MRs, but urine MRs were slightly reduced by voriconazole, probably due to changes in CLr (Table 1).
TABLE 3.
Plasma and urine metabolic ratios of EFV in healthy volunteers after a single 100-mg oral dose of EFV at baseline (control) and after treatment with voriconazole to steady state
| PK parameter | Mean ± SD for group: |
% mean change | P value | |
|---|---|---|---|---|
| Control | Voriconazoleb | |||
| Plasma AUC metabolic ratio (AUC of EFV/AUC of metabolite) | ||||
| 8-OHEFV | ||||
| 0–12 h | 4.6 ± 3.1 | 12.0 ± 6.3 | 189 | <0.001 |
| 0–24 h | 4.4 ± 3.1 | 10.7 ± 5.6 | 171 | <0.001 |
| 0–48 h | 4.1 ± 3.3 | 9.1 ± 5.1 | 146 | <0.001 |
| 7-OHEFV | ||||
| 0–12 h | 156 ± 206 | 133 ± 126 | 18 | 0.09 |
| 0–24 h | 137 ± 209 | 109 ± 101 | 17 | 0.09 |
| 0–48 h | 126 ± 209 | 93 ± 83 | 16 | 0.09 |
| Di-OHEFV | ||||
| 0–12 h | 12.7 ± 10.2 | 56.2 ± 39.9 | 383 | <0.001 |
| 0–24 h | 11.9 ± 10.7 | 48.5 ± 35.7 | 343 | <0.001 |
| 0–48 h | 11.5 ± 12.6 | 40.2 ± 31.5 | 291 | <0.001 |
| 0- to 48-h molar ratio (Ae of metabolite/Ae of EFV)a | ||||
| 8-OHEFV/EFV | 684 ± 386 | 304 ± 158 | −49.1 | <0.001 |
| 7-OHEFV/EFV | 169 ± 119 | 110 ± 62 | −21.4 | <0.001 |
| Di-OHEFV/EFV | 516 ± 285 | 164 ± 90 | −62.1 | <0.001 |
| Di-OHEFV/8-OHEFV | 0.8 ± 0.2 | 0.6 ± 0.5 | −29.5 | <0.001 |
| 8- + di-OHEFV/EFV | 1,200 ± 655 | 468 ± 240 | −55.3 | <0.001 |
Ratios are large because EFV excreted unchanged was relatively small.
Note that the effect of voriconazole on plasma MRs was larger than that on urine MRs.
While the robust effect of voriconazole on MRs and correlation with efavirenz exposure indicate that plasma MRs involving 8-hydroxylation can be reliable markers of CYP2B6, these indices still require sampling at multiple time points after dosing and calculation of the AUCs. Therefore, individual EFV-to-metabolite concentration ratios were calculated and compared between the treatment groups as an easy-to-use marker of CYP2B6 activity (Fig. 3). Voriconazole substantially increased parent to 8- and di-OHEFV and total 8-hydroxylation plasma concentration ratios between 0.5 and 48 h, maximally at about the tmax (2.5 to 3 h) after EFV dosing (Fig. 3A to C), whereas parent to 7-OHEFV ratio did not differ between treatments (Fig. 3D). We then tested whether the 3-hour time point concentration ratios can serve as a surrogate marker of CYP2B6 activity. Compared to the control, voriconazole strikingly increased the 3-hour plasma concentration ratios of EFV to 8-OHEFV, di-OHEFV, and total 8-hydroxylation (see Fig. S3 in the supplemental material). The 3-hour concentration ratios correlated significantly with AUC0–t (r ≥ 0.75, P < 0.0001); the partial plasma AUC MRs involving 8-hydroxylation after 12, 24, and 48 h (r > 0.9; P < 0.00001) (Fig. 4); and the 0- to 48-h urine MRs (r = −0.52, P < 0.001).
FIG 3.
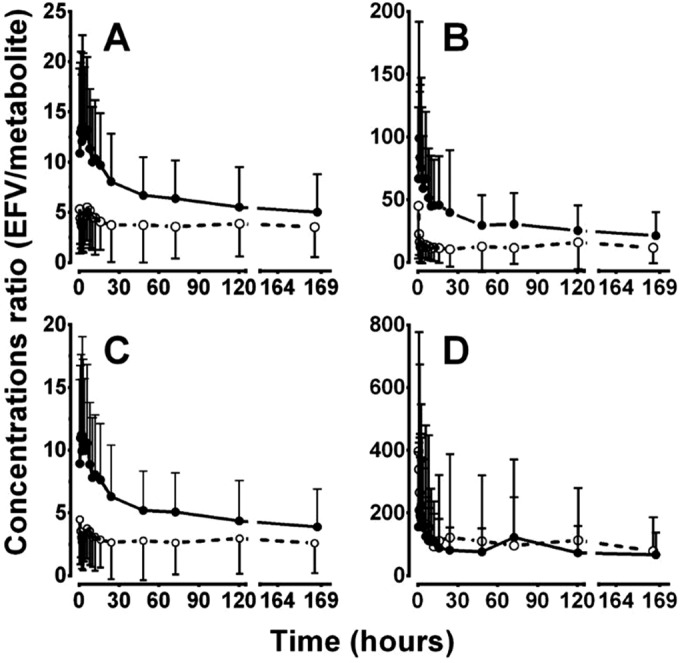
Time course of plasma efavirenz (EFV)-to-metabolite concentration ratios after the administration of a single 100-mg oral dose of EFV at baseline (○) and after treatment with voriconazole (●) to steady state. (A) EFV/8-hydroxy-EFV (8-OHEFV); (B) EFV/di-OHEFV; (C) EFV/(8-OHEFV + di-OHEFV); (D) EFV/7-OHEFV.
FIG 4.
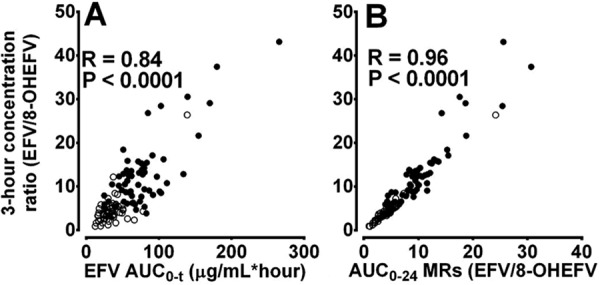
Correlation of the 3-hour concentration ratios of efavirenz (EFV) to 8-hydroxy-EFV (8-OHEFV) with EFV exposure (A) and partial area under the plasma concentration-versus-time curve (AUC) metabolic ratios (MRs) (B) following a single 100-mg oral dose of EFV alone (○) and after treatment with voriconazole to steady state (●).
DISCUSSION
In this in vivo study, we demonstrated for the first time that (i) voriconazole is a potent inhibitor of CYP2B6-mediated EFV 8-hydroxylation (Fig. 5), (ii) the disposition of EFV is a selective marker of CYP2B6 activity, and (iii) the ∼3-h post-EFV plasma concentration metabolic ratio is an easy-to-use, cost-effective index of CYP2B6 activity.
FIG 5.

Proposed mechanisms for increased efavirenz (EFV) exposure by voriconazole (Vori). The di-OHEFV formed from 8-OHEFV represents 8,14-di-OHEFV, while the di-OHEFV formed from 7- and 8-OHEFV represents 7,8-di-OHEFV. OH, hydroxyl.
In humans, EFV is mainly cleared by hepatic metabolism via three primary routes, namely, CYP2A6- and CYP2B6-catalyzed 7- and 8-hydroxylation (9, 28, 29, 37) and direct N-glucuronidation by UGT2B7 (47). The monohydroxymetabolites can be hydroxylated by CYP2B6 to dihydroxymetabolites (29) or conjugated to glucuronide and/or sulfate metabolites (28, 48), with subsequent excretion in urine (37). The present data show that 8-hydroxylation is the major route of elimination, accounting for ∼88% of EFV elimination. Formation of 7-OHEFV represents a minor pathway (<13%), and only trace amounts of unchanged EFV are recovered in urine. EFV N-glucuronide was not measured in this study, but this pathway is considered a minor route (<5%) of EFV elimination (48). Our data broadly concur with the metabolic profiles described previously (37, 49). Together, these data suggest that EFV is predominantly cleared via CYP2B6-mediated 8-hydroxylation.
Voriconazole has been shown to modestly increase steady-state exposure of efavirenz (44) and methadone (45), two drugs known to be cleared by CYP2B6 (9, 11, 28), but the mechanisms involved could not be elucidated as metabolite data were not reported. Voriconazole is a potent inhibitor of CYP2B6 with an in vitro Ki of <0.5 μM (43), which is lower than voriconazole plasma concentrations reported after therapeutic doses (41). Our data demonstrate that voriconazole considerably increases EFV exposure and decreases the Cmax and partial AUCs of 8- and di-OHEFV as well as the amount of these metabolites recovered in urine. Both the 8-hydroxylation of EFV and the conversion of 8-OHEFV to di-OHEFV are primarily catalyzed by CYP2B6 (9, 28, 29, 32, 33, 37). CYP2B6 is also the principal route catalyzing the conversion of 7-OHEFV and 8-OHEFV to 7,8-di-OHEFV (29), which is consistent with the current in vivo observation. Combined with in vitro data (43), the present results strongly support the idea that the mechanism for the effect of voriconazole on EFV elimination is CYP2B6 inhibition (Fig. 5). It is unlikely that inhibition of other competing pathways contributes to the observed effect. Voriconazole did not alter CYP2A6-mediated 7-hydroxylation of EFV in vitro (43) and in vivo (present data). UGT2B7-catalyzed N-glucuronidation represents a minor route of EFV elimination (48) and cannot explain the effect of voriconazole on EFV exposure. Others have suggested that voriconazole may increase EFV exposure through inhibition of CYP3A4 (44). Although voriconazole is a potent inhibitor of CYP3A (43, 50) and this enzyme system shows some activity toward EFV in vitro (28), the in vivo role of CYP3A in the overall elimination of EFV is marginal (49). Overall, the comprehensive analyses of efavirenz and its metabolites in plasma and urine provided a unique insight into the mechanism of the voriconazole-EFV interaction and established that voriconazole slows efavirenz elimination predominantly via inhibition of CYP2B6 in vivo (Fig. 5).
The effect of voriconazole to increase the AUC0–t of EFV by 89% (and AUC0–∞ by 145%) was greater than the previously reported value of 43% (44). Differences in study design (EFV single dose versus steady state) may explain this discrepancy. In our study, the effect of steady-state voriconazole on the metabolism and pharmacokinetics of a single oral dose of EFV (100 mg) was measured. In the other study, the interaction was evaluated after voriconazole (200 mg/day) and efavirenz (400 mg/day) were both administered to steady state (44). Voriconazole steady-state exposure (AUC0–12) was substantially reduced (on average by 80%) in the previous study (44), possibly due to EFV-mediated induction of voriconazole clearance (51) producing a smaller inhibitory effect on CYP2B6. If true, we would expect bidirectional interactions not only between voriconazole and EFV but also with other CYP2B6 substrates that demonstrate autoinduction (e.g., artemisinin, cyclophosphamide, ifosfamide, and nevirapine) (2, 7). It follows that the impact of an inhibitor on the exposure of a CYP2B6 substrate, administered on a one-time basis, would overpredict the expected changes observed after steady-state exposure of the autoinducing substrate. This is consistent with the quantitative differences observed between voriconazole-EFV interactions at a single dose (noninduced state [present data]) and after multiple doses of EFV (induced state [44]). We also expect a robust inhibitory effect of voriconazole with CYP2B6 substrates that do not show induction of metabolism. This can be seen in a study in which steady-state voriconazole markedly increased the steady-state exposure of S-methadone (mean AUC0–24 increased by ∼103%), a CYP2B6 that does not alter voriconazole metabolism upon repeated administration (45). Evaluating steady-state inhibition drug interactions when the victim drug (e.g., EFV) may also be an inducer of the metabolism of the perpetrator drug (e.g., voriconazole) highlights the complexity of using such phenotyping probes to assess the status of hepatic drug metabolizing activity.
The long elimination half-life of EFV (32) limits its routine use as an index of CYP2B6 activity. We sought to test easy-to-use and cost-effective plasma and urine metabolic ratios as phenotyping markers of CYP2B6. These indices are valid because the plasma and urine elimination of 8- and di-OHEFV was formation-rate limited, consistent with our previous studies (29, 32). We identified a number of metabolic ratios involving 8-hydroxylation (Table 3) that were markedly altered by voriconazole and correlated significantly with EFV elimination parameters. However, our approach still entails the collection of serial blood and urine samples, requiring a significant time commitment and cost. To address this limitation, we tested whether a single-time-point determination of plasma EFV and metabolite concentrations could serve as a surrogate for CYP2B6 activity. Our data suggest that the 3-hour plasma concentration ratio of 8-OHEFV, di-OHEFV, or the combination to EFV was markedly increased by voriconazole and correlated with efavirenz exposure and partial AUC metabolic ratios. Thus, this index may represent the most reliable marker of CYP2B6 activity in population studies. The correlations of plasma metabolic ratios with urinary metabolic ratios were less robust.
In summary, the comprehensive analysis of the disposition of EFV and its major metabolites in the plasma and urine with and without voriconazole allowed validation of a low/safe dose of EFV as a selective marker of CYP2B6 activity. Recently, we showed that EFV is a valid probe to assess CYP2B6 induction in vivo and validated the 3-hour metabolic ratio as a marker of this enzyme (36). It is well known that variants in the CYP2B6 gene are associated with EFV metabolism and effect (10, 17, 34, 52–54). Together, EFV is a reliable probe to assess the impact of CYP2B6 genetic polymorphisms and induction and inhibition drug interactions on CYP2B6 activity in vivo. Because the fraction of the EFV dose metabolized via the CYP2B6-mediated 8-hydroxylation is close to unity (37, 49; present data), this probe is superior to other CYP2B6 phenotypic probe drugs used currently (19, 27). The proposed single-point sampling strategy (∼3-hour concentration metabolic ratio), along with the validation of a low dose devoid of common CNS side effects observed at therapeutic doses (40), should facilitate the utility of EFV alone or included in a cocktail as an easy-to-use, safe, and cost-effective CYP2B6 phenotypic probe. Finally, our data suggest that voriconazole would alter the pharmacokinetics of a growing list of CYP2B6 substrates. Thus, monitoring for adverse effects or lack of efficacy is needed when CYP2B6 substrates are coprescribed with voriconazole.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by NIH grants R01GM078501 (Z.D.) and T32GM008425 (Z.D.). The clinical studies were supported by NIH grant UL1TR001108 from the National Center for Advancing Translational Sciences, all from the U.S. Public Health Service, Bethesda, MD. Voriconazole was kindly provided by Pfizer Incorporation through its Investigator-Initiated Research Grant (IIRG) WS669888.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01000-16.
REFERENCES
- 1.Ekins S, Wrighton SA. 1999. The role of CYP2B6 in human xenobiotic metabolism. Drug Metab Rev 31:719–754. doi: 10.1081/DMR-100101942. [DOI] [PubMed] [Google Scholar]
- 2.Wang H, Tompkins LM. 2008. CYP2B6: new insights into a historically overlooked cytochrome P450 isozyme. Curr Drug Metab 9:598–610. doi: 10.2174/138920008785821710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turpeinen M, Zanger UM. 2012. Cytochrome P450 2B6: function, genetics, and clinical relevance. Drug Metabol Drug Interact 27:185–197. doi: 10.1515/dmdi-2012-0027. [DOI] [PubMed] [Google Scholar]
- 4.Zanger UM, Klein K. 2013. Pharmacogenetics of cytochrome P450 2B6 (CYP2B6): advances on polymorphisms, mechanisms, and clinical relevance. Front Genet 4:1–24. doi: 10.3389/fgene.2013.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. 1994. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414–423. [PubMed] [Google Scholar]
- 6.Hodgson E, Rose RL. 2007. The importance of cytochrome P450 2B6 in the human metabolism of environmental chemicals. Pharmacol Ther 113:420–428. doi: 10.1016/j.pharmthera.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Mo SL, Liu YH, Duan W, Wei MQ, Kanwar JR, Zhou SF. 2009. Substrate specificity, regulation, and polymorphism of human cytochrome P450 2B6. Curr Drug Metab 10:730–753. doi: 10.2174/138920009789895534. [DOI] [PubMed] [Google Scholar]
- 8.Stresser DM, Kupfer D. 1999. Monospecific antipeptide antibody to cytochrome P-450 2B6. Drug Metab Dispos 27:517–525. [PubMed] [Google Scholar]
- 9.Desta Z, Saussele T, Ward W, Blievernicht J, Li L, Klein K, Flockhart DA, Zanger UM. 2007. Impact of CYP2B6 polymorphism on hepatic efavirenz metabolism in vitro. Pharmacogenomics 8:547–558. doi: 10.2217/14622416.8.6.547. [DOI] [PubMed] [Google Scholar]
- 10.Zanger UM, Klein K, Saussele T, Blievernicht J, Hofmann H, Schwab M. 2007. Polymorphic CYP2B6: molecular mechanisms and emerging clinical significance. Pharmacogenomics 8:743–759. doi: 10.2217/14622416.8.7.743. [DOI] [PubMed] [Google Scholar]
- 11.Eap CB, Crettol S, Rougier JS, Schlapfer J, Sintra GL, Deglon JJ, Besson J, Croquette-Krokar M, Carrupt PA, Abriel H. 2007. Stereoselective block of hERG channel by (S)-methadone and QT interval prolongation in CYP2B6 slow metabolizers. Clin Pharmacol Ther 81:719–728. doi: 10.1038/sj.clpt.6100120. [DOI] [PubMed] [Google Scholar]
- 12.Peltoniemi MA, Saari TI, Hagelberg NM, Reponen P, Turpeinen M, Laine K, Neuvonen PJ, Olkkola KT. 2011. Exposure to oral S-ketamine is unaffected by itraconazole but greatly increased by ticlopidine. Clin Pharmacol Ther 90:296–302. doi: 10.1038/clpt.2011.140. [DOI] [PubMed] [Google Scholar]
- 13.Kansaku F, Kumai T, Sasaki K, Yokozuka M, Shimizu M, Tateda T, Murayama N, Kobayashi S, Yamazaki H. 2011. Individual differences in pharmacokinetics and pharmacodynamics of anesthetic agent propofol with regard to CYP2B6 and UGT1A9 genotype and patient age. Drug Metab Pharmacokinet 26:532–537. doi: 10.2133/dmpk.DMPK-11-RG-039. [DOI] [PubMed] [Google Scholar]
- 14.Xie H, Griskevicius L, Stahle L, Hassan Z, Yasar U, Rane A, Broberg U, Kimby E, Hassan M. 2006. Pharmacogenetics of cyclophosphamide in patients with hematological malignancies. Eur J Pharm Sci 27:54–61. doi: 10.1016/j.ejps.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Benowitz NL, Zhu AZ, Tyndale RF, Dempsey D, Jacob P III. 2013. Influence of CYP2B6 genetic variants on plasma and urine concentrations of bupropion and metabolites at steady state. Pharmacogenet Genomics 23:135–141. doi: 10.1097/FPC.0b013e32835d9ab0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simonsson US, Jansson B, Hai TN, Huong DX, Tybring G, Ashton M. 2003. Artemisinin autoinduction is caused by involvement of cytochrome P450 2B6 but not 2C9. Clin Pharmacol Ther 74:32–43. doi: 10.1016/S0009-9236(03)00092-4. [DOI] [PubMed] [Google Scholar]
- 17.Rotger M, Colombo S, Furrer H, Bleiber G, Buclin T, Lee BL, Keiser O, Biollaz J, Decosterd L, Telenti A. 2005. Influence of CYP2B6 polymorphism on plasma and intracellular concentrations and toxicity of efavirenz and nevirapine in HIV-infected patients. Pharmacogenet Genomics 15:1–5. doi: 10.1097/01213011-200501000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Faucette SR, Hawke RL, Lecluyse EL, Shord SS, Yan B, Laethem RM, Lindley CM. 2000. Validation of bupropion hydroxylation as a selective marker of human cytochrome P450 2B6 catalytic activity. Drug Metab Dispos 28:1222–1230. [PubMed] [Google Scholar]
- 19.Turpeinen M, Raunio H, Pelkonen O. 2006. The functional role of CYP2B6 in human drug metabolism: substrates and inhibitors in vitro, in vivo and in silico. Curr Drug Metab 7:705–714. doi: 10.2174/138920006778520633. [DOI] [PubMed] [Google Scholar]
- 20.Xu H, Loboz KK, Gross AS, McLachlan AJ. 2007. Stereoselective analysis of hydroxybupropion and application to drug interaction studies. Chirality 19:163–170. doi: 10.1002/chir.20356. [DOI] [PubMed] [Google Scholar]
- 21.Kirchheiner J, Klein C, Meineke I, Sasse J, Zanger UM, Murdter TE, Roots I, Brockmoller J. 2003. Bupropion and 4-OH-bupropion pharmacokinetics in relation to genetic polymorphisms in CYP2B6. Pharmacogenetics 13:619–626. doi: 10.1097/00008571-200310000-00005. [DOI] [PubMed] [Google Scholar]
- 22.Connarn JN, Zhang X, Babiskin A, Sun D. 2015. Metabolism of bupropion by carbonyl reductases in liver and intestine. Drug Metab Dispos 43:1019–1027. doi: 10.1124/dmd.115.063107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Liu HF, Liu L, Nguyen K, Jones EB, Fretland AJ. 2010. The in vitro metabolism of bupropion revisited: concentration dependent involvement of cytochrome P450 2C19. Xenobiotica 40:536–546. doi: 10.3109/00498254.2010.492880. [DOI] [PubMed] [Google Scholar]
- 24.Petsalo A, Turpeinen M, Tolonen A. 2007. Identification of bupropion urinary metabolites by liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom 21:2547–2554. doi: 10.1002/rcm.3117. [DOI] [PubMed] [Google Scholar]
- 25.Gufford BT, Lu JBL, Metzger IF, Jones DR, Desta Z. 2016. Stereoselective glucuronidation of bupropion metabolites in vitro and in vivo. Drug Metab Dispos 44:544–553. doi: 10.1124/dmd.115.068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masters AR, Gufford BT, Lu JB, Metzger IF, Jones DR, Desta Z. 2016. Chiral plasma pharmacokinetics and urinary excretion of bupropion and metabolites in healthy volunteers. J Pharmacol Exp Ther 358:230–238. doi: 10.1124/jpet.116.232876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kharasch ED, Mitchell D, Coles R. 2008. Stereoselective bupropion hydroxylation as an in vivo phenotypic probe for cytochrome P4502B6 (CYP2B6) activity. J Clin Pharmacol 48:464–474. doi: 10.1177/0091270008314254. [DOI] [PubMed] [Google Scholar]
- 28.Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. 2003. The cytochrome P4502B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther 306:287–300. doi: 10.1124/jpet.103.049601. [DOI] [PubMed] [Google Scholar]
- 29.Ogburn ET, Jones DR, Masters AR, Xu C, Guo Y, Desta Z. 2010. Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 (CYP) 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab Dispos 38:1218–1229. doi: 10.1124/dmd.109.031393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu C, Ogburn ET, Guo Y, Desta Z. 2012. Effects of the CYP2B6*6 allele on catalytic properties and inhibition of CYP2B6 in vitro: implication for the mechanism of reduced efavirenz metabolism and other CYP2B6 substrates in vivo. Drug Metab Dispos 40:717–725. doi: 10.1124/dmd.111.042416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu C, Quinney SK, Guo Y, Hall SD, Li L, Desta Z. 2013. CYP2B6 pharmacogenetics-based in vitro-in vivo extrapolation (IVIVE) of efavirenz clearance by PBPK modeling. Drug Metab Dispos 41:2004–2011. doi: 10.1124/dmd.113.051755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang F, Desta Z, Shon JH, Yeo CW, Kim HS, Liu KH, Bae SK, Lee SS, Flockhart DA, Shin JG. 2013. Effects of clopidogrel and itraconazole on the disposition of efavirenz and its hydroxyl metabolites: exploration of a novel CYP2B6 phenotyping index. Br J Clin Pharmacol 75:244–253. doi: 10.1111/j.1365-2125.2012.04314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abdelhady AM, Desta Z, Jiang F, Yeo CW, Shin JG, Overholser BR. 2014. Population pharmacogenetic-based pharmacokinetic modeling of efavirenz, 7-hydroxy- and 8-hydroxyefavirenz. J Clin Pharmacol 54:87–96. doi: 10.1002/jcph.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King J, Aberg JA. 2008. Clinical impact of patient population differences and genomic variation in efavirenz therapy. AIDS 22:1709–1717. doi: 10.1097/QAD.0b013e32830163ad. [DOI] [PubMed] [Google Scholar]
- 35.Meyer zu Schwabedissen HE, Oswald S, Bresser C, Nassif A, Modess C, Desta Z, Ogburn ET, Marinova M, Lütjohann D, Spielhagen C, Nauck M, Kroemer HK, Siegmund W. 2012. Compartment-specific gene regulation of the CAR inducer efavirenz in vivo. Clin Pharmacol Ther 92:103–111. doi: 10.1038/clpt.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho D-Y, Shen JHQ, Lemler SM, Skaar TC, Li L, Blievernicht JK, Zanger UM, Kim K-B, Shin J-G, Flockhart DA, Desta Z. 2016. Rifampin enhances cytochrome P450 (CYP) 2B6-mediated efavirenz 8-hydroxylation in healthy volunteers. Drug Metab Pharmacokinet 31:107–116. doi: 10.1016/j.dmpk.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mutlib AE, Chen H, Nemeth GA, Markwalder JA, Seitz SP, Gan LS, Christ DD. 1999. Identification and characterization of efavirenz metabolites by liquid chromatography/mass spectrometry and high field NMR: species differences in the metabolism of efavirenz. Drug Metab Dispos 27:1319–1333. [PubMed] [Google Scholar]
- 38.US Food and Drug Administration. February 2012. Guidance for industry. Drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. Draft guidance. Center for Drug Evaluation and Research (CDER), US Food and Drug Administration, Rockville, MD. [Google Scholar]
- 39.European Medicines Agency. 21 June 2012. Guideline on the investigation of drug interactions. Committee for Human Medicinal Products (CHMP), European Medicines Agency, London, United Kingdom. [Google Scholar]
- 40.Staszewski S, Morales-Ramirez J, Tashima KT, Rachlis A, Skiest D, Stanford J, Stryker R, Johnson P, Labriola DF, Farina D, Manion DJ, Ruiz NM. 1999. Efavirenz plus zidovudine and lamivudine, efavirenz plus indinavir, and indinavir plus zidovudine and lamivudine in the treatment of HIV-1 infection in adults. Study 006 Team. N Engl J Med 341:1865–1873. [DOI] [PubMed] [Google Scholar]
- 41.Theuretzbacher U, Ihle F, Derendorf H. 2006. Pharmacokinetic/pharmacodynamic profile of voriconazole. Clin Pharmacokinet 45:649–663. doi: 10.2165/00003088-200645070-00002. [DOI] [PubMed] [Google Scholar]
- 42.Bruggemann RJ, Alffenaar JW, Blijlevens NM, Billaud EM, Kosterink JG, Verweij PE, Burger DM. 2009. Clinical relevance of the pharmacokinetic interactions of azole antifungal drugs with other coadministered agents. Clin Infect Dis 48:1441–1458. doi: 10.1086/598327. [DOI] [PubMed] [Google Scholar]
- 43.Jeong S, Nguyen PD, Desta Z. 2009. Comprehensive in vitro analysis of voriconazole inhibition of eight cytochrome P450 (CYP) enzymes: major effect on CYPs 2B6, 2C9, 2C19, and 3A. Antimicrob Agents Chemother 53:541–551. doi: 10.1128/AAC.01123-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu P, Foster G, LaBadie RR, Gutierrez MJ, Sharma A. 2008. Pharmacokinetic interaction between voriconazole and efavirenz at steady state in healthy male subjects. J Clin Pharmacol 48:73–84. doi: 10.1177/0091270007309703. [DOI] [PubMed] [Google Scholar]
- 45.Liu P, Foster G, Labadie R, Somoza E, Sharma A. 2007. Pharmacokinetic interaction between voriconazole and methadone at steady state in patients on methadone therapy. Antimicrob Agents Chemother 51:110–118. doi: 10.1128/AAC.00559-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Huo M, Zhou J, Xie S. 2010. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed 99:306–314. doi: 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 47.Bélanger AS, Caron P, Harvey M, Zimmerman PA, Mehlotra RK, Guillemette C. 2009. Glucuronidation of the antiretroviral drug efavirenz (EFV) by UGT2B7 and an in vitro investigation of drug-drug interaction with zidovudine (AZT). Drug Metab Dispos 37:1793–1796. doi: 10.1124/dmd.109.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho DY, Ogburn ET, Jones D, Desta Z. 2011. Contribution of N-glucuronidation to efavirenz elimination in vivo in the basal and rifampin-induced metabolism of efavirenz. Antimicrob Agents Chemother 55:1504–1509. doi: 10.1128/AAC.00883-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bristol-Myers Squibb Company. April 2016. Sustiva (efavirenz) package insert. Bristol-Myers Squibb Company, Princeton, NJ. [Google Scholar]
- 50.Saari TI, Laine K, Leino K, Valtonen M, Neuvonen PJ, Olkkola KT. 2006. Effect of voriconazole on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Clin Pharmacol Ther 79:362–370. doi: 10.1016/j.clpt.2005.12.305. [DOI] [PubMed] [Google Scholar]
- 51.Michaud V, Ogburn E, Thong N, Aregbe AO, Quigg TC, Flockhart DA, Desta Z. 2012. Induction of CYP2C19 and CYP3A activity following repeated administration of efavirenz in healthy volunteers. Clin Pharmacol Ther 91:475–482. doi: 10.1038/clpt.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yimer G, Amogne W, Habtewold A, Makonnen E, Ueda N, Suda A, Worku A, Haefeli WE, Burhenne J, Aderaye G, Lindquist L, Aklillu E. 2012. High plasma efavirenz level and CYP2B6*6 are associated with efavirenz-based HAART-induced liver injury in the treatment of naive HIV patients from Ethiopia: a prospective cohort study. Pharmacogenomics J 12:499–506. doi: 10.1038/tpj.2011.34. [DOI] [PubMed] [Google Scholar]
- 53.Ribaudo HJ, Haas DW, Tierney C, Kim RB, Wilkinson GR, Gulick RM, Clifford DB, Marzolini C, Fletcher CV, Tashima KT, Kuritzkes DR, Acosta EP, Adult AIDS Clinical Trials Group Study. 2006. Pharmacogenetics of plasma efavirenz exposure after treatment discontinuation: an Adult AIDS Clinical Trials Group Study. Clin Infect Dis 42:401–407. doi: 10.1086/499364. [DOI] [PubMed] [Google Scholar]
- 54.Ngaimisi E, Mugusi S, Minzi O, Sasi P, Riedel KD, Suda A, Ueda N, Janabi M, Mugusi F, Haefeli WE, Bertilsson L, Burhenne J, Aklillu E. 2011. Effect of rifampicin and CYP2B6 genotype on long-term efavirenz autoinduction and plasma exposure in HIV patients with or without tuberculosis. Clin Pharmacol Ther 90:406–413. doi: 10.1038/clpt.2011.129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.