

Abstract
Decreased mortality and disability after traumatic brain injury is a significant medical challenge. Desflurane, a widely used volatile anesthetic has proven to be neuroprotective in a variety of in vitro and in vivo models of ischemic brain injury. The aim of this study was to investigate whether desflurane exhibits neuroprotective properties in an in vitro model of traumatic brain injury. Organotypic hippocampal slice cultures were prepared from brains of 5–7-day-old C57/BL6 mouse pups. After 14 days of culture, the slices were subjected to a focal mechanical trauma and thereafter incubated with three different concentrations of desflurane (2, 4 and 6%) for 2, 24 and 72 hours. Cell injury was assessed with propodium iodide uptake. Our results showed that after 2 hours of desflurane exposure, no significant change in trauma intensity was observed. However, 2% and 4% desflurane could reduce the trauma intensity significantly in the no trauma group than in the no desflurane and trauma group. Incubation with 4% desflurane for 24 hours doubled the trauma intensity in comparison to the trauma control group and the trauma intensity further increased after 72 hours of incubation. Furthermore, a dose-dependent increase of trauma intensity after 24 hours exposure was observed. Our results suggest that a general neuroprotective attribute of desflurane in an in vitro model of traumatic brain injury was not observed.
Keywords: desflurane, neuroprotection, traumatic brain injury, organotypic hippocampal slices
INTRODUCTION
Traumatic brain injury (TBI) is a significant healthcare challenge. Especially young persons (< 30 years) and the elderly are at risk of traumatic brain injury due to falls and traffic accidents. The overall lifetime prevalence of traumatic brain injury with loss of consciousness is estimated at 12 percent (Frost et al., 2013). Annually 1.7 million traumatic brain injuries occur in the United States. About 1.4 million of these patients are treated at an emergency department, 275,000 have to be hospitalized, and 5,200 die as a result of the trauma (Faul et al., 2010). More than 40% of the hospitalized patients, who survived TBI, will suffer long-term disability (Selassie et al., 2008).
There are two main reasons for the cell damage of the brain after a traumatic event:First, the initial lesion of the cells as a result of a mechanical damage through linear translational and rotational Forces as well as a defect caused by the collision to the skull (primary injury) (Feng et al., 2015). Second, the cellular response to an injury (secondary injury) including cell membrane depolarization with a resulting release of neurotransmitters like glutamate and other excitatory amino acids. This depolarization leads to an influx and an additional release of intracellular Ca2+. Furthermore mechanoporation of the cell membrane and the axolemma leads to an increase of intracellular Ca2+ concentration with a following activation of caspases and calpains as well as a generation of free radicals followed by activation of the apoptotic and necrotic pathway (McAllister, 2011).
Desflurane (1,2,2,2-tetrafluoroethyl difluoromethyl ether) is a fluorinated methyl ethyl ether, a commonly used volatile anesthetic. Desflurane acts at the level of the central nervous system interacting with the release of neurotransmitters, influences the reuptake of neurotransmitters, changes the binding of neurotransmitters on the post-synaptic receptor sites, and influences the ionic conductance of the postsynaptic membrane. Further gamma-amonibutyric acid (GABA) receptors are activated, and calcium channels which prevent the release of glutamate are inhibited (Wenker, 1999). Desflurane has proven to be neuroprotective in a variety of in vitro and in vivo models of ischemic brain injury (Haelewyn et al., 2003; Erdem et al., 2005; Wang et al., 2007; Matchett et al., 2009; McAuliff et al., 2009; Yu et al., 2010).
Mainly volatile anesthetics induce neuroapoptosis in several major brain regions during the synaptogenesis (Jevtovic-Todorovic et al., 2003; Bambrink et al., 2010; Briner et al., 2010; Culley et al., 2011; Cao et al., 2014). In a recently performed systematic review, Liu et al. (2014) confirmed a preponderant toxic but also an isolated protective effect of inhaled anesthetics on the developing brain of rodents, piglets and primates. In addition a neurotoxic effect of isoflurane and sevoflurane on hippocampal slice cultures is described (Wise-Faberowski et al., 2005; Brosnan and Bickler, 2013; Wise-Faberowski and Osorio-Lujan, 2013). Here we aimed to reveal the possible neuroprotective or deleterious effects of desflurane in an in vitro model of traumatic brain injury and to further assess how desflurane acts on organotypic hippocampal slices without TBI.
MATERIALS AND METHODS
Ethics statement
All experiments in this study were approved by the animal protection representative at the Institute of Animal Research at RWTH Aachen University Hospital, Germany according to the German animal protection law §4 Section 3.
Hippocampal slice culture
The organotypic hippocampal slice cultures were prepared from brains of 5–7 day old C57/BL6 mouse pups (Charles River Laboratories, Sulzfeld, Germany) using a well-established technique (Stoppini et al., 1991; Loetscher et al., 2009; Rossaint et al., 2009; Schoeler et al., 2012). After extracting the brains quickly out of the skull, the brains were transferred to an ice-cold preparation medium, comprising Grey's balanced salt solution (Sigma Aldrich, Steinheim, Germany), 5 mg/mL D-(+)-glucose (Roth, Karlsruhe, Germany) and 0.1% antibiotic/antimycotic solution (Penicillin G 1,000 U/mL, streptomycin sulphate 10 mg/mL and amphotericin B 25 μg/mL). The hippocampi were prepared under stereomicroscopic vision and cut into 400 μm thick slices using a McIllwain tissue chopper. The slices were gently separated into preparation medium and transferred to the semi-permeable membrane of MilliCell tissue culture inserts (MilliCell-CM, Millipore Corporation, Billerica, MA, USA) which were inserted into 35 mm tissue culture plates (Sarstedt, Newton, MA, USA) and 1 mL of growth medium (50% Eagle minimal essential medium with Earle's salts, 25% Hank's balances salt solution (Sigma-Aldrich, Steinheim, Germany), 25% heat inactivated horse serum, 2 mM L-glutamine, 5 mg/dL D-glucose, 1% antibiotic/antimycotic solution and 50 mM HEPES (hydroxyethyl-piperazine-ethanesulfonic acid) buffer solution, titrated to pH 7.2) infused underneath the membrane. The prepared slices were incubated for 14 days at a humidified atmosphere of 37°C, 95% air and 5% CO2. The growth medium was substituted 24 hours after preparation and on every third day subsequently.
TBI
After 14 days of incubation the growth medium was changed to the experimental medium supplemented with 4.5 μM propodium iodide (PI). The only difference between the growth medium and the experimental medium is the exchange of horse serum for the equal portion of Eagle's minimal essential medium. Afterwards the slices were incubated for 30 minutes at a humidified atmosphere of 37°C, 95% air and 5% CO2 again, before taking a baseline fluorescence image and dividing the slices into groups (TBI positive and TBI negative group). The CA1 region of the hippocampus was identified microscopically and subsequently traumatized with a previously described apparatus (Adamchik et al., 2000; Loetscher et al., 2009; Rossaint et al., 2009). This apparatus allows hitting the CA1 region with a metal stylus under stereomicropscopic control, by deactivating the power of an electromagnet, which retains this stylus at a height of 7 mm. This height and the weight of the stylus correlates to a force of 5.26 μJ. Thereafter, the experimental medium was exchanged for a new experimental medium supplemented with 4.5 μM propodium iodide and the slices were incubated in a pressure chamber for 2, 24 and 72 hours in different atmospheres. The pressure chambers were filled with a vapor of desflurane (D-Vapor, Dräger Medical GmbH, Lübeck, Germany) via an anesthetic machine (Sulla 808V, Dräger Medical GmbH, Lübeck, Germany) at an airflow of 8 L/min for 8 minutes. The carrier gas for desflurane (5% CO2, 21%O2, 74% N2 containing 2 vol%, 4 vol% or 6 vol% desflurane) was mixed by Linde Gas Therapeutics, Unterschleissheim, Germany. A control group (TBI positive) without desflurane was also incubated. The chamber atmosphere was analyzed with an anesthesia monitor system (Datex-Ohmeda AS 3 anesthesia monitor, GE Healthcare, Solingen, Germany) to control the desflurane concentration.
Propodium iodide staining
For staining the slices were stained with propodium iodide (a fluorescence coloring agent which connects to DNA if the cell membrane is disintegrated). Inside the cell propodium iodide becomes highly fluorescent with a main emission spectrum in the red region (Macklis and Madison, 1990). To capture the fluorescence a fluorescence microscope (Zeiss Axioplan, Carl Zeiss MicroImaging GmbH, Jena, Germany) equipped with a rhodamine filter, a low power 4x objective (Zeiss Achroplan 4x/0.10, Carl Zeiss MicroImaging GmbH) and a digital camera with corresponding software (SPOT Pursuit 4 MP Slider, Diagnostic Instruments Inc., Sterling Heights, MI, USA; MetaVue Molecular Devices, Sunnyvale, CA, USA) were used. The exposure time was calculated for each imaging session using a standard fluorescence slice (Fluor-Ref, Omega Optical, Brattleboro, VT, USA) to eliminate any influence of the mercury lamp's fluctuating intensity.
Assessment of cell injury
The fluorescence images were digitalized at 8 bits allowing the distinction between a spectrum of 256 (0–255) gray scale levels. The severity of cells is manifested by a high propodium iodide uptake and a high fluorescence value. Aided by analyzing software (Image J, NIH, USA, http://rsb.info.nih.gov), a histogram of the red pixel value of each slice was created to show the absolute number of pixel with the same grey scale value. A threshold of 100 was set to eliminate the background fluorescence and the pixel value beyond was summarized (Loetscher et al., 2009; Rossaint et al., 2009; Schoeler et al., 2012). Slices with a significant cell death based on the preparation were excluded.
Statistical analysis
Each amount of the pixel values was concluded for each experimental group. The mean ± SEM (standard error of the mean) of the pixel values were calculated using SPSS20 (IBM SPSS Statistics, IBM Cooperation, Somers, NY, USA). The TBI positive group which was incubated without desflurane was set as a reference value. Statistical analyses were performed using an analysis of variance (one-way ANOVA). P-values < 0.05 were considered significant.
RESULTS
A 2-hour incubation with different concentrations of desflurane in TBI organic hippocampal slice cultures resulted in no significant reduction in trauma intensity at concentrations of 6% desflurane (P = 0.384), 4% desflurane (P = 0.273) and 2% desflurane (P = 0.879) (Figure 1). However in the no trauma group a concentration of 2% desflurane (P = 0.033) and 4% desflurane (P = 0.004) could reduce the trauma intensity significantly in comparison to the no desflurane and trauma group (Figure 1).
Figure 1.
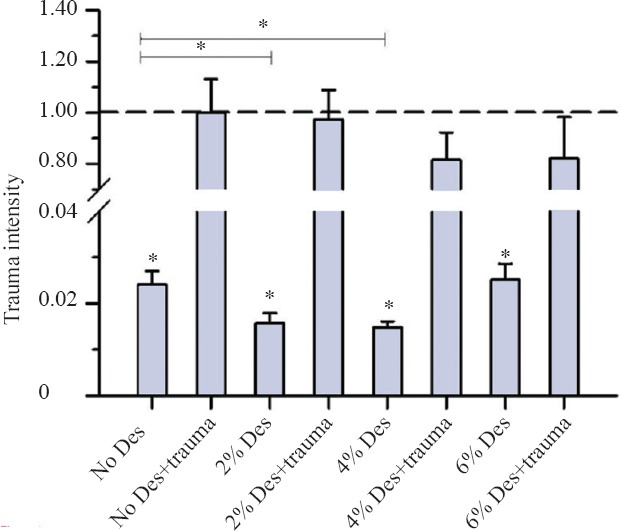
Trauma intensity of TBI organotypic hippocampal slice cultures incubated with various concentrations of desflurane for 2 hours.
Note: After 2-hour incubation, cell death was stained as shown on fluorescence images. Trauma intensity of the different groups is shown in relation to trauma intensity of mechanical traumatized slices (the trauma no desflurane group was set as 1 or 100%) after an incubation with 5% CO2, 21% O2,74% N2 for 2 hours. Significant differences to the trauma no desflurane group are marked with *. There was no significant reduction in trauma intensity at the groups with mechanical trauma (P > 0.05). In the group without TBI, a concentration of 2% desflurane (*P = 0.033) and 4% desflurane (*P =0.004) reduced the trauma intensity significantly in comparison to the group without desflurane and trauma. For each group an average of 80 with a minimum of 56 slices were analyzed.
After 24 hours of incubation with desflurane, an increase of trauma intensity in all groups was observed (Figure 2). In the group without TBI, the increase of trauma intensity rose from 2% deflurane (P = 0.000) to 6% desflurane (P = 0.000). In the trauma group, no significant difference was seen between 2% desflurane and the no desflurane and trauma group (P = 0.837); further an increase of trauma intensity was observed at concentrations of 4% desflurane (P = 0.006) and 6% desflurane (P = 0.000). There was no significant difference between the trauma group and group without trauma at concentration of 6% desflurane (P = 0.385).
Figure 2.
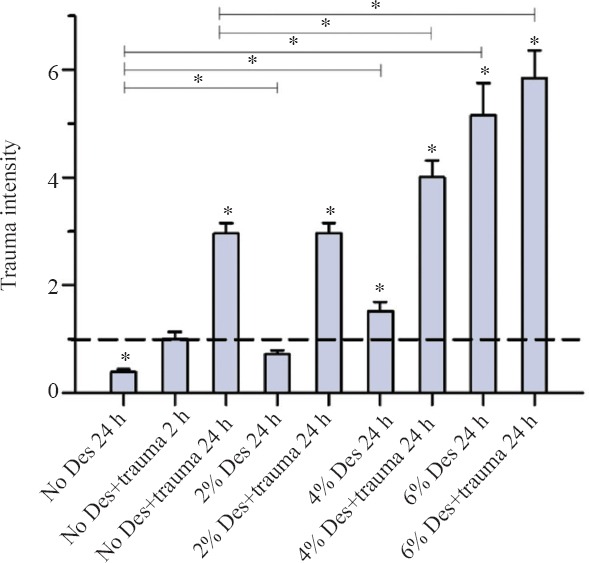
Trauma intensity of TBI organotypic hippocampal slice cultures incubated with various concentrations of desflurane for 24 hours.
Note: After 24-hour incubation, cell death was stained as shown fluorescence images. Trauma intensity of different groups is shown in relation to trauma intensity of traumatized slices (the trauma no desflurane group after 2 hours of incubation was set as 1) after an incubation with 5% CO2, 21% O2,74% N2 for 2 hours. Significant differences compared with the positive control group after two hours of incubation (Trauma without desflurane) are marked with *. Important significant differences between the groups after 24 hour incubation are also marked with * on the top of the figure. After 24 hours of incubation the trauma intensity tripled as compared to the trauma intensity after 2 hours of incubation. In the group without TBI the increase of trauma intensity rises also from 2% desflurane (*P = 0.000) to 6% desflurane (*P = 0.000). For the trauma group a significant increase of trauma intensity was observed at 4% desflurane (*P = 0.006) and 6% desflurane (*P = 0.000). For each group an average of 73 with a minimum of 62 slices were analyzed.
After 72 hours of incubation, the trauma intensity in the trauma and no trauma groups increased continuously especially in the trauma groups to more than the double in comparison to the 24 hours of incubation group (Figure 3).
Figure 3.
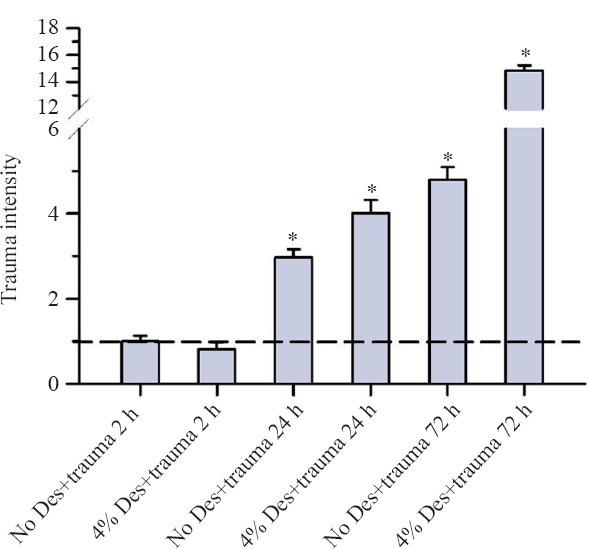
Trauma intensity of TBI organotypic hippocampal slice cultures incubated with concentration of 4% desflurane for 2, 24 and 72 hours.
Note: In summary on this figure slices were exposed to a concentration of 4% desflurane and to 5% CO2, 21% O2, 74% N2 (trauma no desflurane) for 2–72 hours. Trauma intensity of the different groups is shown in relation to trauma intensity of traumatized slices after an incubation without desflurane for 2 hours. Significant differences compared with the trauma no desflurane after two hours of incubation are marked with *. After 2 hours of incubation, there was no significant difference between the trauma no desflurane group and the trauma group with 4% desflurane (P = 0.273). After 24 hours, an increase to the double trauma intensity at the trauma group without desflurane was observed. Incubating with 4% desflurane after trauma led to an increase of trauma intensity to the four-fold. Finally after 72 hours the trauma intensity represents without desflurane and with 4% desflurane in the trauma group. For each group an average of 94 with a minimum of 73 slices were analyzed.
DISCUSSION
We observed no significant change in trauma intensity in TBI organic hippocampal slice cultures after 2 hours of desflurane exposure. After 24 hours the trauma intensity increased up to the fourfold which further increased after 72 hours. Therefore, a general neuroprotective attribute of desflurane in an in vitro model of traumatic brain injury was not observed. After 2 hours of incubation with 2% and 4% desflurane in slices not mechanically damaged a significant reduction of trauma intensity could be observed. It suggests that the absolute value of the trauma intensity is low in these groups, so that the result has to be analyzed critically.
Desflurane has also shown general neuroprotective properties in a variety of in vitro and in vivo models of ischemic brain injury (Haelewyn et al., 2003; Erdem et al., 2005; Wang et al., 2007; Matchett et al., 2009; McAuliffe et al., 2009; Yu et al., 2010). Desflurane preconditioning remarkably reduces the infarct volumes in an in vivo model of middle cerebral artery occlusion of adult rats (Haelewyn et al., 2003). Delayed preconditioning of 3 hours with desflurane, isoflurane and sevoflurane provides neuroprotection in a model of neonatal hypoxia-ischemia in 9-day-old mice. This neuroprotection was assessed by a battery of behavioral tests after a carotid ligation with a resulting hypoxia of 60 minutes. An improved performance in striatal dependent functions was noted in the preconditioned test group. However, performance on the spatial memory-dependent phases which is attributed to the hippocampus function was not improved. Yet a histological evaluation of cell loss of the striatum, the dorsal hippocampus and the ventral hippocampus did not show any significant effect of preconditioning with volatile anesthetics (McAuliffe et al., 2009).
Likewise neuroprotective properties of volatile anesthetics could be observed in an in vitro model with cerebellar slice cultures of 2–3-month-old rats, which were preconditioned for 15 minutes with desflurane, isoflurane, sevoflurane or halothane. 15 minutes after preconditioning this slices where subjected to oxygen glucose deprivation for 10 minutes. A protective character of this preconditioning could be attested by staining the cell damage using spectrophotometric measurement of formazan produced by 2,3,5-triphenyltetrazolium 5 hours after the oxygen glucose deprivation (Wang et al., 2007). Another in vitro model could demonstrate a neuroprotective effect of sevoflurane in a neonatal asphyxia model. Neuronal glia cells were cultured from cerebral neocortices of 1–2-day-old pups and exposed to oxygen-glucose deprivation for 75 minutes. 16 hours thereafter the lactat dehydrogenase (LDH) was analyzed using standardized colorimetric enzyme kit. Preconditioning with sevoflurane at concentrations of 2.7% and 3.3% reduced the LDH release, whereas at concentrations under 2% no effect was seen. A benefit of preconditioning with volatile anesthetics in pure neuronal cultures of 16 day old embryonic was only observed in a combination of sevolfurane and xenon (Luo et al., 2008).
In our model we could notice that desflurane exposure lasting more than two hours in traumatic brain injury model is neurotoxic in the developing brain during the phase of synapthogenesis. Current clinical and preclinical evidence suspects that common anesthetic agents cause an impairment of the developing brain (Jevtovic-Todorovic et al., 2003, 2013; Teng et al., 2005; Satomoto et al., 2009; Briner et al., 2010; Head et al., 2011; Istaphanous et al., 2011; Kodama et al., 2011; Stratmann et al., 2011; Zhang et al., 2012; Sanders et al., 2013). Wise-Faberowski et al. (2005) could confirm a neuronal degeneration of organic hippocampal slice cultures induced by isoflurane which is dependent on the age of the slices and the duration of incubation. This is similar to the effect of desflurane in our research.
The inhibition of N-methyl-D-aspartate (NMDA) receptors and the activation of the γ-aminobutyric-acid (GABA) receptors appear to be responsible for this neurotoxicity (Istaphanous et al., 2011). Moreover halogenated ethers impart their effects on both receptors with a variation in their effects on the NMDA-receptors (Kodama et al., 2011). A combination of an impact on both receptors might induce more neurotoxicity than an activation of only a single one (Fredriksson et al., 2007).
The exact mechanism by which common anesthetics mediate an injuryis not yet clear. One hypothesis is that the anesthetics can activate the intrinsic and extrinsic apoptotic pathway by suppressing neurotrophic synaptic signaling (Sanders et al., 2013). This hypothesis includes the activation of the intrinsic pathway by reducing the release of tissue plasminogen activator (tPA) into the synaptic cleft due to suppressing neuronal activity of the developing neurons by volatile anesthetics. Tissue plasminogen activator converts plasminogen to plasmin which is released from presynaptic vesicles at the time of the depolarization. On his part plasmin converts the neurothrophic factor (BDNF) from its precursor protein proBDNF to the mature BDNF (mBDNF). Mature BDNF enhances neuronal surviving whereas proBDNF induces apoptosis by stimulating the p75 neurotropin receptor (p75NTR). This stimulation of the p75NTR leads to an activation of the Ras homolog gene family, resulting in the depolymerization of the actin cytoskeleton (Teng et al., 2005; Head et al., 2011; Stratmann, 2011). Concurrently the extrinsic pathway is typically activated by external stimuli such as cytokine tumor necrosis factor (TNF-α) which is triggered by pro-inflammatory effects of sedative agents in the young (Sanders et al., 2013).
Furthermore significant changes in the dendritic arbor during the synaptogenesis could be observed in rats after their exposure to volatile anesthetics. These drugs increased dendritic spine density on dendritic shafts during the peak of the synaptogenetic period (Briner et al., 2010). This impact and the change in the dendritic arbor neonatale exposure with common anesthetics may cause learning deficits, deficits in fear conditioning and abnormal social behaviors akin to autism in adulthood (Satomoto et al., 2009). The hippocampus plays an important role in learning and memory processes due to its property of long-term potentiation. It may be particularly harmed by common anesthetics during the synaptogenesis (Stratmann et al., 2011).
The clinical impact of our research is limited as anesthesia duration over 2 hours in the very young is exceptional. Furthermore, the assessment of the cell damage in context to its effect on the living organism is not reproduced in our model. In addition, it is unclear, if the applied cell destruction in the CA1 region of the hippocampus has any effect on the cognition and on the behavior of creatures. We assessed the effect of desflurane in newborn hippocampal slices. The effect in full grown neurons therefore is still to be evaluated. Equally, possible impairment of other brain regions through desflurane is not addressed here.
Adding desflurane to the fixed mixed carrier gas leads to a proportionated decrease of the oxygen concentration. Using 6 % desflurane the oxygen concentration decreases approximately to 19 percent. It remains vague if this minimal reduction of the oxygen concentration leads to an impairment of the organotypic hippocampal slices.
In conclusion, we could not observe a general protective effect of desflurane in an organotypic model of traumatic brain injury. On the contrary, incubation with desflurane for 24 and 72 hours led to a dose- and time-dependent increase in trauma. The exact pathway of how desflurane affects the organotypic slice cultures remains unclear. A potential neuroprotection could be observed in the group without trauma after 2 hours of incubation with 2% and 4% desflurane. This possible neuroprotection has to be evaluated critically due to the low absolute value of the trauma intensity.
Acknowledgments
We would like to thank Rosmarie Blaumeiser-Debarry for her excellent technical assistance.
Footnotes
Funding: The project was partly supported by a grant from Baxter Healthcare Corporation.
Conflicts of interest
None declared.
REFERENCES
- Adamchik Y, Frantseva MV, Weisspapir M, Carlen PL, Perez Velazquez JL. Methods to induce primary and secondary traumatic damage in organotypic hippocampal slice cultures. Brain Res Brain Res Protoc. 2000;5:153–158. doi: 10.1016/s1385-299x(00)00007-6. [DOI] [PubMed] [Google Scholar]
- Bambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang Z, Dissens GA, Creeley CE, Olney JW. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–841. doi: 10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briner A, De Roo M, Dayer A, Muller D, Habre W, Vutskits L. Volatile anesthetics rapidly increase dendritic spine density in the rat medial prefrontal cortex during synaptogenesis. Anesthesiology. 2010;112:546–556. doi: 10.1097/ALN.0b013e3181cd7942. [DOI] [PubMed] [Google Scholar]
- Brosnan H, Bickler PE. Xenon neurotoxicity in rat hippocampal slice cultures is similar to isoflurane and sevoflurane. Anesthesiology. 2013;119:335–344. doi: 10.1097/ALN.0b013e31829417f0. [DOI] [PubMed] [Google Scholar]
- Cao L, Bie X, Huo S, Du J, Liu L, Song W. Effects of diazepam on glutamatergic synaptic transmission in the hippocampal CA1 area of rats with traumatic brain injury. Neural Regen Res. 2014;9:1897–1901. doi: 10.4103/1673-5374.145357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culley DJ, Boyd JD, Palanisamy A, Xie Z, Kojima K, Vacanti CA, Tanzi RE, Crosby G. Isoflurane decreases self-renewal capacity of rat cultured neural stem cells. Anesthesiology. 2011;115:754–763. doi: 10.1097/ALN.0b013e318223b78b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdem AF, Cesur M, Alici HA, Erdogan F, Dogan N, Kursad H, Yuksek MS. Effects of sevoflurane and desflurane in CA1 after incomplete cerebral ischemia in rats. Saudi Med J. 2005;26:1424–1428. [PubMed] [Google Scholar]
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. http://www.cdc.gov/traumaticbraininjury/pdf/blue_book.pdf .
- Feng Z, Zhong YJ, Wang L, Wei TQ. Resuscitation therapy for traumatic brain injury-induced coma in rats: mechanisms of median nerve electrical stimulation. Neural Regen Res. 2015;10:594–598. doi: 10.4103/1673-5374.155433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson A, Pontén E, Gordh T, Eriksson P. Neonatal exposure to a combination of N-methyl-D-aspartate and gamma-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology. 2007;107:427–436. doi: 10.1097/01.anes.0000278892.62305.9c. [DOI] [PubMed] [Google Scholar]
- Frost RB, Farrer TJ, Primosch M, Hedges DW. Prevalence of traumatic brain injury in the general adult population: a meta-analysis. Neuroepidemiology. 2013;40:154–159. doi: 10.1159/000343275. [DOI] [PubMed] [Google Scholar]
- Haelewyn B, Yvon A, Hanouz JL, MacKenzie ET, Ducouret P, Gérard JL, Roussel S. Desflurane affords greater protection than halothane against focal cerebral ischaemia in the rat. Br J Anaesth. 2003;91:390–396. doi: 10.1093/bja/aeg186. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2011;110:813–825. doi: 10.1097/ALN.0b013e31819b602b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Istaphanous GK, Howard J, Nan X, Hughes EA, McCann JC, McAuliffe JJ, Danzer SC, Loepke AW. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology. 2011;114:578–587. doi: 10.1097/ALN.0b013e3182084a70. [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–882. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Absalom AR, Blomgren K, Brambrink A, Crosby G, Culley DJ, Fiskum G, Giffard RG, Herold KF, Loepke AW, Ma D, Orser BA, Planel E, Slikker W, Jr, Soriano SG, Stratmann G, Vutskits L, Xie Z, Hemmings HC., Jr Anaesthetic neurotoxicity and neuroplasticity: an expert group report and statement based on the BJA Salzburg Seminar. Br J Anaesth. 2013;111:143–151. doi: 10.1093/bja/aet177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama M, Satoh Y, Otsubo Y, Araki Y, Yonamine R, Masui K, Kazama T. Neonatal desflurane exposure induces more robust neuroapoptosis than do isoflurane and sevoflurane and impairs working memory. Anesthesiology. 2011;11:979–991. doi: 10.1097/ALN.0b013e318234228b. [DOI] [PubMed] [Google Scholar]
- Liu J, Rossaint R, Sanders RD, Coburn M. Toxic and protective effects of inhaled anaesthetics on the developing animal brain: Systematic review and update of recent experimental work. Eur J Anaesthesiol. 2014;31:669–677. doi: 10.1097/EJA.0000000000000073. [DOI] [PubMed] [Google Scholar]
- Loetscher PD, Rossaint J, Rossaint R, Weis J, Fries M, Fahlenkamp A, Ryang YM, Grottke O, Coburn M. Argon: neuroprotection in in vitro models of cerebral ischemia and traumatic brain injury. Crit Care. 2009;13:R206. doi: 10.1186/cc8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Ma D, Ieong E, Sanders RD, Yu B, Hossain M, Maze M. Xenon and sevoflurane protect against brain injury in a neonatal asphyxia model. Anesthesiology. 2008;109:782–789. doi: 10.1097/ALN.0b013e3181895f88. [DOI] [PubMed] [Google Scholar]
- Matchett GA, Allard MW, Martin RD, Zhang JH. Neuroprotective effect of volatile anesthetic agents: molecular mechanisms. Neurol Res. 2009;31:128–134. doi: 10.1179/174313209X393546. [DOI] [PubMed] [Google Scholar]
- McAllister TW. Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci. 2011;13:287–300. doi: 10.31887/DCNS.2011.13.2/tmcallister. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuliffe JJ, Loepke AW, Miles L, Joseph B, Hughes E, Vorhees CV. Desflurane, isoflurane, and sevoflurane provide limited neuroprotection against neonatal hypoxia-ischemia in a delayed preconditioning paradigm. Anesthesiology. 2009;111:533–546. doi: 10.1097/ALN.0b013e3181b060d3. [DOI] [PubMed] [Google Scholar]
- Noraberg J, Poulsen FR, Blaabjerg M, Kristensen BW, Bonde C, Montero M, Meyer M, Gramsbergen JB, Zimmer J. Organotypic hippocampal slice cultures for studies of brain damage, neuroprotection and neurorepair. Curr Drug Targets CNS Neurol Disord. 2005;4:435–452. doi: 10.2174/1568007054546108. [DOI] [PubMed] [Google Scholar]
- Rossaint J, Rossaint R, Weis J, Fries M, Rex S, Coburn M. Propofol: neuroprotection in an in vitro model of traumatic brain injury. Crit Care. 2009;13:R61. doi: 10.1186/cc7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RD, Hassell J, Davidson AJ, Robertson NJ, Ma D. Impact of anaesthetics and surgery on neurodevelopment: an update. Br J Anaesth. 2013;110(Suppl 1):i53–72. doi: 10.1093/bja/aet054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satomoto M, Satoh Y, Terui K, Miyao H, Takishima K, Ito M, Imaki J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–637. doi: 10.1097/ALN.0b013e3181974fa2. [DOI] [PubMed] [Google Scholar]
- Schoeler M, Loetscher PD, Rossaint R, Fahlenkamp AV, Eberhardt G, Rex S, Weis J, Coburn M. Dexmedetomidine is neuroprotective in an in vitro model for traumatic brain injury. BMC Neurol. 2012;12:20. doi: 10.1186/1471-2377-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selassie AW, Zaloshnja E, Langlois JA, Miller T, Jones P, Steiner C. Incidence of long-term disability following traumatic brain injury hospitalization, United States. J Head Trauma Rehabil. 2008;23:123–131. doi: 10.1097/01.HTR.0000314531.30401.39. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Stratmann G. Review article: Neurotoxicity of anesthetic drugs in the developing brain. Anesth Analg. 2011;113:1170–1179. doi: 10.1213/ANE.0b013e318232066c. [DOI] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Jin Lee J, Zou Z. Pretreatment with volatile anesthetics, but not with the nonimmobilizer 1,2-dichlorohexafluorocyclobutane, reduced cell injury in rat cerebellar slices after an in vitro simulated ischemia. Brain Res. 2007;1152:201–208. doi: 10.1016/j.brainres.2007.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenker O. Review of currently used inhalation anesthetics: Part I. Int J Anesthesiol. 3 http://ispub.com/IJA/3/2/7433 . [Google Scholar]
- Wise-Faberowski L, Osorio-Lujan S. Acute and sustained isoflurane neuroprotection: the effect of culture age and duration of oxygen and glucose deprivation. Brain Inj. 2013;27:444–453. doi: 10.3109/02699052.2012.750755. [DOI] [PubMed] [Google Scholar]
- Wise-Faberowski L, Zhang H, Ing R, Pearlstein RD, Warner DS. Isoflurane-induced neuronal degeneration: an evaluation in organotypic hippocampal slice cultures. Anesth Analg. 2005;101:651–657. doi: 10.1213/01.ane.0000167382.79889.7c. [DOI] [PubMed] [Google Scholar]
- Yu Q, Wang H, Chen J, Gao Y, Liang W. Neuroprotections and mechanisms of inhalational anesthetics against brain ischemia. Front Biosci. 2010;2:1275–1298. doi: 10.2741/e189. [DOI] [PubMed] [Google Scholar]
- Zhang B, Tian M, Zhen Y, Yue Y, Sherman J, Zheng H, Li S, Tanzi RE, Marcantonio ER, Xie Z. The effects of isoflurane and desflurane on cognitive function in humans. Anesth Analg. 2012;114:410–415. doi: 10.1213/ANE.0b013e31823b2602. [DOI] [PMC free article] [PubMed] [Google Scholar]