

Abstract
Background
The search for genetic factors underlying autism spectrum disorders (ASD) has led to the identification of hundreds of genes containing thousands of variants that differ in mode of inheritance, effect size, frequency, and function. A major challenge involves assessing the collective evidence in an unbiased, systematic manner for their functional relevance.
Methods
Here, we describe a scoring algorithm for prioritization of candidate genes based on the cumulative strength of evidence for each ASD-associated variant cataloged in AutDB (also known as SFARI Gene). We retrieved data from 889 publications to generate a dataset of 2187 rare and 711 common variants distributed across 461 genes implicated in ASD. Each individual variant was manually annotated with multiple attributes extracted from the original report, followed by score assignment using a set of standardized parameters yielding a single score for each gene.
Results
There was a wide variation in scores; SHANK3, CHD8, and ADNP had distinctly higher scores than all other genes in the dataset. Our gene scores were significantly correlated with other recently published rankings of ASD genes (R Spearman = 0.40–0.63; p< 0.0001), providing support for our scoring algorithm.
Conclusions
This new resource, which is freely available, for the first time aggregates on one-platform variants identified from various study types (simplex, multiplex, multigenerational, and consanguineous families), from both common and rare variants, and also incorporates their putative functional consequences to arrive at a genetically and biologically driven ranking scheme. This work represents a major step in moving from simply cataloging autism variants to using data-driven approaches to gain insight into their significance.
Electronic supplementary material
The online version of this article (doi:10.1186/s13229-016-0103-y) contains supplementary material, which is available to authorized users.
Keywords: Autistic disorder, Genetic variation, Common variants, Rare variants, Autosomal recessive
Background
Autism spectrum disorder (ASD) is a clinical diagnosis defined by neurodevelopmental impairments in two domains: persistent deficits in social reciprocity and communication across multiple contexts, together with restricted, repetitive patterns of behavior [1]. Individuals with ASD can display a broad clinical profile with varying severity in the core symptoms and often accompanied by medical comorbidities. With onset in the first years of life, ASD entails a life-long condition with diverse outcomes in adulthood [2]. The prevalence of ASD has been estimated as high as 1 in 68 children [3], yet an understanding of the biological mechanisms underlying ASD remains unclear, hampering attempts to develop specific molecular diagnostics or targeted therapeutics.
A multifactorial etiological model for ASD is being increasingly recognized. Several epidemiological studies have firmly established a genetic component underlying ASD with heritability estimates ranging from 50–90 % depending on the study parameters [4–6]. Consequently, numerous efforts to identify genes associated with ASD risk have been undertaken in hopes of inferring molecular pathways or surrogate markers associated with clinical manifestations of ASD. The ability to screen large cohorts using high-throughput genomic technologies has led to the discovery of hundreds of candidate genes containing thousands of variants, highlighting enormous genetic heterogeneity in ASD [7–10]. Although the significance of the vast majority of identified variants remains unresolved, a subset of genes have been found to be highly penetrant for ASD based on recurrent findings of rare, de novo, damaging variants in probands [11]. While initial estimates suggested between 350 and 400 autism susceptibility genes [12], more recent statistical models predict that well over 1000 genes may eventually be associated with ASD [13, 14]. Despite the incredible insight into the molecular genetics of ASD that these studies have provided, the diversity in study design, the significant variance in sample sizes and replication cohorts, and the use of different statistical models have resulted in a large set of candidate genes that are difficult to compare on a single platform. Moreover, within any given ASD candidate gene, multiple variants may be found, each with its own associated risk [15], further complicating a clear understanding of their relevance with respect to autism. To address these challenges, databases of ASD risk genes have been established in attempts to aggregate the ever-increasing number of candidate genes implicated in this disorder [16, 17]. However, only recently have strides been made towards developing methodologies for quantitative assessment of ASD risk genes [13, 18–20]. For example, transmission and de novo association (TADA) analysis was developed to identify risk-conferring genes by integrating rare de novo and inherited genetic variations from high-throughput, whole exome sequencing (WES) studies of large ASD cohorts such as the Autism Sequencing Consortium (ASC) and the Simons Simplex Collection (SSC) [11, 21]. While TADA analysis has proven to be a critical first step, further assessment strategies are required to fully integrate the complete spectrum of ASD genetic variations and consider all potential attributes that are likely to be encountered in patients evaluated in ASD clinics.
The Gene Scoring module (https://gene.sfari.org/autdb/GS_Home.do) of Simons Foundation Autism Research Initiative (SFARI) was created as a means for evaluation of candidate genes on a discrete or categorical scale taking into account the strength of genetic evidence linking a gene to ASD [22]. A set of scoring criteria was developed to assess different types of evidence, methodologies, and variability reported in the genetic studies of ASD [22]. Here, we have extended this initial work to incorporate a systematic evaluation of diverse types of genetic variants implicated in ASD. Our approach is based on assessment of multiple attributes of an ASD variant including mode of inheritance, effect size, and variant frequency in the general population. In this study, we report a consolidated gene score by summing the various evidence scores generated for each individual variant of an ASD-implicated gene. Next, we compared the gene scores generated in this study with the expert-led SFARI Gene Scoring module as well as the top ranking ASD genes identified in simplex families [11, 23]. We found strong concordance between our ASD gene ranking strategy and the other three approaches [11, 23]. Using our model, we prioritized a larger set of genes including SHANK3, CHD8, ADNP, MET, CNTNAP2, and others derived from the most complete collection of genetic variations associated with ASD originating from simplex, multiplex, multigenerational, and consanguineous families.
Methods
AutDB catalog of genetic variations associated with ASD
The AutDB database (also known as SFARI Gene) was first released in 2007 as an online portal providing a comprehensive, up-to-date resource for ASD candidate genes identified in peer-reviewed scientific publications [16]. We have since expanded the ASD gene database to include detailed annotation of both rare and common genetic variations extracted from original scientific reports. In this study, rare variants are defined as those with a population frequency <1 %; common variants are defined as those found in the general population at a frequency of ≥1 %. Annotated variants in AutDB include single nucleotide variants (SNVs), insertion-deletion variants (indels), single gene copy number variants (CNVs occurring within a gene), and chromosomal rearrangements disrupting a single gene thereby capturing the wide range of potentially pathogenic genetic variations identified in ASD cohorts. AutDB is continuously maintained by scheduled quarterly releases in order to provide the most up-to-date resource for the ASD genetics community. The data freeze of January 2015 was used for building the input dataset as described below.
Input dataset
We performed several filtration steps to generate a dataset of ASD-specific variants to use in the scoring process (Additional file 1: Figure S1A). Annotated rare variants were excluded if they were identified in probands lacking a confirmed diagnosis of ASD (e.g., rare variants in ASD-associated genes that had been identified in patients presenting with intellectual disability or epilepsy but without ASD). Next, we excluded rare ASD-associated CNVs lacking statistical significance as determined by either the absence of a reported p value or a p value greater than 0.05. A total of 2187 rare variants remained after variant filtration. Annotated common variants were excluded from variant scoring if they were identified in cohorts lacking a confirmed diagnosis of ASD, such as common variants in ASD-associated genes found associated with other neuropsychiatric disorders such as schizophrenia and bipolar disorder. This filtration process yielded a dataset of 771 common variants associated with ASD.
Variant scoring
Each individual variant included in the Human Gene Module of AutDB was manually annotated with 17 standardized descriptors providing a level of variant-specific detail unique to AutDB. These include significance of genetic association, family structure (simplex, multiplex, multigenerational, or consanguineous), zygosity (heterozygous, homozygous, or hemizygous), inheritance pattern (de novo or transmitted), the type of variant (missense, nonsense, etc.), and the functional effect of the variant (Additional file 2).
The functional effect of each variant represents a scoring criteria shared by both rare and common variants and is based on the reported results from the publication’s experimental analysis or in silico prediction. Then manual curation of these reported results was used to place the variant into one of four functional effect categories, with increasing weight given to variants likely to cause a functional biological effect (Additional file 3: Table S1). Variant Specificity to ASD, and Variant Inheritance and Segregation pattern of rare variants (referred to as scoring terms “R1” and “R2,” respectively) were assigned with scores reflecting the strength of the association of the variant to ASD (Additional file 3: Table S2). For instance, the R1 scoring term provides increasing weight based on the variant’s association with ASD and its absence in control populations included in the study or in large databases of human genetic variations such as dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), the 1000 Genomes Project (http://www.1000genomes.org), or the NHLBI Exome Variant Server (http://evs.gs.washington.edu/EVS/). Similarly, the R2 scoring category gives increasing weight to variants exhibiting a clear segregation pattern in individuals exhibiting ASD symptoms within a pedigree. Finally, “RG5” considers the unique statistical improbability of finding multiple de novo loss-of-function (LOF) variants in a single gene in ASD cases compared to controls [9]. The “RG6” category is a similar scoring factor that adds additional weight to rare bi-allelic LOF variants identified in consanguineous families. Owing to their unique genetic architecture and contributions to ASD as compared to rare variants [4], common variants were annotated using a separate set of scoring criteria. These are summarized by the “CG1” score, which considers the reported odds ratio and associated p value for a variant, and the “CG2” score, which provides increasing weight if the common variant was replicated in a second cohort of ASD individuals within the same study and/or in a new study (Additional file 3: Table S3).
Finally, we developed a comprehensive algorithm that aggregates the sub-scores from each of the categories for every variant in a given gene, weights each scoring sub-category relative to each other based on their importance to ASD linkage (based on published evidence and expert opinion), and compiles a single summary score for each rare and common variant described (Additional file 1: Figure S1B and S1C, respectively). The sum of all scored rare variants and all scored common variants were then calculated to generate a total rare variant score (RVS) and/or total common variant score (CVS) for any given gene.
Results
ASD risk variant distributions
The global landscape of variants associated with ASD analyzed in this study is summarized in Fig. 1. Overall, there were 2187 rare variants and 711 common variants in our dataset. These data were retrieved from 889 scientific publications of which 584 (65.7 %) reported only on rare variants, 266 (29.9 %) reported only on common variants, and only 39 (4.4 %) reported on both rare and common variants associated with ASD (Fig. 1a). The variants were distributed in 461 genes of which 261 (56.6 %) included only rare variants, 120 (26.0 %) included only common variants, and 80 (17.4 %) genes included both rare and common variants (Fig. 1b). Examination of the spectrum of variants in our database revealed that the majority of rare variants associated with ASD are nonsynonymous in the coding region (71.2 %), whereas most common variants are non-coding (88.9 %) (Fig. 1c).
Fig. 1.
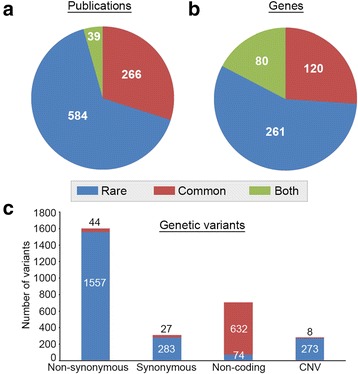
Global overview of the data in this study. a A pie chart showing the number of publications reporting only common (red), rare (blue), or both common and rare (green) genetic variants associated with autism spectrum disorder (ASD) in our database. b A pie chart showing the number of genes including only common (red), rare (blue), or both common and rare (green) genetic variants associated with ASD in our database. c A distribution of the common (red) and rare (blue) genetic variation in our database within four mutation categories: nonsynonymous, synonymous, non-coding, and copy number variations (CNVs)
ASD risk genes scoring
To assess the strength of evidence linking a candidate gene to ASD, we computed two scores for each gene: (1) rare variant score (RVS) that is based on the cumulative evidence of all rare variants within a particular gene, and (2) common variant score (CVS) which is based on the cumulative evidence of all common variants that are associated with a particular gene (Additional file 4: Table S4). Both RVSs and CVSs varied remarkably between genes (Fig. 2a, b) with SHANK3 having the highest RVS (RVS = 346) and MET having the highest CVS (CVS = 85). Notably, both RVS and CVS were significantly correlated with the number of annotated variants per gene (r = 0.84 and r = 0.70, respectively; p < 0.0001; Fig. 2c, d), which were significantly dependent on the number of publications per gene (r = 0.70 and r = 0.69, respectively; p < 0.0001; Fig. 2e, f). No correlation was found between either RVS or CVS and the coding sequence length of the gene. Next, we computed a total gene score (TGS) as the sum of RVS + CVS for each gene. The TGS of all the genes in our database had a log-normal distribution with a geometric mean of TGS = 9.03 ± 3.06 (Fig. 3a). Notably, three genes (SHANK3, CHD8, and ADNP) had distinctively higher scores than other genes in the database.
Fig. 2.
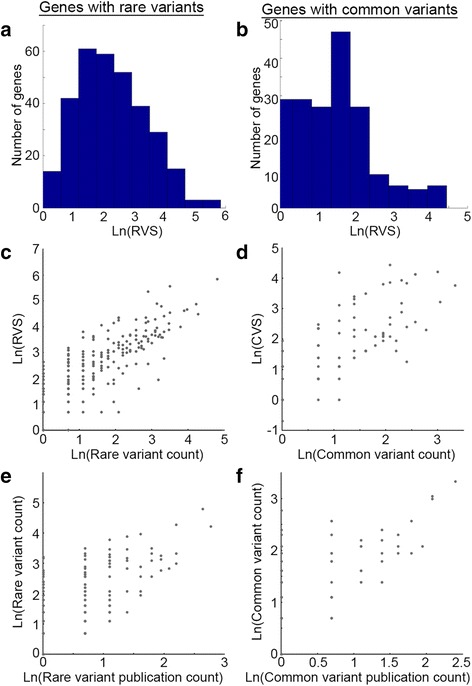
Distributions of rare variant scores (RVS) and common variant scores (CVS). The distributions of the natural logarithm of genes’ RVS and genes’ CVS are depicted as histograms (a, b, respectively), and as a function of variant counts (c, d for rare and common variants, respectively). Both RVS and CVS are significantly correlated with the number of variants per gene (r = 0.84 and r = 0.70, respectively; p < 0.0001). e, f The obvious association between the number of variants and the number of publication per gene (r = 0.70 and r = 0.69, respectively; p < 0.0001)
Fig. 3.
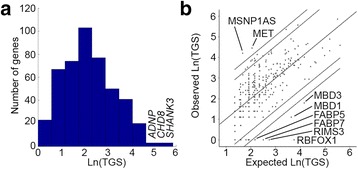
Gene scores distribution. a A histogram of the natural logarithm of the total gene score (LnTGS) of the 461 genes associated with autism spectrum disorder in this study. Three genes (ADNP, CHD8, and SHANK3) have distinctly larger scores than all other genes in the dataset. b A scatterplot of the expected LnTGS according to a linear regression model based on the number of variants per gene vs. the observed LnTGS. 95 and 99 % confidence intervals (CI) of the predicted scores are depicted in dotted and dashed lines, respectively
To examine the effect of variant count on TGS, we ran a multivariate linear regression analysis using both rare and common variant counts as possible predictors. The resulting regression model explained only 50 % (adjusted R 2 = 0.502) of the variation in TGS with both rare and common variants contributing significantly to this variation (B = 0.83 and B = 0.48 for rare and common variants; p< 0.0001). Figure 3b displays the expected ln(TGS) scores of the genes according to this model compared to their observed ln(TGS). Six genes (RBFOX1, RIMS3, MBD1, MBD3, FABP5, and FABP7) had ln(TGS) that was significantly lower than their expected ln(TGS) according to this model (p< 0.01). Only two genes, MET and MSNP1AS, had ln(TGS) that were significantly higher than their expected score (p< 0.01).
Comparison to other ASD risk genes datasets
Next, we compared the results of our gene scoring approach to the results of three other recently published ASD-related gene sets that used other prioritization strategies. Figure 4 summarizes the results of these analyses. Of the 461 genes included in our study, 38 (8.2 %) were also included in all other gene sets and 160 (34.7 %) were exclusively represented in our AutDB (Fig. 4a). Overall, there was a strong agreement between our gene scoring approach and the other three approaches. Specifically, TGSs were significantly correlated with gene categories in the community-based SFARI gene scoring module [22] (Fig. 4b; R Spearman = −0.63; p< 0.0001). Interestingly, there were few genes with TGS that deviated from the distribution of scores in their category. In addition, there were 169 genes not previously scored by the expert-led SFARI Gene Scoring initiative that were included in our analysis (Fig. 4a). These genes had a wide distribution of TGS covering almost the entire spectrum of scores in our database.
Fig. 4.
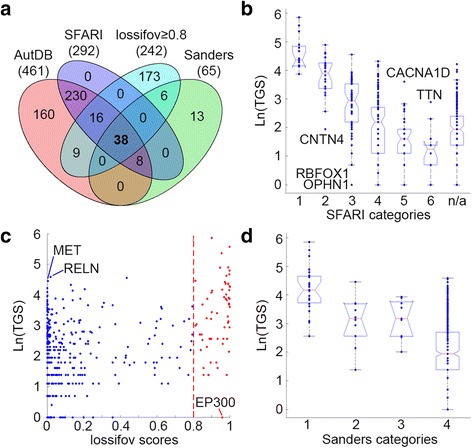
A comparison to other ASD risk genes datasets. a Venn diagram of the 653 ASD risk genes included in AutDB, SFARI gene scoring module [22], Iossifov et al. [23], and Sanders et al. [11]. b A distribution of TGS across the different categories of genes in the SFARI gene scoring module (1 = “high confidence”; 2 = “strong candidate”; 3 = “suggestive evidence”; 4 = “minimal evidence”; 5 = “hypothesized”; 6 = “not supported”). c A scatterplot comparing the TGS of the genes in AutDB to their corresponding posterior probabilities in Iossifov et al. [23]. Genes with posterior probabilities ≥0.8 are highlighted in red. d A distribution of TGS across the different categories of genes reported in Table 4 of Sanders et al. [11]. Categories indicate the probability pf genes to be ASD risk genes (1 = FDR ≤ 0.01; 2 = 0.01 < FDR ≤ 0.05; 3 = 0.05 < FDR ≤ 0.1; 4 = 0.1 < FDR
Finally, we compared our scoring results to those obtained from two recently published studies based on whole exome sequencing of ASD cohorts [11, 23]. We observed a moderate, but highly significant, correlation (R Pearson = 0.4; P = 6.6 × 10−19) between the TGS of the 461 genes in our study and their corresponding posterior probabilities [23] (Fig. 4c). Nevertheless, only 63 (13.7 %) of these genes had a posterior probability of ≥0.8 that was suggested by Iossifov et al. [23] as good candidate ASD genes (Fig. 4a). Furthermore, of the 65 ASD risk genes reported by Sanders et al. [11], 44 (67.7 %) were included in our dataset (Fig. 4a). Here too, we observed strong correlation between the TGS of these genes and their reported ranking in that study (Fig. 4d; R Spearman = −0.41; p= 4.9 × 10−20).
Discussion
Given the accelerated pace of ASD candidate gene discovery, it is critical that resources be available to the research community that not only catalog the identified variants in detail but also provide tools to evaluate the potential risk conferred by each individual variant. In this report, we describe a systematic variant scoring strategy utilizing the autism gene database AutDB that encompasses detailed annotation of both rare and common genetic variants associated with ASD for candidate gene prioritization. The large set of variants analyzed here was extracted from studies that varied in size—from single case reports to analysis of large cohorts such as the Simons Simplex Collection. Additionally, our dataset included variants identified by a variety of methodologies ranging from targeted sequencing to whole genome-based screening. While a number of other recent analyses of ASD genes have focused on rare damaging de novo mutations in simplex ASD cases, our study design also allowed the inclusion of inherited autosomal recessive variants and variants observed in multiplex and multigenerational families.
This scoring approach identified three ASD risk genes (SHANK3, CHD8, and ADNP) that exhibited significantly higher scores than all other genes. SHANK3 was first reported as an ASD candidate gene based on identification of heterozygous mutations in ASD probands from three unrelated families [24]. Subsequently, additional variants in SHANK3 have been identified by targeted sequencing in multiple ASD cohorts [15, 25]. In contrast, the risk conferred by functional variants in CHD8 and ADNP have only recently been described by WES studies of large ASD cohorts [9], followed by smaller studies focused exclusively on the identification of variants within these two genes [25, 26]. However, comparable WES studies of large ASD cohorts have failed to identify a large number of functional variants in SHANK3, due in part to the high GC content of this gene, which complicates WES approaches. These findings clearly indicate the importance of considering genetic evidence from multiple sources and multiple experimental methodologies in accurately prioritizing ASD candidate genes.
A comparison of the prioritized gene list generated by our scoring model with three other recently published ASD-related gene lists [11, 22, 23] demonstrated strong agreement in all three instances, confirming the validity of our approach. Differences in the ranking of autism candidates between our approach and these previous studies are largely due to our exclusive focus on the variant’s/gene’s role in autism, not other neurodevelopmental diseases. For example, in our approach, a candidate gene’s score is entirely dependent on the attributes of the ASD-specific genetic variants; we excluded variants from scoring when associated with a neurodevelopmental disorder without an accompanying diagnosis of ASD. By comparison, SFARI Gene Scoring takes into consideration the broader involvement of an ASD gene in related neurodevelopmental/neuropsychiatric disorders as well as its biological role in relation to ASD. These differences in scoring approaches account at least in part for the discrepancies in scores for genes such as RBFOX1 (Fig. 4b), a gene for which considerable functional evidence exists including its role in regulating other ASD genes [27, 28] and its involvement in ASD pathogenesis as manifested by differential expression in postmortem brain of ASD individuals [29]. As ASD itself is already a heterogeneous diagnosis, we built our model specifically on confirmed cases of ASD only so as to be as stringent as possible to ensure our resultant prioritization scheme is as specific to ASD as possible, as we believe this is critical to the use of such lists both for basic science researchers and especially clinicians.
An important aspect of our study is the inclusion of common variations associated with ASD (Additional file 4: Table S4). As previously indicated, MET had the highest common variant score (CVS = 85) based on replicated genetic association studies. Similarly, a higher evidence category was assigned to MET in the expert-mediated scoring in SFARI Gene. Multiple lines of research indicate an important functional role for MET in ASD [30]. However, the role of common variants with small effect size remains poorly understood in ASD as compared to their role in other neuropsychiatric disorders such as schizophrenia and bipolar disorder. In these other disorders, a number of common variants have reached genome-wide significance across multiple studies; common variants in ASD have by and large failed to show similar replication across independent cohorts [31, 32].
Of note is the concern that more commonly studied genes will have more variants in the database simply by virtue of having been assessed more often and therefore will rank higher in any prioritization scheme. In fact, we did find significant correlations between the total variant scores and the number of publications from which variants for a gene were extracted. This represents somewhat of a “winner’s curse” phenomenon reflecting heightened attention from the ASD research community for select genes. Nevertheless, the number of reported variants per gene (which partially reflects the scientific interest in these genes) explained only ~50 % of the variations in scores—highlighting the comprehensive nature our scoring algorithm. As more unbiased whole exome and whole genome sequencing studies are undertaken and added to this database, this effect should continue to diminish. Furthermore, ongoing future development of our algorithm will attempt to correct for such effects.
Conclusions
In conclusion, we describe the most comprehensive database to date of both common and rare DNA variants associated with ASD. Using our novel scoring and ranking algorithm that considers both genetic and biologic data, we systematically characterized all classes of variants implicated in ASD on one platform and provide a summary score for each ASD-associated genes (Additional file 4: Table S4), which for the first time allows for a fair comparison of ASD-associated gene relevance irrespective of the type, number, or quality of study in which the underlying variant(s) were identified. In addition to strong ASD genes such as CHD8, ADNP, and SCN2A recurrently identified by WES, our prioritized gene set includes SHANK3, MET, and CNTNAP2 supported by multiple lines of genetic evidence, however missed by WES. This database and ranking system represents an important step in moving from simply cataloging ASD genes to using unbiased, data-driven approaches to determine the relative strength of association with ASD of each gene. This resource, which is free to access and will continually be updated, will serve as an important tool to both basic scientists and clinicians working with ASD patients.
Acknowledgements
We thank Catherine C. Swanwick for proof reading the manuscript.
Funding
This research was funded by the Simons Foundation.
Availability of data and materials
All data described in this manuscript will be available in AutDB (http://autism.mindspec.org/autdb/Welcome.do) and SFARI Gene (https://gene.sfari.org/autdb/Welcome.do).
Authors’ contributions
SBB was the principal investigator and conceived this project. EL and MNZ were involved in variant annotation and scoring. EL, AP, and SBB developed the scoring algorithm described in this manuscript. IM and WP performed statistical analysis. EL, IM, MNZ, and SBB were involved in drafting the manuscript. EL, IM, MNZ, WP, AP, and SBB were involved in revising the manuscript. All authors approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- ASC
Autism Sequencing Consortium
- ASD
Autism spectrum disorders
- CNV
Copy number variant
- CVS
Common variant score
- LOF
Loss of function
- RVS
Rare variant score
- SFARI
Simons Foundation Autism Research Initiative
- SNV
Single nucleotide variant
- SSC
Simons Simplex Collection
- TADA
Transmission and de novo association
- TGS
Total gene score
- WES
Whole exome sequencing
Additional files
Gene scoring process. (A) A flowchart describing the curation of both rare and common variants that are used in the overall scores of genes associated with autism spectrum disorder. (B) A formula for gene score calculation based on rare variants. (C) A formula for gene score calculation based on common variants. (TIF 1424 kb)
Supplementary methods. (DOCX 12 kb)
Variant scoring criteria. Tables of scoring criteria common to both rare and common variants (Additional file 3: Table S1), scoring criteria specific for rare variants (Additional file 3: Table S2), and scoring criteria specific for common variants (Additional file 3: Table S3). (DOCX 19 kb)
Scores of ASD candidate genes using the scoring algorithm. Total gene scores were determined from the total score of all rare variants (RVS) and the total score of all common variants (CVS) for a given candidate gene. The coding sequence length of a candidate gene, the number of publications from which rare and common variants were extracted (Pub_Rare and Pub_Common, respectively), and the number of scored rare and common variants (Var_Rare and Var_Comm, respectively) are included for each gene. (XLSX 33 kb)
Footnotes
Eric Larsen and Idan Menashe contributed equally to this work.
Contributor Information
Eric Larsen, Email: eric@mindspec.org.
Idan Menashe, Email: idanmen@bgu.ac.il.
Mark N. Ziats, Email: ziatsm@mail.nih.gov
Wayne Pereanu, Email: wayne@mindspec.org.
Alan Packer, Email: apacker@simonsfoundation.org.
Sharmila Banerjee-Basu, Email: sharmila@mindspec.org.
References
- 1.American Psychiatric Association., American Psychiatric Association. DSM-5 Task Force . Diagnostic and Statistical Manual of Mental Disorders: DSM-5. 5. Washington, D.C.: American Psychiatric Association; 2013. [Google Scholar]
- 2.Anderson DK, Liang JW, Lord C. Predicting young adult outcome among more and less cognitively able individuals with autism spectrum disorders. J Child Psychol Psychiatry. 2014;55(5):485–94. doi: 10.1111/jcpp.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Developmental Disabilities Monitoring Network Surveillance Year Principal I, Centers for Disease C, Prevention Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ. 2014;63(2):1–21. [PubMed] [Google Scholar]
- 4.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–5. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68(11):1095–102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311(17):1770–7. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–73. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–5. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–41. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tammimies K, Marshall CR, Walker S, Kaur G, Thiruvahindrapuram B, Lionel AC, Yuen RK, Uddin M, Roberts W, Weksberg R, et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA. 2015;314(9):895–903. doi: 10.1001/jama.2015.10078. [DOI] [PubMed] [Google Scholar]
- 11.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87(6):1215–33. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74(2):285–99. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He X, Sanders SJ, Liu L, De Rubeis S, Lim ET, Sutcliffe JS, Schellenberg GD, Gibbs RA, Daly MJ, Buxbaum JD, et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013;9(8):e1003671. doi: 10.1371/journal.pgen.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronemus M, Iossifov I, Levy D, Wigler M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet. 2014;15(2):133–41. doi: 10.1038/nrg3585. [DOI] [PubMed] [Google Scholar]
- 15.Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, Giuliano F, Stordeur C, Depienne C, Mouzat K, et al. Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 2014;10(9):e1004580. doi: 10.1371/journal.pgen.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basu SN, Kollu R, Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic Acids Res. 2009;37(Database issue):D832–6. doi: 10.1093/nar/gkn835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu LM, Li JR, Huang Y, Zhao M, Tang X, Wei L. AutismKB: an evidence-based knowledgebase of autism genetics. Nucleic Acids Res. 2012;40(Database issue):D1016–22. doi: 10.1093/nar/gkr1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47(6):582–8. doi: 10.1038/ng.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L, Lei J, Sanders SJ, Willsey AJ, Kou Y, Cicek AE, Klei L, Lu C, He X, Li M, et al. DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol Autism. 2014;5(1):22. doi: 10.1186/2040-2392-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11(3):294–6. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–15. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, Menashe I, Wadkins T, Banerjee-Basu S, Packer A. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs) Mol Autism. 2013;4(1):36. doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iossifov I, Levy D, Allen J, Ye K, Ronemus M, Lee YH, Yamrom B, Wigler M. Low load for disruptive mutations in autism genes and their biased transmission. Proc Natl Acad Sci U S A. 2015;112(41):E5600–7. doi: 10.1073/pnas.1516376112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39(1):25–7. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81(6):1289–97. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J, Schuurs-Hoeijmakers JH, Marcelis CL, Willemsen MH, Vissers LE, et al. A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet. 2014;46(4):380–4. doi: 10.1038/ng.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee JA, Damianov A, Lin CH, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, Martin KC. Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron. 2016;89(1):113–28. doi: 10.1016/j.neuron.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014;6(6):1139–52. doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474(7351):380–4. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng Y, Huentelman M, Smith C, Qiu S. MET receptor tyrosine kinase as an autism genetic risk factor. Int Rev Neurobiol. 2013;113:135–65. doi: 10.1016/B978-0-12-418700-9.00005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Sykes N, Pagnamenta AT, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19(20):4072–82. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curran S, Bolton P, Rozsnyai K, Chiocchetti A, Klauck SM, Duketis E, Poustka F, Schlitt S, Freitag CM, Lee I, et al. No association between a common single nucleotide polymorphism, rs4141463, in the MACROD2 gene and autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(6):633–9. doi: 10.1002/ajmg.b.31201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data described in this manuscript will be available in AutDB (http://autism.mindspec.org/autdb/Welcome.do) and SFARI Gene (https://gene.sfari.org/autdb/Welcome.do).