

Abstract
We used reversed-phase liquid chromatography to separate the yeast proteome into 23 fractions. These fractions were then analyzed using capillary zone electrophoresis (CZE) coupled to a Q-Exactive HF mass spectrometer using an electrokinetically pumped sheath flow interface. The parameters of the mass spectrometer were first optimized for top-down proteomics using a mixture of seven model proteins; we observed that intact protein mode with trapping pressure of 0.2 and normalized collision energy of 20% produced the highest intact protein signals and most protein identifications. Then, we applied the optimized parameters for analysis of the fractionated yeast proteome. 580 proteoforms and 180 protein groups were identified via database searching of the MS/MS spectra. This number of proteoform identifications is two times larger than previous CZE-MS/MS studies. An additional 3,243 protein species were detected based on the parent ion spectra. Post-translational modifications including N-terminal acetylation, signal peptide removal, and oxidation were identified.
Keywords: Capillary zone electrophoresis, top-down proteomics, yeast, electrokinetically pumped nano-electrospray, post-translational modifications, proteoforms, RPLC fractionation
Graphical Abstract
INTRODUCTION
Top-down proteomics has emerged as an interesting alternative to bottom-up proteomics for the identification and localization of post-translational modifications (PTMs) and sequence variants.1,2 Recent advances in instrumentation and separation have extended the application of top-down proteomics from analysis of standard proteins or simple protein mixtures3–5 to large scale and in depth analysis of complex biological samples.6–9 Kelleher’s group has published a series of manuscripts describing the implement of GELFrEE10 separation and nano LC-MS/MS for large scale human proteome characterization. 1,043 gene products and over 3,000 protein species were identified from a human cell lysate with a three-stage separation system8, while in another experiment, 347 human mitochondrial proteins were identified and over 5,000 proteoforms were observed.7 Ansong et al. employed a four-hour UPLC separation of intact proteins from Salmonella typhimurium and identified 563 unique proteins and 1,665 proteoforms.9
Top-down proteomics primarily couples reverse phase liquid chromatography (RPLC) to a mass spectrometer. Capillary zone electrophoresis (CZE) is an orthogonal separation mode to RPLC, offering a separation based on size-to-charge rather than hydrophobic interactions.11–13 CZE can resolve a different set of proteoforms than RPLC because proteins with minor sequence variations or PTMs often have similar hydrophobicities but different charges. CZE-ESI-MS/MS has been employed to analyze single protein proteoforms14–15 including pharmaceutical protein glycoform profiling,14 and more recently, complex biological samples.16–18 CZE can be interfaced with mass spectrometers via electrospray ionization (ESI). Our group developed an electrokinetically pumped sheath-flow nanospray CZE-ESI-MS interface that transfers the analyte from the separation capillary to a glass emitter filled with sheath liquid for nanospray.19,20 Its stability and sensitivity has been greatly improved with our third generation interface21 and has been effective for various bottom-up22–28 and top down proteomics analyses.16–18,29,30 For example, 58 proteoforms were identified by a Q Exactive mass spectrometer from the Mycobacterium marinum secretome in a single shot experiment.17 Another study coupled CZE to an Orbitrap Elite mass spectrometer through a prototype sheathless CESI interface31 to characterize the Pyrococcus furiosus proteome and identified 134 protein groups and 291 proteoforms from three SPE column fractions.32
The performance of CZE can be degraded due to both the small sample injection volume that is typically employed33 and sample adsorption to inner capillary wall.34 Sample injection volume can be increased by the use of dynamic pH junction based CZE,35,36 which has been recently applied to bottom-up proteomics.37 Sample adsorption has been partially solved by the use of linear polyacrylamide (LPA)-coated capillaries. However, commercially available LPA-coated capillaries often provide poor performance.30 A thermally-initiated coating protocol developed by this group improved the uniformity and stability of this coating for peptides and standard proteins.38 These two techniques were combined in this study to improve the overall performance of CZE for top-down proteomics.
In this work, we prefractionated the yeast proteome by RPLC and explored the potential of CZE-ESI-MS/MS for intact protein characterization. A Q Exactive HF mass spectrometer was first optimized for intact protein detection and fragmentation. CZE-ESI-MS/MS was then employed to characterize 23 fractions produced by RPLC. Dynamic pH junction and an LPA-coated capillary were applied to improve the loading amount and peak capacity of CZE. 580 proteoforms and 180 protein groups were identified from the 23 fractions. To our knowledge, this is the largest top-down proteome dataset based on CZE-ESI-MS/MS reported to date.
EXPERIMENTAL SECTION
Materials and Reagents
All reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless stated otherwise. Formic acid (FA) and glacial acetic acid (HOAc) were purchased from Fisher Scientific (Pittsburgh, PA). Methanol was purchased from Honeywell Burdick & Jackson (Wicklow, Ireland). Water was deionized by a NanoPure system from Thermo Scientific (Marietta, OH). Fused silica capillary tubing was purchased from Polymicro Technologies (Phoenix, AZ). The RPLC column (Jupiter 5 μm C5 300 Å, LC Column 250 × 4.6 mm) was purchased from Phenomenex (Torrance, CA).
Sample Preparation
Cytochrome c, myoglobin, carbonic anhydrase, β-casein, insulin, superoxide dismutase and ubiquitin were dissolved in 0.07% formic acid and 30% acetonitrile in water with concentrations of 0.1 mg/mL, 0.2 mg/mL, 0.2 mg/mL, 0.8 mg/mL, 0.1 mg/mL and 0.1 mg/mL, respectively. For the yeast proteome, a small portion (~ 0.2 g) of commercial baker’s yeast (S. cerevisiae) (Red Star Active Dry Yeast) was added to 150 mL of yeast extract peptone dextrose (YPD) medium (50 g Difco™ YPD in 150 mL distilled water, autoclaved at 120 °C for 15 min) and grown overnight at 37 °C on a shaker. Yeast lysis was performed as described.39 Briefly, a yeast suspension was centrifuged at 4000 g for 5 min and washed with PBS for 3 times. After adding lysate buffer (100 mM DTT, 5% SDS), the cell suspension was heated at 95 °C for 5 min, followed by sonication for 15 min at the maximum power. Finally, the lysate was centrifuged at 16000 g for 5 min and the supernatant was collected. Next, 100 μL of 1 M iodoacetamide were added to 200 μL of yeast protein extract and reacted for 20 min at room temperature. Cold acetone precipitation was performed by adding 1.2 mL of cold acetone (−20 °C) to 300 uL of sample, incubating at −20 °C overnight, centrifuging at 18000 g for 15 min, washing with cold acetone, and centrifuging again. An ~800 μg of protein pellet was resuspended in 400 μL of a solution containing 8 M urea and 100 mM NH4HCO3. The sample solution was passed through a 30 kDa centrifugal filter (Millipore, MA). The protein was extracted from the filter membrane with 8 M urea and 100 mM NH4HCO3 to form a final protein solution of 500 μL (~1.6 mg/mL based on BSA analysis of the sample before acetone precipitation).
RPLC Fractionation
Intact protein fractionation was performed on a Waters e2695 HPLC system with a C5 stationary phase Jupiter column, 5 μm particle diameter, 300 Å pore size, 250 × 4.6 mm column size. Mobile phase A was composed of water with 0.1% FA. Mobile phase B was composed of acetonitrile with 0.1% FA. The operating flow rate was 0.8 mL/min. The RPLC system was activated with 80% mobile phase B for 10 min, and then equilibrated with 5% mobile phase B for 10 min. A ~320 μg yeast protein sample was injected, followed by 10 min of washing with 5% of mobile phase B. A 70 min linear gradient was set from 5% mobile phase B to 80% mobile phase B. A total of 46 fractions were collected from 15 min to 61 min (one fraction per minute). The fractions collected were then lyophilized and suspended in 5 mM NH4HCO3. The 46 RPLC fractions were combined to 23 fractions according to Table S1 with a final volume of 5 μL and were subjected to CZE-ESI-MS/MS analysis.
CZE-ESI-MS/MS Analysis
The preparation of a LPA-coated capillary was described elsewhere.38 Briefly, the silica capillary was pretreated with gamma-methacryloxypropyl-trimethoxysilane, and then the monomer mixture and ammonium persulfate initiator were introduced into the capillary without TEMED initiator. The filled capillary was heated in a water bath to initiate polymerization to form LPA coatings. CZE was coupled to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Electrospray was generated using an electrokinetically pumped sheath flow through a nanospray emitter.19 The borosilicate glass emitter (1.0 mm o.d. × 0.75 mm i.d., 10 cm length) was pulled with a Sutter P-1000 flaming/brown micropipet puller. The emitter inner diameter was 15–20 μm. Separation was performed in 60 cm and 80 cm long, 50 μm ID, 150 μm OD LPA-coated fused silica capillaries for standard proteins and yeast fractions. The separation buffer was 5% (v/v) HOAc. The electrospray sheath liquid was 10% (v/v) methanol and 0.5% (v/v) FA. About 20 nL standard protein solution or 120 nL (240 nL for fractions 2–8) yeast fraction solution was injected for each CE-MS experiment. The separation voltages were 18 kV and 24 kV for standard proteins and yeast fractions, respectively. The electrospray voltages were 1.5 kV and 1.8 kV for standard proteins and yeast fractions, respectively.
The inlet ion transfer tube was held at 300°C and the S-lens rf level was set at 60%. For standard proteins, full MS scans were acquired in the Orbitrap over the m/z 400–1800 range with a resolving power of 60,000 at m/z 200. The three most intense peaks with charge state ≥5 were selected in data-dependent fashion for fragmentation. Detection for all tandem mass spectra was performed in the Orbitrap with a resolving power of 60,000 at m/z 200. The MS1 AGC target value was 1,000,000 with a maximum injection time of 100 ms, while MS/MS scans have an AGC target value of 500,000 and a maximum injection time of 200 ms. Seven and three microscans were used in MS1 and MS2 scans, respectively. An exclusion window of ±10 ppm was constructed around the monoisotopic peak of each selected precursor for 5 seconds. For yeast fractions, most of the mass spectrometer parameters were the same and the exceptions were listed below. Full MS scans were acquired over the m/z 600–2000 range. MS/MS scans have an AGC target value of 100,000 and a maximum injection time of 300 ms. Dynamic exclusion time was set at 20 seconds.
Data Analysis
The tandem spectra were decharged and deisotoped by MS-Deconv (version 0.8.0.7370),40 followed by database searching with TopPIC software (version 0.9.1).41 Raw files from Q Exactive HF were first converted to mzXML files with ReAdW (version 4.3.1). Then, MS-Deconv (v 0.8.0.7370) was used to generate msalign files with mzXML files as the input. Finally, TopPIC software (http://proteomics.informatics.iupui.edu/software/toppic/) was used for database searching with msalign files as the input. Uniprot protein database for yeast (reviewed, 23,525 entries) was used for database searching. The parameters for database searching included N-terminal variable PTM as methionine exclusion and acetylation, number of the unexpected PTMs as 2, mass error tolerance as 10 ppm, cysteine protecting group as carbamidomethylation, cutoff type as EVALUE, and cutoff value as 0.001. The results of 23 fractions were combined manually and all the proteoforms with mass shift of 1 Da were counted as the same proteoforms.
RESULTS AND DISCUSSION
Q Exactive HF Mass Spectrometer Intact Protein Mode Optimization
The Q Exactive HF mass spectrometer combines a state-of-art segmented quadrupole with a high-resolution ultra-field Orbitrap mass analyzer. An intact protein MS mode is included in the instrument setting; this setting is designed for analysis of intact proteins with optimized pressure and ion optic parameters. Besides turning on/off the intact protein mode, the trapping gas pressure can be manipulated; the optimized value is 1.0 for non-intact and 0.2 for intact protein modes. In this experiment, we first investigated the effect of this intact protein mode and the trapping gas pressure for intact protein detection, fragmentation and identification. Figure 1A shows the extracted ion electropherogram for a seven-protein mixture detected under intact protein mode with trapping pressure of 0.2. All seven proteins were separated with reasonable peak shape in 20 min. Compared to other settings, intact protein mode with trapping pressure of 0.2 detected proteins with significantly higher signal intensities for all seven proteins except myoglobin which showed comparable signal intensity to the result of intact mode with 0.3 trapping pressure (Figure 1B).
Figure 1. Results from analysis of standard proteins.

A. Extracted ion electropherogram for the seven-protein mixture. The signal for each protein was amplified to the same height as shown in the graph. The inserted graphs represent the averaged mass spectra for each protein. Top spectrum: intact mode with pressure at 0.2. Bottom spectrum: non-intact mode.
B. The signal intensities for each protein with different instrument settings. Non-intact mode with trapping gas pressure at 1.0 and intact protein mode with trapping gas pressure at 0.1, 0.2, 0.3, and 0.5 were listed here. Duplicate runs were performed for all conditions, except non-intact mode where triplicate runs were performed.
The inserted spectra in Figure 1A are butterfly plots of the averaged MS1 spectra for each protein, with the intact mode spectra on top and the non-intact mode spectra on the bottom. With the intact protein mode, spectra with higher charge ions were generated. This effect is increasingly significant as the size of the protein increases. For example, the signal of higher charged ions was only slightly enhanced for ubiquitin and insulin, and was moderately enhanced for cytochrome c, myoglobin and superoxide dismutase. When the protein sizes were greater than 20 kDa (i.e. carbonic anhydrase and β-casein), the signal intensities for higher charged ions as well as the overall signal intensity were significantly enhanced. The fragmentation efficiency for higher charged ions is generally better than lower charged ions due to their higher charge densities, and thus resulted in higher quality tandem spectra and improved identification rate.
To further evaluate the intact protein mode, the protein identification rate for both intact protein and non-intact mode were evaluated. First, the normalized collision energy (NCE) was optimized for the best identification results. NCE of 15, 20 and 25% were applied under intact protein mode and as listed in Table 1, where 20% NCE produced the most identifications. Six out of seven proteins were identified with intact protein mode and 20% NCE. Table 1 also lists the identification results for non-intact mode with 20% NCE, where only three proteins were identified. Although ubiquitin was not identified by tandem spectra either in intact mode or non-intact mode, it can be easily identified based on its parent ion mass. Similarly, all proteins were identified by their masses even though some of them were not identified by tandem spectra. Therefore, intact protein mode with NCE of 20% was employed in the following characterization of yeast fractions.
Table 1.
The standard protein identifications with different modes and NCE values shown in the bracket. Non-intact mode was performed with NCE of 20%. “○” means that the protein was identified and “×” means that the protein was not identified.
| Intact Mode (20%) | Intact Mode (15%) | Intact Mode (25%) | Non-intact Mode | |
|---|---|---|---|---|
| Cytochrome C | ○ | × | × | × |
| Ubiquitin | × | × | × | × |
| Myoglobin | ○ | ○ | × | ○ |
| Carbonic Anhydrase | ○ | ○ | ○ | ○ |
| Superoxide Dismutase | ○ | ○ | ○ | × |
| Insulin | ○ | × | × | × |
| β-casein | ○ | ○ | ○ | ○ |
RPLC-CZE-ESI-MS/MS
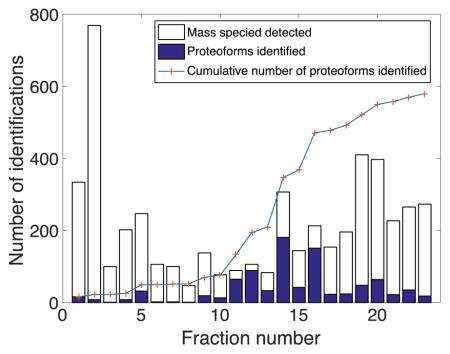
By employing dynamic pH junction and LPA-coated capillary, we not only obtained a high peak capacity for CZE but also generated high protein signals to ensure quality tandem spectra for identifications. The average peak capacity for CZE separation was about 50 (excluding 6 fractions in which no useful electrophoretic peaks were generated), and the average separation window was ~20 min. The average base peak signal intensity for 23 fractions was about 3×107. The detailed peak capacities and signal intensities are listed in Figure S1. The best identification result was generated from fraction 14, which identified 180 proteoforms within a 40 min CZE-ESI-MS/MS run and increased the number of total identifications by 137 (Table 2, Figure S1 fraction 14), demonstrating good performances for both the CZE-ESI-MS/MS system and RPLC fractionation. The peak capacity was about 75, and the total separation window was about 20 min, generating over four identifications per minute for this run.
Table 2.
Mass species detected from MS1 spectra (> 5 kDa) and proteoforms identified for the fractionated yeast proteome corresponding to the values shown in Figure 2.
| CZE Fraction Number | Number of Mass Species | Proteoforms identified | Cumulative number of proteoform identifications |
|---|---|---|---|
| 1 | 334 | 16 | 16 |
| 2 | 769 | 8 | 22 |
| 3 | 100 | 1 | 22 |
| 4 | 202 | 8 | 26 |
| 5 | 247 | 32 | 49 |
| 6 | 106 | 2 | 50 |
| 7 | 100 | 2 | 51 |
| 8 | 47 | 1 | 52 |
| 9 | 138 | 19 | 70 |
| 10 | 77 | 13 | 76 |
| 11 | 89 | 65 | 133 |
| 12 | 106 | 89 | 194 |
| 13 | 83 | 33 | 210 |
| 14 | 307 | 181 | 347 |
| 15 | 144 | 42 | 368 |
| 16 | 213 | 151 | 471 |
| 17 | 154 | 23 | 478 |
| 18 | 196 | 24 | 492 |
| 19 | 410 | 48 | 521 |
| 20 | 397 | 64 | 550 |
| 21 | 227 | 22 | 558 |
| 22 | 265 | 35 | 570 |
| 23 | 273 | 18 | 580 |
| Total Number of Mass Species Detected 3243 | |||
| Total Number of Protein Groups 180 | |||
In total, 580 proteoforms and 180 protein groups were identified from 23 CZE-ESI-MS/MS runs of the fractionated proteome. The combined identified protein groups and the masses of their proteoforms are listed in the supporting materials. C5 RPLC column provides efficient fractionation, so the overlaps were reasonably small between fractions as shown in Figure 2 and Table 2. All fractions (except fraction 3 with only one identification) contributed to unique identifications when combined with the previous fractions. Later fractions generated more identifications compared to the earlier ones. One possible explanation is that the earlier fractions may contain more impurities that resulted in the large peaks shown in Figure S2 Fraction 4&5, which suppressed the protein signal and possibly reduced the separation efficiency of CZE. The increases in total identifications were less for the last few fractions compared to the middle fractions, indicating that the number of identifications would not be improved if more fractions were generated in the same dimension. A second dimension of fractionation (i.e. size-based separation) before CZE separation would help further improve the identification result. Despite the limited number of proteoforms being identified from tandem spectra, the number of detected species from MS1 spectra was much larger. A total of 3243 species with masses greater than 5 kDa were detected from the 23 fractions. Therefore, there is potential for CZE-ESI-MS/MS to identify more proteoforms given a higher resolution and sensitivity mass spectrometer.
Figure 2.

Summary of the identification results from CZE-ESI-MS/MS top-down analysis of 23 fractions isolated from the yeast proteome. The number of mass species detected only includes the mass species greater than 5 kDa. The number of total identifications is the cumulative number of produced by combining the new fraction to all the previous ones.
When examining the molecular weight (MW) distribution of the identifications, we found a MW bias towards the low MW region, Figure 3A. Most of the proteoforms identified were small, truncated proteins (~5 kDa), which were preferentially identified by the Orbitrap detector and more effectively fragmented. Only 1/10 of the proteoforms have MW close to or larger than 10 kDa. This result was not surprising because the Orbitrap generally shows decreased sensitivity for larger proteins that have more charge states and isotopic peaks, which resulted in poorly resolved isotopic envelope and low quality tandem spectra. For example, higher concentrations were used for larger proteins such as β-casein in the standard protein experiment and signal intensities of 107 were reached for efficient fragmentations. In contrast, we found that larger MW species in the yeast sample typically have signals as low as 105, which decreases successful deconvolution and tandem spectra identification. There were more large mass species detected in MS1 but not identified with tandem spectra, Figure 3A. As a result, incorporating a size-based fractionation dimension prior to RPLC fractionation would greatly improve the number of identifications by concentrating large proteins and optimizing fragmentation conditions for narrower mass ranges.
Figure 3.

Summary of identifications. (A) Molecular weight distributions of yeast proteoforms identifications and mass species detected (> 5 kDa) in MS1 combined from 23 CZE-ESI-MS/MS experiments. (B) Protein abundance distributions of identified protein groups. X-axis is plotted on a log10 scale.
The integrated protein abundances listed in PaxDb (http://pax-db.org/) were used to evaluate the protein abundance distribution of identified protein groups, Figure 3B. All the protein groups identified fell in the middle- to high-region of the yeast total protein abundance distribution. Most of them have abundance at around 103 ppm, and the total identifications spanned a range from 10 ppm to 105 ppm, indicating that our CZE-ESI-MS/MS system is able to identify a reasonably wide dynamic range in a biological sample.
The identification of proteoforms and PTMs are the most valuable advantages of top-down proteomics. In this study, an average of three proteoforms was identified for each protein group. PTMs including N-terminal acetylation, signal peptide removal, and oxidation were identified for a number of proteins. Since the protein sample were reduced and alkylated, the fixed modification, carbamidomethylation, was also successfully identified on cysteine residues.
Supplementary Material
Acknowledgments
We thank Joseph Ong in the Goodson lab at Notre Dame for the kind donation of the yeast sample. We also express our gratitude to Dr. William Boggess in the Notre Dame Mass Spectrometry and Proteomics Facility for his help with this project. This work was funded by a grant from the National Institutes of Health (R01GM096767).
Footnotes
The following files are available free of charge via the Internet at http://pubs.acs.org:
Supporting Information.docx – fraction pooling scheme and base peak electropherograms.
SI_revised.xlsx – list of proteoforms and protein groups.
References
- 1.Kelleher NL. Top-down proteomics. Anal Chem. 2004;76:196a–203a. [PubMed] [Google Scholar]
- 2.Chait BT. Mass spectrometry: Bottom-up or top-down? Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- 3.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J Am Chem Soc. 1999;121:806–812. [Google Scholar]
- 4.Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP, McLafferty FW. Top down characterization of larger proteins (45 kDa) by electron capture dissociation mass spectrometry. J Am Chem Soc. 2002;124:672–678. doi: 10.1021/ja011335z. [DOI] [PubMed] [Google Scholar]
- 5.Han X, Jin M, Breuker K, McLafferty FW. Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons. Science. 2006;314:109–112. doi: 10.1126/science.1128868. [DOI] [PubMed] [Google Scholar]
- 6.Kellie JF, Catherman AD, Durbin KR, Tran JC, Tipton JD, Norris JL, Witkowski CE. 2nd; Thomas, P. M., Kelleher, N. L. Robust Analysis of the Yeast Proteome under 50 kDa by Molecular-Mass-Based Fractionation and Top-Down Mass Spectrometry. Anal Chem. 2012;84:209–215. doi: 10.1021/ac202384v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catherman AD, Li M, Tran JC, Durbin KR, Compton PD, Early BP, Thomas PM, Kelleher NL. Top down proteomics of human membrane proteins from enriched mitochondrial fractions. Anal Chem. 2013;85:1880–1888. doi: 10.1021/ac3031527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–258. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, Nakayasu ES, Cort JR, Pevzner P, Smith RD, Heffron F, Adkins JN, Pasa-Tolic L. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella Typhimurium in response to infection-like conditions. Proc Natl Acad Sci USA. 2013;110:10153–10158. doi: 10.1073/pnas.1221210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran JC, Doucette AA. Gel-eluted liquid fraction entrapment electrophoresis: An electrophoretic method for broad molecular weight range proteome separation. Anal Chem. 2008;80:1568–1573. doi: 10.1021/ac702197w. [DOI] [PubMed] [Google Scholar]
- 11.Volkman HE, Clay H, Beery D, Chang JC, Sherman DR, Ramakrishnan L. Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol. 2004;2:e367. doi: 10.1371/journal.pbio.0020367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simo C, Herrero M, Neususs C, Pelzing M, Kenndler E, Barbas C, Ibáñez E, Cifuentes A. Characterization of proteins from Spirulina platensis microalga using capillary electrophoresis-ion trap-mass spectrometry and capillary electrophoresis-time of flight-mass spectrometry. Electrophoresis. 2005;26:2674–2683. doi: 10.1002/elps.200500055. [DOI] [PubMed] [Google Scholar]
- 13.Jorgenson JW, Lukacs KD. Capillary zone electrophoresis. Science. 1983;222:266–272. doi: 10.1126/science.6623076. [DOI] [PubMed] [Google Scholar]
- 14.Haselberg R, de Jong GJ, Somsen GW. Low-flow sheathless capillary electrophoresis-mass spectrometry for sensitive glycoform profiling of intact pharmaceutical proteins. Anal Chem. 2013;85:2289–2296. doi: 10.1021/ac303158f. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Barendregt A, Kamerling JP, Heck AJ. Analyzing protein micro-heterogeneity in chicken ovalbumin by high-resolution native mass spectrometry exposes qualitatively and semi-quantitatively 59 proteoforms. Anal Chem. 2013;85:12037–12045. doi: 10.1021/ac403057y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Riley NM, Sun L, Hebert AS, Yan X, Westphall MS, Rush MJ, Zhu G, Champion MM, Mba Medie F, Champion PA, Coon JJ, Dovichi NJ. Coupling Capillary Zone Electrophoresis with Electron Transfer Dissociation and Activated Ion Electron Transfer Dissociation for Top-Down Proteomics. Anal Chem. 2015;87:5422–5429. doi: 10.1021/acs.analchem.5b00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y, Sun L, Champion MM, Knierman MD, Dovichi NJ. Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry for Top-Down Characterization of the Mycobacterium marinum Secretome. Anal Chem. 2014;86:4873–4878. doi: 10.1021/ac500092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Compton PD, Tran JC, Ntai I, Kelleher NL. Optimizing capillary electrophoresis for top-down proteomics of 30–80 kDa proteins. Proteomics. 2014;14:1158–1164. doi: 10.1002/pmic.201300381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wojcik R, Dada OO, Sadilek M, Dovichi NJ. Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis. Rapid communications in mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:2554–2560. doi: 10.1002/rcm.4672. [DOI] [PubMed] [Google Scholar]
- 20.Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ. Ultrasensitive and Fast Bottom-up Analysis of Femtogram Amounts of Complex Proteome Digests. Angew Chem Int Edit. 2013;52:13661–13664. doi: 10.1002/anie.201308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun L, Zhu G, Zhang Z, Mou S, Dovichi NJ. Third-Generation Electrokinetically Pumped Sheath-Flow Nanospray Interface with Improved Stability and Sensitivity for Automated Capillary Zone Electrophoresis-Mass Spectrometry Analysis of Complex Proteome Digests. J Proteome Res. 2015;4:2312–2321. doi: 10.1021/acs.jproteome.5b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wojcik R, Zhu G, Zhang Z, Yan X, Zhao Y, Sun L, Champion MM, Dovichi NJ. Capillary zone electrophoresis as a tool for bottom-up protein analysis. Bioanalysis. 2016;8:89–92. doi: 10.4155/bio.15.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun L, Hebert AS, Yan X, Zhao Y, Westphall MS, Rush MJ, Zhu G, Champion MM, Coon JJ, Dovichi NJ. Over 10000 Peptide Identifications from the HeLa Proteome by Using Single-Shot Capillary Zone Electrophoresis Combined with Tandem Mass Spectrometry. Angew Chem Int Edit. 2014;53:13931–13933. doi: 10.1002/anie.201409075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun L, Zhu G, Mou S, Zhao Y, Champion MM, Dovichi NJ. Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry for quantitative parallel reaction monitoring of peptide abundance and single-shot proteomic analysis of a human cell line. J Chromatogr A. 2014;1359:303–308. doi: 10.1016/j.chroma.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Champion MM, Sun L, Champion PA, Wojcik R, Dovichi NJ. Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry as an alternative proteomics platform to ultraperformance liquid chromatography-electrospray ionization-tandem mass spectrometry for samples of intermediate complexity. Anal Chem. 2012;84:1617–1622. doi: 10.1021/ac202899p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mou S, Sun L, Wojcik R, Dovichi NJ. Coupling immobilized alkaline phosphatase-based automated diagonal capillary electrophoresis to tandem mass spectrometry for phosphopeptide analysis. Talanta. 2013;116:985–990. doi: 10.1016/j.talanta.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan X, Essaka DC, Sun L, Zhu G, Dovichi NJ. Bottom-up proteome analysis of E. coli using capillary zone electrophoresis-tandem mass spectrometry with an electrokinetic sheath-flow electrospray interface. Proteomics. 2013;13:2546–2551. doi: 10.1002/pmic.201300062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu G, Sun L, Yan X, Dovichi NJ. Single-Shot Proteomics Using Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry with Production of More than 1 250 Escherichia coli Peptide Identifications in a 50 min Separation. Anal Chem. 2013;85:2569–2573. doi: 10.1021/ac303750g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L, Knierman MD, Zhu G, Dovichi NJ. Fast Top-Down Intact Protein Characterization with Capillary Zone Electrophoresis-Electrospray Ionization Tandem Mass Spectrometry. Anal Chem. 2013;85:5989–5995. doi: 10.1021/ac4008122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Sun L, Knierman MD, Dovichi NJ. Fast separation and analysis of reduced monoclonal antibodies with capillary zone electrophoresis coupled to mass spectrometry. Talanta. 2016;148:529–533. doi: 10.1016/j.talanta.2015.11.020. [DOI] [PubMed] [Google Scholar]
- 31.Moini M. Simplifying CE-MS operation. 2. Interfacing low-flow separation techniques to mass spectrometry using a porous tip. Anal Chem. 2007;79:4241–4246. doi: 10.1021/ac0704560. [DOI] [PubMed] [Google Scholar]
- 32.Han X, Wang Y, Aslanian A, Bern M, Lavallee-Adam M, Yates JR. 3rd. Sheathless Capillary Electrophoresis-Tandem Mass Spectrometry for Top-Down Characterization of Pyrococcus furiosus Proteins on a Proteome Scale. Anal Chem. 2014;86:11006–11012. doi: 10.1021/ac503439n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng YF, Wu SL, Chen DY, Dovichi NJ. Interaction of Capillary Zone Electrophoresis with a Sheath Flow Cuvette Detector. Anal Chem. 1990;62:496–503. [Google Scholar]
- 34.Righetti PG, Sebastiano R, Citterio A. Capillary electrophoresis and isoelectric focusing in peptide and protein analysis. Proteomics. 2013;13:325–340. doi: 10.1002/pmic.201200378. [DOI] [PubMed] [Google Scholar]
- 35.Britz-McKibbin P, Chen DDY. Selective focusing of catecholamines and weakly acidic compounds by capillary electrophoresis using a dynamic pH junction. Anal Chem. 2000;72:1242–1252. doi: 10.1021/ac990898e. [DOI] [PubMed] [Google Scholar]
- 36.Aebersold R, Morrison HD. Analysis of Dilute Peptide Samples by Capillary Zone Electrophoresis. J Chromatogr. 1990;516:79–88. doi: 10.1016/s0021-9673(01)90206-7. [DOI] [PubMed] [Google Scholar]
- 37.Zhu G, Sun L, Yan X, Dovichi NJ. Bottom-Up Proteomics of Escherichia coli Using Dynamic pH Junction Preconcentration. Capillary Zone Electrophoresis-Electrospray Ionization-Tandem Mass Spectrometry. Anal Chem. 2014;86:6331–6336. doi: 10.1021/ac5004486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu G, Sun L, Dovichi NJ. Thermally-initiated free radical polymerization for reproducible production of stable linear polyacrylamide coated capillaries, and their application to proteomic analysis using capillary zone electrophoresis-mass spectrometry. Talanta. 2016;146:839–843. doi: 10.1016/j.talanta.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagaraj N, Kulak NA, Cox J, Neuhauser N, Mayr K, Hoerning O, Vorm O, Mann M. System-wide Perturbation Analysis with Nearly Complete Coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol Cell Proteomics. 2012;11:M111.013722. doi: 10.1074/mcp.M111.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Inbar Y, Dorrestein PC, Wynne C, Edwards N, Souda P, Whitelegge JP, Bafna V, Pevzner PA. Deconvolution and database search of complex tandem mass spectra of intact proteins: a combinatorial approach. Mol Cell Proteomics. 2010;9:2772–2782. doi: 10.1074/mcp.M110.002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu XW, Sirotkin Y, Shen YF, Anderson G, Tsai YS, Ting YS, Goodlett DR, Smith RD, Bafna V, Pevzner PA. Protein Identification Using Top-Down Spectra. Mol Cell Proteomics. 2010;11:M111.008524. doi: 10.1074/mcp.M111.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.