

Summary
Humans differ in the outcome that follows exposure to life-threatening pathogens, yet the extent of population differences in immune responses and their genetic and evolutionary determinants remain undefined. Here, we characterized, using RNA sequencing, the transcriptional response of primary monocytes from Africans and Europeans to bacterial and viral stimuli—ligands activating Toll-like receptor pathways (TLR1/2, TLR4, and TLR7/8) and influenza virus—and mapped expression quantitative trait loci (eQTLs). We identify numerous cis-eQTLs that contribute to the marked differences in immune responses detected within and between populations and a strong trans-eQTL hotspot at TLR1 that decreases expression of pro-inflammatory genes in Europeans only. We find that immune-responsive regulatory variants are enriched in population-specific signals of natural selection and show that admixture with Neandertals introduced regulatory variants into European genomes, affecting preferentially responses to viral challenges. Together, our study uncovers evolutionarily important determinants of differences in host immune responsiveness between human populations.
Keywords: population genetics, immune response, eQTL mapping, transcriptional responses, monocytes, evolution, natural selection, Neandertal admixture
Graphical Abstract
Highlights
-
•
Human populations differ in their transcriptional responses to immune challenges
-
•
Cis- and trans-eQTLs contribute to population differences in immune responses
-
•
Immune-responsive regulatory variants have participated in human adaptation
-
•
Neandertals introduced variants affecting immune responses into European genomes
Genetic variants enriched in population-specific signals of natural selection and, among Europeans, of Neandertal ancestry play a major role in the differences in transcriptional responses to inflammatory and infectious challenges observed between human populations.
Introduction
The immune response to stress is a highly complex phenotype. Inappropriate immune activity can increase susceptibility to infectious, inflammatory, and autoimmune diseases, the clinical manifestations of which vary considerably between individuals and populations (Brinkworth and Barreiro, 2014, Casanova et al., 2013). The contribution of host genetic factors in explaining such heterogeneity is increasingly documented by genome-wide association studies (GWASs), which have identified variants, often located in non-coding regions, associated with disease risk (Parkes et al., 2013, Schaub et al., 2012). However, it remains unknown how these variants functionally impact immune responses across populations.
Genetic variants exerting regulatory effects on gene expression, known as expression quantitative trait loci (eQTLs), have proven to be of significant biomedical interest (Montgomery and Dermitzakis, 2011), as they help to establish links between intermediate phenotypes and organismal traits, such as immunity to infection (Fairfax and Knight, 2014). While eQTL studies have mostly focused on steady-state expression measurements (Lappalainen et al., 2013, Montgomery et al., 2010, Pickrell et al., 2010, Spielman et al., 2007, Stranger et al., 2012), recent work has characterized response eQTLs in human cells exposed to various immune or infectious challenges (Barreiro et al., 2012, Çalışkan et al., 2015, Fairfax et al., 2014, Lee et al., 2014). However, the extent and genetic determinants of inter-population transcriptional differences upon immune stimulation remain largely unexplored, yet this is critical to increase knowledge on the varying susceptibility to immune disorders observed at the population level.
Understanding how natural selection has shaped genome variability represents a powerful approach to identify genes that have played a major role in host survival, complementing immunological as well as clinical and epidemiological genetic studies (Casanova et al., 2013, Quintana-Murci et al., 2007). Indeed, microorganisms are among the strongest selective pressures encountered by humans, and multiple host genes and variants associated with immune functions and diseases are reported to evolve adaptively (Fumagalli and Sironi, 2014, Karlsson et al., 2014, Quintana-Murci and Clark, 2013). Furthermore, there is growing evidence that regulatory variants play a major role in population adaptation and contribute to the diversity of complex phenotypes (Fraser, 2013, Pickrell, 2014, Schaub et al., 2012).
Besides the occurrence of new advantageous mutations, functional variants can be introduced in human populations through interbreeding with now-extinct lineages (Vattathil and Akey, 2015). Recent data showed that 1%–6% of the genome of modern Eurasians derives from Neandertals or Denisovans (Prüfer et al., 2014, Reich et al., 2010, Sankararaman et al., 2014, Vernot and Akey, 2014). Furthermore, humans appear to have acquired genetic diversity from archaic hominins at several immune genes, such as HLA, TLR1, or the OAS cluster (Abi-Rached et al., 2011, Dannemann et al., 2016, Deschamps et al., 2016, Mendez et al., 2013). Despite these findings, the impact of selection and archaic admixture on driving population differences in immune responses remains to be investigated.
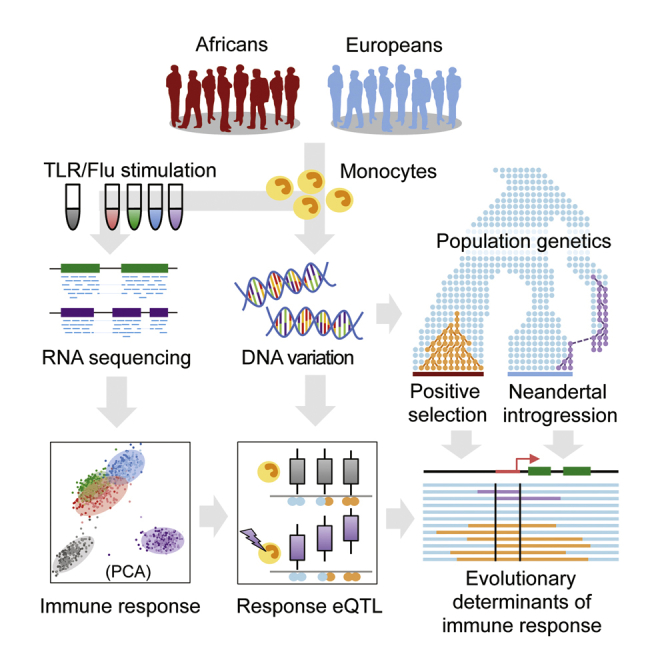
Here, we determined the degree of immune response variation, and of its genetic and evolutionary sources, within and between human populations. We used RNA sequencing (RNA-seq) to characterize the responses of primary monocytes, from individuals of European and African descent, to various Toll-like receptor (TLR) ligands and influenza A virus, and we mapped eQTLs. We found that marked differences in immune responses exist between populations due to the contribution of cis- and trans-acting regulatory variants. We also show that natural selection has contributed to the observed population differences of immune responses and establish that admixture with Neandertals introduced regulatory variants affecting responsiveness to immune stimuli into European genomes.
Results
An Experimental and Computational Approach to Understand Immune Response Variation
Population variation in immune responses was characterized in primary monocytes, as a model of an innate immune cell type, from 200 healthy individuals of self-reported African and European ancestry (100 individuals of each population) (see STAR Methods; Figure S1). Cells were exposed, for 6 hr, to ligands activating TLR4 (bacterial lipopolysaccharide [LPS]) and TLR1/2 (Pam3CSK4, a synthetic triacylated lipopeptide), responsible principally for sensing bacterial components, TLR7/8 (R848, an imidazoquinoline compound), responsible predominantly for sensing viral nucleic acids, and to a human seasonal influenza A virus (IAV). RNA-seq data were collected from unstimulated and stimulated monocytes, yielding a final dataset of 970 transcriptional profiles with ∼34 million single-end reads per sample. High-density genome-wide genotyping and whole-exome sequencing data were generated for all individuals and used to characterize their genetic ancestry, map eQTLs, explore patterns of allele-specific expression (ASE), and perform population and evolutionary genetic analyses.
Figure S1.

Overview of the Experimental Setting, Related to STAR Methods
The transcriptional response of primary monocytes from 200 healthy donors of European and African descent to various immune stimulations was dissected to pinpoint molecular phenotypes differing between populations and under genetic control.
Context-Specific Transcriptional Signatures of Monocyte Activation
To assess the transcriptional responses of CD14+ monocytes, we first processed and normalized the RNA-seq data and accounted for potential batch effects and confounding factors that could impact gene expression variation (Figures S2 and S3; see STAR Methods). We obtained a final set of 12,578 expressed genes, 6,752 of which were differentially expressed upon stimulation in at least one condition (|log2(fold change [FC])| > 1) at a false discovery rate (FDR) < 0.05 (Table S1A). Using adjusted expression data, principal component (PC) analysis revealed that PC1 and PC2, accounting for 65% of total variation, corresponded primarily to IAV infection and TLR activation, respectively (Figure 1A).
Figure S2.

Processing of RNA-Sequencing Data, Related to STAR Methods
(A) Read counts across the 970 RNA samples (in millions of reads). (B) Alignment of reads with various genomic features. Reads aligning with splice junctions are counted as many times as the number of genomic features they overlap. TSS: transcription start site, UTR: untranslated region, CDS: coding sequence, TES: transcription end site, up: upstream, down: downstream. (C) Distribution of GC content across samples. (D) Correlations between gene expression and global GC content. The expression of a large proportion of high-GC content genes was positively correlated with GC content (3rd quartile of GC content, in red), whereas the expression of low-GC content genes tended to be negatively correlated to GC content (1st quartile of GC content, in blue). The correlation distribution for total genes is shown in gray. Expectations were calculated by permutation (black). (E) Illustration of the effect of global GC content on gene expression. UFM1 (GC content: 33.9%) expression is plotted for all RNA samples, ordered by global GC content, for each condition and population. (F) Effect of various technical batches and confounding factors on gene expression. The proportion of genes whose expression levels are associated, for different levels of significance, with each factor is presented before and after correction of the data (up and bottom panels, respectively). The association of each gene with each cofactor was assessed by determining the proportion of the variance of gene expression explained by the cofactor under consideration, after adjustment for all other cofactors. (G) Boxplots of coefficient of variation (CV) in technical and biological replicates across conditions. CV distributions of technical replicates are smaller in magnitude and less variable compared to distributions of pairwise biological replicates (Wilcoxon Rank-Sum Test, ∗∗∗p < 0.001). (H) Boxplots of correlation coefficient (r) between technical and biological replicates. r calculated between technical replicates (red circles) are significant outliers to the r distributions of pairwise biological replicates (boxplots).
Figure S3.

Overview of the Pipeline Used for RNA-Seq Data Pre-processing, Related to STAR Methods
We filtered out samples with irregular gene body coverage and used Cufflinks/CuffDiff to estimate transcript level FPKM. We removed all transcripts for which the total gene FPKM was less than 1 in all conditions. We then log-transformed the data and performed adjustments for GC content, 3′/5′ bias, date of experiment and library preparation batch. We carried out kNN imputation before ComBat, to handle missing values. Batch covariates were treated sequentially, as ComBat can only handle one batch variable at a time. The corrected FPKM values were then transformed back to the normal scale and forced to positive values to calculate gene-level FPKM. Gene expression was considered on the log scale for all subsequent analyses.
Figure 1.

Transcriptional Response of Primary Monocytes to TLR Activation and Influenza A Virus Infection
(A) PC analysis of adjusted RNA-sequencing expression profiles in the five conditions tested in Africans (AFB) and Europeans (EUB).
(B) Weighted correlation network analysis. Relative size of the modules (left), expression patterns of genes in modules that are upregulated after stimulation (1–4) with boxplots representing relative expression based on PC1 (middle), and most associated transcription factor binding motifs in genes within modules (right).
(C) Most significant GO biological process enrichments of genes in modules 1–4.
We next used weighted correlation network analysis (WGCNA) (Langfelder and Horvath, 2008) to define modules of genes that showed similar behaviors (up-/downregulation) upon immune stimulation and identified ten modules, each comprising 257–4,070 genes (Figures 1B and S4). The gene modules upregulated upon stimulation (modules 1–4) were enriched in diverse Gene Ontology (GO) functions related to host defense, including an inflammatory response restricted to TLR activation and a global antiviral response exacerbated upon IAV infection (Figure 1C; Table S1B). The gene modules downregulated (modules 5–9), or containing similarly expressed genes across conditions (module 10), were enriched in functions such as RNA processing and translational termination (Table S1B).
Figure S4.

Weighted Correlation Network Analysis, Related to Figure 1
The relative expression of each gene module is based on the first principal component for the expression of genes present in the corresponding module.
Using the transcription factor affinity prediction (TRAP) method (Thomas-Chollier et al., 2011), we found that the annotated promoter regions of the genes within each module were enriched in binding motifs for specific transcription factors, such as nuclear factor κB (NF-κB), IRF1, and GATA2 (Table S1C). These analyses show that cellular context is the major determinant of transcriptional variability and provide a genome-wide view of common and specific responses of CD14+ monocytes to activation by TLR ligands and IAV infection.
Transcriptional Responses to Immune Stimulation Differ between Populations
We investigated how Africans and Europeans differ in transcriptional responses to immune stimulation. The estimated genetic ancestry of individuals accurately reflected self-reported ethnicity, indicating negligible levels of admixture between the two groups (Figures S5A and S5B). We searched for genes that show population differences in expression (popDEGs; FDR < 0.05) and further considered the magnitude of such differences by setting different thresholds of fold change between populations (FCpop). We identified 5,501 popDEGs with a |log2(FCpop)| > 0.2 in at least one condition, a figure that dropped to 821 and 70 when increasing the magnitude of fold change (|log2(FCpop)| > 0.5 and > 1, respectively; Table S1D). Among genes displaying the largest population differences (Table 1), we observed the scavenger receptor MARCO, involved in early inflammatory responses to influenza (Areschoug and Gordon, 2009); the chemokine receptor CX3CR1, mediating skin wound healing (Ishida et al., 2008); and, more generally, several interferon-stimulated genes.
Figure S5.

Genetic Structure of the Population Samples, Related to STAR Methods
(A) Genetic ancestry of African-descent Belgians (AFB) and European-descent Belgians (AFB), estimated by ADMIXTURE. Each vertical line represents an individual genome, which is partitioned into K different genetic clusters. This analysis was performed on 229,320 independent SNPs and 789 individuals from 22 populations, including EUB and AFB, together with a selection of representative populations from sub-Saharan Africa, North Africa, the Near East and Europe (Behar et al., 2010, Patin et al., 2014). We made K vary from 2 to 10, and ran five iterations with different random seeds for each K value. The run with the lowest cross-validation error rate for each K value is shown for K = 2 to 5. (B) Cross-validation (CV) error rates of ADMIXTURE results for 5 different iterations and K prior values. Minimum CV values for each K are in red. CV values start increasing at K = 6. (C) Local genetic sub-structure in the AFB population sample, estimated by principal component analysis (PCA). This analysis was performed on 341,593 independent SNPs and 511 individuals from 7 western and central African populations (Patin et al., 2014). (D) Local genetic sub-structure in the EUB population, estimated by PCA. This analysis was performed on 182,572 independent SNPs and 220 individuals from 13 European populations (Behar et al., 2010). (C-D) PC1 and PC2 are shown, together with the proportion of variance explained.
Table 1.
Genes Displaying the Highest Degree of Differential Expression or Differential Immune-Induced Responses between Africans and Europeans
| Condition | Africans | Europeans |
|---|---|---|
| Resting cells (NS) | CCL3L1, CCL3L3, CX3CR1, LPL, TMEM14C, TREML4, VNN1 | HTRA3, MARCO, MT1X, PADI4, RP11-105C19.1, RP11-645C24.5, S100P, TMEM176A, TMEM176B, USP32P1 |
| TLR4 (LPS) | AC131056.3, CEP128, LPL, RP11-1143G9.4, RP11-7F17.7, TREML3P, TREML4, VNN1 | AC004988.1, APOBEC3A, BATF2, CCL13, CCL15, CCL23, CCL8, CMPK2, CXCL10, DHX58, DNAAF1, ETV7, GBP4, HERC5, IFIT1, IFIT2, IFIT3, MARCO, NCOA7, PLXNA3, RP11-105C19.1, RP11-645C24.5, RSAD2, SIGLEC1, TMEM176A, TMEM176B, TNFSF10, U1, USP18, USP32P1 |
| TLR1/2 (Pam3CSK4) | AC131056.3, C2CD4B, CCL3L1, CCL3L3, CEP128, CPXM1, CSF3, GBA3, IL12B, IRG1, LPL, NKX3-1, SLC25A37, SNORD3B-1, SUCNR1, TREML4, VNN1 | CCL15, HMOX1, IFIT1, IFIT2, IFIT3, PLXNA3, RP11-105C19.1, RP11-645C24.5, RSAD2, TMEM176A, TMEM176B, U1, USP32P1 |
| TLR7/8 (R848) | AC131056.3, LPL, RP11-7F17.7, SUCNR1, TREML3P, TREML4 | PAM, PLXNA3, RP11-105C19.1, RP11-128M1.1, RP11-645C24.5, TMEM176A, TMEM176B, U1 |
| Influenza A virus (IAV) | CCL3L1, CCL3L3, CTSC, HS3ST3B1, IL6, LGALS17A, NUPR1, RP11-1143G9.4, SLC25A37, TREML4 | J01415.23, MARCO, MDGA1, PADI4, PAM, RASD2, RP11-105C19.1, RP11-105C19.2, RP11-645C24.5, S100P, SNHG5, TMEM176A, TMEM176B, U1 |
The genes listed are divided according to the population where they present the highest expression. All genes reported are differentially expressed between populations in the various cellular conditions (popDEGs, |log2(FCpop)| > 1), while those presented as underlined are further characterized by their stronger population differences in response to treatment, with respect to the non-stimulated condition (popDRGs). Underlined genes in the non-stimulated (NS) condition correspond to those that are differentially expressed between populations only in that condition. Genes presenting a |log2(FCpop)| > 1 at FDR of 5% are presented.
We next searched for genes presenting population differences in their response to treatment, relative to non-stimulated cells (popDRGs). We found 3,841 popDRGs (FDR < 0.05, 70% of popDEGs), the majority of which were treatment specific (2,687 popDRGs; Table S1E). popDRGs displaying stronger responses in Africans were enriched in GO functions from metabolic processes to defense responses, while popDRGs responding more strongly in Europeans were essentially restricted to defense functions in the TLR conditions and enriched in translational processes upon IAV infection (Table S1F). popDRGs showing the greatest population differences (|log2(FCpop)| > 1) were enriched in cytokines and chemokines (Fisher’s exact test, odds ratio [OR] = 36.7, p < 10−8), including IL12B and CSF3, responding more strongly to Pam3CSK4 in Africans, and CCL8, CCL13, CCL15, CCL23 and CXCL10, being more responsive to LPS in Europeans (Table 1). These results indicate that while population transcriptional differences of moderate effect are widespread, strong differences predominantly affect antiviral and inflammation-related genes that differ markedly in responsiveness between Africans and Europeans.
Detecting Local Immune-Responsive Regulatory Variation
We next mapped eQTLs by testing for associations between 10,278,745 SNPs (the set of genotyped and imputed SNPs presenting a minor allele frequency [MAF] > 0.05) and gene expression phenotypes. We first mapped local, likely cis-acting eQTLs within 1 Mb of each gene in Africans and Europeans separately. We used an additive linear model (Shabalin, 2012) that included the first two PCs of the genetic data (Figures S5C and S5D) to account for possible population substructure. Considering only eQTLs having an effect size of |βeQTL| > 0.2 at a FDR of 5%, we found 2,665 genes with an eQTL in at least one condition (Figure S6A; Table S2A). Of these, 917 genes presented a response eQTL (reQTL), an eQTL with a significantly larger effect size after treatment than at the basal state (Δ|βeQTL| > 0 and p < 10−3, Figure 2A). Consistent with data for other cell types or stimuli (Fairfax et al., 2014, Lee et al., 2014); most reQTLs were treatment specific (62%, 570 genes), indicating strong context specificity of the genetic regulation of immune responses.
Figure S6.

eQTL and aseQTL Mapping, Related to Figures 2 and 3
(A) Number of genes associated with an eQTL across conditions. (B) Enrichment of (r)eQTLs in regulatory elements. CD14+ cell regulatory elements from Ensembl Regulatory Build were considered for the analysis. Significance was assessed relative to the genome-wide distribution of SNPs overlapping regulatory elements within 1Mb of the gene transcription start site. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (C) General rationale of allele-specific eQTL mapping. We identified eQTLs leading to allelic imbalance (i.e., aseQTLs) by determining the ratio of reads from the two alleles of a heterozygous exonic SNP, according to their phase with the genotypes of the regulatory SNP that affects a transcription factor binding site. (D) Allele-specific expression in the context of cell stimulation. The aseQTL is detectable only after stimulation (TLR/IAV) by the presence of a transcription factor that is not expressed at the basal state (NS). (E) Power to detect aseQTLs as a function of eQTL effect size, number of observations and number of reads per exonic SNP. (F-G) Effect of regulatory variants on allele-specific expression, with (F) distribution of aseQTLs among local eQTLs and (G) distribution of asrQTLs among local reQTLs.
Figure 2.

Genetic Determinants of Population Differences in Immune Response
(A) Number of genes harboring reQTLs in single conditions or combinations of stimulations (∗∗∗p < 0.001, significance of overlap between stimulation conditions).
(B) Enrichment of (r)eQTLs in transcription factor (TF) binding sites. The five TFs presenting the strongest enrichments are shown for basal state eQTLs and reQTLs in different sets of stimulated conditions. Dots show the estimated odds ratio for the presence of binding sites of the TFs under consideration among (r)eQTLs, and horizontal lines show the 95% confidence interval of the odds ratio.
(C) Proportion (in percentage) of popDRGs harboring a local reQTL. Within each condition, popDRGs of different strengths (light color, |log2FCpop| > 0.2; dark color, |log2FCpop| > 0.5), as well as the proportion of reQTLs expected at the genome-wide level (in gray), are represented (∗∗∗p < 0.001, significance of enrichment).
(D) Fraction of population differences attributable to (r)eQTLs among popDEGs and popDRGs. (r)eQTLs are sorted by increasing effect size and divided into quintiles. Dark color bars indicate the fraction of |log2FCpop| of popDRGs attributable to reQTLs for each stimulation, and the light color bars represent the fraction that is not explained by reQTLs. For the non-stimulated (NS) condition, the fraction of |log2FCpop| of popDEGs attributable to eQTLs is reported.
(E) TLR-induced reQTL at NCF2 in both Africans and Europeans (left), and mean population expression of NCF2 (right).
(F) European-specific reQTL at ST3GAL6 induced by R848 (left), and mean population expression of ST3GAL6 (right).
To investigate the functional features of (r)eQTLs, we used the predicted regulatory elements of CD14+ monocytes (Zerbino et al., 2015) and identified a strong enrichment in such elements, particularly in promoter sequences (OR > 10.4, p < 10−16; Figure S6B). Furthermore, we observed strong enrichments of basal eQTLs and reQTLs in binding sites for several transcription factors (TFs), including KDM5A and THAP1 at the basal state, TBP and STAT3 after TLR activation, and STAT2, HMGN3, and IRF1 following R848 and IAV treatments (Figure 2B), highlighting mediators of cellular responses to immune activation.
Uncovering the Genetic Basis of Population Differences in Immune Response
We subsequently investigated the contribution of genetic variants to population differences in immune responses. We found that popDRGs were enriched in reQTLs in all conditions (OR > 2.6, p < 10−14), an enrichment that increased with the magnitude of the population fold change in gene expression (|log2FCpop|; Figure 2C). This suggests that differences in transcriptional responses between populations are, at least partially, under genetic control. To test this hypothesis, we evaluated the fraction of population transcriptional differences that can be explained by genetics (see STAR Methods). We found that (r)eQTLs account on average for ∼50% of such expression differences and for up to 70% when focusing on (r)eQTLs of strong effect size (i.e., fifth quintile; Figure 2D). Furthermore, reQTLs associated with popDRGs showed a stronger degree of population differentiation (mean difference in derived allele frequency |ΔDAF| = 0.24 for popDRGs versus 0.16 for non-popDRGs, p < 2.2 × 10−16), suggesting that differences in transcriptional responses are mainly accounted for by population variation in allele frequency of reQTLs. An example is provided by NCF2, which is downregulated specifically in Africans upon TLR activation, due to the higher DAF of reQTL rs2274065, with respect to Europeans (DAFAFB = 0.50 versus DAFEUB = 0.07) (Figure 2E).
We next searched for population-specific (r)eQTLs, i.e., SNPs present at similar population frequencies (MAF > 0.05) but having a regulatory effect in one population only. We found 16 eQTLs presenting significant differences in effect size between populations (pinteraction < 0.001), 5 of which were reQTLs (Table S2B). For example, rs1051712 was associated with decreased ST3GAL6 expression upon R848 stimulation in Europeans only (Figure 2F). Our analyses suggest that while population-specific gene regulation can occur, population differences in immune responses are mostly the result of regulatory variants presenting different allele frequencies between Africans and Europeans.
Allele-Specific Expression Reveals cis-Regulatory Effects on the Immune Response
To provide a more accurate evaluation of cis effects affecting immune response variation, we mapped allele-specific expression QTLs (aseQTLs) (Figures S6C and S6D). aseQTL mapping is constrained by not only the availability of heterozygotes and read depth but also by effect size, which strongly impacts the power of detection (Figure S6E). To ensure sufficient power, we focused on the 233 genes with large-effect eQTLs (|βeQTL| > 0.5) that could be tested and found 200 with an aseQTL (86%), including 160 assessed with high confidence (paseQTL < 10−3) (Figure S6F; Table S2C). Similarly, among the 42 reQTL genes that could be tested, we detected 33 (78%) with a stimulus-induced allele-specific response QTL (asrQTL), including 20 assessed with high confidence (Figures 3A and S6G; Table S2D). Among these, we found the TLR-induced NCF2 and PCID2 and the IAV-induced ARL5B (Figure 3B), which regulates the RIG-I-like receptor MDA5 (Kitai et al., 2015).
Figure 3.

Allele-Specific Expression upon Immune Stimulation
(A) Strongest allele-specific response QTLs (asrQTLs) for each stimulation condition and population.
(B) asrQTL of ARL5B. Individual allelic ratios, in non-stimulated and IAV-infected conditions, are grouped by reQTL rs2130531 genotype and phase with exonic variants, with color-coding for population (dark and light colors for Africans and Europeans, respectively). Vertical bars show the 95% binomial confidence interval of the allelic ratio.
(C) Distribution of allelic ratios across genes harboring aseQTLs. Ratios are grouped by eQTL genotype (HMZ, homozygous; HTZ, heterozygous) for basal state or stimulated conditions.
(D) Percentage of ASE events observed for different categories of rare exonic variants. Vertical bars indicate 95% confidence intervals for the estimated percentage (∗∗∗p < 0.001).
We next assessed the contribution of common regulatory variants (MAF > 0.05) to ASE at the individual level. Out of 5,889 genes for which ASE could be tested, we identified 1,942 genes with at least one ASE event (|log2(Nalternative/Nreference)| > 0.2, FDR < 0.05), yielding an average of ∼188 ASE events per individual (Table S2E). Of these, 275 genes presented evidence of allele-specific responses (i.e., significant differences in ASE before and after stimulation), suggesting G × E interactions (Table S2F). Focusing on the 160 aseQTLs detected at the population level, we consistently observed stronger allelic imbalance in heterozygous individuals, ∼70% of whom displayed ASE in both the presence and absence of stimulation (p < 2.2 × 10−16; Figure 3C). Our results indicate that, upon immune stimulation, a large fraction of ASE events can be accounted for by common regulatory variants, as shown for steady-state expression (Battle et al., 2014, Martin et al., 2014, Montgomery et al., 2011).
Finally, we evaluated whether rare coding variants, presenting a frequency ≤1% and characterized through whole-exome sequencing, impact ASE upon immune stimulation. A significant increase in ASE was observed in individuals carrying rare missense variants in stimulated conditions (OR = 1.34, p < 5.0 × 10−8; Figure 3D). Notably, nonsense variants contributed to the strong increase in ASE in both basal and stimulated states (OR = 6.8, p < 5.9 × 10−6 and OR = 10.6, p < 2.0 × 10−31, respectively). This is consistent with a role of rare coding variants in the generation of allelic imbalance in monocytes, particularly premature stop variants, possibly through nonsense-mediated decay, as reported for other cell types and tissues (Kukurba et al., 2014, Lappalainen et al., 2013, MacArthur et al., 2012).
Besides the contribution of common regulatory variants and rare coding mutations, our results identified a fraction of ASE events that are not explained by nearby eQTLs (i.e., ∼17% of homozygotes display ASE). This suggests the occurrence of secondary mechanisms regulating ASE, including undetected eQTLs of small effect size or epigenetic effects.
Trans Regulation Affects the Population Differentiation of Immune Responses
To detect master regulators underlying population differences in immune responses, we mapped trans-eQTLs, i.e., SNPs regulating gene networks over long distances. Our genome-wide mapping across stimulations, correcting for multiple testing, resolved a total of 42 trans-eQTLs regulating 165 genes at an FDR of 5% (p < 2.7 × 10−12; Figure 4A; Table S3A). Of these, 62% (103 genes) were trans-regulated in one condition only, highlighting the high degree of context specificity. We assessed the contribution of trans regulation to population differences in immune responses, and found that trans-regulated genes, upon TLR4 and TLR1/2 treatments, were strongly enriched in popDRGs (OR > 10.3, p < 1.6 × 10−16; Figure 4B).
Figure 4.

Identification of Master Regulatory Loci of Immune Responses
(A) Genome-wide distribution of trans-eQTLs. For each locus, the number of associated genes identified at a genome-wide FDR of 5% or using an SNP-based Bonferroni correction is represented by black and gray bars, respectively.
(B) Enrichment of popDRGs in genes regulated by trans-eQTLs. Within each condition of stimulation, popDRGs of different strengths (light color, |log2FCpop| > 0.2; dark color, |log2FCpop| > 0.5) are represented (∗p < 0.05, ∗∗∗p < 0.001).
(C) Fine mapping of the Pam3CSK4-induced trans-eQTL at TLR1. The significance of SNP associations with the expression patterns (PC1) of the 432 trans-regulated genes is shown for basal and Pam3CSK4 conditions (gray and green dots, respectively). Only the gene overlapping the strongest trans-eQTL signal is represented.
(D) TLR1 trans-associated genes at pBonferroni < 0.05. The size of the circles reflects the proportion of the variance of gene expression explained by rs5743618, and colors indicate the direction of the change in expression associated with the derived allele. Only the 100 most significant genes are shown.
(E) Fraction of population differences in gene expression (|log2FCpop|) attributable to rs5743618 among popDRGs regulated by the TLR1 locus (in dark green). Only the tail distribution of popDRGs with the largest population differences is represented. Genes involved in the GO biological process “response to molecule of bacterial origin” are reported.
(F) Impact of the derived allele of TLR1 rs5743618 (C allele) on the expression patterns (PC1) of the 81 inflammatory genes from module 1.
See also Table S3.
To decrease the multiple testing burden of detecting trans-associations, we further interrogated the 42 trans-eQTLs on a single SNP basis (see STAR Methods). This enabled the detection of 794 trans-regulated genes (p < 4.4 × 10−6, Bonferroni-corrected p < 0.05), the large majority of which (98%) were associated to a single trans-eQTL. Furthermore, we observed that only two loci, IFNB1 and TLR1, account for 88% of these associations (Figure 4A; Table S3A). The IFNB1 locus, previously reported upon LPS treatment for 24 hr in Europeans (Fairfax et al., 2014), was the strongest trans-regulatory hotspot. We found that this locus controlled, in both populations, a TLR4- and TLR1/2-mediated antiviral gene network (Table S3B), corresponding mostly to genes belonging to module 2 (96% of overlap). Genes in this network were enriched in popDRGs (OR > 9.2, p < 10−38), owing to population differences in IFNB1 response. Local IFNB1 regulatory variants had similar population frequencies (maximum |ΔDAF| = 0.1) and explained only up to 9% of the differences in IFNB1 response. Thus, the population differences observed for IFNB1 trans-regulated genes are not due to variation in the cis regulation of IFNB1 itself but instead are due to yet-unidentified genetic and non-genetic factors.
A TLR1 Master Regulator Modulates the Inflammatory Response in Europeans
We identified a Pam3CSK4-induced gene network that is trans-regulated by the TLR1 missense variant rs5743618 (I602S). This European-specific trans-eQTL (DAFEUB = 0.71, DAFAFB = 0.01) was also associated with the expression of one of the largest networks (432 genes, Bonferroni-corrected p < 0.05; Figures 4A, 4C, and 4D). Genes downregulated by the rs5743618-derived variant were enriched in genes belonging to module 1 (67% of genes) and module 4 (18% of genes) (OR > 11.5, p < 2.5 × 10−19; Figure 1B). These genes were preferentially involved in responses to bacterial infection (OR = 6.3, p = 6.5 × 10−9; Table S3C) and included regulators of inflammation, such as CCL5, IL10, IL12B, and PTGS2 (Figure 4D). Among upregulated genes, signaling-related functions were overrepresented and included BID, IKBKE, and PAK1, involved in TNFR1 signaling (Figure 4D). Remarkably, TLR1 trans-associated genes displayed strong enrichment in popDRGs (OR = 8.6, p < 10−28), and, contrary to IFNB1, such population transcriptional differences were largely explained by genetics (i.e., the rs5743618 variant; Figure 4E).
We then investigated the effects of this trans-regulatory variant on the inflammatory response, as we previously showed its functional impact on NF-κB activity (Barreiro et al., 2009), and assessed the correlation of rs5743618 genotypes with the expression of the 81 inflammatory response genes of module 1 (Figures 1B and 1C; Table S1B). The derived C allele (602S) was associated with a significant overall decrease in the expression of inflammatory response genes (p = 1.2 × 10−13; Figure 4F). These results reveal major population differences in TLR1/2-mediated responses, which are largely explained by a European-specific TLR1 trans-regulatory hotspot that contributes significantly to differences in the strength of the inflammatory response between Africans and Europeans.
Natural Selection Targeted Immune-Responsive Regulatory Variation
We next assessed how natural selection, as opposed to genetic drift, has contributed to differences in immune responses between populations. We computed two metrics—FST, based on the degree of population differentiation (Holsinger and Weir, 2009), and iHS, based on haplotype homozygosity (Voight et al., 2006)—to detect signals of old and recent events of positive selection, respectively. After matching for MAF and linkage disequilibrium (LD) patterns, we found that basal eQTLs and reQTLs were enriched in stronger values of FST (p < 0.005 for eQTLs and p < 1 × 10−4 for reQTLs, respectively) and iHS (p < 0.002 and p < 1 × 10−4, respectively), relative to genome-wide expectations, in Africans and Europeans (Figure 5A). Significant enrichments in selection signals were also obtained using a composite selection score (CSS) combining FST and iHS, which detects signals of recent, strong positive selection, and the XP-CLR method, which uses allele frequency differentiation at linked loci to detect selective sweeps (Chen et al., 2010). Among reQTLs, the strongest enrichments were observed for the IAV condition in both Africans (iHS p = 0.04, XP-CLR p < 10−4) and Europeans (FST p < 10−4, CSS p = 0.002) (Table S4A). This supports a history of positive selection targeting immune-responsive regulatory variants, particularly those involved in responses to viral infection.
Figure 5.

Natural Selection Driving Population Differences of Immune Response
(A) Distribution of neutrality statistics among local (r)eQTLs. For each locus, the maximum FST or |iHS| across all SNPs in high LD (r2 > 0.8) is considered, focusing on selection signals targeting the derived allele. The genome-wide distribution of these statistics (after pruning for LD, to avoid overweighting long haplotypes) is provided as a reference (all). Significance was assessed by resampling random SNPs from the genome, matched for MAF and LD (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). Top: Africans. Bottom: Europeans.
(B) Fine mapping of the selection signal detected at the CCR1 reQTL in Africans. Top: SNP associations with CCR1 expression (-log10(peQTL)) are represented for the basal (gray) and Pam3CSK4 (green) conditions. SNPs located in the CCR1 reQTL peak region are in high LD (r2 > 0.8) with the reQTL peak-SNP. Middle and bottom: FST and |iHS| values for SNPs with a DAF ≥ 0.2, respectively. For each SNP, the size of the dots is proportional to the association with CCR1 expression, with blue and red indicating selection on derived and ancestral alleles, respectively. The blue line represents the recombination rate at the locus. Only the gene for which the eQTL was detected is represented.
See also Table S4.
To highlight specific (r)eQTL candidates that may have participated in population adaptation at different timescales, we considered loci presenting extreme values of FST or iHS at the genome-wide level (>99th percentile; Tables S4B and S4C). Among these, we conservatively retained (r)eQTLs that were located in genomic regions presenting a significant enrichment in selection signals and deviated from neutral expectations based on validated demographic scenarios (see STAR Methods). Among local eQTLs, the strongest signal detected by FST involved an eQTL associated with stronger expression of the methyltransferase gene PCMTD1 in Europeans (FST = 0.8, iHS = −3), while the strongest signal of iHS involved a reQTL associated with a reduced expression of CCR1 following TLR1/2 activation in Africans (iHS = −4; FST = 0.4) (Figure 5B). With respect to trans-eQTLs, the master regulatory SNP rs5743618 at TLR1 also presented a strong signal of local adaptation in Europeans (FST = 0.7, iHS = −1.5, pempirical-FST = 0.002, psim-FST = 0.007).
Together, our results provide genome-wide support for the important role of regulatory variants affecting basal gene expression and responses to immune stimuli in driving human adaptation. This, together with the enrichments in genes showing differential expression between populations (OR = 2.1, p = 1.1 × 10−11) among (r)eQTLs with selection signatures (Tables S4B and S4C), emphasizes the contribution of natural selection to the differences in immune responses observed between human populations.
Neandertal Contribution to Transcriptional Responses to Immune Challenges
We investigated the impact of admixture between Neandertals and the ancestors of Europeans on genome-wide expression profiles (see STAR Methods). We first defined a set of 197,959 variants as of putative Neandertal ancestry (archaic SNPs [aSNPs]) if the Neandertal allele was present in Europeans and absent in Africans and located in genomic regions with a high probability of Neandertal ancestry (Sankararaman et al., 2014). We identified a total of 52 loci harboring at least one aSNP overlapping a local eQTL (archaic eQTL). Interestingly, relative to genome-wide expectations, an enrichment in aSNPs was observed for basal eQTLs (p < 0.003) and reQTLs in R848 and IAV conditions (p < 0.014 and p < 10−3, respectively; Figure 6A). To identify archaic eQTLs with high-confidence, we next focused on those located in haplotypes longer than expected under a scenario of incomplete lineage sorting (Figure S7A). Among the 19 eQTLs presenting strong evidence of Neandertal origin (Table S5), 9 corresponded to R848- and IAV-induced reQTLs, implicating genes encoding Ras GTPases such as RAB3IP and RAPGEF3.
Figure 6.

Neandertal Introgression of Immune Regulatory Variants in Europeans
(A) Enrichment of (r)eQTLs in archaic SNPs. The observed number of archaic eQTLs is presented for each condition (colored dots) in Europeans with respect to the expected distribution of archaic eQTLs under the assumption of independence (gray bars) (see STAR Methods) (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(B) Fine mapping of the archaic reQTL at PNMA1 in Europeans. SNP associations with PNMA1 expression (−log10(peQTL)) for the basal (gray) and stimulated (in colors) conditions (top). European individuals, and their corresponding archaic and modern haplotypes at the PNMA1 locus, are sorted by increasing levels of PNMA1 expression. Red dots represent archaic SNPs, and red lines represent the largest consecutive stretch of archaic alleles associated with PNMA1 expression (middle). Frequency distribution of archaic SNPs at the locus is shown (bottom). Only the gene for which the eQTL was observed is represented.
Figure S7.

Introgression of Regulatory Variants from Neandertals, Related to Figure 6
(A) Models of incomplete lineage sorting and introgression from Neandertals. Incomplete lineage sorting (ILS) scenario (left panel). An ancient variant predating the split between humans and Neandertals was retained in both lineages, but lost from the African population. Haplotypes carrying this ancient allele in Europeans are expected to be short because the time window allowed multiple recombination events to occur. Scenario of introgression from Neandertals (right panel). An archaic allele from Neandertals has been introgressed in Europeans and haplotypes containing this allele are expected to be longer than those resulting from ILS, due to the more recent occurrence of the admixture event. (B) Frequency of the archaic haplotype of PNMA1, tagged by SNP rs12436322, in different world-wide populations. Pie size is proportional to the number of individuals. The non-archaic allele is reported in violet (1000 Genomes data) and in dark blue (Simons Genome Diversity Project Dataset), and the archaic allele is presented in orange. (C) SNP associations, [-log10(peQTL)], with PNMA1 expression in Africans and Europeans are shown (upper panel). Archaic haplotypes at the PNMA1 locus in Europeans (lower panel) are ordered by increasing levels of PNMA1 expression, and archaic SNPs are represented in red for each haplotype. Red lines indicate the core archaic haplotype, defined as the haplotype within the eQTL carrying the largest number of archaic alleles. The eQTL identified in Europeans spans a region of 102 kb surrounding the gene, and overlaps an eQTL present in Africans. (D) Dissection of the PNMA1 core haplotype. Haplotypes at the PNMA1 locus are represented by horizontal lines showing, for SNPs with a MAF ≥ 5% in either population, the ancestral allele in white and the derived allele in color. The black horizontal line separates European haplotypes (top) from African haplotypes (bottom). Within each population, SNPs associated with PNMA1 expression are highlighted in blue for those specific to the Neandertal lineage (i.e. archaic SNPs) and in red for the others. In total, 12 archaic alleles at the locus tag the archaic core haplotype associated with an upregulation of PNMA1 expression, either in the basal condition or in response to R848 and IAV. This haplotype is longer than expected under the ILS scenario, suggesting that introgression occurred in Europeans through admixture with Neandertals. This archaic haplotype, tagged by the aSNP rs12436322, has re-introduced the ancestral allele of rs6574138 in Europeans (i.e. all individuals not carrying the archaic haplotypes have the derived allele), which is also present in Africans and associated with PNMA1 expression. SNP rs6574138 overlaps with ENCODE binding sites, consistent with its putative functional role in the regulation of PNMA1 expression in both Europeans and Africans. (E) Inferred history of the ancestral and derived alleles at rs6574138 in modern human populations, up to their most recent common ancestor. Our data suggest that rs6574138 predates the split between Neandertals and modern humans and that the derived allele of this variant was fixed in early Europeans. The ancestral allele of rs6574138 was then reintroduced in Europeans by introgression of the Neandertal haplotype tagged by rs12436322, and is responsible for the variability in PNMA1 expression in the contemporary European population.
Some of these (r)eQTLs carry archaic alleles that are at appreciable frequencies in Europeans, suggestive of adaptive introgression. To test this hypothesis, we reasoned that an archaic allele that introgressed into Europeans and East Asians and was advantageous in one population only should present today unusually high levels of genetic differentiation, relative to genome-wide expectations (Vernot and Akey, 2014). When comparing FST between Europeans and East Asians at archaic (r)eQTLs against the genome-wide distribution of aSNPs (Table S5), we identified a haplotype that regulates the response of PNMA1 to R848 and IAV as a significant genomic outlier (FST = 0.28; pemp = 0.01; Figures 6B and S7B–S7E). This archaic haplotype is present at very high frequency in Europeans (33.5%), while it is absent in East Asians (Figure S7B). Using simulations that make conservative assumptions about the past frequency spectrum of archaic alleles (see STAR Methods), we found that the high frequency of the PNMA1 haplotype in Europeans is not compatible with neutral expectations (psim < 0.05; Table S5), providing support to the adaptive nature of this introgression event.
Collectively, these results indicate that regulatory variants affecting steady-state gene expression and transcriptional responsiveness to immune challenges, particularly those that are viral related, were preferentially introduced into European genomes via admixture with Neandertals, of which some may have conferred a selective advantage to modern populations.
Discussion
Recent studies have offered proof of concept that eQTL mapping detects key genetic variants relevant to immunity and infection (Fairfax and Knight, 2014). Here, using RNA-seq data, we characterized, at an unprecedented level of resolution, the transcriptional response of primary monocytes to inflammatory and infectious cues. We defined the respective contributions of natural selection and archaic admixture to differences in immune response regulation between populations. In doing so, we identify regulatory variants and molecular phenotypes that have been important to human survival and that are of biomedical interest for the understanding of genetic susceptibility to immune-related diseases.
Our analyses uncovered extensive variation, globally of moderate effect, in transcriptional responses to immune challenges between individuals of African and European descent, with the strongest differences being observed for genes with antiviral and inflammatory-related functions. These genes are enriched in associations with cis- and trans-eQTLs, and regulatory variants presenting different allele frequencies between populations account for a large fraction of the population differences in immune responses observed. Highlighting one pertinent example, we identify a reQTL (rs2274065), whose cis-action was supported by our analyses of ASE, leading to TLR-mediated NCF2 downregulation in Africans, where this variant is present at high frequency (∼50%). That this mutation has been associated with systemic lupus erythematosus (Jacob et al., 2007) suggests that lower levels of NCF2 expression may contribute to the higher prevalence and severity of this disease in Africans (Fernández et al., 2007). This example illustrates the value of mapping response eQTLs across populations to uncover mechanisms that might explain ethnic disparities in the clinical manifestation of immune disorders.
This study also establishes that natural selection has contributed to the differences in immune responses observed between populations by providing genome-wide support that regulatory variants associated with different responsiveness to immune challenges have been targeted by positive selection. In doing so, we identify multiple regulatory variants showing signatures of population local adaptation. For example, selection appears to have increased the frequency of the African-specific reQTL rs7426702 (39%), leading to stronger CCR1 downregulation following TLR1/2 activation. Interestingly, the inhibition of CCR1 limits leukocyte recruitment and prevents inflammatory responses in experimental settings (Gladue et al., 2006). Our results thus suggest that CCR1 downregulation has conferred a selective advantage in Africans, likely to favor diminished inflammation.
Further support for this concept is provided by the strong selection signature detected for the European trans-eQTL at TLR1, spanning a region shown to have evolved adaptively (Barreiro et al., 2009, Deschamps et al., 2016, Mathieson et al., 2015, Pickrell et al., 2009). The TLR1 variant is a strong trans-regulatory hotspot associated with a gene network presenting marked population differences in the response to immune activation. We also found that the advantageous rs5743618 allele, which impairs NF-κB activity (Barreiro et al., 2009), is associated with a global decreased expression of inflammatory response genes, consistent with an attenuated TLR1-mediated signaling beneficial to Europeans. Together, our findings highlight the evolutionary tradeoff between activating efficient responses to sense microorganisms, both pathogenic and commensal, while avoiding aberrant, deleterious inflammation.
Genetic variation transmitted through admixture with Neandertals can also represent a source of functional, potentially advantageous variants (Vattathil and Akey, 2015). Relative to genome-wide expectations, we show that genetic segments introgressed from Neandertals have preferentially introduced regulatory variants into European genomes, affecting steady-state expression and responses to TLR7/8 stimulation and IAV. Furthermore, we report several loci presenting strong evidence of archaic ancestry that exert a regulatory effect in cis. Among these, we find the IAV-induced reQTL of PNMA1, which encodes a protein that physically interacts with the IAV protein PB2 and stimulates interferon production (Shapira et al., 2009). That the PNMA1 haplotype presents a frequency in Europeans that is not compatible with neutral evolution, together with its strong levels of population differentiation between modern Europeans and East Asians, supports its contribution to European adaptation and provides a case of adaptive introgression. The functional roles of the introgressed regulatory variants require further investigation, but our results clearly establish that archaic admixture, whether adaptive or not, has increased the diversity of the immune repertoire of contemporary Europeans.
Collectively, our analyses provide a comprehensive view of the impact of population genetic differences on transcriptional responses to innate immunity activation and highlight evolutionarily important determinants of host immune responsiveness. The regulatory variants identified here constitute a useful resource for evaluating the role of such variants in the molecular and cellular mechanisms underlying host immunity to infection and susceptibility to disease, both at the individual and population levels.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Magnetic CD14 microbeads, human | Miltenyi Biotec | Cat#130-050-201 |
| CD14-APC, human | Miltenyi Biotec | Cat#130-091-243 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ficoll-Paque | GE Healthcare | Cat#17-1440-03 |
| Fetal calf serum | PAA Laboratories | Cat#A15-502 |
| Dimethyl sulfoxide | Sigma Aldrich | Cat#D2438 |
| Penicillin/streptomycin | Life Technologies | Cat#15140-122 |
| Propidium iodide | Miltenyi Biotec | Cat#130-093-233 |
| LPS | Invivogen | Cat#tlrl-3pelps |
| Pam3CSK4 | Invivogen | Cat#tlrl-pms |
| R848 | Invivogen | Cat#tlrl-r848-5 |
| Critical Commercial Assays | ||
| Nucleospin miRNA kit | Macherey Nagel | Cat#740971.250 |
| RNA 6000 nano kit | Agilent Technologies | Cat#5067-1511 |
| Quant-iT PicoGreen dsDNA Assay Kit | Life Technologies | Cat#P7589 |
| TruSeq RNA Sample Prep Kit v2 | Illumina | Cat# RS-122-2001 |
| TruSeq SR Cluster Kit v3-HS | Illumina | Cat# GD-401-3001 |
| TruSeq SBS kit v3-HS | Illumina | Cat# FC-401-3001 |
| HumanOmni5-Quad BeadChips | Illumina | Cat#WG-311-5001 |
| Nextera Rapid Capture Expanded Exome kit | Illumina | Cat#FC-140-1006 |
| Deposited Data | ||
| Genotyping, exome and RNA sequencing data | European Genome-phenome Archive (EGA) | EGAS00001001895 |
| Experimental Models: Organisms/Strains | ||
| Human primary monocytes | This paper | N/A |
| Influenza A virus, strain A/USSR/90/1977 | (Pothlichet et al., 2013) | N/A |
| Software and Algorithms | ||
| FlowJo vX.0.6 | FlowJo, LLC | N/A |
| TopHat | (Kim et al., 2013) | https://ccb.jhu.edu/software/tophat/index.shtml |
| RSeQC package | (Wang et al., 2012) | http://rseqc.sourceforge.net |
| Cufflinks/CuffDiff (v2.0.2) | (Trapnell et al., 2012) | http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/ |
| ComBat, sva R package | (Johnson et al., 2007) | https://www.bioconductor.org/ |
| WGCNA | (Langfelder and Horvath, 2008) | https://www.bioconductor.org/ |
| PASTAA | (Roider et al., 2009) | http://trap.molgen.mpg.de/cgi-bin/pastaa.cgi |
| TRAP | (Thomas-Chollier et al., 2011) | http://trap.molgen.mpg.de/cgi-bin/download.cgi |
| fdrtool, R package | (Strimmer, 2008) | http://cran.r-project.org/ |
| GOSeq, R package | (Ashburner et al., 2000) | https://www.bioconductor.org/ |
| PLINK v1.9 | (Chang et al., 2015) | http://pngu.mgh.harvard.edu/∼purcell/plink/download.shtml#download |
| KING | (Manichaikul et al., 2010) | http://people.virginia.edu/∼wc9c/KING/index.html |
| ADMIXTURE | (Alexander et al., 2009) | https://www.genetics.ucla.edu/software/admixture/ |
| BWA v.0.7.7 | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| Picard Tools v.1.94 | N/A | http://broadinstitute.github.io/picard |
| GATK v.3.2.2 | (DePristo et al., 2011) | https://software.broadinstitute.org/gatk/ |
| SHAPEIT2 | (Delaneau et al., 2013) | http://www.shapeit.fr |
| IMPUTE v.2 | (Howie et al., 2009) | http://mathgen.stats.ox.ac.uk/impute/impute_v2.1.0.html |
| EIGENSTRAT | (Patterson et al., 2006) | http://genepath.med.harvard.edu/∼reich/EIGENSTRAT.htm |
| MatrixEqtl, R package | (Shabalin, 2012) | http://www.bios.unc.edu/research/genomic_software/Matrix_eQTL/ |
| WASP | (van de Geijn et al., 2015) | https://github.com/bmvdgeijn/WASP |
| SAMtools mpileup | (Li, 2011) | http://samtools.sourceforge.net/ |
| Variant Effect Predictor | (McLaren et al., 2010) | http://www.ensembl.org/info/docs/tools/vep/index.html |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Lluis Quintana-Murci (quintana@pasteur.fr).
Experimental Model and Subject Details
We recruited 100 healthy, male donors of self-reported European descent (EUB) and 100 of self-reported African descent (AFB), all living in Belgium, at the Center for Vaccinology (CEVAC) of Ghent University Hospital (Ghent, Belgium). Samples were collected after written informed consent had been obtained, and the study was approved by the local ethics committee (Ethics Committee of the Ghent University), the Ethics Board of Institut Pasteur (EVOIMMUNOPOP-281297) and the relevant French authorities (CPP, CCITRS and CNIL). Inclusion was restricted to donors between 19 and 50 years of age, nominally healthy at the time of sample collection. A case report form was obtained for all donors, including information on vital sign measurements, medication, medical history and travel. No overrepresentation of any particular disease was observed relative to official report statistics published by the World Health Organization or in epidemiological studies. Serological testing was performed for all donors at the CEVAC, and those with serological signs of past or ongoing infection with human immunodeficiency virus (HIV), hepatitis B virus (HBV) or hepatitis C virus (HCV) were excluded.
Method Details
PBMC Isolation
For each participant, we collected 300 ml of whole blood into anticoagulant EDTA-blood collection tubes and peripheral blood mononuclear cells (PBMCs) were isolated on Ficoll-Paque density gradients. PBMCs were frozen in 90% fetal calf serum (FCS) and 10% dimethyl sulfoxide, at a density of 50 × 106 PBMCs/ml and transported in dry shipper from CEVAC to Institut Pasteur. Vials were then cryopreserved in liquid nitrogen until use.
Monocyte Separation
For each donor, 300 × 106 PBMCs were thawed, washed twice and resuspended in pre-warmed RPMI-1640 Glutamax medium, supplemented with 10% FCS and penicillin/streptomycin (complete medium). Monocytes were then positively selected with magnetic CD14 microbeads, according to the manufacturer’s instructions. The number of monocytes was determined with a Kova Glasstic Slide 10 with a grid in the presence of trypan blue. For each donor, 30 × 106 monocytes were split between five 25 cm2 non-treated flasks (i.e. one flask per condition and five conditions per donor), each containing 6 × 106 monocytes in 9 ml of complete medium. Monocytes were allowed to rest for one hour at 37°C under 5% CO2 before stimulation.
Monocyte Purity and Cell Death Assessment
Purity and cell death of the isolated monocytes were assessed for all donors on a fraction of 105 CD14+ monocytes stained, according to the manufacturer’s instructions, with fluorescent APC-conjugated anti-CD14 antibodies and propidium iodide, respectively. Samples were then analyzed on a MACSQuant Analyzer 10 benchtop flow cytometer (Miltenyi Biotec) and using FlowJo vX.0.6 software. The mean values obtained for all samples were 96.8% for monocyte purity and 2.1% for initial cell death rates.
TLR Stimulation and Influenza A Virus Assays
Monocytes were exposed to five different conditions for 6 hr, in order to capture transcriptional signatures from both an early response and the beginning of a late response, i.e., an “intermediate response” (Huang et al., 2001). The choice of this time point was also based on a pilot study on the kinetics of gene expression of several key inflammatory and antiviral response genes (IL1A, IL23A, IL6, IL8, TNF, IRF1 and STAT2) upon immune activation. Our results showed that 6 hr of stimulation was the best time point to capture simultaneously expression signals from early, intermediate and late response genes, with respect to other time points at 2, 4, 8 and 24 hr (data not shown). One monocyte flask was left untreated as a baseline control, while the others were each exposed to one of four different immune stimuli. These stimuli included synthetic ligands specifically activating three Toll-like receptor (TLR) signaling pathways: 1 ng/ml ultrapure LPS from E. coli, 0.2 μg/ml synthetic triacylated lipoprotein Pam3CSK4, and 0.3 μg/ml imidazoquinoline compound R848. Monocytes were also infected with strain A/USSR/90/1977(H1N1) of the human seasonal influenza A virus (IAV) at a MOI = 1, and IAV particles were produced as previously described (Pothlichet et al., 2013). After stimulation, cells were collected by centrifugation, lysed in a guanidinium thiocyanate solution provided in the Nucleospin miRNA kit, according to the manufacturer’s instructions, and stored at −80°C until RNA extraction. Cellular assays were performed per batch of 30 samples from 6 individuals, including 3 Africans and 3 Europeans, across all 5 conditions.
RNA Extraction
Total RNA was extracted with the Nucleospin miRNA kit from Macherey Nagel, including the enzymatic digestion of genomic DNA. Extractions were performed in batches of 30 samples (i.e. 5 conditions for 3 Africans and 3 Europeans), and RNA quality and quantity were assessed with a Nanodrop spectrometer and the Agilent Bioanalyzer RNA 6000 nano kit. We generated a final set of 978 samples from the 200 donors fulfilling the quality and quantity criteria (RIN > 7, quantity > 2.5 μg) for high-throughput RNA-sequencing, including 200, 188, 197, 193 and 200 samples for the non-simulated, LPS, Pam3CSK4, R848 and IAV conditions, respectively.
RNA Sequencing
RNA was obtained from 978 of the 1000 samples, and was sequenced on an Illumina HiSeq2000. The quality and quantity of all samples was reassessed before sequencing. Samples were then randomized before library preparation in order to obtain a balanced number of samples across ethnicity and cellular conditions per sequencing batch/lane/machine/index. Standard reagents were used for transcriptome sequencing: TruSeq RNA Sample Prep Kit v2 for mRNA library construction, TruSeq SR Cluster Kit v3-HS for cluster generation and TruSeq SBS kit v3-HS for sequencing. We pooled six samples per lane to generate outputs of around 30 million 101-bp single-end reads per sample (ranging from 27.7 to 94.8 million reads, mean 34.4) (Figure S2A).
DNA Extraction
Genomic DNA was extracted from the CD14-negative cell fraction (i.e. non-monocyte cells) by a standard phenol/chloroform protocol followed by ethanol precipitation. The DNA was quantified by Nanodrop spectrometry and with the Quant-iT PicoGreen dsDNA Assay Kit.
SNP Genotyping and Whole-Exome Sequencing
The 200 subjects studied were genotyped for a total of 4,301,332 SNPs on the Illumina HumanOmni5-Quad BeadChips. Whole-exome sequencing was carried out for the same individuals with the Nextera Rapid Capture Expanded Exome kit, on the Illumina HiSeq 2000 platform, with 100-bp paired-end reads. This kit delivers 62 Mb of genomic content per individual, including exons, untranslated regions (UTR), and microRNAs.
Quantification and Statistical Analysis
RNA-Sequencing Analysis
Reads were assessed for multiple quality metrics, including number of reads, nucleotide distribution and sequencing quality, and the last base of all reads was trimmed due to a fall in sequencing quality. RNA reads were then mapped onto the human GRCh37 genome with TopHat (Kim et al., 2013), resulting in the successful mapping of 89.9% of reads per sample on average (minimum 67.3%; maximum 93.7%). We used the RSeQC package to assess the alignment of reads with various genomic features, GC content, and gene body coverage (Figures S2B–S2E). Samples with uneven gene body coverage were found to be more likely to be outliers. We used gene body coverage regularity as an indicator of library quality, removing eight samples due to irregular gene body coverage. The remaining 970 samples were used for subsequent analyses and consisted of 200 non-stimulated (EUB: 100, AFB: 100), 184 LPS (EUB: 96, AFB: 88), 196 Pam3CSK4 (EUB: 100, AFB: 96), 191 R848 (EUB: 98, AFB: 93), and 199 IAV samples (EUB: 99, AFB: 100).
Cufflinks/CuffDiff (v2.0.2) (Trapnell et al., 2012) was used to quantify expression levels in FPKM (fragments per kilobase of transcript per million mapped reads) for each annotated transcript of the genome in Ensembl (v.70), and FPKM values for which Cufflinks returned FAIL status (< 0.5% of quantified transcripts) were set to missing values. Gene expression data were filtered to remove genes with low levels of expression (mean FPKM < 1 in all conditions) and their quality was checked by principal component analysis (PCA). PCA captured differences between conditions and populations on the first two axes, but we tested for additional causes of technical variability, by fitting, for each gene, a mixed model of gene expression as a function of condition, population, and technical covariates, including total RNA concentration, RIN, percentage of high-quality bases (Q30), mean GC content, library concentration, 5′/3′ coverage bias (measured as the mean difference in coverage between the 5′ and 3′ ends of the gene) as continuous covariates, and date of experiment, library preparation batch, sequencing batch, sequencer used, sequencing index, and sequencing lane as putative batch effects. Putative batch effects were modeled as random effects to prevent the loss of degrees of freedom, whereas all other covariates (condition, population and continuous covariates) were included as fixed effects, giving the following model for gene i and sample j:
where is the intercept, and are the fixed effect of the condition and population on sample j, are the fixed effect of continuous covariates on sample j, the are the random effects of batch covariate k, on sample j, and are the residuals.
The proportions of genes affected by each factor are reported in Figure S2F for various levels of explained variance. We observed that GC content, 5′/3′ bias, date of the experiment and library batch were among the strongest confounding factors, and accordingly corrected the data for these factors before analysis, following the pipeline detailed in Figure S3. First, we adjusted the data for GC content and 5′/3′ bias using linear models. Then, we imputed missing values by K-nearest neighbor imputation, and adjusted for experiment date and library batch by sequentially running ComBat (Johnson et al., 2007) for each batch effect, with condition and population as covariates. Batch-corrected gene expression levels, in FPKM, were then recalculated from the adjusted transcript level estimates. Refitting our linear mixed model confirmed that correction was satisfactory for most of the technical covariates (Figure S2F).
Assessment of Technical and Biological Variability
The reproducibility of our RNA-Seq experiments was assessed by performing technical and biological replicates on seven independent donors (4 AFB and 3 EUB) across the five experimental conditions. We showed that (i) the coefficients of variation of genes within technical replicates were consistently, and significantly, smaller in magnitude and less variable than those within biological replicates (Wilcoxon Rank-Sum Test, p < 10−16; Figure S2G), and (ii) technical replicates exhibit higher correlation coefficients (r) between samples with respect to the distribution of r values calculated from pairwise comparisons between biological replicates (Figure S2H).
Modules of Correlated Genes
Modules of genes presenting correlated expression patterns, extracted from log-transformed FPKM data, were defined by weighted correlation network analysis (WGCNA) (Langfelder and Horvath, 2008). In our setting of immune response activation, this analysis detects modules of correlated genes that can reflect either shared regulation by common transcription factors, or regulation by independent transcription factors with similar patterns of activation upon stimulation. Tukey’s biweight correlation was used as a measure of gene relatedness to reduce the influence of outliers, and correlations were measured across all 970 samples. The scale-free topology of the networks was assessed for various values of the β shrinkage parameter, according to WGCNA user manual, and the default value of β = 6 appeared to give a satisfactory fit to scale-free topology. Signed clustering of genes (grouping only positively correlated genes) was used to simplify the interpretation of the extracted modules. We also found that varying the level of shrinkage (β = 5 or 6) or the depth of the clustering (deepsplit parameter set to 3 or 4) had only a mild impact on the number of clusters or the enrichments obtained, confirming the robustness of these analyses.
For each module, we used PASTAA (Roider et al., 2009) to identify transcription factor binding site motifs overrepresented within the annotated proximal promoters of the genes within each module. We first defined the proximal promoter region for each gene as the region extending 200 bp on either side of the transcription start site (TSS) of the most abundant transcript on the basis of Cufflinks FPKM estimates. We then used the transcription factor affinity prediction (TRAP) method (Thomas-Chollier et al., 2011) to measure the binding affinities of each transcription factor present in the Jaspar core vertebrate database (Mathelier et al., 2014) with the proximal promoters of the 12,578 expressed genes, and these affinities were then used as the input for PASTAA enrichment analysis. We reported only enrichments significant at a false discovery rate (FDR) of 0.05 with a fold-change (i.e. observed/expected) greater than 1.2. For each module, we represent the transcription factor binding sites with the highest value for the lower limit of the odds ratio confidence interval.
Differential Expression Analysis
Differential expression was assessed directly from log-transformed FPKM, using t tests for each condition. FDR was then calculated jointly for all conditions, with the R package fdrtool, and genes differentially expressed between populations (popDEGs) were defined as genes presenting an absolute log2 fold change between populations – – greater than 0.2 and at FDR < 0.05. We then calculated the fold-change in expression after stimulation relative to the basal state, and used t tests to determine whether there was a differential response. Population differential response genes (popDRGs) were then defined as popDEGs for which there was a differential response between populations under stimulated conditions, at FDR < 0.05 (i.e. the transcriptional response to treatment, relative to the basal state, differed between populations), resulting in a larger difference in expression after stimulation.
Gene Ontology Enrichment Analysis
All Gene Ontology (GO) enrichment analyses were performed with GOSeq package (Ashburner et al., 2000), using the default settings, with the 12,578 expressed genes as the background set. Only enrichments significant at FDR of 0.05 and with a fold-change (i.e. observed/expected) greater than 1.2 are reported.
SNP Genotyping Data Analysis
Using PLINK v1.9 (Chang et al., 2015), we removed SNPs that: (i) were typed with probes mapping to several genomic locations (N = 12,440), (ii) presented a poor genotype clustering (GenTrain score < 0.35; N = 809), (iii) had the same chromosomal position as another SNP in dbSNP b138 (N = 6,968), (iv) were not reported in dbSNP b138 (N = 5,311), (v) presented a call rate < 95% (N = 79,310), (vi) were monomorphic in our sample (N = 652,385), (vii) were located on the sex chromosomes (N = 50,994), and (viii) presented a Hardy-Weinberg p < 10−3 in AFB or EUB populations (N = 4,007). After quality-control filtering, we retained a total of 3,489,108 SNPs. The SNP call rate for the 200 individuals was 99.8% on average, ranging from 98.89% to 99.99%. No evidence was found for 2nd-degree cryptic relatedness (kinship coefficient > 0.07) in KING (Manichaikul et al., 2010), or for sex mismatch, for any of the individuals. Two AFB individuals presented an excess of heterozygosity (< ± 3SD from the population average), as a result of their moderate levels of non-African ancestry, as estimated using ADMIXTURE.
Whole-Exome Data Analysis
Read-pairs were processed according to the GATK Best Practice recommendations. Read-pairs were first mapped onto the human GRCh37 genome with BWA v.0.7.7 (Li and Durbin, 2009), and reads duplicating the start position of another read were marked as duplicates with Picard Tools v.1.94 (http://broadinstitute.github.io/picard). We used GATK v.3.2.2 (DePristo et al., 2011) for base quality score recalibration (“BaseRecalibrator”), insertion/deletion realignment (“IndelRealigner”), and SNP and insertion/deletion discovery for each sample (“Haplotype Caller”). Individual variant files were combined with “GenotypeGVCFs” and filtered with “VariantQualityScoreRecalibration.” Individual coverage was 52.32 × on average, ranging from 33.84 to 100.59 ×, and individual breadth of coverage at 5 × was 92.42%, ranging from 83.5% to 95.0%. We removed those of the 540,990 SNPs obtained that: (i) were triallelic (N = 11,925), (ii) presented a call rate < 95% (N = 44,716), (iii) were located on the sex chromosomes (N = 8,369), and (iv) presented a Hardy-Weinberg p < 10−3 in AFB or EUB populations (N = 4,510). The application of these quality-control filters resulted in the retention of 471,740 SNPs.
Imputation of Genome-wide SNP and Exome Data
Before merging the Omni5 and exome datasets, we first checked genotype concordance for 169,406 SNPs common to the two platforms. We flipped alleles for 8,025 SNPs with incompatible allelic states, and removed 119 SNPs with alleles that remained incompatible after allele flipping. The total concordance rate was 99.66%. The concordance rates for each of the 200 individuals exceeded 99%, confirming an absence of errors during DNA sample processing. Of the 8,155 SNPs with discordance rates > 1%, 296 were due to C/G or A/T SNPs, and high genotype concordance between the two DNA typing technologies was restored by allele flipping. The remaining 7,881 SNPs were removed. The entire Omni5 and exome datasets (3,489,108 and 471,740 SNPs, respectively) were then merged, yielding a final concordance rate of 99.93%, for a total of 3,782,260 SNPs.
Before imputation, we phased the data with SHAPEIT2 (Delaneau et al., 2013), using 500 conditioning haplotypes, 50 MCMC iterations, 10 burn-in and 10 pruning iterations. SNPs and allelic states were then aligned with the 1,000 Genomes Project imputation reference panel (Phase 1 v3.2010/11/23). We removed 8,705 SNPs with identical positions in our data and in the reference panel but incompatible alleles, even after allele flipping, and 4,137 SNPs with C/G or A/T alleles. Genotype imputation was performed with IMPUTE v.2 (Howie et al., 2009), considering 1-Mb windows and a buffer region of 1 Mb.
Of the 38,098,530 SNPs obtained after imputation, we removed SNPs that: (i) presented an information metric below 0.8 (N = 18,085,215), (ii) had a duplicate (N = 59,914), (iii) presented a call rate < 90% (N = 329,910), and (iv) were monomorphic (N = 4,053). The final imputed dataset included 19,619,457 SNPs.
To evaluate imputation accuracy, we estimated correlation coefficients R2 between true genotypes (i.e., obtained by Illumina array genotyping or exome sequencing) and imputed genotypes for the same SNPs (i.e., obtained by artificially removing genotyped SNPs from the data before imputation and then imputing them). In very good agreement with recent studies (Auton et al., 2015), the average correlation coefficient was 95.6% across all genotyped SNPs with information metric > 0.8 (93.6% for SNPs with MAF < 0.10 and 97.7% for SNPs with MAF > 0.10). This shows that our stringent quality filters ensure that only accurately imputed SNPs are analyzed.
Populations Genetic Structure
Two methods were used to infer the genetic structure of our population set of 100 African-descent and 100 European-descent Belgians (AFB and EUB, respectively). Because both methods assume linkage equilibrium among SNPs, we pruned the datasets for SNPs in linkage disequilibrium (LD), using PLINK v1.9 (Chang et al., 2015). Specifically, we removed SNPs in 50-SNP windows that present LD r2 > 0.5 (“–indep-pairwise 1000 10 0.5” option). The first model-based approach, ADMIXTURE (Alexander et al., 2009), estimates the proportions of each individual’s genome originating from K ancestral populations, K being specified a priori. This analysis was performed on 229,320 independent SNPs and 789 individuals from 22 populations, including EUB and AFB, together with a selection of representative populations from sub-Saharan Africa, North Africa, the Near East and Europe (Altshuler et al., 2010, Behar et al., 2010, Patin et al., 2014). We made K vary from 2 to 10. To obtain the most supported results and test for their stability, all ADMIXTURE analyses were run five times with different random seeds, for each K value. We kept results providing the lowest cross-validation error (CV) among iterations. The second model-free approach is the principal component analysis (PCA) implemented in EIGENSTRAT (Patterson et al., 2006). We used this approach to describe the local genetic sub-structure of AFB and EUB separately. The analysis for AFB was performed on 341,593 independent SNPs and 511 individuals from 7 western and central African populations, while the analysis for EUB was performed on 182,572 independent SNPs and 220 individuals from 13 European populations (Altshuler et al., 2010, Behar et al., 2010, Patin et al., 2014).
eQTL Mapping
For expression quantitative trait loci (eQTL) mapping, only variants with a minor allele frequency (MAF) ≥ 0.05 in the population studied were retained in the analysis, resulting in a set of 10,278,745 SNPs (i.e., corresponding to the merged genotyping and imputed SNP dataset; 8,913,090 SNPs in Africans and 6,178,808 SNPs in Europeans,). We mapped eQTLs with the MatrixEQTL R package (Shabalin, 2012). PC1 and PC2 of the genotype matrix were included in the model to account for possible population stratification. The inclusion of additional PC in the model (up to PC6) was tested and showed highly consistent results (i.e., correlation of -log10 p-values of eQTL > 0.95). For each gene, SNPs were considered “local,” likely cis-acting, if they were located less than 1Mb away from the gene transcription start or end site. They were otherwise considered to be trans-acting. eQTL mapping was performed separately for each population and condition, and false-positives due to outliers were prevented by discarding, from the analysis, eQTL associations that did not pass a p-value threshold of 10−3 for local eQTLs, and 10−5 for trans-eQTLs in Kruskal-Wallis rank tests.
For both cis- and trans-eQTLs, FDR was computed by mapping eQTLs on 100 datasets with genotypes permuted within each population. We then kept, after each permutation, the most significant p-value per gene, across all conditions and populations. Finally, we computed the false discovery rate associated with each p-value threshold in cis or in trans, and subsequently selected the p-value threshold that provided a 5% FDR, leading to p = 7.67 × 10−7 and p = 2.7 × 10−12 for cis- and trans-eQTLs, respectively. For local eQTLs, we report only the SNP at which the strongest association was observed (i.e., eQTL peak-SNP). When multiple SNPs in perfect LD fell within the peak, only one SNP is reported. eQTLs for which the eQTL peak-SNP had an allelic effect size (|βeQTL|) below 0.2 were discarded from further analysis. We next mapped fold-changes between the basal and stimulated states using MatrixEQTL, and defined response eQTLs (reQTLs) as stimulated eQTLs associated to a significant difference in response to stimulation (p < 10−3, ). For trans-eQTLs, we reported, within each 1Mb window, the SNP for which we observed both the largest number of trans-associated genes and strongest p-value of association. Furthermore, for each trans-eQTL that passed genome-wide significance at p = 2.7 × 10−12 (FDR of 5%), we performed a SNP-based analysis to identify genes regulated in trans by the eQTL at a Bonferroni p < 0.05, correcting for the 12,578 genes tested within the condition where the eQTL was found.
Population Differences Attributable to Genetics
To estimate the fraction of population differences in gene expression that can be attributed to genetic variants, we used a two-step strategy. First, we consider the set of all SNPs in LD (r2 > 0.5) with the eQTL peak-SNP, in the population where the eQTL was discovered and fine map the eQTL signal by fitting across populations the following linear model:
where SNPj is the genotype of the individual j for the variant under study, Popj is a binary variable indicating the population origin (0 for Europeans and 1 for Africans), and εj is a random, normally distributed residual. In this model, α is the intercept, β reflects the effect of the derived allele of the SNP on gene expression, and γ estimates the fold change in expression between populations observed for individuals with identical genotype (i.e. gene expression differences that are not explained by genetics). We next focused on the SNP showing the strongest association p-value with gene expression across populations, and estimated the difference in population expression that is attributable to the SNP as:
with γ’ representing γ set to ensure that the ratio of FCSNP/FCpop is between 0 and 1, i.e. γ’ = 0, if the sign of γ differs from that of FCpop; γ’ = FCpop, if |γ| > |FCpop|; and γ’ = γ otherwise. The percentage of population differences in expression that is attributable to genetics is then given by the ratio FCSNP/FCpop.
Defining Population-Specific eQTLs
We aimed at distinguishing population specific eQTLs (i.e., SNPs present at similar frequencies in both populations but having an effect on gene expression in one population only) from eQTLs detected in one population only due to population differences in allelic frequencies. To do so, we first focused on the 1,109 genes associated with an eQTL (including 363 genes associated with a reQTL) where all SNPs in LD (r2 > 0.5) with the eQTL peak-SNP were present at frequency > 5% in both populations. We then tested these eQTLs for replication at a relaxed threshold of p < 0.05 across all SNPs at the locus, to decrease the false negative rate, and focused on the 127 genes for which the eQTL was not replicated (including 28 genes with a reQTL).
Finally, we considered as population-specific, eQTLs whose effect size was significantly different between populations. To do so, we fit, for each SNP at the locus (r2 > 0.5 with the eQTL peak-SNP), the following linear model:
where SNPj is the genotype of the individual j for the variant under study, Popj is a binary variable indicating the population origin (0 for Europeans and 1 for Africans), and εj is a random, normally distributed residual. In this model, β reflects the effect of the derived allele of the SNP on gene expression, γ estimates the fold change in expression between populations observed for individuals with identical genotype, and δ captures the differences in eQTL effect size between populations. Such a model allows to test for a difference in eQTL effect size between populations by testing the null hypothesis, δ = 0 (interaction test).
To be conservative and to account for the uncertainty in detecting the causal variant at the eQTL, we considered an eQTL as population specific if all SNPs in LD (r2 > 0.5) with the eQTL peak-SNP presented a significant interaction p-value.
We then considered eQTLs (or reQTLs) as being population specific when the interaction p-value at the locus was lower than 10−3 (corresponding to FDR < 0.01), leading to a final set of 16 population-specific eQTLs (including 5 reQTLs).
Regulatory Elements and Transcription Factor Binding Sites
Regulatory features were extracted from Ensembl Regulatory Build v80 (Zerbino et al., 2015), which contains regulatory element predictions based on open chromatin regions and histone marks from ENCODE and the Roadmap Epigenomics datasets (Ernst and Kellis, 2015, Kundaje et al., 2015). SNPs overlapping a regulatory element were then classified into four categories: promoter, promoter flanking, enhancer, and CTCF binding sites. Similarly, ENCODE uniformly processed transcription factor binding site (TFBS) clusters (V3) (Ernst and Kellis, 2015) were downloaded from UCSC, and their overlap with the physical position of all SNPs was determined. We then used Fisher’s exact test to assess the eQTL enrichment of specific TFBS or regulatory elements, considering the peak-SNP at each locus, or a randomly selected SNP if multiple SNPs in perfect LD were found. All SNPs with a MAF ≥ 0.05 located less than 1Mb away from an expressed gene were used to constitute the background set. In each condition (or combination of conditions), only the TFBS with the highest values for the lower limit of the odds ratio confidence intervals are reported.
Quantification of Allelic Imbalance
For the quantification of allele-specific imbalance, we focused on exonic SNPs genotyped as heterozygous in our exome data, excluding SNPs with discordant genotypes in the Omni5 data. We used BWA mem (v.0.7.7) (Li and Durbin, 2009) to remap RNA-seq reads onto the hg19 genome for all 970 samples, and extracted all reads aligned with a genetic variant. We reduced mapping bias, by using WASP (van de Geijn et al., 2015) to exclude reads overlapping with known variants (based on dbSNP138) likely to alter the read mapping location. Briefly, for each read overlapping one or more dbSNP variants, WASP creates alternative reads consisting of all possible combinations of reads given these SNPs. It then remaps the alternative reads to the genome, and keeps the original read only if all alternative versions of the read map to the same position. Finally, SAMtools mpileup (Li, 2011), with option -d 10000, was used to count the number of reads mapping to each allele at heterozygous loci. The allelic ratio (AR) was defined for each site as the proportion of minor alleles among all reads, and the allelic imbalance (AI) was defined as the absolute deviation from a balanced ratio of 0.5 (i.e. AI = |AR-0.5|).
aseQTL and asrQTL Mapping
We mapped allele-specific expression QTLs (aseQTLs), by estimating the allelic ratio on the subset of eQTL-genes with sufficient expression coverage at heterozygous exonic SNPs (N ≥ 10 reads) in at least five individuals of each eQTL genotype (heterozygous/homozygous). We extracted the phase information between the strongest local eQTL and the exonic SNP, and tested the correlation between the AR and the phased eQTL genotypes (coded 0 for homozygous, and ± 1 for heterozygotes with variants in phase or in the opposite phase), in a gene-, condition- and population-specific manner. Each exonic SNP was considered as an independent observation. Similarly, allele-specific response QTLs (asrQTLs) were mapped by assessing the correlation between the phased reQTL genotypes and the change in AR at the exonic site after stimulation. The power to detect aseQTLs was computed for various eQTL effect sizes |β|, number of observations n and number of reads per exonic SNP N. We assumed the same number of observations for heterozygous and homozygous genotypes at the eQTL, and equal coverage across all exonic SNPs. Power was then computed for a standard t-test assuming a mean allelic ratio Nalternative/Nreference of 0.5 in homozygous individuals and 2β/(1+ 2β) in heterozygous individuals. Residual variance was set to 0.25/N to match that of a binomial distribution with parameters (0.5, N).
ASE Analysis at the Individual Level
To ensure sufficient power when exploring ASE within single individuals, we considered a higher coverage of heterozygous exonic SNPs (N ≥ 30 reads), and used a binomial test to evaluate allelic imbalance. We also excluded sites at which one allele accounted for less than 2% of the reads or less than 3 reads in total, as such sites might be subject to genotyping errors or systematic mapping biases. The FDR was first calculated across all SNPs, individuals and conditions, using fdrtool, and ASE was defined as the combination of significance at FDR = 0.05 and an absolute log2 fold change of expression between alleles of more than 0.2 (|log2(Nalt/Nref)| > 0.2). For each significant ASE event in stimulated conditions, we checked for differences in allelic imbalance relative to the non-stimulated condition, and defined allele-specific response as the subset of ASE displaying significantly higher allelic imbalance (p < 10−3, Fisher’s exact test) after stimulation with respect to the basal state. Finally, we used simulations to evaluate FDR among the set of genes with at least one ASE/ASR event. We generated 1,000 null datasets, by randomly reassigning reads to the alternative and reference allele with equal probability, and estimated the number of genes with at least one significant ASE or ASR event at each p-value threshold. We then computed FDR as the ratio of the average number of genes with ASE in our resampling to the observed number of genes with ASE at the same p-value threshold.
ASE Enrichment in Rare Coding Variants
For each exonic SNP for which we quantified ASE, we used Variant Effect Predictor (VEP) (McLaren et al., 2010) with Ensembl v.70 Transcript Annotation to identify the set of transcripts overlapping the variant, and Cufflinks FPKM to identify the most strongly expressed overlapping transcript in the individual/condition concerned. VEP annotation was then used to classify variants, according to the most abundant transcript, as synonymous (synonymous_variant/ non_coding exon_variant), missense (missense_variant) or nonsense (stop_gained, stop_lost, initiator_codon_variant). Enrichment in rare coding variants was then assessed using Fisher’s exact test comparing each category with synonymous variants.
Natural Selection Analysis: Neutrality Statistics
We used two metrics, FST and iHS, which detect signals of population-specific positive selection, i.e., mutations that provided a selective advantage to a specific human population. FST measures population differentiation by comparing the variance of allele frequencies within and between populations (Holsinger and Weir, 2009), as local positive selection tends to increase allele frequency differences between populations. As FST is a population pairwise comparison, we derived a directional FST, equal in absolute value to the pairwise FST but with a positive sign if the derived allele was more frequent in the population studied, and a negative sign otherwise. This enables to distinguish selection events that likely occurred in Africans from those that likely occurred in Europeans. The integrated haplotype score (iHS) measures the degree of extended haplotype homozygosity of the putatively selected allele over that of the putatively neutral allele (Voight et al., 2006), as the long-range associations of the selected mutation with neighboring SNPs are not disrupted by recombination.
Furthermore, we used a composite selection score (CSS) allowing to capture signals of recent, strong selective events, by combining FST and iHS. The CSS was designed to identify variants with both a higher derived allele frequency in one population (positive value of directional FST), and a longer haplotype length around the derived allele of the variant in that population (characterized by a negative iHS value). It was computed for all variants with derived allele frequency 0.2 ≤ DAF ≤ 0.95 from genome-wide ranks of both directional FST and iHS attributing the highest rank to positive values of FST and negative values of iHS, respectively. We defined the CSS as following:
with being the total number of variants with 0.2 ≤ DAF ≤ 0.95 in the population studied. CSS ranges from 0 to 1, and increases with the strength of positive selection targeting the derived allele.
Finally, we used the cross-population composite likelihood ratio score, XP-CLR, a region-based metric detecting extended regions where the allele frequencies of multiple contiguous markers are distorted from the prediction under neutrality (Chen et al., 2010). XP-CLR detects classical selective sweeps as well as selection events on pre-existing alleles (standing variation). XP-CLR was scored every 2,000 bp, using windows of 0.2 cM and downsampling to 200 SNPs per window.
Enrichment Tests for Natural Selection Signals
To map selection signals at haplotypes containing eQTLs, we determined, for each statistic (iHS, FST, or CSS), the strongest signal of selection on derived alleles of all SNPs in high LD (r2 > 0.8) with the eQTL peak-SNP. To assess significance, we then compared, for each population and condition, the mean of these values across all eQTLs/reQTLs, with the expected distribution obtained from resampling 10,000 sets of random SNPs matched for MAF (using bins of MAF of 0.05) and the number of SNPs in LD (r2 > 0.8, using bins of 0-2, 3-5, 6-10, 11-20, 21-50, and > 50 SNPs in LD). Similarly, for XP-CLR, we compared the mean of XP-CLR scores at eQTLs/reQTLs (considering the region that contains the eQTL peak-SNP), to the expected distribution obtained from resampling 10,000 sets of random SNPs matched for MAF and LD patterns.
Detection of Candidate eQTLs under Selection
To identify candidate eQTLs under selection, we used an outlier approach where we computed the top 1% values of FST and iHS at the genome-wide level, focusing on signals consistent with selection on derived alleles, within each population separately. To support the adaptive nature of candidate eQTLs, we computed neutral p values for each statistic using simulations based on validated demographic models of Africans and Europeans (Grossman et al., 2013). Furthermore, we tested for local enrichment of outliers (top 1% signals) within a 100kb-window around each eQTL (50kb on each side), similarly to previous work (Grossman et al., 2013, Voight et al., 2006). The proportion of outliers of FST or iHS (1% threshold) was computed from SNPs with DAF ≥ 0.2 in a 100kb window around each putatively selected locus. Significance was assessed from a beta binomial distribution fitted, in each population separately, to the observed genome-wide distribution of the proportion of outliers, to account for variations in the number of SNPs at each locus.
Archaic eQTLs and Enrichment Analyses
We determined the level of Neandertal ancestry of the detected eQTLs, by first defining an “archaic eQTL” as an eQTL for which regulatory variants were introduced into European genomes by introgression from archaic hominins. We identified such eQTLs using the complete genome sequence of Neandertal from Altai (Prüfer et al., 2014). Briefly, the 1000 Genomes phase 3 variants (Auton et al., 2015) were considered as of putative archaic origin (archaic SNPs, or aSNPs) if the Neandertal allele was present in at least one non-African individual and absent from the Yoruba population. According to this definition, 230,779 aSNPs were detected in the 100 European individuals analyzed here. We rendered the analysis more conservative, by further restricting the definition of aSNPs to those in regions of the modern human genome for which Neandertal ancestry has been predicted with a high degree of confidence (marginal probability of Neandertal ancestry ≥ 0.9 and a genetic length ≥ 0.02cM) (Sankararaman et al., 2014). This resulted in a final set of 197,959 aSNPs, of which 77,823 presented a MAF > 0.05. More than 96% of these aSNPs had an archaic allele frequency below 1% in our African samples (who are slightly differentiated from the Yoruba of 1000 Genomes phase 3), consistent with a strong enrichment in true archaic variants. To account for LD between aSNPs and characterize haplotypes that were inherited from Neandertal, we used PLINK (Chang et al., 2015) to extract a set of 924,362 genome-wide SNPs tagging all European variants at an r2 > 0.8. Among these, 9,677 tagged all aSNPs in Europeans and are referred to here as “archaic tagging SNPs.” They were not necessarily aSNPs themselves, reflecting the fact that haplotypes inherited from Neandertals can harbor a mixture of different variants (i.e. variants that appeared in the Neandertal lineage, and ancient variants pre-existing in both lineages before admixture, but for which one allele is carried almost exclusively by Neandertal haplotypes in modern Europeans).
We explored the effect of introgression from Neandertals on the immune repertoire of Europeans, by counting, for each condition, the number of eQTLs overlapped by at least one archaic tagging SNP (or for which the archaic tagging SNP overlapped the reQTL in stimulated conditions), referred to here as archaic eQTLs. We then compared the number of archaic eQTLs detected with the number of SNPs expected to overlap the eQTL, when resampling SNPs tagging non-archaic haplotypes, at random from genic regions (< 1 Mb from a gene). We resampled 1,000 sets of 9,677 tagSNPs with the same allele frequency spectrum as the 9,677 archaic tagging SNPs found in Europeans, and determined their overlap with (r)eQTLs, to assess the significance of our observations. We finally report only the archaic (r)eQTLs for which at least 2 aSNPs were found to be in high LD with the eQTL peak-SNP, and (ii) the haplotype containing the largest number of archaic alleles within the eQTL was sufficiently long for the formal exclusion of incomplete lineage sorting.
The presence of aSNPs in present-day humans can be explained either by introgression or by incomplete lineage sorting (ILS). ILS occurs when an ancestral variant predating the split between humans and Neandertals is retained in both lineages, but lost from a specific human population (i.e. the African population; Figure S7A). Given the time since introgression (47,000-65,000 years ago), the haplotypes containing alleles resulting from ILS would be expected to be shorter than those containing an aSNP introgressed from Neandertals. We distinguished between these two scenarios by first defining the core archaic haplotype for each eQTL as the haplotype within the eQTL carrying the longest stretch of archaic alleles, and then determining whether its size exceeded the expected length of haplotypes assuming an ILS model. We used the approach described by (Huerta-Sánchez et al., 2014) and the most conservative parameters for the age of Altai Neandertal and Denisovan bones reported by (Dannemann et al., 2016). We used the mean recombination rate calculated for a region composed of the core archaic haplotype in a region of 1Mb surrounding the haplotypes (500 kb on either side of the eQTL) in the 1000 Genomes CEU individuals (phase 1). p values were adjusted for multiple testing with the Benjamini-Hochberg procedure.
Adaptive Introgression at Archaic eQTLs
To test if archaic eQTLs result from adaptive introgression, we used both empirical and simulation-based approaches. The empirical approach tests if archaic alleles at eQTLs are more frequent in Europeans than expected, by comparing their levels of genetic differentiation between European and East Asian populations with respect to the genome-wide distribution of aSNPs, similarly to a recent study (Vernot and Akey, 2014). The rationale is that an archaic allele that introgressed into Europeans and East Asians ∼40,000-50,000 years ago and was advantageous in one population only should present today unusually high levels of genetic differentiation, relative to genome-wide expectations. We thus computed the genome-wide distribution of FST between European and East Asian populations at archaic SNPs, using the 1000 Genomes Project phase 3 (Auton et al., 2015), and estimated the empirical p value for candidate archaic eQTL SNPs by dividing their rank by the total number of archaic SNPs.
We next tested if the high frequency of archaic alleles at candidate (r)eQTLs is compatible with a neutral model of evolution, using simulations. Importantly, while a detailed demographic model of Neandertals is not specifically required for such simulations, we need an estimated site frequency spectrum (SFS) of Neandertal alleles in Europeans at the time of their introgression, which is unknown. We thus used three different approximated SFS that rely on different assumptions, detailed below. In each case, the simulation-based p value for candidate archaic eQTL SNPs was obtained by comparing observed frequencies to the neutral simulated SFS in the current generation. Although each of the simulations used present different limitations related to the SFS of archaic alleles at the time of introgression, they can collectively provide information about the expected fate of introgressed alleles under simple scenarios.
The first approximated SFS of Neandertal alleles in present-day European populations, called here “Sankararaman SFS,” was retrieved from a previous study (Sankararaman et al., 2014). This SFS was obtained assuming that archaic alleles in Europeans 40,000 YA (i) evolve under neutrality, and (ii) could not have a frequency larger than 4%. For the second approximated SFS, called here “Fixed-in-Neandertal SFS,” we relaxed the assumption that archaic ancestral frequencies were lower than 4% and used 100,000 forward simulations based on the Wright-Fisher model to simulate frequency changes of archaic alleles in Europeans since Neandertal introgression, 1,440-2,200 generations ago (Vernot and Akey, 2015). We used the best-fit demographic model of Tennessen and colleagues (Tennessen et al., 2012) to model changes in the effective population size of Europeans (i.e., from an ancestral Ne of 1,032, two successive exponential growths with rate 0.31% and 1.95% occur 920 and 205 generations ago, respectively). We assumed here that (i) archaic alleles evolve under neutrality, and (ii) Neandertal alleles that segregate today in Europeans were most likely fixed in Neandertals at the time they were introgressed. This second assumption is conservative, as we neglect all rare Neandertal alleles that had a higher probability to be lost by genetic drift. We modeled the SFS of archaic alleles in Europeans 40,000 YA by a Gaussian distribution with average 5% (i.e., the estimated Neandertal ancestry in European ancient DNAs from this period; (Fu et al., 2016)) and 1% standard deviation. For the third approximated SFS, called here “ancient DNA-based SFS,” we sought to circumvent the uncertainty inherent to the estimation of past Neandertal allele frequencies, by retrieving them from maximum likelihood estimates in ancient DNAs of European hunter-gatherers, early farmers and steppe herders (Mathieson et al., 2015). We computed the SFS of Neandertal alleles in European populations ∼8,000 YA based on the 5,900 SNPs that were detected in EUB as aSNPs and that were covered in the Mathieson’s study, to approximate the SFS of archaic alleles ∼320 generations ago. For convenience, we fitted to this observed SFS a beta distribution (α = 1.21, β = 10.23). We then used 100,000 forward simulations under the same Wright-Fisher model with two exponential growths, to simulate the fate of neutral alleles during the last 320 generations. This simulation analysis only tests if archaic alleles at candidate eQTLs have been under positive selection in the last 8,000 years.
Data and Software Availability
Data Resources
Genome-wide SNP genotyping, whole exome sequencing and RNA-sequencing data generated in this study have been deposited in the European Genome-phenome Archive (EGA) under accession code EGA: EGAS00001001895.
Author Contributions
H.Q. and J.P designed and conducted experiments. M.R. and Y.-H.E.L designed and performed computational analysis. M. Dannemann and J.K. designed and conducted the Neandertal analysis. N.Z. performed flow cytometry analysis, with contributions from M. Deschamps. G.L., E.P., and M.L. assisted in computational analysis. N.N., D.D., and M.L.A. advised on experiments and data interpretation. A.C., G.L.-R., and F.C. managed the clinical protocol and recruited patients. A.B. and J.-F.D. generated genotyping data. H.Q. oversaw all aspects of the project. H.Q., M.R., and L.Q.-M. analyzed and interpreted results and wrote the paper with input from all authors. L.Q.-M. conceived and supervised the research and obtained the funding.
Acknowledgments
This project was funded by the European Research Council under the European Union’s Seventh Framework Programme (FP/2007–2013)/ERC grant agreement 281297 (to L.Q.-M.). We thank Macrogen Inc. for the use of their RNA-sequencing facilities. M.R. was supported by a Marie Skłodowska-Curie fellowship (DLV-655417). M. Dannemann and J.K. are supported by the Max Planck Society and a grant from the Deutsche Forschungsgemeinschaft (SFB 1052, project A02).
Published: October 20, 2016
Footnotes
Supplemental Information includes seven figures, and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.09.024.
Supplemental Information
Mean gene expression (FPKM) across populations, conditions and their associated co-expression modules (A). GO enrichments of modules of correlated genes (B). Enrichment in Jaspar transcription factor binding motifs among genes within each module (C). List of population differentially expressed genes (popDEG) in all cellular conditions (D). List of population differential response genes (popDRG) in stimulated conditions (E). GO enrichments of population differential response genes (popDRG) (F).
List of local eQTL and reQTL (A). Population-specific eQTL and reQTL (B). List of eQTL having a true cis-effect on gene expression (aseQTL) (C). List of reQTL having a true cis effect on gene expression in response to cellular stimulations (asrQTL) (D). List of genes displaying allele-specific expression events (ASE) (E). List of genes displaying allele-specific response (ASR) (F).
List of trans-eQTL at FDR of 5% (A). GO enrichments of IFNB1 trans-associated genes (B). GO enrichment of genes associated with the derived allele of the TLR1 trans-eQTL SNP rs5743618 (C).
Significance of eQTL and reQTL enrichments in signals of positive selection, with respect to genome-wide expectations (A). Signals of positive selection based on empirical FST outliers (B). Signals of positive selection based on empirical iHS outliers (C).
References
- Abi-Rached L., Jobin M.J., Kulkarni S., McWhinnie A., Dalva K., Gragert L., Babrzadeh F., Gharizadeh B., Luo M., Plummer F.A. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science. 2011;334:89–94. doi: 10.1126/science.1209202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Areschoug T., Gordon S. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell. Microbiol. 2009;11:1160–1169. doi: 10.1111/j.1462-5822.2009.01326.x. [DOI] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., Abecasis G.R., 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro L.B., Ben-Ali M., Quach H., Laval G., Patin E., Pickrell J.K., Bouchier C., Tichit M., Neyrolles O., Gicquel B. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet. 2009;5:e1000562. doi: 10.1371/journal.pgen.1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro L.B., Tailleux L., Pai A.A., Gicquel B., Marioni J.C., Gilad Y. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA. 2012;109:1204–1209. doi: 10.1073/pnas.1115761109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A., Mostafavi S., Zhu X., Potash J.B., Weissman M.M., McCormick C., Haudenschild C.D., Beckman K.B., Shi J., Mei R. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar D.M., Yunusbayev B., Metspalu M., Metspalu E., Rosset S., Parik J., Rootsi S., Chaubey G., Kutuev I., Yudkovsky G. The genome-wide structure of the Jewish people. Nature. 2010;466:238–242. doi: 10.1038/nature09103. [DOI] [PubMed] [Google Scholar]
- Brinkworth J.F., Barreiro L.B. The contribution of natural selection to present-day susceptibility to chronic inflammatory and autoimmune disease. Curr. Opin. Immunol. 2014;31:66–78. doi: 10.1016/j.coi.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çalışkan M., Baker S.W., Gilad Y., Ober C. Host genetic variation influences gene expression response to rhinovirus infection. PLoS Genet. 2015;11:e1005111. doi: 10.1371/journal.pgen.1005111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova J.L., Abel L., Quintana-Murci L. Immunology taught by human genetics. Cold Spring Harb. Symp. Quant. Biol. 2013;78:157–172. doi: 10.1101/sqb.2013.78.019968. [DOI] [PubMed] [Google Scholar]
- Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Patterson N., Reich D. Population differentiation as a test for selective sweeps. Genome Res. 2010;20:393–402. doi: 10.1101/gr.100545.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannemann M., Andrés A.M., Kelso J. Introgression of Neandertal- and Denisovan-like haplotypes contributes to adaptive variation in human Toll-like receptors. Am. J. Hum. Genet. 2016;98:22–33. doi: 10.1016/j.ajhg.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O., Zagury J.F., Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps M., Laval G., Fagny M., Itan Y., Abel L., Casanova J.L., Patin E., Quintana-Murci L. Genomic signatures of selective pressures and introgression from archaic hominins at human innate immunity genes. Am. J. Hum. Genet. 2016;98:5–21. doi: 10.1016/j.ajhg.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat. Biotechnol. 2015;33:364–376. doi: 10.1038/nbt.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfax B.P., Knight J.C. Genetics of gene expression in immunity to infection. Curr. Opin. Immunol. 2014;30:63–71. doi: 10.1016/j.coi.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfax B.P., Humburg P., Makino S., Naranbhai V., Wong D., Lau E., Jostins L., Plant K., Andrews R., McGee C., Knight J.C. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. 2014;343:1246949. doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández M., Alarcón G.S., Calvo-Alén J., Andrade R., McGwin G., Jr., Vilá L.M., Reveille J.D., LUMINA Study Group A multiethnic, multicenter cohort of patients with systemic lupus erythematosus (SLE) as a model for the study of ethnic disparities in SLE. Arthritis Rheum. 2007;57:576–584. doi: 10.1002/art.22672. [DOI] [PubMed] [Google Scholar]
- Fraser H.B. Gene expression drives local adaptation in humans. Genome Res. 2013;23:1089–1096. doi: 10.1101/gr.152710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q., Posth C., Hajdinjak M., Petr M., Mallick S., Fernandes D., Furtwängler A., Haak W., Meyer M., Mittnik A. The genetic history of Ice Age Europe. Nature. 2016;534:200–205. doi: 10.1038/nature17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M., Sironi M. Human genome variability, natural selection and infectious diseases. Curr. Opin. Immunol. 2014;30:9–16. doi: 10.1016/j.coi.2014.05.001. [DOI] [PubMed] [Google Scholar]
- Gladue R.P., Cole S.H., Roach M.L., Tylaska L.A., Nelson R.T., Shepard R.M., McNeish J.D., Ogborne K.T., Neote K.S. The human specific CCR1 antagonist CP-481,715 inhibits cell infiltration and inflammatory responses in human CCR1 transgenic mice. J. Immunol. 2006;176:3141–3148. doi: 10.4049/jimmunol.176.5.3141. [DOI] [PubMed] [Google Scholar]
- Grossman S.R., Andersen K.G., Shlyakhter I., Tabrizi S., Winnicki S., Yen A., Park D.J., Griesemer D., Karlsson E.K., Wong S.H., 1000 Genomes Project Identifying recent adaptations in large-scale genomic data. Cell. 2013;152:703–713. doi: 10.1016/j.cell.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger K.E., Weir B.S. Genetics in geographically structured populations: defining, estimating and interpreting F(ST) Nat. Rev. Genet. 2009;10:639–650. doi: 10.1038/nrg2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B.N., Donnelly P., Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Liu D., Majewski P., Schulte L.C., Korn J.M., Young R.A., Lander E.S., Hacohen N. The plasticity of dendritic cell responses to pathogens and their components. Science. 2001;294:870–875. doi: 10.1126/science.294.5543.870. [DOI] [PubMed] [Google Scholar]
- Huerta-Sánchez E., Jin X., Asan, Bianba Z., Peter B.M., Vinckenbosch N., Liang Y., Yi X., He M., Somel M. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature. 2014;512:194–197. doi: 10.1038/nature13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y., Gao J.L., Murphy P.M. Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. J. Immunol. 2008;180:569–579. doi: 10.4049/jimmunol.180.1.569. [DOI] [PubMed] [Google Scholar]
- Jacob C.O., Reiff A., Armstrong D.L., Myones B.L., Silverman E., Klein-Gitelman M., McCurdy D., Wagner-Weiner L., Nocton J.J., Solomon A., Zidovetzki R. Identification of novel susceptibility genes in childhood-onset systemic lupus erythematosus using a uniquely designed candidate gene pathway platform. Arthritis Rheum. 2007;56:4164–4173. doi: 10.1002/art.23060. [DOI] [PubMed] [Google Scholar]
- Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Karlsson E.K., Kwiatkowski D.P., Sabeti P.C. Natural selection and infectious disease in human populations. Nat. Rev. Genet. 2014;15:379–393. doi: 10.1038/nrg3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitai Y., Takeuchi O., Kawasaki T., Ori D., Sueyoshi T., Murase M., Akira S., Kawai T. Negative regulation of melanoma differentiation-associated gene 5 (MDA5)-dependent antiviral innate immune responses by Arf-like protein 5B. J. Biol. Chem. 2015;290:1269–1280. doi: 10.1074/jbc.M114.611053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukurba K.R., Zhang R., Li X., Smith K.S., Knowles D.A., How Tan M., Piskol R., Lek M., Snyder M., Macarthur D.G. Allelic expression of deleterious protein-coding variants across human tissues. PLoS Genet. 2014;10:e1004304. doi: 10.1371/journal.pgen.1004304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M.J., Roadmap Epigenomics Consortium Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., Geuvadis Consortium Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.N., Ye C., Villani A.C., Raj T., Li W., Eisenhaure T.M., Imboywa S.H., Chipendo P.I., Ran F.A., Slowikowski K. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science. 2014;343:1246980. doi: 10.1126/science.1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A.R., Costa H.A., Lappalainen T., Henn B.M., Kidd J.M., Yee M.C., Grubert F., Cann H.M., Snyder M., Montgomery S.B., Bustamante C.D. Transcriptome sequencing from diverse human populations reveals differentiated regulatory architecture. PLoS Genet. 2014;10:e1004549. doi: 10.1371/journal.pgen.1004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42:D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I., Lazaridis I., Rohland N., Mallick S., Patterson N., Roodenberg S.A., Harney E., Stewardson K., Fernandes D., Novak M. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez F.L., Watkins J.C., Hammer M.F. Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol. Biol. Evol. 2013;30:798–801. doi: 10.1093/molbev/mst004. [DOI] [PubMed] [Google Scholar]
- Montgomery S.B., Dermitzakis E.T. From expression QTLs to personalized transcriptomics. Nat. Rev. Genet. 2011;12:277–282. doi: 10.1038/nrg2969. [DOI] [PubMed] [Google Scholar]
- Montgomery S.B., Sammeth M., Gutierrez-Arcelus M., Lach R.P., Ingle C., Nisbett J., Guigo R., Dermitzakis E.T. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery S.B., Lappalainen T., Gutierrez-Arcelus M., Dermitzakis E.T. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 2011;7:e1002144. doi: 10.1371/journal.pgen.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes M., Cortes A., van Heel D.A., Brown M.A. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat. Rev. Genet. 2013;14:661–673. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- Patin E., Siddle K.J., Laval G., Quach H., Harmant C., Becker N., Froment A., Régnault B., Lemée L., Gravel S. The impact of agricultural emergence on the genetic history of African rainforest hunter-gatherers and agriculturalists. Nat. Commun. 2014;5:3163. doi: 10.1038/ncomms4163. [DOI] [PubMed] [Google Scholar]
- Patterson N., Price A.L., Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J.K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 2014;94:559–573. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J.K., Coop G., Novembre J., Kudaravalli S., Li J.Z., Absher D., Srinivasan B.S., Barsh G.S., Myers R.M., Feldman M.W., Pritchard J.K. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J.K., Marioni J.C., Pai A.A., Degner J.F., Engelhardt B.E., Nkadori E., Veyrieras J.B., Stephens M., Gilad Y., Pritchard J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothlichet J., Meunier I., Davis B.K., Ting J.P., Skamene E., von Messling V., Vidal S.M. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013;9:e1003256. doi: 10.1371/journal.ppat.1003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K., Racimo F., Patterson N., Jay F., Sankararaman S., Sawyer S., Heinze A., Renaud G., Sudmant P.H., de Filippo C. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505:43–49. doi: 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana-Murci L., Clark A.G. Population genetic tools for dissecting innate immunity in humans. Nat. Rev. Immunol. 2013;13:280–293. doi: 10.1038/nri3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana-Murci L., Alcaïs A., Abel L., Casanova J.L. Immunology in natura: clinical, epidemiological and evolutionary genetics of infectious diseases. Nat. Immunol. 2007;8:1165–1171. doi: 10.1038/ni1535. [DOI] [PubMed] [Google Scholar]
- Reich D., Green R.E., Kircher M., Krause J., Patterson N., Durand E.Y., Viola B., Briggs A.W., Stenzel U., Johnson P.L. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roider H.G., Manke T., O’Keeffe S., Vingron M., Haas S.A. PASTAA: identifying transcription factors associated with sets of co-regulated genes. Bioinformatics. 2009;25:435–442. doi: 10.1093/bioinformatics/btn627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankararaman S., Mallick S., Dannemann M., Prüfer K., Kelso J., Pääbo S., Patterson N., Reich D. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;507:354–357. doi: 10.1038/nature12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub M.A., Boyle A.P., Kundaje A., Batzoglou S., Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira S.D., Gat-Viks I., Shum B.O., Dricot A., de Grace M.M., Wu L., Gupta P.B., Hao T., Silver S.J., Root D.E. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman R.S., Bastone L.A., Burdick J.T., Morley M., Ewens W.J., Cheung V.G. Common genetic variants account for differences in gene expression among ethnic groups. Nat. Genet. 2007;39:226–231. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger B.E., Montgomery S.B., Dimas A.S., Parts L., Stegle O., Ingle C.E., Sekowska M., Smith G.D., Evans D., Gutierrez-Arcelus M. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24:1461–1462. doi: 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M., Hufton A., Heinig M., O’Keeffe S., Masri N.E., Roider H.G., Manke T., Vingron M. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat. Protoc. 2011;6:1860–1869. doi: 10.1038/nprot.2011.409. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geijn B., McVicker G., Gilad Y., Pritchard J.K. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods. 2015;12:1061–1063. doi: 10.1038/nmeth.3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattathil S., Akey J.M. Small Amounts of Archaic Admixture Provide Big Insights into Human History. Cell. 2015;163:281–284. doi: 10.1016/j.cell.2015.09.042. [DOI] [PubMed] [Google Scholar]
- Vernot B., Akey J.M. Resurrecting surviving Neandertal lineages from modern human genomes. Science. 2014;343:1017–1021. doi: 10.1126/science.1245938. [DOI] [PubMed] [Google Scholar]
- Vernot B., Akey J.M. Complex history of admixture between modern humans and Neandertals. Am. J. Hum. Genet. 2015;96:448–453. doi: 10.1016/j.ajhg.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang S., Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- Zerbino D.R., Wilder S.P., Johnson N., Juettemann T., Flicek P.R. The ensembl regulatory build. Genome Biol. 2015;16:56. doi: 10.1186/s13059-015-0621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mean gene expression (FPKM) across populations, conditions and their associated co-expression modules (A). GO enrichments of modules of correlated genes (B). Enrichment in Jaspar transcription factor binding motifs among genes within each module (C). List of population differentially expressed genes (popDEG) in all cellular conditions (D). List of population differential response genes (popDRG) in stimulated conditions (E). GO enrichments of population differential response genes (popDRG) (F).
List of local eQTL and reQTL (A). Population-specific eQTL and reQTL (B). List of eQTL having a true cis-effect on gene expression (aseQTL) (C). List of reQTL having a true cis effect on gene expression in response to cellular stimulations (asrQTL) (D). List of genes displaying allele-specific expression events (ASE) (E). List of genes displaying allele-specific response (ASR) (F).
List of trans-eQTL at FDR of 5% (A). GO enrichments of IFNB1 trans-associated genes (B). GO enrichment of genes associated with the derived allele of the TLR1 trans-eQTL SNP rs5743618 (C).
Significance of eQTL and reQTL enrichments in signals of positive selection, with respect to genome-wide expectations (A). Signals of positive selection based on empirical FST outliers (B). Signals of positive selection based on empirical iHS outliers (C).