

Abstract
One of the greatest challenges with lithium-oxygen batteries involves identifying catalysts that facilitate the growth and evolution of cathode species on an oxygen electrode. Heterogeneous solid catalysts cannot adequately address the problematic overpotentials when the surfaces become passivated. However, there exists a class of biomolecules which have been designed by nature to guide complex solution-based oxygen chemistries. Here, we show that the heme molecule, a common porphyrin cofactor in blood, can function as a soluble redox catalyst and oxygen shuttle for efficient oxygen evolution in non-aqueous Li-O2 batteries. The heme's oxygen binding capability facilitates battery recharge by accepting and releasing dissociated oxygen species while benefiting charge transfer with the cathode. We reveal the chemical change of heme redox molecules where synergy exists with the electrolyte species. This study brings focus to the rational design of solution-based catalysts and suggests a sustainable cross-link between biomolecules and advanced energy storage.
 Natural biomolecules including heme-like porphyrins can be applied as sustainable chemical catalysts in lithium-oxygen batteries. Here, the authors show that the heme molecule can function as a soluble redox catalyst and oxygen shuttle for efficient oxygen evolution in non-aqueous lithium-oxygen cells.
Natural biomolecules including heme-like porphyrins can be applied as sustainable chemical catalysts in lithium-oxygen batteries. Here, the authors show that the heme molecule can function as a soluble redox catalyst and oxygen shuttle for efficient oxygen evolution in non-aqueous lithium-oxygen cells.
The goal of developing next-generation energy storage systems ‘beyond lithium-ion' will be achieved by employing novel, efficient and sustainable battery chemistries1,2. Lithium-oxygen (Li-O2) batteries utilizing lightweight and abundant reactants have drawn significant interest due to their high-energy density, two to three times greater than lithium-ion cells3,4,5,6. The most common, non-aqueous variety of Li-O2 battery operates via surface reactions, which involve the formation and evolution of solid lithium oxides (2Li++O2+2e−↔Li2O2, E°=2.96 V versus Li/Li+)7. However, the evolution reaction, which dictates cell reversibility, suffers from large (∼1 V) overpotentials and impeded kinetics due to poor electron conduction in the solid products8. Efforts have been made to develop various solid-state catalysts (that is, noble metals, metal oxides) decorated on oxygen electrodes, yet catalytic sites are easily deactivated by the precipitation of solid products and consequently electron transfer at the solid/solid interface is too slow9,10,11,12,13. In this regard, new catalyst chemistries should be considered to alleviate electrode deactivation and offer continuous catalytic function14,15. Incorporating catalytic molecules directly into electrolytes can improve the accessibility of these species to interfacial products and the electrode surface16,17. Moreover, redox molecules can mediate the efficient transport of electrons between the insulating products and the oxygen electrode during oxygen evolution, reducing ohmic losses18,19,20. Potential redox molecules should offer: (i) good solubility and facile diffusion in Li+ containing electrolytes, (ii) fast electron transfer with the oxygen electrode, (iii) oxygen-complexing ability to increase electrolyte oxygen solubility, and (iv) natural abundance with a low environmental impact. A few redox molecules (for example, Li iodide (LiI), tetrathiafulvalene (TTF), iron phthalocyanine (FePc), (2,2,6,6-tetramethylpiperdin-1-yl)oxyl (TEMPO), and tris[4-(diethylamino)phenyl]amine (TDPA)) have been investigated for Li-O2 systems and have shown promise for this approach16,18,21,22,23. However, most of these redox molecules only operate via simple electron transfer reactions and do not have an established oxygen-complexing ability.
Intermittent attempts to introduce biomolecules into energy storage and conversion devices have demonstrated some success with flow batteries, fuel cells and solar cells24,25,26. Using biomolecules extracted from natural supplies can theoretically address the global demand for sustainable and ubiquitous chemical catalysts. Recyclable bio-wastes (for example, blood waste, dietary trash, natural enzymes), may be employed for the environmentally friendly and cost-effective production of catalysts27. The heme molecule, a porphyrin with an Fe ion centre and a cofactor in the blood protein hemoglobin, has been highlighted in diverse applications involving oxygen transport28,29,30. The heme metalloenzyme, acting as an oxygen shuttle and reservoir, exhibits auto-oxy/deoxygenation properties, responding to external environments where its Fe ion centre promotes chemical and electrochemical redox31,32,33,34. The Fe ion centre of the heme is bound to four pyrrole nitrogens, leaving an open axial coordination site that can bind dioxygen (Fig. 1a)35. The reversible redox characteristics (Fe3+/Fe2+ couple) facilitate catalysis by rapid electron transfer to/from Fe ions in heme36,37,38. While the redox activity of heme has been widely established in aqueous environments (∼1 V)34,37, the behaviour of heme molecules in non-aqueous lithium electrolytes remains poorly understood. Unlike the conventional oxygen-evolving reaction in water, which is a liquid to gas phase process, oxygen evolution in Li-O2 cells occurs via a solid (that is, LiO2, Li2O2) to gas phase transformation. This reaction may make particular use of intermediate heme redox processes at electrochemical interfaces.
Figure 1. Heme redox properties.

(a) Structure of heme molecule. (b) Schematic illustration of oxygen electrode charged in the heme-containing Li-O2 cell; CV curves in various electrolyte media: (c) 1 M LiClO4+TEGDME and (d) 1 M LiClO4+TEGDME+Heme after He and O2 purging at scanning rate of 5 mV s−1. CV curves in the electrolyte of (e) 1 M LiClO4+TEGDME, (f) 1 M LiClO4+TEGDME+Heme after O2 purging at different scanning rates. All CV curves were collected using glassy carbon electrodes in the voltage window between 2 and 4.5 V. The concentration of heme molecule is 2.3 mM.
Here, we present heme as a bifunctional redox biomolecule with charge transfer and oxygen-shuttling properties that improves the energy efficiency of Li-O2 battery systems. We elucidate the mechanism of heme redox during Li-O2 reactions by studying electron transfer, oxygen binding and the effect of salt anions on these processes. We find a synergistic effect between the heme molecule and LiClO4 that demonstrates the significance of selecting optimal material combinations. As illustrated in Fig. 1b, the electrochemical properties of the heme-assisted Li-O2 cell are evaluated to verify the effect on oxygen evolution and cell efficiency. The chemistry and reversibility of discharge products in the cell comprising the heme molecules are investigated by ex-situ characterizations. Biomolecules based on similar porphyrin structures can serve as a platform to further improve Li-O2 cell efficiency with soluble catalysts. The introduction of eco-friendly biomolecules hints at a simple and sustainable path to develop battery systems that merge biochemistry with advanced energy storage.
Results
Heme electrochemistry
To understand the catalytic function of heme for simple electron transfer, oxygen complexation, or a combination thereof, we investigated its electrochemical behaviour in a typical Li-O2 cell environment. We show cyclic voltammograms (CVs) to demonstrate the electrochemical properties of hemin (heme with a Cl− anion) dissolved in 1 M LiClO4 in tetraethylene glycol dimethyl ether (TEGDME) (Fig. 1c,d). No significant reaction features are seen in the CV curve for LiClO4+TEGDME under an inert (He purging) atmosphere (Fig. 1c). However, after O2 purging, broad peaks appear in both cathodic and anodic regions near 2.2 V (Ec,1) and 3.2 V (Ea,1), respectively (Supplementary Table 1). These peaks correspond to the formation and evolution of Li2O2 products, respectively8,39. With the addition of heme, a reduction feature appears even under He purging, indicating that this reaction at 2.94 V (Ec,3) primarily involves electron transfer (heme(Fe3+)+e−→heme(Fe2+)) when no O2 is available (Fig. 1d)40. The oxidation feature (Ea,2) with heme at 3.9 V is likely related to the cathodic reaction (heme(Fe2+)→e−+heme(Fe3+)). We also observe under He purging a peak at 4.2 V, which could relate to the irreversible oxidation of heme(Fe3+) to heme(Fe4+) in the absence of O2 (refs 41, 42). When under an O2 atmosphere, additional features are present at 2.5 V (Ec,2) and 2.2 V (Ec,1) for the cathodic regime (Fig. 1d). While the 2.2 V (Ec,1) peak is consistent with O2 reduction in the LiClO4+TEGDME electrolyte without heme, the feature at 2.5 V (Ec,2) with heme suggests the formation of an intermediate superoxide: O2− or heme(Fe2+)–O2. We show in the anodic region that features at 3.2 V (Ea,1) and 4 V (Ea,2) are present, implying the reverse of the oxygen reduction processes. This result indicates that the heme participates in evolving the superoxide intermediate and facilitating the oxidation of Li2O2 species.
To individually investigate the heme redox peaks and possible interaction between Li salt and heme molecules, tetraethylammonium perchlorate (TEAClO4) was added instead of LiClO4 to identify any changes to the electrochemical redox peaks in O2 purged solutions (Supplementary Fig. 1). Although the current signal is relatively small due to the low solubility of TEAClO4 in TEGDME, we observe no peaks similar to those attributed to the formation and evolution of lithium-oxide products (Ec,1,Ea,1) in TEAClO4+TEGDME or TEAClO4+TEGDME+heme. Nevertheless, redox peaks associated with heme electron transfer (Ec,2, Ec,3, Ea,2) do appear near their expected positions, consistent with CV curves for LiClO4+TEGDME+heme. We conclude that Li+ cations have no specific interaction with the heme molecule and the heme redox reactions occur independent of Li salt. To disentangle the electrochemical reactions further, we observe the scan rate effect on the electrochemical features (Fig. 1e,f). From the Randles–Sevcik equation, the peak current density is proportional to the square root of the scan rate (Ip=(2.69 × 105)n3/2ACD1/2v1/2, where n is the number of electrons transferred, A is the surface area of the electrode, C is the concentration of reactant, D is the diffusion coefficient of the reactant and v is the scan rate)43. We find that the peak current of the heme redox at∼3 V (cathodic) and∼4 V (anodic) is less affected by increases in the scan rate than the features related to lithium-oxide product formation and evolution (∼2.3 V (cathodic) and 3.2 V (anodic)). This finding suggests that the O2 redox processes are more kinetically limited than those of heme. With a heme concentration of 2.3 mM, significantly less than the concentrations of dissociated LiClO4 (1 M) or dissolved O2 in TEGDME (4.43 mM)44, it is reasonable to attribute the benefit of a small amount of heme to its efficacy as an intermediate in O2 redox processes.
Li-O2 battery performance
We evaluate the electrochemical functioning of full Li-O2 cells with the heme catalyst to assess its practical advantages (Fig. 2). To simplify and focus only on the function of the heme, a multi-walled carbon nanotube (MWCNT) oxygen electrode was used without loading of other catalysts (Supplementary Fig. 2)14. We present the first charge/discharge profiles of MWCNT electrodes cycled between 4.35 and 2.35 V at a current density of 100 mA g−1carbon in a LiClO4+TEGDME electrolyte with and without heme (Fig. 2a). While the cells without and with heme exhibit similar discharge capacities of 2,830 and 2,871 mAh g−1carbon, the charge capacity of the cell cycled with heme (2,665 mAh g−1carbon) was higher than that without (2,390 mAh g−1carbon), corresponding to improved coulombic efficiency (84→93%). In addition, a lower voltage profile was seen in the charging region. To further study the benefit to cell voltage offered by the heme molecule, galvanostatic intermittent titration (GITT) curves during the 1st charge cycle were acquired (Fig. 2b). The difference between constant current voltage (Ion) and open circuit voltage (Ioff) in this experiment reflects the overpotential (related to ohmic loss) at each point during charging. Interestingly, the heme-containing electrolyte shows a significantly lower overpotential (∼0.3 V) for charging, compared to the electrolyte without heme (∼0.8 V). While the open circuit voltage in the electrolyte without heme gradually increased, the open circuit voltage in the heme-containing electrolyte quickly reached 3.5 V and was stably maintained for 70 h. The lower charge voltage is related to facile evolution of discharge products promoted by charge transfer between insulating products, heme molecules and the electrode. The charge potential (∼3.5 V) of the Li-O2 cell with heme is lower than the CV peak potential (∼4 V) shown in Fig. 1d. The apparent difference of 0.5 V corresponds to an overpotential induced by a high scan rate with CV and by insulating discharge products on the flat glassy carbon disc. A similar voltage difference is evident between the cathodic peak potential in CVs (2.2 V) and the discharge plateau in charge–discharge profiles (2.7 V), which is consistent with the effects of overpotential.
Figure 2. Li-O2 cell performance in heme-containing electrolyte.

(a) Initial charge/discharge curves of the MWCNT electrode in 1 M LiClO4+TEGDME and 1 M LiClO4+TEGDME+Heme solutions in a voltage window between 4.3 and 2.35 V at a current density of 100 mA g−1carbon; (b) galvanostatic intermittent titration curves of the MWCNT electrode in 1 M LiClO4+TEGDME and 1 M LiClO4+TEGDME+Heme solutions, which were acquired with a current density of 50 mA g−1 for 24 min and a 120 min time interval during the 1st charging; (c) Voltage versus time curves of the MWCNT electrodes in 1 M LiClO4+TEGDME+Heme solution at various cycles under a specific capacity limit of 600 mAh g−1 between 4.5 and 2.3 V at a current density of 200 mA g−1carbon; (d) Electrochemical impedance spectroscopy (EIS) spectra of the MWCNT electrode in 1 M LiClO4+TEGDME and 1 M LiClO4+TEGDME+Heme solutions after the 1st, 5th, and 10th discharge cycle.
We show the cycle stability of the cells with and without heme between 4.5 and 2.3 V at a current density of 200 mA g−1carbon (Fig. 2c). Voltage versus time profiles show the terminal voltages after capacity-limited discharge and charge cycling; a metric related to cell efficiency over multiple cycles. While the terminal charge voltage for a cell without heme quickly reached the top of the voltage window of 4.5 V at the beginning of cycling, a cell with heme exhibited a stable terminal charge voltage of only 4.2 V. For the cell without heme, a voltage drop consistent with cell death was observed after only 150 h. On the other hand, the heme-containing cell maintained its voltage two times longer (∼300 h). These results demonstrate that the heme molecule effectively catalyzes oxygen evolution, improving the efficiency and cycle life of Li-O2 batteries.
The electrochemical resistance of a Li-O2 cell employing the heme catalyst was evaluated with electrochemical impedance spectroscopy after the 1st, 5th and 10th discharge cycles to discern the cause of decreased overpotentials (Fig. 2d). The 1st semicircle in the Nyquist diagram at high frequencies (ca. 1 MHz through 1 Hz) corresponds to the Li anode/electrolyte interface and the 2nd semicircle at low frequencies (ca. 1 Hz through 1 mHz) is associated with the discharge products and oxygen electrode interface12,45,46,47. While the low frequency resistance (1,854 Ω) of the oxygen electrode in the cell without heme increased with increasing cycle number, the oxygen electrode with heme exhibited a lower and relatively stable cell resistance (1,273 Ω) over 10 cycles. The increase of the 2nd semicircle for the cell without heme is likely related to the incomplete evolution and consequent accumulation of discharge products. Continuous and efficient oxygen evolution assisted by heme molecules prevents a substantial increase in cell resistance, consistent with the cycle performance data shown in Fig. 2c.
Electrochemical product characterization
Ex-situ characterizations were performed to confirm the chemistry of discharge products formed on the electrode surface and verify the reversibility of heme-containing cells. We present ex-situ X-ray photoelectron spectra (XPS) of electrode surfaces collected in the C 1s, O 1s, Fe 2p regions (Fig. 3a–c, respectively). C–H and COOH features related to pristine MWCNT and seen in pristine electrodes (Supplementary Fig. 3), were diminished after discharge and retrieved after charge, consistent with the formation and subsequent evolution of a layer of discharge products (Fig. 3a). Li2O2 products were detected on discharged electrodes and the related feature (531.4 eV) was indeed diminished after charging (Fig. 3b). Product chemistry was similarly examined by the ex-situ Raman spectroscopy of these electrodes (Fig. 3d). After discharge, both LiO2 and Li2O2 species were detected at 1,121 and 791 cm−1, respectively14,48,49,50,51, and these product species appear to be fully reversible. Features related to CO32− and C–O are apparent on several electrodes, but these likely result from carbon electrode degradation52, electrolyte decomposition53 or minor surface contamination by limited exposure to ambient air. In the O 1s XPS spectrum, strong peaks related to LiClO4 salt were found for all samples, suggesting that the electrolyte is integrated into the electrochemical products. The products on discharged electrodes show no XRD peaks verifying that the lithium-oxide species are not well crystallized (Supplementary Fig. 4). To investigate the possibility that the heme molecule could be incorporated into the discharge products, we show the Fe 2p XPS region of a discharged and charged electrode after 20 cycles (Fig. 3c). We observe no Fe peaks from either electrode, demonstrating that the heme molecule only facilitates the Li-O2 cell reaction as a soluble catalyst and does not form a component of the product as a result of heme decomposition and side reactions. Heme components were also not observed on discharged or charged electrodes using ex-situ Raman spectroscopy (Supplementary Fig. 5).
Figure 3. Ex-situ measurements on oxygen electrodes.

Ex-situ X-ray photoelectron spectra obtained from 1 st discharged, 1st charged, 20th discharged, and 20th charged electrodes collected in the (a) C 1s, (b) O 1s; (c) Ex-situ X-ray photoelectron spectra obtained from 20th discharged and 20th charged electrodes collected in the Fe 2p; (d) Ex-situ Raman spectra obtained from pristine, 1st discharged, 1st charged, 20th discharged, and 20th charged electrodes. (blue star: Li2O2, black star: TEGDME, black cross: LiClO4, red star: LiO2).
We show the surface morphologies of the MWCNT electrodes loaded on Ni-mesh at various charge–discharge states (1st and 20th cycles). (Supplementary Fig. 6) Cell tests were performed in 1 M LiClO4+TEGDME+Heme solutions to collect electrodes at different electrochemical states for ex-situ SEM analysis. When the cells were first discharged, discharge products were precipitated and covered the electrode (Supplementary Fig. 6a,b). When cells were subsequently charged, the products were decomposed and disappeared from the electrode (Supplementary Fig. 6c). After the 20th cycle, results similar to those of the 1st cycle were observed (Supplementary Fig. 6d,e). These results verify the excellent reversibility of the Li-O2 cell employing a heme biomolecular catalyst. From the magnified SEM images (Supplementary Fig. 6f), it can be seen that the products were fully covered on the electrode and the shape looks like a mixture of particulate and amorphous products.
Heme–oxygen interaction
We utilized ultraviolet–vis absorption to explore how the chemical states of the heme molecule change in the presence of oxygen (Fig. 4). Potassium oxide (KO2), which generates the superoxide ion (O2−) in solution, was introduced into various electrolyte media and the absorbance of the heme was monitored over time. No particular absorbance feature was observed for TEGDME and LiClO4+TEGDME solutions in the visible range, but a feature <300 nm is indicative of TEGDME (Supplementary Fig. 7). Superoxide is known to absorb at ∼253 nm and a modest increase in the absorbance at <300 nm after injection of KO2 indicates its presence54,55. When the heme molecule is introduced into the solvent, a strong peak near 400 nm (Soret band) and sub-peaks between 450 and 700 nm (Q-bands) were identified, corresponding to typical heme absorbance (Fig. 4; Supplementary Table 2)56. The Soret band indicates a strong electronic transition of the metalloporphyrin and the Q-band is associated with a weak transition in the porphyrin ring. For a heme-containing solution without LiClO4, a red shift of the Soret and Q-bands were observed after KO2 injection (Fig. 4a,b). A red shift of the Soret band of heme molecules has been documented previously as relating to the transition of the Fe ions from high- to a low-spin state due to coordination of species with the Fe d orbital (eg orbital)56,57. In the case of the red shift caused by adding KO2, this suggests a change in state from heme(Fe3+) to heme(Fe2+)–O2 by coordination with a soluble superoxide58. When LiClO4 is included in the solution with heme, the addition of KO2 to the solution demonstrates a similar red shift in the Soret band, but is stabilized quickly (Fig. 4c,d). In this case, we accept that two phenomena may occur, which prevents further coordination between the heme and the superoxide. First, the presence of Li+ may initiate precipitation of LiO2 or disproportionation to form Li2O2 so that superoxide is quickly removed from solution. Second, the ClO4− anion may competitively bind to the heme(Fe3+) site due to its negative charge and delocalized electron. There is evidence for anion coordination from the slight red shift (384→394 nm) in the Soret band with LiClO4 even before KO2 injection. To investigate the Li salt anion effect on coordination chemistry, experiments were repeated with LiPF6 salt (Supplementary Fig. 8). We find that the PF6− anion does not influence the Soret band and Q-band of heme when KO2 is added. Thus, we suggest that the superoxide cannot bind with heme in this electrolyte environment. There is some evidence to support the decomposition of PF6− by the superoxide59, which could prevent heme-superoxide coordination. In addition, we note the potential effect of a higher donor number for the ClO4− anion (8.4) compared with PF6− (2.5), which may help to stabilize superoxide species and form a superoxide complex with heme molecules60.
Figure 4. Chemical states of heme molecule by binding with oxygen species.

Ultraviolet–vis spectra of heme-containing electrolytes with increasing time after the injection of KO2 solution; (a) TEGDME+Heme (Soret band region), (b) TEGDME+Heme (Q-band region), (c) 1 M LiClO4+TEGDME+Heme (Soret band region), (d) 1 M LiClO4+TEGDME+Heme (Q-band region).
Heme structural change during reaction
To better elucidate chemical changes in the heme molecule at different electrochemical states during electroreduction and oxidation, we conducted in situ spectroelectrochemical measurements on a heme-containing electrolyte. We present CV curves (Fig. 5a,b) and the corresponding ultraviolet–vis spectra collected at different voltages (Fig. 5c,d). The spectroelectrochemical cells were designed with symmetric Au working and counter electrodes. While additional redox peaks related to Au oxidation/reduction near 3.67 V were found, the CV features (Fig. 5a) are similar to those in Fig. 1d, which was obtained with a glassy carbon working electrode, Pt counter electrode and Li/Li+ reference electrode. For an Ar purged cell, the heme Soret band exhibits a red shift during the cathodic scan and its position is maintained for the reverse scan up to 4.5 V versus Li/Li+ (Supplementary Fig. 9). As we previously observed in CV results, this shift corresponds to direct electron transfer to heme(Fe3+) without oxygen (heme(Fe3+)+e−→ heme(Fe2+)). In an O2 atmosphere, a red shift is similarly evident following reduction, indicating the formation of either heme(Fe2+) or heme(Fe2+)–O261. A blue shift at 4.2 V occurs during oxidation, highlighting the reversibility of the reduction process in the presence of O2 (Fig. 5c; Supplementary Fig. 10). The heme is therefore thought to bind O2− ions during the discharge reaction and release O2 and e− (to the electrode) during the charge reaction (Fig. 5e). On charging near 3.9 V, any existing O2− ions, potentially generated from oxide products (LiO2 or Li2O2) are bound with available heme(Fe3+), itself regenerated by heme(Fe2+)–O2 oxidation (Fig. 5e). The heme(Fe2+)–O2 complex is mobile in the electrolyte, unlike Li-O2 solid products, and thus may travel to and from the electrode surface. When most oxide products are decomposed, the heme(Fe2+) molecules are oxidized to heme(Fe3+) themselves at high anodic potential. Redox mediation by heme therefore involves a complexation mechanism, unlike conventional mediators, which only function by electron transfer.
Figure 5. In situ observation of heme molecule at different electrochemical states.

Spectroelectrochemical data of the different electrolytes (1 M LiClO4+TEGDME+Heme and 1 M LiPF6+TEGDME+Heme); CV curves of (a) 1 M LiClO4+TEGDME+Heme and (b) 1 M LiPF6+TEGDME+Heme and corresponding ultraviolet–vis spectrum of (c) 1 M LiClO4+TEGDME+Heme and (d) 1 M LiPF6+TEGDME+Heme. The spectroelectrochemical tests were performed with a commercial Au honeycomb working electrode and Au counter electrode in the voltage range of −1.5 to 1.5 V versus Pt pseudo-reference in an O2 atmosphere. Voltage values were converted to reflect a Li/Li+ reference; (e) Schematic illustration of chemical states of the heme molecule in Li-O2 cell.
Discussion
Li-O2 cell function with heme can be summarized by some representative steps for discharge and charge (Fig. 5e). Our findings suggest a reaction mechanism involving the coordination of heme molecules during cell operation:
Oxygen reduction reactions (discharge)
 |
 |
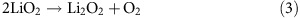 |
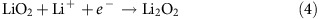 |
 |
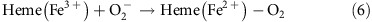 |
Oxygen evolution reactions (charge)
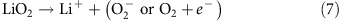 |
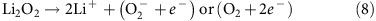 |
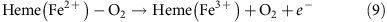 |
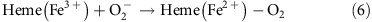 |
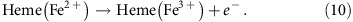 |
During the discharge reaction, superoxides (O2−) are generated by oxygen reduction (equation (1)) and they form lithium oxides such as LiO2 and Li2O2 (equations (2, 3 and 4)). Heme (Fe3+) may accept an electron from the electrode or bind with available O2−, forming heme(Fe2+) or the heme(Fe2+)–O2 complex (equations (5 and 6)). On charging, the heme(Fe2+)–O2 complex evolves oxygen and transfers an electron directly to the electrode (redox mediator), thereby reverting back to the heme(Fe3+) state (equation (9)). Heme(Fe3+) may accept any available superoxides generated from the oxidation of LiO2 or Li2O2 (equations (7 and 8)) and form the heme(Fe2+)–O2 complex once again (equation (6)). These charge processes may circulate until most of the available oxide species has been evolved. At high potential where Li-O2 products are fully evolved, the heme(Fe3+) remains in its original state (equation 10). In this scheme, we may consider the heme molecule both a ‘sink' and ‘transporter' for superoxide and electrons during charging. Its influence on the kinetics of LiO2 or Li2O2 evolution makes the designation of soluble catalyst appropriate.
Interestingly, the heme molecule exhibits this redox function in a LiClO4 containing electrolyte but not one with LiPF6. We carried out spectroelectrochemical measurements in a LiPF6 containing electrolyte with the same concentration of heme to verify the Li salt effects (Fig. 5b,d). Although a large reduction peak in the CV near 1.8 V corresponds to discharge product formation there are no oxidation peaks in the charge region. The ultraviolet–vis spectra indicate no significant shifts in the Soret band during reduction or oxidation with a LiPF6 electrolyte, demonstrating that the heme chemical structure is not changed even in an O2-containing environment (Fig. 5d). This result is consistent with the time-course ultraviolet–vis experiment as shown in Supplementary Fig. 8 and provides additional evidence that PF6− anions not only inhibit oxygen coordination but also prevent electron transfer with heme. Together, these results indicate that the heme undergoes several charge transfer processes including electron transfer, O2 coordination and anion inhibition. We highlight the need to select appropriate combinations of electrolyte and redox biomolecule to achieve electrochemical synergies.
In summary, we report the use of heme as an abundant and eco-friendly biomolecular catalyst to facilitate Li-O2 oxidation and improve battery function. The soluble heme molecule enables charge transfer between insulating Li-O2 discharge products and the electrode by engaging in electron transfer and coordinating with superoxide intermediates. The oxygen species bind with the Fe3+ heme centre and release oxygen while transferring charges to the electrode. The reversible chemical transitions of the heme under various electrochemical conditions were investigated by in situ spectroelectrochemical measurements. Interestingly, the heme molecules exhibit their function synergistically with ClO4− anions in the electrolyte and not with PF6−, indicating that pairing the Li salt and redox molecules is necessary to promote effective catalytic function. The Li-O2 cell with heme catalysis achieves a lower polarization and longer cycle life, compared with the control. We also verify the reversible formation and decomposition of LiO2 and Li2O2 on the oxygen electrode by ex-situ characterization and show that heme is not incorporated into solid products, but remains a mobile electrolyte species. Indeed, redox biomolecules with complexing catalytic functions present a new path to improve electrochemical storage efficiency using sustainable materials.
Methods
Materials and chemicals
High purity multi-walled carbon nanotubes (MWCNT, SWeNT SMW100) were donated by SouthWest Nanotechnologies. Hemin (from bovine, ≥90%), lithium perchlorate (LiClO4, battery grade, 99.99%), tetraethylene glycol dimethyl ether (TEGDME, anhydrous, ≤80 p.p.m. H2O), tetraethylammonium perchlorate (TEAClO4, for electrochemical analysis, ≥99%), poly(vinylidene fluoride) (PVDF, Mw ∼180,000) and 1-methyl-2-pyrrolidinone (NMP, anhydrous, 99.5%) were purchased from Sigma-Aldrich (St Louis, USA). Potassium superoxide (KO2, 96.5%) was purchased from Alfa Aesar (Ward Hill, USA). All chemicals were used without further purification.
Electrochemical characterization
Electrochemical characterization was performed in sealed purged Swagelok cells with gas inlet and outlet valves on the oxygen electrode side. The oxygen electrode was fabricated by casting a slurry, consisting of 90 wt% of oxygen electrode materials (MWCNT) and 10 wt% PVDF binder in NMP solvent, on a flattened Ni-mesh (diameter: 12.7 mm). The electrode was dried under vacuum at 75 °C before cell assembly in an argon-filled glove box. For electrolyte preparation, LiClO4 was dried for 24 h in a vacuum oven and then stored in the glove box for use. The electrolytes were prepared by mixing the LiClO4 (1 M), TEGDME and hemin (2.3 mM) in the Ar glove box and stirring overnight. Li-metal foil (11.1 mm diameter) was used as the counter electrode and the separator was a Whatman GF/A glass fibre (13 mm diameter). To eliminate the concentration effect of heme in electrolyte, the 2.3 mM concentration of heme we have used in this study is close to the solubility limit in TEGDME. A Bio-Logic VSP potentiostat with impedance function was used for galvanostatic cycling, galvanostatic intermittent titration and electrochemical impedance spectroscopy measurements of cells. All electrochemical experiments were performed at room temperature.
Cyclic voltammetry tests
For CV experiments, a three-electrode cell was used with 50 ml of electrolytes and a sealed Li/Li+ reference electrode. All electrolytes and fresh reference electrodes were prepared inside an isolated glove box. Due to low solubility of the TEAClO4 salt in TEGDME, the solution was centrifuged after stirring to remove residual undissolved salts. TEAClO4 containing electrolytes are considered to be saturated solutions. A Pt mesh and a 5 mm diameter glassy carbon electrode were used as the counter electrode and the working electrode, respectively. On removal from the glove box, solutions were immediately purged with dry He gas for 30 min to ensure an inert atmosphere. CV experiments were carried out under an He blanket in a voltage window between 2 and 4.5 V at different scan rates. Solutions were then purged with dry O2 for 30 min and CV experiments were repeated. The glassy carbon electrode was cleaned with ethanol and acetone after each CV test and dried completely.
Time-course ultraviolet–vis spectroscopy tests
The concentration of hemin molecules was diluted to 0.38 × 10−4 M in TEGDME and 1 M LiClO4/TEGDME for time-course ultraviolet–vis spectra. Overall, 5 mM KO2 was mixed with TEGDME for 24 h in an Ar-filled glove box to saturate the precursor solution. The actual concentration of KO2 in TEGDME is <5 mM due to reaction with residual water and incomplete dissolution. Electrolyte solutions (2.5 ml) were added into a quartz cuvette and the cuvettes were tightly sealed with a septum cap. A syringe injected 0.5 ml of the KO2 supernatant into the cuvettes and ultraviolet–vis spectra were measured for 2 h (Varian Cary 3E, Agilent Technologies, USA). The nominal ratio of KO2 to hemin in the mixture is ∼27:1, but due to the poor solubility of KO2 in TEGDME, the true ratio is significantly less.
Spectroelectrochemical measurement
The spectroelectrochemical measurement was carried out to monitor the chemical change of the heme molecule during discharge and charge. The electrolytes were transferred into a quartz cuvette with a commercial Au honeycomb electrode (Pine Research Instrumentation, NC, USA). The working electrode is perforated with a honeycomb pattern of holes that allow light to pass through the electrode. The active surface of the working electrode includes Au coating along the inner walls of the holes. As the light beam from the spectrometer passes through the holes, the beam grazes the walls of each hole. An Au counter electrode and Pt pseudo-reference electrode were used to construct the 3-electrode cell. The electrode potential was controlled with a potentiostat (Princeton Applied Research, VersaSTAT 4) and ultraviolet–vis spectra were collected at varying potentials on a Varian Cary 50. The electrolytes were purged with dry Ar or dry O2 for at least 20 minutes before each experiment. All potential values for CVs were calibrated by voltage versus Li/Li+ (E°=−3.04 V).
Ex-situ characterization
The crystal structures and bonding within the electrode samples after cycling were analyzed by X-ray photoelectron spectroscopy (XPS), Raman spectroscopy (T64000, Horiba Jobin-Yvon, excitation at 633 nm) and X-ray diffraction (XRD, RIGAKU, D/MAX-RC). Raman spectra were recorded with a CCD detector that was cooled to −70 °C and a silicon wafer was used to calibrate the Raman shift. XPS measurements were performed at Brookhaven National Laboratory on a SPECS GmbH instrument under ultrahigh vacuum (UHV) conditions. The X-ray source was Al Kα at a power of 300 W.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
Additional information
How to cite this article: Ryu, W.-H. et al. Heme biomolecule as redox mediator and oxygen shuttle for efficient charging of lithium-oxygen batteries. Nat. Commun. 7, 12925 doi: 10.1038/ncomms12925 (2016).
Supplementary Material
Supplementary Figures 1-10 and Supplementary Tables 1-2
Acknowledgments
We gratefully acknowledge Dr Lisa Pfefferle for the use of Raman equipment and Dr Menachem Elimelech for the use of the ultraviolet–vis spectrophotometer and Dr Xiao Tong for assistance with XPS measurements. W-H.R. acknowledges support from The NatureNet Program of the Nature Conservancy. We are grateful for the support from NSF under Grant MRSEC DMR 1119826 (CRISP) and A.D.T acknowledges NSF-CBET-0954985 PECASE Award for providing partial support of this work. Electrochemical and ultraviolet–vis spectroscopic studies were supported by a grant from the Department of Energy, Office of Basic Energy Sciences, Divisions of Chemical Sciences (DE-FG02-07ER15909 to G.W.B.). The Yale Institute for Nanoscience and Quantum Engineering (YINQE) and NSF MRSEC DMR 1119826 (CRISP) provided facility support. This research used resources of the Centre for Functional Nanomaterials, which is a U.S. DOE Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704. This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2016R1C1B2011442). This Research was supported by the Sookmyung Women's University Research Grants 1-1603-2014. Chasm Technologies are acknowledged for their kind supply of multi-walled carbon nanotubes. Sandia National Laboratories is a multiprogram laboratory managed and operated by Sandia Corporation, a wholly owned subsidiary of Lockheed Martin Corporation, for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000.
Footnotes
Author contributions W-H.R. and A.D.T. conceptualized the project. W-H.R. and F.S.G. designed and performed the electrochemical experiments and data analysis. W-H.R. performed the TEM experiments and data analysis. W-H.R. and F.S.G. performed the ultraviolet–vis absorption and XPS experiments and resulting data analysis. W-H.R., J.M.T. and G.W.B. performed in situ spectroelectrochemical measurement experiments. W-H.R., F.S.G. and J.L. designed the mechanism of heme redox and drew reaction schematics. W-H.R. and M.J.S. performed the Raman experiments and resulting data analysis. A.D.T, W-H.R. and F.S.G. laid out the design of the manuscript. W-H.R. wrote the original manuscript. All authors discussed the results and commented on the manuscript.
References
- Chu S. & Majumdar A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012). [DOI] [PubMed] [Google Scholar]
- Dunn B., Kamath H. & Tarascon J. M. Electrical energy storage for the grid: a battery of choices. Science 334, 928–935 (2011). [DOI] [PubMed] [Google Scholar]
- Gallagher K. G. et al. Quantifying the promise of lithium-air batteries for electric vehicles. Energ. Environ. Sci. 7, 1555–1563 (2014). [Google Scholar]
- Girishkumar G., McCloskey B., Luntz A. C., Swanson S. & Wilcke W. Lithium—air battery: promise and challenges. J. Phys. Chem. Lett. 1, 2193–2203 (2010). [Google Scholar]
- Kwabi D. G. et al. Materials challenges in rechargeable lithium-air batteries. MRS Bull. 39, 443–452 (2014). [Google Scholar]
- Bruce P. G., Freunberger S. A., Hardwick L. J. & Tarascon J. M. Li-O2 and Li-S batteries with high energy storage. Nat. Mater. 11, 19–29 (2012). [DOI] [PubMed] [Google Scholar]
- Lu J. et al. Aprotic and aqueous Li-O2 batteries. Chem. Rev. 114, 5611–5640 (2014). [DOI] [PubMed] [Google Scholar]
- Lu Y. C. et al. Lithium-oxygen batteries: bridging mechanistic understanding and battery performance. Energ. Environ. Sci. 6, 750–768 (2013). [Google Scholar]
- Ryu W. H. et al. Bifunctional composite catalysts using Co3O4 nanofibers immobilized on nonoxidized graphene nanoflakes for high-capacity and long-cycle Li-O2 batteries. Nano Lett. 13, 4190–4197 (2013). [DOI] [PubMed] [Google Scholar]
- Black R., Lee J. H., Adams B., Mims C. A. & Nazar L. F. The role of catalysts and peroxide oxidation in lithium-oxygen batteries. Angew. Chem. Int. Ed. 52, 392–396 (2013). [DOI] [PubMed] [Google Scholar]
- Xie J. et al. Three dimensionally ordered mesoporous carbon as a stable, high-performance Li-O2 battery cathode. Angew. Chem. Int. Ed. 54, 4299–4303 (2015). [DOI] [PubMed] [Google Scholar]
- Gittleson F. S., Ryu W. H. & Taylor A. D. Operando observation of the gold-electrolyte interface in Li-O2 batteries. ACS Appl. Mater. Interfaces 6, 19017–19025 (2014). [DOI] [PubMed] [Google Scholar]
- Gittleson F. S., Sekol R. C., Doubek G., Linardi M. & Taylor A. D. Catalyst and electrolyte synergy in Li-O2 batteries. Phys. Chem. Chem. Phys. 16, 3230–3237 (2014). [DOI] [PubMed] [Google Scholar]
- Ryu W. H., Gittleson F. S., Schwab M., Goh T. & Taylor A. D. A mesoporous catalytic membrane architecture for lithium-oxygen battery systems. Nano Lett. 15, 434–441 (2015). [DOI] [PubMed] [Google Scholar]
- Ryu W. H., Gittleson F. S., Li J., Tong X. & Taylor A. D. A new design strategy for observing lithium oxide growth-evolution interactions using geometric catalyst positioning. Nano Lett. 16, 4799–4806 (2016). [DOI] [PubMed] [Google Scholar]
- Bergner B. J., Schurmann A., Peppler K., Garsuch A. & Janek J. TEMPO: a mobile catalyst for rechargeable Li-O2 batteries. J. Am. Chem. Soc. 136, 15054–15064 (2014). [DOI] [PubMed] [Google Scholar]
- Sun D. et al. A solution-phase bifunctional catalyst for lithium-oxygen batteries. J. Am. Chem. Soc. 136, 8941–8946 (2014). [DOI] [PubMed] [Google Scholar]
- Chen Y. H., Freunberger S. A., Peng Z. Q., Fontaine O. & Bruce P. G. Charging a Li-O2 battery using a redox mediator. Nat. Chem. 5, 489–494 (2013). [DOI] [PubMed] [Google Scholar]
- Feng N., He P. & Zhou H. Enabling catalytic oxidation of Li2O2 at the liquid-solid interface: the evolution of an aprotic Li-O2 battery. ChemSusChem 8, 600–602 (2015). [DOI] [PubMed] [Google Scholar]
- Wang Y. G. & Xia Y. Y. Li-O2 batteries an agent for change. Nat. Chem. 5, 445–447 (2013). [DOI] [PubMed] [Google Scholar]
- Lim H. D. et al. Superior rechargeability and efficiency of lithium-oxygen batteries: hierarchical air electrode architecture combined with a soluble catalyst. Angew. Chem. Int. Ed. 53, 3926–3931 (2014). [DOI] [PubMed] [Google Scholar]
- Yu M. Z., Ren X. D., Ma L. & Wu Y. Y. Integrating a redox-coupled dye-sensitized photoelectrode into a lithium-oxygen battery for photoassisted charging. Nat. Commun. 5, 5111 (2014). [DOI] [PubMed] [Google Scholar]
- Kundu D., Black R., Adams B. & Nazar L. F. A highly active low voltage redox mediator for enhanced rechargeability of lithium–oxygen batteries. ACS Cent. Sci. 1, 510–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda M. K., Ladomenou K. & Coutsolelos A. G. Porphyrins in bio-inspired transformations: light-harvesting to solar cell. Coordin. Chem. Rev. 256, 2601–2627 (2012). [Google Scholar]
- Huskinson B. et al. A metal-free organic-inorganic aqueous flow battery. Nature 505, 195–198 (2014). [DOI] [PubMed] [Google Scholar]
- Willner I. Biomaterials for sensors, fuel cells, and circuitry. Science 298, 2407–2408 (2002). [DOI] [PubMed] [Google Scholar]
- Bah C. S. F., Bekhit A. E. A., Carne A. & McConnell M. A. Slaughterhouse blood: an emerging source of bioactive compounds. Compr. Rev. Food Sci. F 12, 314–331 (2013). [Google Scholar]
- Maines M. D. Heme oxygenase—function, multiplicity, regulatory mechanisms, and clinical-applications. Faseb J. 2, 2557–2568 (1988). [PubMed] [Google Scholar]
- Poulos T. L. Heme enzyme structure and function. Chem. Rev. 114, 3919–3962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que L. & Tolman W. B. Biologically inspired oxidation catalysis. Nature 455, 333–340 (2008). [DOI] [PubMed] [Google Scholar]
- Yin L. et al. Rev-erb alpha, a heme sensor that coordinates metabolic and circadian pathways. Science 318, 1786–1789 (2007). [DOI] [PubMed] [Google Scholar]
- Liu X. Q., Freeman R., Golub E. & Willner I. Chemiluminescence and chemiluminescence resonance energy transfer (CRET) aptamer sensors using catalytic hemin/G-quadruplexes. ACS Nano. 5, 7648–7655 (2011). [DOI] [PubMed] [Google Scholar]
- Auwarter W., Ecija D., Klappenberger F. & Barth J. V. Porphyrins at interfaces. Nat. Chem. 7, 105–120 (2015). [DOI] [PubMed] [Google Scholar]
- Collman J. P., Boulatov R., Sunderland C. J. & Fu L. Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem. Rev. 104, 561–588 (2004). [DOI] [PubMed] [Google Scholar]
- Sugimoto H. et al. Crystal structure of human indoleamine 2,3-dioxygenase: catalytic mechanism of O2 incorporation by a heme-containing dioxygenase. Proc. Natl Acad. Sci. USA 103, 2611–2616 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagi-Damodaran A., Petrik I. D., Marshall N. M., Robinson H. & Lu Y. Systematic tuning of heme redox potentials and its effects on O2 reduction rates in a designed oxidase in myoglobin. J. Am. Chem. Soc. 136, 11882–11885 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue T. et al. Graphene-supported hemin as a highly active biomimetic oxidation catalyst. Angew. Chem. Int. Ed. 51, 3822–3825 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. F. et al. Direct electrochemistry and electrocatalysis of heme proteins entrapped in agarose hydrogel films in room-temperature ionic liquids. Langmuir 21, 9260–9266 (2005). [DOI] [PubMed] [Google Scholar]
- Li F. J., Zhang T. & Zhou H. S. Challenges of non-aqueous Li-O2 batteries: electrolytes, catalysts, and anodes. Energ. Environ. Sci. 6, 1125–1141 (2013). [Google Scholar]
- Battistuzzi G., Borsari M., Ranieri A. & Sola M. Redox thermodynamics of the Fe3+/Fe2+ couple in horseradish peroxidase and its cyanide complex. J. Am. Chem. Soc. 124, 26–27 (2002). [DOI] [PubMed] [Google Scholar]
- Nam W. High-valent iron(IV)-oxo complexes of heme and non-heme ligands in oxygenation reactions. Acc. Chem. Res. 40, 522–531 (2007). [DOI] [PubMed] [Google Scholar]
- Johnson C. S. et al. Li2O removal from Li5FeO4: a cathode precursor for lithium-ion batteries. Chem. Mater. 22, 1263–1270 (2010). [Google Scholar]
- Laoire C. O., Mukerjee S., Abraham K. M., Plichta E. J. & Hendrickson M. A. Elucidating the mechanism of oxygen reduction for lithium-air battery applications. J. Phys. Chem. C 113, 20127–20134 (2009). [Google Scholar]
- Laoire C. O., Mukerjee S., Abraham K. M., Plichta E. J. & Hendrickson M. A. Influence of nonaqueous solvents on the electrochemistry of oxygen in the rechargeable lithium-air battery. J. Phys. Chem. C 114, 9178–9186 (2010). [Google Scholar]
- Rinaldi A., Wijaya O., Hoster H. E. & Yu D. Y. W. History effects in lithium-oxygen batteries: how initial seeding influences the discharge capacity. ChemSusChem 7, 1283–1288 (2014). [DOI] [PubMed] [Google Scholar]
- Laoire C. O., Mukerjee S., Plichta E. J., Hendrickson M. A. & Abraham K. M. Rechargeable lithium/TEGDME-LiPF6/O2 battery. J. Electrochem. Soc. 158, A302–A308 (2011). [Google Scholar]
- Landa-Medrano I. et al. In situ monitoring of discharge/charge processes in Li-O2 batteries by electrochemical impedance spectroscopy. J. Power Sources 249, 110–117 (2014). [Google Scholar]
- Gittleson F. S. et al. Raman spectroscopy in lithium–oxygen battery systems. ChemElectroChem 2, 1446–1457 (2015). [Google Scholar]
- Lu J. et al. A lithium-oxygen battery based on lithium superoxide. Nature 529, 377–382 (2016). [DOI] [PubMed] [Google Scholar]
- Zhai D. et al. Raman evidence for late stage disproportionation in a Li–O2 battery. J. Phys. Chem. Lett. 5, 2705–2710 (2014). [DOI] [PubMed] [Google Scholar]
- Gittleson F. S., Ryu W. H., Schwab M., Tong X. & Taylor A. D. Pt and Pd catalyzed oxidation of Li2O2 and DMSO during Li-O2 battery charging. Chem. Commun. 52, 6605–6608 (2016). [DOI] [PubMed] [Google Scholar]
- Thotiyl M. M. O., Freunberger S. A., Peng Z. Q. & Bruce P. G. The carbon electrode in nonaqueous Li-O2 cells. J. Am. Chem. Soc. 135, 494–500 (2013). [DOI] [PubMed] [Google Scholar]
- Younesi R., Hahlin M., Bjorefors F., Johansson P. & Edstrom K. Li-O2 battery degradation by lithium peroxide (Li2O2): a model study. Chem. Mater. 25, 77–84 (2013). [Google Scholar]
- Vasudevan D. & Wendt H. Electroreduction of oxygen in aprotic media. J. Electroanal. Chem. 392, 69–74 (1995). [Google Scholar]
- Schwenke K. U., Meini S., Wu X. H., Gasteiger H. A. & Piana M. Stability of superoxide radicals in glyme solvents for non-aqueous Li-O2 battery electrolytes. Phys. Chem. Chem. Phys. 15, 11830–11839 (2013). [DOI] [PubMed] [Google Scholar]
- Moreira L. M., Poli A. L., Costa A. J. & Imasato H. Pentacoordinate and hexacoordinate ferric hemes in acid medium: EPR, UV–vis and CD studies of the giant extracellular hemoglobin of Glossoscolex paulistus. Biophys. Chem. 124, 62–72 (2006). [DOI] [PubMed] [Google Scholar]
- Karow D. S. et al. Characterization of functional heme domains from soluble guanylate cyclase. Biochemistry 44, 16266–16274 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Yeung N., Wang Z. L., Guo Z. J. & Lu Y. Effects of metal ions in the Cu-B center on the redox properties of heme in heme-copper oxidases: spectroelectrochemical studies of an engineered heme-copper center in myoglobin. Biochemistry 44, 1210–1214 (2005). [DOI] [PubMed] [Google Scholar]
- Black R. et al. Screening for superoxide reactivity in Li-O2 batteries: effect on Li2O2/LiOH crystallization. J. Am. Chem. Soc. 134, 2902–2905 (2012). [DOI] [PubMed] [Google Scholar]
- Abraham K. M. Electrolyte-directed reactions of the oxygen electrode in lithium-air batteries. J. Electrochem. Soc. 162, A3021–A3031 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Yeung N., Wang Z., Guo Z. & Lu Y. Effects of metal ions in the CuB center on the redox properties of heme in heme-copper oxidases: spectroelectrochemical studies of an engineered heme-copper center in myoglobin. Biochemistry 44, 1210–1214 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-10 and Supplementary Tables 1-2
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.